

Abstract
Background/aims
To evaluate the safety and feasibility of immunotherapy based on autologous dendritic cells (DC) for patients with unresectable primary liver cancer (PLC).
Methods
A total of ten patients were enrolled and immunized with DCs. Autologous DCs were generated ex vivo in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin-4 (IL-4). Cells were then pulsed with tumor lysate (TL), tumor necrosis factor-α (TNF-α) and keyhole limpet hemocyanin (KLH). Non-adherent cells were collected on day 9 and cells were administered into the inguinal lymph node. Each patient received 1–10×106 cells four times at weekly intervals.
Results
Immunization was well tolerated in all patients without significant toxicity. DC vaccination induced delayed-type hypersensitivity (DTH) against KLH in seven out of ten patients. In one patient, one of the two liver tumors (tumor in segment 7, 13 mm in diameter) decreased in size to 7 mm and showed necrotic change on computed tomography examination after eight immunizations. In two patients, serum levels of tumor markers decreased after vaccination.
Conclusion
The present clinical trial suggested that immunization by TL-pulsed DCs is feasible in patients with unresectable PLC without any toxicity. Further improvement in the clinical results of immunotherapy might be expected by modifying the therapeutic protocol.
Keywords: Hepatocellular carcinoma, Cholangiocellular carcinoma, Cell therapy, Delayed-type hypersensitivity, Keyhole limpet hemocyanin
Introduction
Hepatocellular carcinoma (HCC) accounts for 95% of primary liver cancer (PLC) in Japan and is one of the most frequently occurring malignancies in the East Asian area. The incidence exceeds 30 cases/100,000 per year [31]. Worldwide, it accounts for almost 1 million deaths per year. Resection, transplantation, chemoembolization, alcohol injection and cryoablation are potentially curative treatments for HCC, but are only effective in small and localized tumors [15, 16, 25]. Unfortunately, advanced disease is observed upon diagnosis in most HCC patients and current systemic therapies are largely ineffective. Cholangiocellular carcinoma (CCC) is a highly malignant tumor of the bile duct with no effective therapy except surgical resection and a poor long-term prognosis [1]. Currently, there are also no effective treatments for unresectable CCC. Therefore, the development of novel treatment strategies for unresectable PLC is urgently required.
DCs are the most potent antigen-presenting cells (APCs) and are clearly central to the regulation, maturation and maintenance of a cellular immune response to cancer [28]. DC-based cancer immunotherapies are currently being tested in clinical trials. As a source of antigen, whole protein [12], tumor lysate (TL) [19], HLA-restricted peptides [21], RNA [18] or hybrid cells [10, 14] could be presented to T lymphocytes by DCs. There are some published clinical studies of immunotherapy with TL-pulsed autologous DCs in the treatment of melanoma [19], renal cell carcinoma [11], pediatric solid tumor [8, 9], and ovarian cancer [34]. A major drawback of unfractionated tumor antigens is the possibility of inducing an autoimmune reactivity to epitopes that are shared by normal tissues [17]. However, in these clinical trials, no relevant autoimmune responses have been detected.
There are several advantages of using TL. First, it contains multiple known as well as unknown antigens that can be presented to T cells by both MHC class I and class II pathways [4, 26]. Second, it can be used independently of the HLA type of the patient [24]. Furthermore, the generation of cytotoxic T lymphocyte (CTL) clones with multiple specificities may be an advantage in heterogeneous tumors and could also reduce the risk of tumor escape variants [3]. DC-based immunotherapies against HCC have not yet been reported as clinical pilot studies, although Takayama et al. [30] have demonstrated that adoptive immunotherapy can improve postoperative recurrence-free outcomes after surgery for HCC. This indicates the possibility of DC-based immunotherapies as a novel treatment strategy against HCC. Therefore, in this study, we performed a phase I trial to assess the safety and feasibility of TL-pulsed autologous DC therapy against PLC.
