

Abstract
Head and neck squamous cell carcinoma (HNSCC) is an aggressive epithelial malignancy that is the sixth most common neoplasm in the world. Despite numerous advances in treatments involving surgery, radiation, and chemotherapy, the 5-year survival has remained at less than 50% for the last 30 years primarily due to local recurrences [66]. Consequently, the possibility of developing immunotherapeutic approaches as a treatment for HNSCC has gained interest. The present review has 3 objectives pertaining to immunotherapeutic means to treat HNSCC patients: (1) to summarize the feasibility of such approaches, (2) to provide an overview of the obstacles to attaining protective immune reactivity, and (3) to consider how these obstacles can be overcome to stimulate immune reactivity to HNSCC. These objectives will also be considered in the context of what lessons have been learned from immunotherapeutic trials for other solid malignancies and the applicability of this information to HNSCC.
Keywords: Head and neck cancer, HNSCC, Immune, Immunotherapy, Immunosuppression
Feasibility of immunotherapeutic means for the treatment of HNSCC patients
Cancers are vulnerable to immune effector cells such as macrophages, natural killer (NK) cells, and cytotoxic T lymphocytes (CTL) [16, 35, 40, 72]. Anticancer immune responses include presentation of cancer antigens, production of Th1 cytokines, and activation of CTL. HNSCC cells can also be killed through antibody-dependent cell-mediated cytotoxicity (ADCC), although NK cells have been shown to be the prominent cells mediating such cytotoxicity [10].
Many, although not all, HNSCC preferentially express tumor antigens, including members of the families of the MAGE, RAGE, and GAGE antigens, making these tumor antigens possible targets for stimulating reactivity to HNSCC tumors [29, 37, 53]. Also, carcinoembryonic antigen, which is a known tumor antigen for gastrointestinal cancers, is expressed on cancers of the head and neck [36]. Cells that overexpress carcinoembryonic antigen can become targets of CTL. Other embryonic antigens that are expressed on HNSCC and whose inhibition with antibody has antitumor effects include Wnt and frizzled (Fz) family members [61]. To reinforce the concept of immune recognition of tumor antigens, a study with a murine HNSCC model demonstrated that tumor cells expressing alloantigens were able to stimulate immunity that was then protective to the wild-type tumor [28].
Monoclonal antibodies to tumor surface antigens have been used in several studies with animal tumor models and cancer patients to localize as well as treat tumors [46, 62]. Such studies have shown the feasibility of using tumor antigens as therapeutic targets. In vitro studies with human HNSCC cells showed that antibodies to HER2 could augment the growth-inhibitory effects of irradiation [71]. In vivo studies with adenocarcinoma patients showed some clinical responses to treatment with antibody to the carcinoma surface adhesion molecule 17-1A [46]. Using a nude mouse xenograft model, antibody to this adhesion molecule was shown to inhibit growth of human colon carcinomas through its facilitation of ADCC [54]. Antibody conjugates have also been tested to target delivery of the conjugate to tumor. For example, administration of a chimeric variant of anti-Lewisy monoclonal antibody conjugated to doxorubicin, to patients with Lewisy-expressing gastric carcinomas resulted in stabilization of disease in one third of the patients [1]. Among the few antibody trials conducted with head and neck cancer patients, a phase I study showed the safety of a monoclonal antibody to the EGF receptor in patients with HNSCC of the larynx and hypopharynx [11]. In separate studies, the combination of EGR receptor antibodies with cisplatin showed major responses in HNSCC patients [67]. These studies show that tumors express tumor antigens that can be targets of immune recognition.
The clinical role of active immune reactivity, rather than passively administered antibody treatments, to achieve recognition of tumor has been suggested in several different studies. For example, improved prognosis of patients with esophageal squamous cell carcinoma was associated with an increase in intratumoral CD4+ and CD8+ T cells, although not with the intratumoral content of natural killer cells [14]. Increased expression of class I MHC antigens on HNSCC cells coincided with reduced lymph node metastasis [30]. Overall, these studies indicate the feasibility of immune recognition and immune reactivity to HNSCC and raise the question of why the immune recognition does not result in a protective immune response.
