

Abstract
A Wilms’ tumor gene WT1 is expressed at high levels not only in most types of leukemia but also in various types of solid tumors, including lung and breast cancer. WT1 protein has been reported to serve as a target antigen for tumor-specific immunotherapy both in vitro in human systems and in vivo in murine models. We have shown that mice immunized with WT1 peptide or WT1 cDNA could reject a challenge from WT1-expressing tumor cells (a “prophylactic” model). However, it was not examined whether WT1 peptide vaccination had the potency to reject tumor cells in a “therapeutic” setting. In the present study, we demonstrated for the first time that WT1 peptide vaccination combined with Mycobacterium bovis bacillus Calmette-Guérin cell wall skeleton (BCG-CWS) was more effective for eradication of WT1-expressing tumor cells that had been implanted into mice before vaccination (a “therapeutic” model) compared with WT1 peptide vaccination alone. An intradermal injection of BCG-CWS into mice, followed by that of WT1 peptide at the same site on the next day, generated WT1-specific cytotoxic T lymphocytes (CTLs) and led to rejection of WT1-expressing leukemia or lung cancer cells. These results showed that BCG-CWS, which was well known to enhance innate immunity, could enhance WT1-specific immune responses (acquired immunity) in combination with WT1 peptide vaccination. Therefore, WT1 peptide vaccination combined with BCG-CWS may be applied to cancer immunotherapy in clinical settings.
Keywords: BCG-CWS, Immunotherapy, Wilms’ tumor gene, WT1
Introduction
The Wilms’ tumor gene, WT1, was first reported as a gene responsible for Wilms’ tumor, a pediatric renal cancer [5, 8]. This gene encodes a zinc finger transcription factor involved in tissue development, in cell proliferation and differentiation, and in apoptosis, and is categorized as a tumor suppressor gene [21]. The WT1 gene product regulates the expression of various genes either positively or negatively depending upon how it combines with other regulatory proteins in different types of cells.
We [13, 14] and others [3, 22, 23, 24] have identified high expression levels of the wild-type WT1 gene in leukemic cells regardless of the types of disease. On the basis of accumulated evidence [14, 15, 34, 38], we have proposed that the wild-type WT1 gene performs an oncogenic rather than a tumor suppressor gene function in hematopoietic progenitor cells. Moreover, we found that various types of solid tumors, including lung, gastric, colon, and breast cancer cell lines, expressed the wild-type WT1 gene [27]. Cancer cells of lung and breast cancer patients also expressed the WT1 gene at high levels [19, 27, 28]. Growth of WT1-overexpressing tumor cells was specifically inhibited by WT1 antisense oligodeoxynucleotides [38], thus suggesting a close relationship between WT1 overexpression and tumorigenesis. These results indicate that the WT1 gene product could be a promising tumor-specific antigen not only for leukemia but also for various types of solid tumors. In fact, we [29, 32] and others [9, 26] have generated human WT1-specific CTLs in vitro. Furthermore, we have shown that mice immunized with WT1 peptides with anchor motifs needed for binding to MHC class I molecules, or with WT1 plasmid DNA, elicited WT1-specific CTLs and rejected challenge of WT1-expressing tumor cells, indicating that WT1 protein can serve as a tumor rejection antigen in vivo [30, 32, 35]. However, these murine systems are “prophylactic” models, in which tumors were transplanted after vaccination of the mice with WT1 peptide or cDNA. It has not been examined whether WT1 peptide vaccination has the potency to reject tumor cells in a “therapeutic” model.
Mycobacterium bovis bacillus Calmette-Guérin cell wall skeleton (BCG-CWS) is being used for cancer immunotherapy in clinical settings [1, 10, 16, 17, 18, 25, 37, 39, 40], and the mechanism of the enhancement of immunity against tumor was investigated in some reports [11, 20, 33, 36]. BCG-CWS was shown to enhance innate immunity by the activation of dendritic cells (DCs), followed by activation of NK cells. The ability of BCG-CWS to activate DCs led us to an idea that we could use BCG-CWS as adjuvant to enhance tumor antigen-specific immune response.