Patients and methods
Patients
The study protocol was reviewed and approved by the Ethical Committee of Oita Medical University, and all the patients provided signed informed consent. According to the protocol, patients were required to have unresectable PLC, which other standard treatments had failed to cure, with an ECOG performance status of 0 to 2 and a life expectancy of at least 16 weeks. Furthermore, patients who had received radiation therapy, chemotherapy or immunotherapy within the previous 8 weeks or those who were pregnant were excluded. Treatment was carried out at the Department of Surgery I, Oita Medical University from May 2000 through December 2001. Ten patients were enrolled and their clinical characteristics are summarized in Table 1.
Table 1.
Patient characteristic and status before DC vaccination (HCC hepatocellular carcinoma, CCC cholangiocellular carcinoma; LC liver cirrhosis, CH chronic hepatitis; HBV hepatitis B virus positive, HCV hepatitis C virus positive; TAI transarterial injection, TAE transarterial embolization, MCT microwave coagulation therapy, PEI percutaneous ethanol injection, RFA radiofrequency ablation)
| Patient | Age (years) | Sex | Diagnosis | Background liver | Metastasis | Previous therapies |
|---|---|---|---|---|---|---|
| 1 | 64 | M | HCC | LC (HCV) | Bone | TAI, MCT, radiation |
| 2 | 48 | M | CCC | Normal | – | – |
| 3 | 70 | M | HCC | LC (HBV) | – | TAI |
| 4 | 65 | M | HCC | LC (HCV) | Lung | TAI, TAE, PEI, hepatectomy |
| 5 | 54 | M | HCC | CH (HBV) | – | TAI |
| 6 | 65 | M | HCC | CH (HBV) | – | TAI, chemotherapy |
| 7 | 63 | M | HCC | LC (virus-free) | – | Hepatectomy, TAI, TAE |
| 8 | 83 | F | CCC | Normal | – | Hepatectomy, TAI, Chemotherapy |
| 9 | 75 | F | HCC | LC (HCV) | – | TAI, TAE, PEI, RFA |
| 10 | 63 | M | HCC | LC (HCV) | Bone, adrenal gland | Hepatectomy, Chemotherapy |
Preparation of tumor lysate
Fine-needle biopsy under sonographic guidance was performed in all patients using an 18-gauge biopsy needle (Majima needle; Top Company, Tokyo, Japan) to obtain autologous TL. Tumor biopsies from each patient were immediately placed in phosphate-buffered saline (PBS). Subsequently, tumor cells were dispersed to create a single-cell suspension. Cells were lysed by three freeze-thaw cycles. Larger particles were removed by centrifugation (10 min, 600 rpm). The supernatant was collected and passed through a 0.2-μm filter. The protein concentration of the lysate was determined using a commercial assay (BCA Protein Assay; Pierce, Rockford, Ill.).
In vitro generation of DCs
DCs were generated from peripheral blood mononuclear cells (PBMCs) from each PLC patient as described previously [23] with some modifications. Briefly, PBMCs were isolated from 60 ml peripheral blood using a Ficoll-Hypaque gradient (Amersham, Uppsala, Sweden). Two patients (Nos. 9 and 10) underwent leukapheresis using a cell separator (Kobe Spectra, Japan) in order to obtain a sufficient number of PBMCs. The cells were resuspended in culture medium comprising RPMI-1640 supplemented with 2 mM l-glutamine, 50 μg/ml streptomycin, 50 U/ml penicillin, and 1% autologous serum, and allowed to adhere to the plastic culture flask (Becton Dickinson, Franklin Lakes, N.J.). After 2 h at 37°C, non-adherent cells were removed and adherent cells were cultured in medium supplemented with 50 ng/ml GM-CSF (kindly provided by Novartis Pharmaceutical Corporation, Tokyo, Japan) and 50 ng/ml IL-4 (purchased from Pepro Tech, Rocky Hill, N.J.). Fresh medium was added and the medium was supplemented with cytokines every other day. On day 6 of culture, cells were pulsed with prepared TL (100 μg/ml) for 12 h. Then TNF-α (100 ng/ml; R&D Systems, Minneapolis, Minn.) and keyhole limpet hemocyanin (KLH) (50 μg/ml; Calbiochem, Bad Soden, Germany) were added to the culture on day 7. Cultured DCs were harvested by vigorous washing with culture medium on day 9. Aliquots were taken for cell counting and viability staining by trypan blue. Cell differentiation was monitored by light microscopy.