Obstacles to protective immune reactivity to tumor
In contrast to the potential antitumor responses that can be generated, growth of a wide variety of solid cancers leads to alterations in immunologic parameters [20]. For example, lymphocyte functions are suppressed in cancer patients and in animal tumor models [2, 52, 58, 59, 79]. Tumor-infiltrating T cells from patients with melanoma or with colorectal carcinoma have reduced expression of T-cell activation markers [19]. Among the immunologic alterations that have been described in cancer patients and in animals bearing tumors are reductions in CD4+ cell numbers, responsiveness to cytokines, and proliferation in response to stimuli [32, 60, 87]. This depressed immune function is seen in vivo as well, such as by the reduced skin test reactivity to recall antigens [39].
HNSCC patients and mouse models of HNSCC are particularly deficient in their immune responsiveness [59, 69, 81]. Lymph nodes of HNSCC patients have been shown to be reduced in size and to have diminished T-cell content [49]. In one study, T cells from about one third of HNSCC patients were shown to be unresponsive to stimulation through the CD3/T-cell receptor [65]. The impact of this immune depression in HNSCC patients on the clinical course of disease is indicated by the association between reduced T-cell function and poorer disease-specific survival [32]. Efforts to enhance immune competence have resulted in tumor regression and, in preliminary analyses, have suggested enhanced survival [9, 31].
The immune defects of cancer patients are most likely due to a multiplicity of mechanisms, making the reversal of this immune inhibition more difficult. These include mechanisms by which the tumor directly and indirectly inhibits immune reactivity (Table 1) (Fig. 1). Direct inhibition of immune function can occur by HNSCC production of immune inhibitory factors such as prostaglandin E2 (PGE2) and transforming growth factor β (TGF-β) [44, 59, 63, 84]. In fact, blocking the inhibitory effects of TGF-β has been suggested as a means to enhance immune function of HNSCC patients [38, 77]. Similarly, the effectiveness of reducing PGE2 levels to lessen the immune inhibitory effects that are mediated by tumor-derived prostaglandins has long been known [80]. Treatment of HNSCC patients with cyclooxygenase-2 inhibitors to block production of PGE2 has been shown to result in restoration of immune functions and increased T-cell infiltration into the tumor mass, suggesting this to be a mechanism that contributes to the clinical responses to cyclooxygenase inhibitors [17, 41].
Table 1.
Obstacles to protective immune reactivity to tumor
| Direct immune inhibition by tumor: | References | |
|---|---|---|
| PGE2 | [63, 84] | |
| TGF-β | [44, 59, 84] | |
| IL-6 | [48] | |
| IL-10 | [68] | |
| Indirect immune inhibition | References | |
| Blockage of dendritic cell maturation | [3, 24, 56] | |
| Skewing toward Th2 reactivity | [55, 64, 76] | |
| Th2-skewing monocytes / dendritic cells | [34, 91] | |
| Mobilization of CD34+ precursor cells | [57, 74, 83, 85] | |
Fig. 1.