In the present study, we demonstrated for the first time that WT1 peptide vaccination combined with BCG-CWS was efficient for rejection of WT1-expressing tumor cells that had been implanted before vaccination and that BCG-CWS could enhance WT1 protein (tumor antigen)-specific immune responses in combination with WT1 peptide.
Materials and methods
Mice
Male C57BL/6 (H-2Db) mice were obtained from Clea Japan (Tokyo, Japan) and were used at 6-8 weeks of age and maintained in a specific pathogen-free (SPF) containment facility.
WT1 peptide and BCG-CWS
Db126 peptide (a.a.126-134 RMFPNAPYL), MHC class I (H-2Db)-binding peptide was synthesized with an ABI430A peptide synthesizer (Applied Biosystems, Foster City, CA, USA) using Fmoc chemistry. It was then purified by PR-HPLC with a C18 Microbondasphere column (Waters Japan, Osaka, Japan). Synthesis of the correct peptide was confirmed with the aid of an API IIIE triple quadrupole mass spectrometer (Sciex, Thorinhill, Toronto, Canada), and concentrations of the peptide were determined by means of MicroBCA assay (Pierco, Rockford, IL, USA) using bovine serum albumin as the standard. The peptide was dissolved in PBS. Aliquots were stored frozen at –20°C.
Oil-in-water emulsion of BCG-CWS was prepared as described elsewhere [1]. BCG-CWS (2 mg) and 9.6 μl of Squalane (Wako Pure Chemical Industries, Osaka, Japan) were mixed and homogenized for 1 min. Then the mixture was homogenized with 660 μl of 1.1% Tween 80 in PBS for 8 min.
Cells
C1498, WT1-nonexpressing murine leukemia cell line of C57BL/6 origin was obtained from American Type Culture Collection (ATCC, Rockville, MD, USA) [30, 35]. The WT1-nonexpressing murine lung cancer cell line of C57BL/6 origin, 3LL, was obtained from ATCC. WT1-expressing murine WT1-C1498 (mWT1-C1498) and 3LL (mWT1-3LL) were established by transfection of C1498 cells or 3LL cells with murine WT1 cDNA (a kind gift from Dr D. Housman, Massachusetts Institute of Technology, via Dr H. Nakagama, National Cancer Center Research Institute, Japan), respectively [30, 35]. Both the mWT1-C1498 and mWT1-3LL expressed WT1 protein at levels similar to those in naturally WT1-overexpressing murine leukemia cell line, FBL3.
In vivo tumor challenges and vaccination schedule
The inoculated dose of the tumor cells was optimized by preliminary experiments. Tumor inoculation and vaccination schedule are shown in Fig. 1. Mice were intraperitoneally (i.p.) implanted with 5×105 mWT1-C1498 cells in 100 μl of PBS, or subcutaneously (s.c.) in the abdomen with 2.5×104 mWT1-3LL cells in 100 μl of PBS on day 0. Then 100 μg of BCG-CWS in 100 μl of PBS was intradermally (i.d.) injected in the abdomen on days 1, 8, 15, and 22, followed by i.d. injection of 100 μg of WT1 peptide in 100 μl of PBS at the same site as that of BCG-CWS injection on days 2, 9, 16, and 23. In control groups, mice were i.d. injected with either 100 μg of BCG-CWS in 100 μl of PBS, 100 μg of WT1 peptide in 100 μl of PBS, or 100 μl PBS alone on days 1, 8, 15, and 22. Tumor growth was monitored by measuring the longest diameter of the palpable mass.
Fig. 1.