Phenotypic characterization of DCs
Phenotypic characterization of cultured DCs was performed using a FACSCalibur system (Becton Dickinson) with mouse antibody against human HLA-DR, CD40, CD83, CD80 (all purchased from Immunotech, Marseilles, France) and CD86 (Serotec, Oxford, UK). FITC-conjugated rabbit antimouse IgG was used as the second antibody (Dako Japan, Japan). In order to evaluate the degree of maturation of DC from the PLC patients, the expression of CD83 and CD86 was compared with that of five healthy volunteers. Fluorescence staining was analyzed using CellQuest software. The results are presented as means±SD and analyzed statistically using the Mann-Whitney U-test and Wilcoxon's rank-sum test. P values <0.05 were considered to be significant.
Patient treatment
Eligible patients received four vaccinations of 1×106 to 1×107 TL-pulsed DCs at weekly intervals. Immunization was subsequently continued at monthly intervals for up to 12 vaccinations, depending on clinical response. Before injection, DCs were washed three times in sterile PBS and resuspended in a total volume of 0.5 ml PBS. DC preparations were confirmed to be endotoxin-free (<10 pg/ml in the supernatant). DCs were then administered immediately into an inguinal lymph node under ultrasound control.
Evaluation
Adverse effects were recorded using common WHO toxicity criteria during the protocol. Tumor markers such as alpha-fetoprotein (AFP), protein induced by vitamin K absence or antagonist II (PIVKA-II) and CA19-9 were determined and imaging studies (CT scans) of the tumor sites were reviewed before treatment and after the fourth immunization. Standard definitions of major objective responses (CR, complete response; PR, partial response) were used. Stable disease (SD) was defined as less than a 50% decrease or not more than a 25% increase in the diameters of the largest tumor nodules. Progressive disease (PD) was defined as more than a 25% increase in the diameters of the largest tumor nodule or one of the measurable lesions, or the appearance of new lesions.
Delayed-type hypersensitivity
DTH skin tests were performed with KLH (10 μg/0.1 ml saline) before and after the fourth immunization. A positive skin-test reaction was defined as erythema of >5 mm diameter and/or induration 48 h after intradermal injection.
Results
Patient characteristics
The characteristics of the ten patients enrolled in the study are summarized in Table 1. There were eight men and two women, with a median age of 65 (range 48–83 years). Eight of the ten patients had HCC and the remainder CCC. All had unresectable liver tumors with or without extrahepatic lesions. The virus markers of each patient are also shown in Table 1. These patients had already been treated intensively with surgery, chemotherapy, radiotherapy, transarterial injection/embolization and/or percutaneous ethanol injection.
Characteristics of DCs
In eight patients (Nos. 1–8), the yields of DCs were less than expected. The use of leukapheresis for the last two patients (Nos. 9 and 10) provided a sufficient number of DCs (Table 2).
Table 2.
Summary of treatments and toxicity
| Patient | Number of DC vaccinations | Total number of injected DCs (×106) | Toxicity |
|---|---|---|---|
| 1 | 4 | 5.2 | − |
| 2 | 12 | 18.1 | − |
| 3 | 5 | 7.9 | − |
| 4 | 4 | 5.7 | − |
| 5 | 8 | 6.3 | − |
| 6 | 4 | 7.8 | − |
| 7 | 12 | 21.4 | − |
| 8 | 8 | 21.3 | − |
| 9 | 4 | 29.4 | − |
| 10 | 8 | 30.3 | − |
Cultured DCs were monitored by light microscopy (Fig. 1a, b) before administration. Their morphological features were those of immature DCs with short, blunt prolongations, despite the use of TNF-α. FACS analysis (Fig. 2, Table 3) revealed that the harvested population of cells consisted of 50% DCs with high levels of expression of CD40 and HLA-DR (more than 80% positive). The percentages of cells expressing CD1a (21.2±18% positive cells) and CD80 (37.0±25% positive cells) were not uniform. However, the percentages of cells expressing mature DC markers were significantly (P<0.05) reduced (13.1±15.4% CD83-positive cells, 47.7±28.5% CD86 positive cells) compared with those from the healthy volunteers (56.0±29.1% CD83-positive cells, 77.6±14.7% CD86-positive cells).