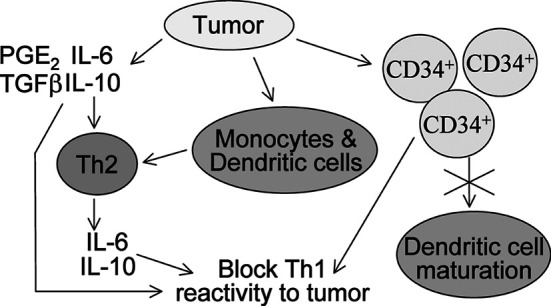
Schematic of the pathways by which tumors block antitumor immune reactivity. Direct blockage of immune reactivity occurs through production of cytokines that are inhibitory to Th1 antitumor responses. Tumor-derived cytokines can skew responses to Th2 cytokine production, which also blocks Th1 reactivity. Finally, tumors can mobilize immune inhibitory CD34+ precursor cells and block maturation of dendritic cells, both of which result in the failure to stimulate Th1 antitumor activity
Although tumors can inhibit immune responses, their presence can lead to a shift from responses that are beneficial for antitumor reactivity to responses that are less effective. Thus, the absence of antitumor reactivity cannot necessarily be attributed to immune depression. For example, melanoma patients who are undergoing immunization to tumor antigen have CD4+CD25+ T cells that can restrict effectiveness of the immunization [33]. Immune responses in tumor bearers have been shown to be biased toward a Th2 cytokine profile, which prevents the antitumor reactivity that can be induced through Th1 responses [64]. Th1 responses include the production of interferon γ (IFN-γ) and interleukin 2 (IL-2). Th2 responses are classified as those involving increased production of IL-4, IL-6, and IL-10. Primary cultures of head and neck squamous cell carcinoma cells have been shown to produce IL-4, IL-6, and granulocyte-macrophage colony-stimulating factor (GM-CSF) [48]. Once triggered, production of Th2 cytokines can be escalated, for example, by the capacity of IL-6 to stimulate production of IL-10 by tumor cells such as human colon carcinoma cells [68]. IL-10 diminishes production of Th1 cytokines, further exaggerating the shift toward a Th2 immune profile [15, 21]. The Th2 cytokine bias in tumor bearers is also reflected by the cytokine profiles of intratumoral T cells. For example, T cells that infiltrate cervical cancers are skewed toward the Th2 phenotype, with increased production of IL-4 and IL-6 [64]. The biological impact of the Th2 bias was shown with a mouse melanoma model in which tumor presence elicited a Th2 cytokine response that resulted in failure of protective immunity to a tumor vaccine strategy [76]. However, a shift to a Th1 cytokine bias allowed for therapeutic responsiveness to tumor. Studies of patients with non–small cell lung cancer showed that reduced survival correlated with a reduced capacity to produce the Th1 cytokine IL-2 [55]. While HNSCC patients have been noted to have pronounced immune dysfunctions, their immune parameters have not been defined to the extent that has been accomplished for patients with other malignancies. Our studies have suggested a prominent shift to a Th2 bias in the HNSCC patients and, to a lesser extent in patients with non-HNSCC malignancies. However, the Th2 shift in HNSCC patients appears to be incomplete since levels of only select Th1 cytokines were reduced. This may suggest an arrest at the Th0 stage without full commitment to Th2 skewing. What was surprising was that the skewing toward a Th2 cytokine profile was dependent on tumor presence and, for most cytokines, was not associated with either the extent of tumor burden or extent of metastasis (unpublished data).
Tumor-induced bias of immune responses toward a Th2 cytokine profile may also be mediated through the tumor’s effects on the monocyte/macrophage or dendritic cell populations. For example, gliomas can change monocyte cytokine expression to increase their production of IL-10, which leads to the secretion of a Th2 cytokine repertoire from T cells [91]. That this is relevant to the tumor in vivo is suggested in breast cancer by the association of an increased intratumoral macrophage content with inhibited T-cell activity [70]. However, this has not been determined for HNSCC tumors and was shown not to be the case in colorectal tumors. What has been known is that monocytes of HNSCC patients can inhibit T-cell reactivity through their production of PGE2 [6, 73].
The Th2 skewing of immune responses of cancer patients is not only mediated by T cells, but can be induced by antigen-presenting cells. For example, dendritic cells of cancer patients can also skew T-cell cytokine profiles toward Th2 responses. Dendritic cells that are exposed to IL-10 or PGE2, both being factors that are produced by multiple tumor types, produce diminished levels of IL-12 and, in turn, preferentially stimulate Th2 responses [34]. The maturation and differentiation of dendritic precursors into cells that can trigger antitumor reactivity is also diminished in tumor bearers as well as in cancer patients, including those with HNSCC [3, 24, 56]. Tumor-derived VEGF contributes to this blockage of dendritic cell development. Consistent with this blockage in maturation of dendritic cells is an increased appearance of immature myeloid precursor cells [4, 25, 83, 87, 90].