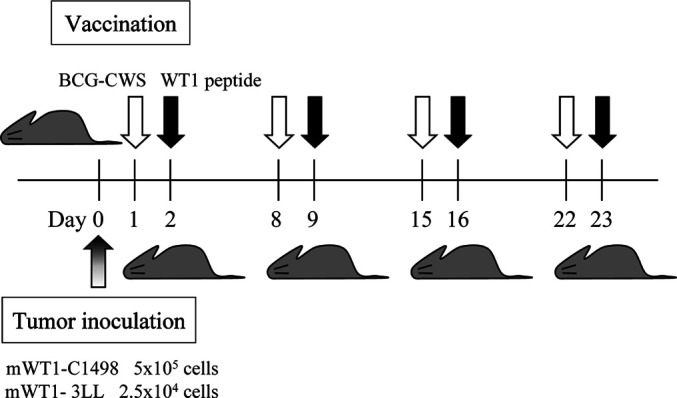
In vivo tumor challenges and vaccination schedule. Mice were intraperitoneally (i.p.) inoculated with 5×105 mWT1-C1498 cells, or subcutaneously (s.c.) in the abdomen with 2.5×104 mWT1-3LL cells on day 0. In BCG+WT1 group, 100 μg of BCG-CWS was intradermally (i.d.) injected in the abdomen on days 1, 8, 15, and 22, followed by i.d. injection of 100 μg of WT1 peptide at the same site as that of BCG-CWS injection on days 2, 9, 16, and 23. In control groups, either 100 μg of BCG-CWS or 100 μg of WT1 peptide was i.d. injected in the abdomen on days 1, 8, 15, and 22. The white and black arrows indicate BCG-CWS and WT1 peptide injection, respectively
51Cr-release cytotoxity assay
Spleens were resected from the BCG-CWS+WT1 peptide-immunized mice 7 days after the fourth WT1 peptide injection (day 30) or from nontreated mice, and the splenocytes were stimulated with irradiated syngenic splenocytes pulsed with the WT1 peptide. After 5 days of culture, 51Cr-release cytotoxicity assay was performed against mWT1-C1498 or C1498 cells, as described previously. Target cells (1×104 cells in 100 μl) labeled with 51Cr were added to wells containing varying numbers of effector cells (100 μl) using U-bottomed 96-well plates. After 4 h of incubation at 37°C, cells were centrifuged and 100 μl of supernatant was collected and measured for radioactivity. Percentage of specific lysis (% specific lysis) was calculated as follows: percentage lysis = (cpm experimental release – cpm spontaneous release) / (cpm maximal release – cpm spontaneous release)×100. Fluorescence emission of culture supernatant of target cells alone or of target cells that were lysed completely by the treatment with 1% Triton X-100 was used as a spontaneous and a maximal release, respectively.
Histology
Physiologically WT1-expressing organs such as kidney and bone marrow were removed from the surviving mice that had rejected the tumor challenges as a result of BCG+WT1 immunization and fixed in Bouin’s solution. Paraffin sections of 8-mm thickness were stained with hematoxylin eosin by standard methods.
Colony assay
The bone marrow cells were seeded at 3×104 cells / 35-mm dish in α-MEM containing 20% fetal bovine serum, 1% bovine serum albumin Fr 5, 1.5% methylcellulose, and cytokines (100 ng/ml SCF, 10 ng/ml IL-3, 10 ng/ml IL-6, 3,000 U/ml EPO, 10 ng/ml GM-CSF, and 10 ng/ml G-CSF) and cultured for 14 days. Colonies with more than 50 cells were counted.
Statistical analysis
Significant differences in tumor sizes, disease-free survival, or overall survival between experimental groups were evaluated using the log-rank test.
Results
Effect of WT1 peptide vaccination combined with BCG-CWS on rejection of transplanted tumors
To investigate the in vivo effect of vaccination with WT1 peptide combined with BCG-CWS (BCG+WT1), two murine models, in which mice transplanted with either WT1-expressing leukemia or lung cancer cells were treated with the BCG+WT1, were developed (Fig. 1). In both models, the days when tumor cells were implanted were defined as day 0. BCG-CWS was i.d. injected at the abdomen on day 1, followed by i.d. injection of WT1 peptide at the same site as that of BCG-CWS injection on day 2. The BCG+WT1 vaccination was repeated four times at weekly intervals. In control groups, either WT1 peptide or BCG-CWS was injected on day 1, and the injection was repeated four times at weekly intervals.