Fig. 1a, b.
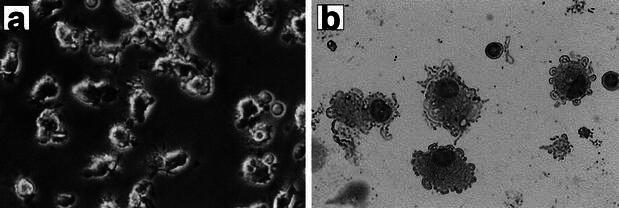
Morphological features of DCs derived from PLC patients. Cultured DCs were monitored by light microscopy before administration (a). DCs from PLC patients morphologically appeared to be immature with short, blunt prolongations, despite stimulation with TNF-α(b) (May-Giemsa staining, ×200)
Fig. 2.
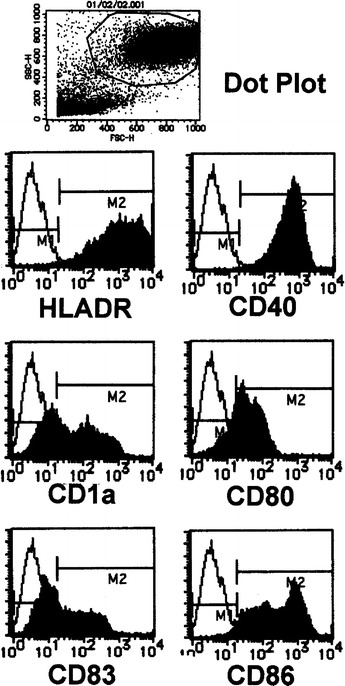
FACS analysis of DCs derived from PLC patients. The harvested population of cells consisted of 50% DCs. Representative phenotyping analysis of a patient is shown
Table 3.
Phenotypic analysis of cultured DCs (% positive) (n.d. not done)
| Patient | HLA-DR | CD40 | CD1a | CD80 | CD83 | CD86 |
|---|---|---|---|---|---|---|
| 1 | 89.6 | 92.2 | n.d. | 41.6 | 3.2 | 13.5 |
| 2 | 95.4 | 94.3 | 56.0 | 86.3 | 44.5 | 75.6 |
| 3 | 71.2 | 88.6 | 4.9 | 9.9 | 2.8 | 24.1 |
| 4 | 82.6 | 88.1 | 13.6 | 46.7 | 12.3 | 43.7 |
| 5 | 67.3 | 29.2 | 0.5 | 5.8 | 0.2 | 14.6 |
| 6 | 87.9 | 80.0 | 20.6 | 32.5 | 4.2 | 43.7 |
| 7 | 97.3 | 98.5 | 44.2 | 61.4 | 36.8 | 96.0 |
| 8 | 81.2 | n.d. | 8.3 | 11.7 | 1.5 | 38.5 |
| 9 | 89.1 | 92.4 | 16.7 | 40.0 | 14 | 43.9 |
| 10 | 94.5 | 90 | 26.2 | 34.3 | 11.5 | 82.9 |
| Mean | 85.6 | 83.7 | 21.2 | 37.0 | 13.1 | 47.7 |
Toxicity
All the patients received at least four immunizations (Table 2). The protocols were well tolerated. Because autologous TL was used as the antigen target, autoimmune adverse effects were carefully assessed. However, no hematological, hepatic, pulmonary or renal toxicities related to the treatment were observed in any of the patients.
Immunological and clinical response
Table 4 summarizes the responses to the treatment. KLH-specific DTH reactions were not observed before vaccination in any patient. However, seven of the ten immunized patients developed a positive DTH response, and five had tumor diameters more than 50 mm. In one patient, one of the two liver tumors (tumor in segment 7, 13 mm in diameter) decreased in size to 7 mm and showed necrotic change on CT examination after eight immunizations (Fig. 3). No viable cells were recognized in the biopsy specimen taken from the decreased tumor. In two patients (Nos. 8 and 9), serum levels of tumor markers (AFP, PIVKA-II or CA19-9) decreased after treatment.