Our studies with HNSCC patients and with the murine Lewis lung carcinoma (LLC) model have shown the appearance of immune inhibitory cells that are distinct from mature lymphoid or monocytic cells and which can be recognized by intense surface expression of CD34 [57, 83, 85]. This suppressor cell population is an immature progenitor cell having clonogenic capabilities in soft agar and whose mobilization results from tumor production of GM-CSF [83]. In fact, studies with the murine lung cancer model showed a shift in immune suppressor cells from PGE2-secreting monocytes/macrophages during early periods of tumor growth, to the less mature CD34+ precursor cells during later phases of tumor progression [82]. The levels of the immune suppressive CD34+ progenitor cells are increased in the peripheral blood of HNSCC patients and in the bone marrow, spleen, and blood of LLC-bearing mice. Of greater importance is that the immune inhibitory CD34+ cells are also present within the HNSCC and LLC tumor tissue where they inhibit the activity of intratumoral T cells [57, 74]. These tumor-mobilized CD34+ cells inhibit T-cell reactivity through their production of TGF-β and, to a lesser extent, nitric oxide [78, 83, 85]. Of additional interest is that the CD34+ cells from tumor bearers are more resistant to apoptosis following cytokine withdrawal than are CD34+ cells from normal animals, and that they persist in increased numbers in mice and patients whose cancers have been surgically removed [88]. That tumor production of GM-CSF and the increased appearance of these cells could impact on antitumor immune reactivity in HNSCC patients and on progression of HNSCC disease was suggested by the reduced activity levels of intratumoral T cells and the reduced 2-year survival of HNSCC patients whose primary tumors released high levels of GM-CSF and which contained high levels of CD34+ progenitor cells [84, 86].
Clearly tumors, including HNSCC, are either inhibitory or divert immune responses to those that are less effective against tumors through multiple means. These include direct inhibition of immune reactivity by their production of immune inhibitory products and indirect inhibition by modifying immune cell function to, in turn, minimize immune reactivity toward tumor. In this sense, HNSCC tumors are similar to other solid malignancies, and it is very likely HNSCC are altering reactivity through multiple concurrent means. However, there have been no comprehensive studies that have tested the spectrum of approaches that are utilized by HNSCC to subvert immune antitumor defenses.
Overcoming obstacles to stimulate immune reactivity to cancer
Studies with a variety of solid cancers have shared the disappointment that no individual immunotherapy has become the means by which to stimulate curative responses against solid tumors. This has led to the increased testing of multimodality treatment approaches (Table 2), although such studies have lagged behind with HNSCC patients. Combining chemotherapy with immunotherapy, such as administering 5-fluorouracil/leucovorin and tumor cell / bacille Calmette-Guérin (BCG) vaccines to patients with stage III colon carcinoma has shown some success at improving effectiveness of immunotherapy [5]. Realizing the immune inhibitory effects of HNSCC presence, an alternative approach that was tested was to incorporate treatment that can diminish inhibitory effects of T cells, treatment to block the inhibitory effect of prostaglandins, along with a natural cytokine mixture containing IL-2 activity. Such treatment was shown to restore the depleted T-cell content of lymph nodes, increase immune infiltration into tumor, reduce tumor mass, and increase mean survival time of HNSCC patients [31, 49]. Using the SCC VII/SF immune competent murine HNSCC model, cyclophosphamide treatment was also tested together with IL-12. While the tumors were shown to be nonimmunogenic, the sequential administration of IL-12 followed by cyclophosphamide plus IL-12 converted them into immunogenic tumors and resulted in a high number of tumor cures [47]. Combination cytokine gene therapeutic approaches consisting of IL-2 with either GM-CSF or IL-12 in a murine HNSCC model resulted in increased immune reactivity and clinical antitumor responses [18, 44].
Table 2.