In the leukemia model (Fig. 2A), 9 of the 13 mice immunized with BCG+WT1 developed tumor and died, while the remaining 4 mice survived without development of tumors until sacrifice for colony assay on day 65. On the other hand, all of the BCG-CWS-immunized, WT1-immunized, and nonimmunized mice developed tumors and died, except 3 tumor-bearing mice immunized with BCG-CWS, which were sacrificed for colony assay on day 65. Disease-free survival of BCG+WT1-immunized mice was 31% and significantly longer in comparison with that of the other three groups (p<0.05) (Fig. 2B).
Fig. 2A,B.

Therapeutic effect of WT1 peptide vaccination combined with BCG-CWS on rejection of transplanted leukemia cells. A Tumor size: after implantation with 5×105 mWT1-C1498 cells, mice were four times immunized with 100 μg of BCG-CWS + 100 μg of WT1 peptide, 100 μg of BCG-CWS alone, or 100 μg of WT1 peptide alone. The tumor growth curves represent tumor size of individual mice. Tumor size shows the longest diameter. Asterisks indicate sacrifice for colony assay on day 65. B Disease-free survival: solid line immunized with 100 μg of BCG-CWS + 100 μg of WT1 peptide, dotted line 100 μg of BCG-CWS alone, dash-dotted line 100 μg of WT1 peptide alone, double-dash-dotted line nonimmunized
In the solid tumor model (Fig. 3A), tumor growth was observed in 7 of 10 mice immunized with BCG+WT1, but the remaining 3 mice survived until they were sacrificed on day 132. On the other hand, tumor growth was observed in 8 of the 9 BCG-CWS-immunized, all of the 10 WT1-immunized, and 9 of the 10 nonimmunized mice. Disease-free survival for 4 months was 30% in the mice immunized with BCG+WT1, while it was 10%, 0%, and 10% in BCG-immunized, WT1-immunized, or nonimmunized mice, respectively. The disease-free survival of BCG+WT1-immunized mice was significantly longer than that of the other groups (p<0.05) (Fig. 3B).
Fig. 3A,B.

Therapeutic effect of WT1 peptide vaccination combined with BCG-CWS on rejection of transplanted lung cancer cells. A Tumor size: after implantation with 2.5×104 mWT1-3LL cells, mice were four times immunized with 100 μg of BCG-CWS + 100 μg of WT1 peptide, 100 μg of BCG-CWS alone, or 100 μg of WT1 peptide alone. The tumor growth curves represent tumor size of individual mice. Tumor size shows the longest diameter. B Disease-free survival: solid line immunized with 100 μg of BCG-CWS + 100 μg of WT1 peptide, dotted line 100 μg of BCG-CWS alone, dash-dotted line 100 μg of WT1 peptide alone, double-dash-dotted line nonimmunized
WT1-specifc CTL induction by immunization with BCG-CWS+WT1 peptide
Splenocytes from the mice immunized four times with BCG-CWS+WT1 peptide or from nonimmunized mice were in vitro restimulated with WT1 peptide-pulsed splenocytes and assayed for cytotoxic activity against WT1-expressing mWT1-C1498, and WT1-nonexpressing C1498 (Fig. 4). The splenocytes from the immunized mice showed a significant specific lysis against WT1-expressing mWT1-C1498 compared with that against WT1-nonexpressing C1498. The splenocytes from the nonimmunized mice did not show a significant specific lysis against the target cells. These results demonstrated that BCG-CWS+WT1 immunization could induce WT1-specific CTL responses, resulting in suppression of the growth of the implanted tumor cells.
Fig. 4.