Table 4.
Response to DC therapy (SD stable disease, PD progressive disease, MR mixed response)
| Patient | DTHa | Tumor marker | Before | After | Clinical response | ||
|---|---|---|---|---|---|---|---|
| First | Second | Third | |||||
| 1 | − | AFP (ng/ml) | 3797 | 6446 | SD | ||
| 2 | ++ | None | SD | ||||
| 3 | − | PIVKA-II (ng/ml) | 14.8 | 113.3 | SD | ||
| 4 | − | AFP (ng/ml) | 5576 | 10154 | PD | ||
| 5 | + | PIVKA-II (ng/ml) | <10 | 125.8 | 400.2 | SD | |
| 6 | + | AFP (ng/ml) | 5.3 | 7.1 | PD | ||
| 7 | ++ | AFP (ng/ml) | 50.7 | 78.2 | 148 | 293.4 | MR |
| PIVKA-II (ng/ml) | 1350 | 894.4 | 1718 | >2000 | |||
| 8 | ++ | CA19-9 (U/ml) | 116.3 | 121.7 | 85.3 | SD | |
| 9 | ++ | AFP (ng/ml) | 128 | 94.5 | SD |
||
| PIVKA-II (ng/ml) | 325.4 | 76.2 | |||||
| 10 | ++ | None | PD | ||||
aDTH to KLH. Erythema and/or induration after 48 h: − <5 mm diameter, + 5–50 mm, ++ >50 mm
Fig. 3a, b.
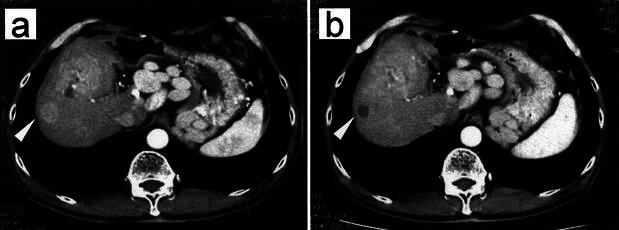
Clinical response to DC vaccination. CT scan of patient no. 7 with recurrent tumors located at S5 and S7 (white arrow) before (a) (enhanced area 13 mm in diameter) and after (b) (low density area 7 mm in diameter) eight immunizations with tumor lysate and KLH-pulsed DCs
Discussion
As far as we know, there are no reports of a clinical trial of DC therapy in PLC patients. Therefore, the main purpose of this study was not to evaluate the effectiveness of this therapy but to assess the safety and feasibility of TL-pulsed DC immunotherapy for patients with unresectable PLC. Because autologous TL from individual PLC patients was used as the antigen target, autoimmune adverse effects were carefully assessed. No toxicity such as aggravation of hepatitis was observed in any of the patients. This finding is consistent with those of other clinical trials targeting TL of other types of tumor [8, 9, 11, 19], in which autoimmune responses also were not seen. Therefore, our results were felt to be satisfactory for a phase I trial of DC immunotherapy in PLC patients. However, further modification and improvement of the DC maturation methods in HCC patients are required to achieve clinical effectiveness of this therapy.
Hepatitis viral infection may be involved in poor maturation of DC derived from HCC patients. Kakumu et al. have recently demonstrated that the DC maturation is disturbed in HCC patients with hepatitis B virus (HBV) or hepatitis C virus (HCV) [13]. HCC often follows HCV and/or HBV virus infection. In our series of ten enrolled patients, the DC maturation of HCC patients positive for HBV or HCV was extremely poor as compared with that of virus-free HCC patients. In one virus-free HCC patient (no. 7), the maturation of DC was not as impaired as it was in others and in this patient we observed regression of one of two liver tumors. Inhibition of viral activity or a decrease in viral load may improve DC therapy in HCC patients. Indeed, in one study an improvement in impaired DC maturation was found in HCV-positive patients when HCV was resolved by antiviral therapy [2]. Therefore, interferon α (IFN-α) therapy may be a useful candidate to combine with DC-based immunotherapy because of its antiviral and immunomodulatory effects. On the other hand, the remaining tumor in segment 6 in the same patient, which was larger than the other, was not reduced after the treatment and gradually increased in volume. These results suggest that immunotherapy might be more effective for tumors that are small and at an early stage.