Overcoming obstacles to stimulate immune reactivity to cancer
| In vivo immune stimulatory approaches | References | |
|---|---|---|
| ■ Combination chemoimmunotherapy | [5, 47] | |
| ■ Block production of prostaglandins and other inhibitory factors | [31, 49] | |
| ■ Cytokine therapies | [18, 44] | |
| ■ Tumor vaccines | [12, 45] | |
| ■ Differentiation of mobilized immune inhibitory CD34+ precursor cells | [4, 8, 43] | |
| Ex vivo preparatory strategies prior to in vivo administration | References | |
| ■ Dendritic cell maturation and pulsing | [7, 12, 22, 27] | |
| ■ T-cell expansion and activation | [13, 65] | |
The immune stimulatory activity of dendritic cells is the basis for their use in cancer immunotherapy and several dendritic cell–based cancer vaccine trials have been initiated with patients having various cancers [12, 45]. To capitalize on the potent antigen-stimulatory capabilities of dendritic cells and to overcome the tumor-induced blockades in their differentiation, a vaccine of dendritic cells that were infected with adenovirus was tested in a murine tumor model, with the rationale that this infection can facilitate dendritic cell differentiation [50]. Alternative approaches to enhancing the functional competence of dendritic cells have included use of GM-CSF to enhance their development [51]. Since HNSCC tumors have also been shown to hamper dendritic cell maturation and function, such approaches could be highly relevant to approaches in the treatment of HNSCC cancers.
Recognizing that the tumor environment may be detrimental to the effective generation of antitumor immune reactivity, immune activation in vitro has been tested in animal tumor models as well as in patients. A number of trials have tested dendritic cell vaccines in which the dendritic cells were generated and pulsed with tumor antigens in vitro and then administered to patients (Table 2). Administration of such CD34+ cell–derived dendritic cells that were pulsed in vitro with a panel of melanoma antigens resulted in varied degrees of immune responses, but clinical responses coincided with the number of tumor antigens of the tumor vaccine to which melanoma patients mounted immunologic responses [7]. In a study with prostate cancer patients, dendritic cells that were pulsed in vitro with murine or human prostatic acid phosphatase were able to break patient unresponsiveness to the human tumor antigen [12, 22]. Dendritic cells that were generated by in vitro culture from monocytes and pulsed with tumor lysates were effective at stimulating delayed-type hypersensitivity responses and stabilization of disease in a number of pediatric malignancies [27].
In a different approach to avoid the immune inhibitory effects of the tumor environment, HNSCC patients were immunized with irradiated autologous tumor cells admixed with BCG, their primed lymph node cells were collected, more fully activated in vitro and reinfused [13]. While clinical responses were not observed, these studies demonstrate that removing primed T cells from the tumor environment can allow for the successful activation of the T cells to tumor. In contrast, studies with lymph node and peripheral blood mononuclear cells of HNSCC patients have shown T cells of some patients to be nonresponders to stimulation through the CD3/T-cell receptor complex, even in vitro and removed from the tumor environment [65]. These nonresponders could, however, become responsive upon exposure to anti-CD3/anti-CD28 coated beads, with responsiveness being seen as a Th1 response and a cytolysis of autologous tumor.
Several approaches have been used to capitalize on the increase in immature precursor cells that accumulate in tumor bearers, including in HNSCC patients [3, 23, 83, 84]. Treatment of these immature cells with all-trans-retinoic acid facilitates their differentiation into dendritic cells, and restores that capacity of the resulting dendritic cells to stimulate antigen-specific T-cell responses [4]. The CD34+ cells that are mobilized by tumors, including in HNSCC patients, can also be directed to differentiate into dendritic cells, with active vitamin D3 analogs [26, 42, 43, 75, 89]. In a phase 1B study, treatment of HNSCC patients with 25-hydroxyvitamin D3 resulted in not only a reduction in the frequency of immune inhibitory CD34+ cells in the peripheral blood, but also an increase in expression of activation markers on blood antigen-presenting cells and an increase in T-cell responsiveness to activation [43]. In a mouse tumor model, the increased presence of immature CD34+ precursor cells was capitalized on by recruiting the immature CD34+ cells to the tumor surgical excision site as a first step to differentiate them in situ into mature dendritic cells capable of stimulating immune reactivity to residual tumor cells [8]. Whether or not the resulting dendritic cells will maintain the functional capability to stimulate protective immune reactivity to tumor within the environment of residual tumor cells has yet to be tested.