Induction of WT1-specifc CTLs by immunization with BCG-CWS+WT1 peptide. Splenocytes from the mice immunized four times with BCG-CWS+WT1 peptide or from nonimmunized mice were in vitro restimulated with WT1 peptide-pulsed splenocytes. Their cytotoxic activities against WT1-expressing mWT1-C1498, or WT1-nonexpressing C1498, were tested by 51Cr-release cytotoxicity assay at the indicated E/T ratios in triplicate. Closed circles and squares represent cytotoxic activities of splenocytes from the immunized mice against WT1-expressing mWT1-C1498 and WT1-nonexpressing C1498, respectively. Open circles and squares represent cytotoxic activities of splenocytes from the nonimmunized mice against WT1-expressing mWT1-C1498 and WT1-nonexpressing C1498, respectively. Values shown are the means of the results from three mice (bars indicate standard error)
No evidence of autoimmunity in surviving mice that rejected tumors
WT1 is expressed in some normal tissues of adult mice, including podocytes of kidney glomeruli, bone marrow CD34+ cells, gonads, and mesothelial structures [4, 6, 31]. To evaluate the risk of autoimmunity by immunization with BCG-CWS+WT1 peptide, the whole tissues of the immunized mice were pathologically examined 1 week after the fourth immunization. All of the organs, including kidney and bone marrow, showed normal structure and cellularity in all the mice examined, and no pathological changes caused by immune response such as lymphocyte infiltration and tissue destruction and repair were observed (Fig. 5). Furthermore, colony assay was performed for bone marrow cells from either the immunized or nonimmunized mice in the presence of IL-3, IL-6, SCF, EPO, G-CSF, and GM-CSF (Fig. 6). However, no differences in the colony number of CFU-GEMM, CFU-GM, CFU-G, CFU-M, and BFU-E were found between the immunized and nonimmunized mice. These results showed that WT1-specific CTLs were ignorant of normal self-cells that expressed WT1 at physiological levels.
Fig. 5.

No pathological changes of kidney and bone marrow in the immunized mice that rejected tumors. Kidney glomeruli and bone marrow stained with hematoxylin and eosin are shown. No pathological changes such as lymphocyte infiltration and tissue destruction and repair are observed
Fig. 6.

Colony assay of bone marrow cells from mice immunized with BCG-CWS+WT1 peptide. Closed and open columns represent the colony number of bone marrow cells from immunized or nonimmunized mice, respectively. CFU-GEMM colony-forming-unit granulocyte-erythroid-macrophage-megakaryocyte, CFU-GM colony-forming-unit granulocyte-macrophage, CFU-G colony-forming-unit granulocyte, CFU-M colony-forming-unit macrophage, BFU-E burst-forming-unit erythroid
Discussion
We and others have previously shown that WT1 protein can become a target for cancer immunotherapy. MHC class I-restricted, WT1-specific CTLs were generated from human peripheral blood mononuclear cells by in vitro stimulation with WT1 peptide [9, 26, 29, 32]. Furthermore, mice immunized with WT1 peptide or cDNA rejected challenges of WT1-expressing tumor cells, indicating that WT1 protein can serve as a tumor rejection antigen in vivo [30, 32, 35]. However, this murine system is a “prophylactic” model, in which WT1-expressing tumors were transplanted after the vaccination of mice. In the present study, we demonstrated for the first time a “therapeutic” model in which BCG+WT1 peptide vaccination was effective for rejection of hematopoietic or solid tumors that had already been transplanted before vaccination.
In preliminary experiments the immunization was started 2 days after tumor transplantation, but no significant difference in survival was observed between the BCG+WT1-immunized and control groups (data not shown). Immunization with BCG+WT1 might not be strong enough to eradicate numerous tumor cells that had expanded in vivo during the 2 days after transplantation.
Although immunization with BCG+WT1 peptide was the most effective for rejection of tumor cells, immunization with BCG-CWS alone showed better efficacy on tumor rejection and growth inhibition than immunization with WT1 peptide alone in the leukemia model. Innate immunity activated by injection of BCG-CWS might be useful for rejection of tumor cells, as several investigations reported [11, 20, 33, 36] that BCG-CWS could activate innate immunity by activation and maturation of DCs, followed by activation of NK cells. WT1-specific CTLs were generated from the splenocytes of the mice immunized with WT1 peptide combined with BCG-CWS, indicating that the WT1-specific immune responses were also generated effectively by the vaccination. A speculated scenario of activation of WT1-specific CTLs is as follows: BCG-CWS activates DCs into mature form to have highly-expressed MHC and costimulatory molecules [33, 36]. The matured DCs, whose MHC class I molecules are bound with injected WT1 peptides, may subsequently activate and expand WT1-specific CTL precursors which exist with low frequencies in the mice. Taken together, it was suggest that BCG-CWS activated DCs, which led to activation of innate immunity including NK cells and acquired immunity, namely, antigen (WT1)-specific CTLs.