As shown in Table 2, immature DCs of patients with PLC did not respond appropriately to stimulation with TNF-α. In contrast to immature DCs, mature DCs have the ability to prime and activate T cells by expression of many accessory molecules that interact with receptors on T cells to enhance signaling (costimulation), and secrete high levels of IL-12 that enhances both innate and acquired immunity [28]. According to our results, maturation as well as the number of injected DCs might be correlated with induction of DTH reactivity against KLH. Efficient maturation of DCs might be necessary to establish more effective DC-based immunotherapy against HCC. In order to induce maturation, we and others [2] have been using TNF-α as the stimulus. This widely accepted method was able to mature DCs purified from healthy donors. However, TNF-α might be insufficient and even inadequate as a stimulus to induce maturation of DCs derived from PLC patients. As Schnurr et al. [24] have demonstrated, potent stimuli combined with TNF-α and prostaglandin E2 may improve immunotherapy against HCC by increasing the maturation and subsequent immune response of the DCs. Further studies are required to establish a novel protocol for DC-based immunotherapy for PLC patients.
Although clinical efficacy is the final goal of cancer immunotherapies, what is needed is evaluation of immunological responses of intermediate markers for the most likely candidate for success. For in vivo detection of the immune response, we performed a DTH skin test to KLH (which was used as a marker antigen). DTH remains one of the most frequently used immune tests performed in immunotherapy studies [22, 27]. Immune reactivity to KLH may be a reflection of the underlying immune competence of the subjects. In this study, seven of the ten immunized patients developed a positive DTH response to KLH, and five had a reaction diameter of more than 50 mm, and three of these patients showed tumor regression or a decreases expression of tumor markers, as described above. On the other hand, three patients who had a negative response to KLH showed a poor outcome (they died within 4 months after the therapy). Thus, the DTH test to KLH might be useful as a surrogate marker for the adaptability to immunotherapy.
In the present study, we used lysate from the autologous tumor as a target because it could be used independently of the HLA type of the patient. However, in future, we should choose the available antigens properly, as described by Nestle et al. [19]. Antigens such as MAGE (melanoma antigen) [29], p53 [32], and AFP [5, 6] may become potential targets for the immunotherapy of HCC. The MAGE genes are expressed in a significant proportion of malignant tumors of various histological origins, whereas no expression has been observed in normal tissues except the testis [7]. MAGE has not been used for DC-based immunotherapy for PLC, although a number of clinical trials for various tumors have been performed using a MAGE-peptide as the target antigen, with promising results [20, 21]. We have previously demonstrated that MAGE gene expression is also frequent in HCC [29]. These results indicate that MAGE-specific cancer immunotherapy might be one of the more attractive and effective strategies for the treatment of HCC. The majority of human HCC overexpress the oncofetal antigen AFP [33]. Butterfield et al. [5, 6] have recently shown that HLA-A*0201-restricted peptides derived from human AFP can be recognized by human T cells and subsequently induce a CTL response. Therefore, AFP could be another potential target for HCC immunotherapy.
In conclusion, immunization by TL-pulsed DCs was feasible in unresectable PLC patients without any toxicity. Further improvement in the clinical results of immunotherapy can be expected by modifying the therapeutic protocol.
Acknowledgements
We thank Dr. Roger Lord (University of Tasmania, Australia) for his critical review of the manuscript, Dr. Hiroshi Kikuchi (Blood Transfusion Center of Oita Medical University, Japan) for kindly providing advice on leukapheresis, and Ms. Michiyo Hisaka and Ms. Masae Hikida for their excellent technical support.