With the realization that tumors inhibit antitumor immune defenses through a multitude of mechanisms, approaches to overcome this immune inhibition and to generate protective immune reactivity have become increasingly more complex. What has become most apparent is the importance of not only attempting to stimulate immune reactivity, but to first overcome the immune suppression or immune unresponsiveness that is induced by tumor. Many of these lessons have been learned from studies of patients with cancers other than HNSCC. However, the highly immune inhibitory nature of HNSCC emphasizes the importance of applying these lessons of how to overcome the immune inhibitory properties of HNSCC and how to then stimulate the restored immune capability so as to confer protective immune reactivity in HNSCC patients toward autologous cancer.
References
- 1.Ajani Cancer J. 2000;6:78. [Google Scholar]
- 2.Alexander Cancer Res. 1993;53:1380. [Google Scholar]
- 3.Almand Clin Cancer Res. 2000;6:1755. [PubMed] [Google Scholar]
- 4.Almand J Immunol. 2001;166:678. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 5.Baars Br J Cancer. 2002;86:1230. doi: 10.1038/sj.bjc.6600254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balch Ann Surg. 1982;196:645. doi: 10.1097/00000658-198212001-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banchereau Cancer Res. 2001;61:6451. [PubMed] [Google Scholar]
- 8.Banich J Immunother. 2003;26:31. doi: 10.1097/00002371-200301000-00004. [DOI] [PubMed] [Google Scholar]
- 9.Barrera Arch Otolaryngol Head Neck Surg. 2000;126:345. doi: 10.1001/archotol.126.3.345. [DOI] [PubMed] [Google Scholar]
- 10.Bier Cancer Immunol Immunother. 1998;46:167. doi: 10.1007/s002620050475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bier Cancer Chemother Pharmacol. 2001;47:519. doi: 10.1007/s002800000270. [DOI] [PubMed] [Google Scholar]
- 12.Burch Clin Cancer Res. 2000;6:2175. [Google Scholar]
- 13.Chang Head Neck. 2003;25:198. doi: 10.1002/hed.10195. [DOI] [PubMed] [Google Scholar]
- 14.Cho Cancer Res. 2003;63:1555. [Google Scholar]
- 15.Corinti J Immunol. 2001;166:4312. doi: 10.4049/jimmunol.166.7.4312. [DOI] [PubMed] [Google Scholar]
- 16.Cox J Immunol. 1992;149:3290. [PubMed] [Google Scholar]
- 17.Cross Arch Otolaryngol Head Neck Surg. 1992;118:526. doi: 10.1001/archotol.1992.01880050080019. [DOI] [PubMed] [Google Scholar]
- 18.Day Laryngoscope. 2001;111:801. doi: 10.1097/00005537-200105000-00009. [DOI] [PubMed] [Google Scholar]
- 19.De Br J Cancer. 2003;88:320. doi: 10.1038/sj.bjc.6600679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finke Immunol Today. 1999;20:158. doi: 10.1016/s0167-5699(98)01435-2. [DOI] [PubMed] [Google Scholar]
- 21.Fiorentino J Immunol. 1991;146:3444. [Google Scholar]
- 22.Fong J Immunol. 2001;167:7150. doi: 10.4049/jimmunol.167.12.7150. [DOI] [PubMed] [Google Scholar]
- 23.Gabrilovich Cell Immunol. 1996;170:111. doi: 10.1006/cimm.1996.0140. [DOI] [PubMed] [Google Scholar]
- 24.Gabrilovich Blood. 1998;92:4150. [PubMed] [Google Scholar]
- 25.Gabrilovich J Immunol. 2001;166:5398. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 26.Garrity Int J Cancer. 1997;73:663. doi: 10.1002/(sici)1097-0215(19971127)73:5<663::aid-ijc9>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 27.Geiger Cancer Res. 2001;61:8513. [PubMed] [Google Scholar]