In this study, WT1 peptide was injected 1 day after injection of BCG-CWS for the following reasons: (1) It takes some time for BCG-CWS to activate DCs from immature to mature forms that have higher potency to induce antigen-specific immune responses [33, 36], and (2) the activated DCs are required to stay at the site of BCG-CWS injection [2, 7] until the injection of WT1 peptide. In fact, immunization with a mixture of BCG-CWS and WT1 peptide did not give better effects on tumor rejection and growth inhibition compared to immunization with WT1 peptide alone in preliminary studies of ours (data not shown). Elucidation of mechanisms of activation of DCs by BCG-CWS, which leads to further enhancement of innate immunity and activation of antigen-specific acquired immunity, would lead to optimization of administration of the BCG-CWS and WT1 peptide (optimal doses and administration intervals), followed by enhancement of efficacy of the vaccination for tumor rejection.
The surviving mice that rejected tumors by immunization with BCG+WT1 peptide did not have damage to organs, including kidney and bone marrow which physiologically expressed WT1. Furthermore, in colony assay of bone marrow hematopoietic cells, which also expressed WT1, no differences in the colony numbers were found between the immunized and nonimmunized mice. These results demonstrated that the WT1-specific CTLs generated in vivo in this murine model could discriminate between WT1-expressing tumor cells and physiologically WT1-expressing normal cells, resulting in the killing of tumor cells alone with no damage to normal tissues. These findings support the previous reports [9, 26, 29] that demonstrated that WT1-specific CTLs could give rise to damage to tumor cells, but not to normal cells. At first, preferred killing of tumor cells by the WT1-specific CTLs was considered to be due to difference in the WT1 expression levels between tumor and normal cells. However, we have recently reported [12] that WT1 expression levels in normal CD34+ BM cells were similar to those in K562 leukemia cells. Furthermore in this study, it was shown that WT1 mRNA and protein expression levels of mWT1-expressing C1498 and 3LL were as high as in normal kidney. The mechanism involved in processing of WT1 protein and/or presentation of WT1 peptide may be different between tumor and normal cells, resulting in poor presentation of the WT1 antigen onto the cell surface in normal cells. Further studies are needed to address this issue.
Phase I clinical trials of WT1 peptide cancer vaccine against leukemia, lung, and breast cancer had been already started. The WT1 peptide vaccination was shown to be effective in some patients. In this trial, Montanide ISA 51 is being used as an adjuvant. Since BCG-CWS has already been used in clinical settings, and since its toxicity and safety have been evaluated to a considerable extent, WT1 peptide vaccination combined with BCG-CWS, which can enhance innate immunity as well as acquired immunity (namely, tumor antigen-specific immune response), may be also applied to clinical trials in the near future.