References
- 1.Ahrendt Clin Liver Dis. 2001;5:191. doi: 10.1016/s1089-3261(05)70161-6. [DOI] [PubMed] [Google Scholar]
- 2.Auffermann-Gretzinger Blood. 2001;97:3171. doi: 10.1182/blood.V97.10.3171. [DOI] [PubMed] [Google Scholar]
- 3.Bennett J Exp Med. 1997;186:65. doi: 10.1084/jem.186.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brossart Blood. 1997;90:1594. [PMC free article] [PubMed] [Google Scholar]
- 5.Butterfield Cancer Res. 1999;59:3134. [PubMed] [Google Scholar]
- 6.Butterfield J Immunol. 2001;166:5300. doi: 10.4049/jimmunol.166.8.5300. [DOI] [PubMed] [Google Scholar]
- 7.De Proc Natl Acad Sci U S A. 1996;93:7149. doi: 10.1073/pnas.93.14.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geiger Lancet. 2000;356:1163. doi: 10.1016/S0140-6736(00)02762-8. [DOI] [PubMed] [Google Scholar]
- 9.Geiger Cancer Res. 2001;61:8513. [PubMed] [Google Scholar]
- 10.Guo Science. 1994;263:518. [Google Scholar]
- 11.Holtl J Urol. 1999;161:777. [PubMed] [Google Scholar]
- 12.Hsu Nat Med. 1996;2:52. [Google Scholar]
- 13.Kakumu J Gastroenterol Hepatol. 2000;15:431. doi: 10.1046/j.1440-1746.2000.02161.x. [DOI] [PubMed] [Google Scholar]
- 14.Kugler Nat Med. 2000;6:332. doi: 10.1038/73193. [DOI] [PubMed] [Google Scholar]
- 15.Levin N Engl J Med. 1995;332:1294. doi: 10.1056/NEJM199505113321910. [DOI] [PubMed] [Google Scholar]
- 16.Liu Am J Surg. 1997;173:358. doi: 10.1016/S0002-9610(96)00384-4. [DOI] [PubMed] [Google Scholar]
- 17.Ludewig J Exp Med. 2000;191:795. doi: 10.1084/jem.191.5.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nair Int J Cancer. 1999;82:121. doi: 10.1002/(sici)1097-0215(19990702)82:1<121::aid-ijc20>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 19.Nestle Nat Med. 1998;4:328. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 20.Nishiyama Clin Cancer Res. 2001;7:23. [Google Scholar]
- 21.Sadanaga Clin Cancer Res. 2001;7:2277. [PubMed] [Google Scholar]
- 22.Salgaller Prostate. 1998;35:144. doi: 10.1002/(SICI)1097-0045(19980501)35:2<144::AID-PROS8>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 23.Sallusto J Exp Med. 1994;179:1109. [Google Scholar]
- 24.Schnurr Cancer Res. 2001;61:6445. [PubMed] [Google Scholar]
- 25.Schuster Gastroenterology. 1997;112:656. doi: 10.1053/gast.1997.v112.agast970656. [DOI] [PubMed] [Google Scholar]
- 26.Shen J Immunol. 1997;158:2723. [PubMed] [Google Scholar]
- 27.Simons Cancer Res. 1999;59:5160. [PubMed] [Google Scholar]
- 28.Steinman Annu Rev Immunol. 1991;9:271. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 29.Tahara Cancer. 1999;85:1234. doi: 10.1002/(SICI)1097-0142(19990315)85:6<1234::AID-CNCR4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 30.Takayama Lancet. 2000;356:802. doi: 10.1016/S0140-6736(00)02654-4. [DOI] [PubMed] [Google Scholar]
- 31.Teo Dig Dis. 2001;19:263. doi: 10.1159/000050692. [DOI] [PubMed] [Google Scholar]
- 32.Theobald Proc Natl Acad Sci U S A. 1995;92:11993. doi: 10.1073/pnas.92.26.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vollmer Cancer Res. 1999;59:3064. [PubMed] [Google Scholar]
- 34.Zhao Immunol Invest. 2001;30:33. doi: 10.1081/IMM-100103689. [DOI] [PubMed] [Google Scholar]