- 28.Gleich Head Neck. 2003;25:274. doi: 10.1002/hed.10258. [DOI] [PubMed] [Google Scholar]
- 29.Gotte Acta Otolaryngol. 2002;122:546. [Google Scholar]
- 30.Grandis Clin Cancer Res. 2000;6:2794. [PubMed] [Google Scholar]
- 31.Hadden Int Immunopharmacol. 2003;3:1073. doi: 10.1016/S1567-5769(03)00029-8. [DOI] [PubMed] [Google Scholar]
- 32.Heimdal Acta Otolaryngol. 1999;119:281. doi: 10.1080/00016489950181828. [DOI] [PubMed] [Google Scholar]
- 33.Javia J Immunother. 2003;26:85. doi: 10.1097/00002371-200301000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalinski Immunology Today. 2000;20:561. doi: 10.1016/S0167-5699(99)01547-9. [DOI] [PubMed] [Google Scholar]
- 35.Karagiannis Eur J Immunol. 2003;33:1030. doi: 10.1002/eji.200323185. [DOI] [PubMed] [Google Scholar]
- 36.Kass Cancer Res. 2002;62:5049. [PubMed] [Google Scholar]
- 37.Kienstra Head Neck. 2003;25:457. doi: 10.1002/hed.10223. [DOI] [PubMed] [Google Scholar]
- 38.Kobie Cancer Res. 2003;63:1860. [PubMed] [Google Scholar]
- 39.Kondo Am J Clin Oncol. 1996;19:159. doi: 10.1097/00000421-199604000-00014. [DOI] [PubMed] [Google Scholar]
- 40.Kuge J Immunol. 1995;154:1777. [PubMed] [Google Scholar]
- 41.Lang FASEB J. 2003;17:286. [Google Scholar]
- 42.Lathers J Leuko Biol. 1999;65:623. doi: 10.1002/jlb.65.5.623. [DOI] [PubMed] [Google Scholar]
- 43.Lathers Human Immunol. 2001;62:1282. doi: 10.1016/S0198-8859(01)00317-2. [DOI] [PubMed] [Google Scholar]
- 44.Li Arch Otolaryngol Head Neck Surg. 2001;127:1319. doi: 10.1001/archotol.127.11.1319. [DOI] [PubMed] [Google Scholar]
- 45.Lodge Cancer Res. 2000;60:829. [PubMed] [Google Scholar]
- 46.Makower Cancer Invest. 2003;21:177. doi: 10.1081/CNV-120016413. [DOI] [PubMed] [Google Scholar]
- 47.Mandpe Arch Otolaryngol Head Neck Surg. 2003;129:786. doi: 10.1001/archotol.129.7.786. [DOI] [PubMed] [Google Scholar]
- 48.Mann Am J Surg. 1992;164:567. doi: 10.1016/s0002-9610(05)80708-1. [DOI] [PubMed] [Google Scholar]
- 49.Meneses Int Immunopharmacol. 2003;3:1083. doi: 10.1016/S1567-5769(03)00017-1. [DOI] [PubMed] [Google Scholar]
- 50.Miller Cancer Res. 2002;62:5260. [PubMed] [Google Scholar]
- 51.Miller J Immunol. 2002;169:2875. doi: 10.4049/jimmunol.169.6.2875. [DOI] [PubMed] [Google Scholar]
- 52.Mizoguchi Science. 1992;258:1795. doi: 10.1126/science.1465616. [DOI] [PubMed] [Google Scholar]
- 53.Monji Biochem Biophys Res Commun. 2002;294:734. doi: 10.1016/S0006-291X(02)00543-0. [DOI] [PubMed] [Google Scholar]
- 54.Naundorf Int J Cancer. 2002;100:101. doi: 10.1002/ijc.10443. [DOI] [PubMed] [Google Scholar]
- 55.Neuner Int J Cancer. 2002;101:287. doi: 10.1002/ijc.10604. [DOI] [PubMed] [Google Scholar]
- 56.Ohm J Immunol. 1999;163:3260. [PubMed] [Google Scholar]
- 57.Pandit Ann Otol Rhinol Laryngol. 2000;109:749. [Google Scholar]
- 58.Prechel Cancer Lett. 1995;92:235. doi: 10.1016/0304-3835(95)03804-6. [DOI] [PubMed] [Google Scholar]
- 59.Qin Mol Ther. 2001;4:551. doi: 10.1006/mthe.2001.0493. [DOI] [PubMed] [Google Scholar]
- 60.Reichert Cancer Res. 1998;58:5344. [PubMed] [Google Scholar]