Footnotes
H. Nakajima and K. Kawasaki contributed equally to this study.
References
- 1.Azuma Cancer Res. 1979;24:121. [Google Scholar]
- 2.Banchereau Annu Rev Immunol. 2000;18:767. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 3.Briegar Leukemia. 1994;8:2138. [PubMed] [Google Scholar]
- 4.Buckler Mol Cell Biol. 1991;11:1707. doi: 10.1128/mcb.11.3.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Call Cell. 1990;60:509. [Google Scholar]
- 6.Davies R, Moore A, Schedl A, Bratt E, Miyahawa K, Ladomery M, Mile C, Menke A, van Heyningen V, Hastie N (1999) Multiple roles for the Wilms’ tumor suppressor, WT1. Cancer Res 59[Suppl 7]:1747, 1751 [PubMed]
- 7.Fausch Cancer Res. 2003;63:3478. [PubMed] [Google Scholar]
- 8.Gessler Nature. 1990;343:774. doi: 10.1038/343774a0. [DOI] [PubMed] [Google Scholar]
- 9.Gao Blood. 2000;95:2198. [PubMed] [Google Scholar]
- 10.Hayashi Proc Jpn Acad. 1998;70:205. [Google Scholar]
- 11.Hirahashi Int Immunopharmacol. 2002;2:423. doi: 10.1016/S1567-5769(01)00166-7. [DOI] [PubMed] [Google Scholar]
- 12.Hosen Br J Haematol. 2002;116:409. doi: 10.1046/j.1365-2141.2002.03261.x. [DOI] [PubMed] [Google Scholar]
- 13.Inoue Blood. 1994;84:3071. [PubMed] [Google Scholar]
- 14.Inoue Blood. 1997;89:1405. [PubMed] [Google Scholar]
- 15.Inoue Blood. 1998;91:2969. [PubMed] [Google Scholar]
- 16.Lamm N Engl J Med. 1991;325:1205. doi: 10.1056/NEJM199110243251703. [DOI] [PubMed] [Google Scholar]
- 17.Lipton Cancer. 1983;51:57. [Google Scholar]
- 18.Lipton J Clin Oncol. 1991;9:1551. [Google Scholar]
- 19.Loeb Cancer Res. 2001;61:921. [PubMed] [Google Scholar]
- 20.Matsumoto Int Immunophamacol. 2001;1:1559. doi: 10.1016/S1567-5769(01)00071-6. [DOI] [Google Scholar]
- 21.Menke Int Rev Cytol. 1998;181:151. doi: 10.1016/s0074-7696(08)60418-0. [DOI] [PubMed] [Google Scholar]
- 22.Menssen Leukemia. 1995;9:1060. [Google Scholar]
- 23.Miwa Leukemia. 1992;6:405. [PubMed] [Google Scholar]
- 24.Miyagi Leukemia. 1993;7:970. [PubMed] [Google Scholar]
- 25.Ochiai Cancer Immunol Immunother. 1983;14:167. doi: 10.1007/BF00205355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohminami Blood. 2000;95:286. [PubMed] [Google Scholar]
- 27.Oji Jpn J Cancer Res. 1999;90:194. doi: 10.1111/j.1349-7006.1999.tb00733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oji Int J Cancer. 2002;100:297. doi: 10.1002/ijc.10476. [DOI] [PubMed] [Google Scholar]
- 29.Oka Immunogenetics. 2000;51:99. [Google Scholar]
- 30.Oka J Immunol. 2000;164:1873. [Google Scholar]
- 31.Park Nature Genet. 1993;4:415. doi: 10.1038/ng0893-415. [DOI] [PubMed] [Google Scholar]
- 32.Sugiyama Int J Hematol. 2002;76:127. doi: 10.1007/BF02982574. [DOI] [PubMed] [Google Scholar]
- 33.Thurnher Int J Cancer. 1997;70:128. doi: 10.1002/(sici)1097-0215(19970106)70:1<128::aid-ijc19>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 34.Tsuboi Leuk Res. 1999;23:499. doi: 10.1016/s0145-2126(99)00037-5. [DOI] [PubMed] [Google Scholar]
- 35.Tsuboi J Clin Immunol. 2000;20:195. doi: 10.1023/a:1006637529995. [DOI] [PubMed] [Google Scholar]
- 36.Tsuji Infect Immun. 2000;68:6883. doi: 10.1128/IAI.68.12.6883-6890.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Veronesi N Engl J Med. 1982;307:913. doi: 10.1056/NEJM198210073071503. [DOI] [PubMed] [Google Scholar]
- 38.Yamagami Blood. 1996;87:2878. [PubMed] [Google Scholar]
- 39.Yamamura Cancer. 1979;43:1314. doi: 10.1002/1097-0142(197904)43:4<1314::aid-cncr2820430420>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 40.Yasumoto Cancer Res. 1979;39:3262. [PubMed] [Google Scholar]