- 61.Rhee Oncogene. 2002;21:6598. doi: 10.1038/sj.onc.1205920. [DOI] [PubMed] [Google Scholar]
- 62.Schechter Anticancer Drugs. 2003;14:49. doi: 10.1097/00001813-200301000-00007. [DOI] [PubMed] [Google Scholar]
- 63.Scioscia Am J Otolaryngol. 1997;18:1. doi: 10.1016/s0196-0709(97)90041-7. [DOI] [PubMed] [Google Scholar]
- 64.Sheu J Immunol. 2001;167:2972. doi: 10.4049/jimmunol.167.5.2972. [DOI] [PubMed] [Google Scholar]
- 65.Shibuya Arch Otolaryngol Head Neck Surg. 2000;126:473. doi: 10.1001/archotol.126.4.473. [DOI] [PubMed] [Google Scholar]
- 66.Shin Semin Oncol. 1999;26:100. [Google Scholar]
- 67.Shin Clin Cancer Res. 2001;7:1204. [PubMed] [Google Scholar]
- 68.Suzuki Int J Oncol. 2001;18:581. [PubMed] [Google Scholar]
- 69.Thomas Int J Cancer. 2000;86:368. doi: 10.1002/(sici)1097-0215(20000501)86:3<368::aid-ijc11>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 70.Toomey Immunol Invest. 1999;28:29. doi: 10.3109/08820139909022721. [DOI] [PubMed] [Google Scholar]
- 71.Uno Int J Cancer. 2001;94:474. doi: 10.1002/ijc.1493. [DOI] [PubMed] [Google Scholar]
- 72.Vujanovic J Immunol. 1995;154:281. [PubMed] [Google Scholar]
- 73.Wanebo Cancer. 1988;61:462. doi: 10.1002/1097-0142(19880201)61:3<462::aid-cncr2820610310>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 74.Wiers Clin Exp Med. 1998;16:275. doi: 10.1023/A:1006501110857. [DOI] [PubMed] [Google Scholar]
- 75.Wiers J Immunother. 2000;23:115. doi: 10.1097/00002371-200001000-00014. [DOI] [PubMed] [Google Scholar]
- 76.Winter Immunology. 2003;108:409. doi: 10.1046/j.1365-2567.2003.01596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wojtowiczpraga J Immunother. 1997;20:165. [Google Scholar]
- 78.Wright Cancer Immunol Immunother. 1998;46:253. doi: 10.1007/s002620050485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yoshino Cancer Res. 1992;52:775. [PubMed] [Google Scholar]
- 80.Young Immunol Comm. 1983;12:11. [Google Scholar]
- 81.Young Int J Immunopharmacol. 1999;21:241. doi: 10.1016/S0192-0561(99)00008-9. [DOI] [PubMed] [Google Scholar]
- 82.Young Cancer Res. 1989;49:1931. [PubMed] [Google Scholar]
- 83.Young Clin Cancer Res. 1995;1:95. [PubMed] [Google Scholar]
- 84.Young Int J Cancer. 1996;67:333. doi: 10.1002/(SICI)1097-0215(19960729)67:3<333::AID-IJC5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 85.Young J Immunol. 1996;156:1916. [PubMed] [Google Scholar]
- 86.Young Int J Cancer. 1997;74:69. doi: 10.1002/(sici)1097-0215(19970220)74:1<69::aid-ijc12>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 87.Young J Immunol. 1997;159:990. [Google Scholar]
- 88.Young Int J Cancer. 1999;82:609. doi: 10.1002/(SICI)1097-0215(19990812)82:4<609::AID-IJC23>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 89.Young MRI, Wright MA, Vellody K, Lathers DMR (1999) Skewed differentiation of bone marrow CD34+ cells of tumor bearers from dendritic toward monocytic cells, and the induction of differentiation toward dendritic cells by 1α,25-dihydroxyvitamin D3. Int J Immunopharmacol 675–688 [DOI] [PubMed]
- 90.Young Human Immunol. 2001;62:332. doi: 10.1016/S0198-8859(01)00222-1. [DOI] [PubMed] [Google Scholar]
- 91.Zou J Immunol. 1999;162:4882. [PubMed] [Google Scholar]