

Abstract
It is still not clear why some tumours will be recognized and destroyed by the immune system, and others will persist, grow, and eventually kill the host. It has been hypothesized that tumour cells might evade immunological destruction by expressing Fas ligand (FasL), a molecule which induces apoptosis in Fas+ target cells. However, the role of FasL in creating an immune privileged status within a tumour remains controversial. To determine whether FasL is associated with skin tumour progression, we developed a tumour model enabling us to compare two squamous cell carcinomas (SCC). One is a regressor SCC which spontaneously regresses after injection into syngeneic mice. The other is a progressor SCC which evades immunological destruction. Detailed flow cytometric analysis was used to study tumour cell expression of FasL, Fas, CD80, CD86 and MHC class II. We also analysed the percentage of apoptotic tumour cells in vivo using annexin V and correlated skin tumour progression with CD4 and CD8 T cell infiltration. Progressor tumours expressed high levels of FasL in vivo, which was virtually absent from regressor tumours. The percentage of progressor tumours expressing MHC II was significantly greater than regressor tumours, while neither tumour expressed CD80 or CD86 costimulatory molecules. Consistent with a regressor phenotype, the percentage of viable tumour cells was significantly lower for regressor compared to progressor tumours which coincided with a significantly larger CD4+ T cell infiltrate into the tumour mass. The results suggest that progression of skin tumours occurs if tumour cells express high levels of MHC II but not costimulatory molecules such as CD80 or CD86. This implies that tumours may induce anergy in CD4+ T cells via MHC II antigen presentation in the absence of costimulation. To ensure escape from the immune system, tumours may then kill these T cells via a FasL-dependent mechanism.
Keywords: Tumour immunity, Tumour progression, CD4 T cells, FasL, Costimulation
Introduction
Expression of activation markers on the surface of tumour cells and their role in tumour regression has been a point of contention for many years. MHC class II expressed by professional antigen-presenting cells (APC) is necessary for the effective activation of antigen-specific T cells. However, activation is only possible when antigen is presented by APC in conjunction with costimulatory molecules such as CD80, CD86 and/or CD40. When these are absent, T-cell anergy or tolerance may develop [1]. Indeed, tumours which are found to express MHC II in the absence of CD80 are unable to induce antitumour immunity or immunize naive hosts against tumour challenge [2]. In contrast, when MHC II and CD80 are present on tumour cells, regression is observed and effective immunization against tumour challenge is possible [3]. In other studies, aggressive tumours were transfected with the genes encoding MHC II and CD80 or CD86 and it was found that these previously non-immunogenic tumours now underwent regression [4, 5, 6]. Supporting the theory that MHC II expression by tumours in the absence of costimulatory molecules fails to activate antitumour immunity is the strong correlation between MHC II expression and degree of malignancy [7]. Indeed, it has been known for many years that malignant and metastatic melanoma cells express higher levels of MHC II than non-neoplastic melanocytes in naevi. More importantly, this expression has been associated with a poor prognosis [8]. However, the precise role of these surface markers in mediating skin tumour regression is currently unknown.
T cells are crucial to a successful antitumour immune response. Activated by APC in the draining lymph nodes, antigen-specific T cells migrate to the periphery where they participate in tumour destruction. Both CD4+ and CD8+ T cells are important in antitumour immunity. One of the proposed mechanisms by which T cells carry out tumour destruction is through the expression of FasL (CD95L) on their surface. When FasL comes into contact with its receptor Fas (CD95), apoptosis is induced in the Fas+ target cell. This process is also important in T-cell activation-induced cell death (AICD). FasL is also expressed by cells in the testes and eye, explaining their immune privileged status. It has been hypothesized that if tumour cells are able to express FasL on their surface, then a tumour immune privileged site might be created, one which is free from T cell-mediated destruction [9]. Indeed, there have been numerous reports correlating FasL expression with tumour progression and immune evasion. In fact, when renal tumour cells are transfected with genes encoding FasL, they progress [10]. Despite these reports, controversy still surrounds the role of this molecule in tumour progression, with many conflicting studies showing that malignant tumours do not express FasL, and that this is not a mechanism of tumour immune evasion [11]. Indeed, FasL on the surface of tumour cells may actually enhance antitumour immunity [12, 13].
Previous tumour studies have used only progressor tumours in an in vitro assay. Rather, we have developed an animal tumour model that enables us to compare two types of squamous cell carcinomas (SCC) in vivo. The first is a regressing SCC which, when injected subcutaneously into syngeneic mice will initially grow progressively, before undergoing spontaneous regression. We have previously shown that this regression is immunologically mediated [14]. The second is a progressing SCC which, when injected subcutaneously into syngeneic mice, continues to grow progressively with time, effectively evading immunological detection and destruction. Therefore, using this model, we present new evidence of the importance of MHC II, CD80, CD86, Fas and FasL in tumour progression and correlate this with tumour cell apoptosis and infiltration of the tumours with CD4+ T cells.
Methods
Tumour growth in mice
Female C3H/HeN (H-2k) mice aged 8–14 weeks were used throughout these experiments (Bosch Animal House, University of Sydney, Australia) in accordance with animal ethics guidelines.
The UV13-1 progressor skin tumour is a malignant epidermally derived tumour cell line that does not show obvious carcinomatous differentiation. It was derived from a skin tumour which arose in a C3H/HeN mouse irradiated with ultraviolet (UV) (provided by Prof. Margaret Kripke; MD Anderson Cancer Center, Houston, Tx.). LK-2 is an epidermally derived regressor SCC line derived from a skin tumour that arose in a UV-irradiated C3H/HeN mouse in our department [14]. The cell lines were maintained by tissue culture in Dulbecco's modified Eagle's medium (DMEM, Trace Biosciences, Melbourne, Australia) supplemented with 10% fetal calf serum (FCS) (GibcoBRL Life Technologies, Auckland, New Zealand) and 8 mM l-glutamine (Trace Biosciences). After four to six passages, 50 μl of each tumour cell line (4×107 cells/ml phosphate-buffered saline, PBS) was injected subcutaneously into the left and right flanks of syngeneic C3H/HeN mice. Only one type of tumour was injected into the same mouse. Tumour diameter was measured using engineer's callipers (Mitutoyo Corporation, Japan) 5–7 days after tumour inoculation and then every 2 days until the tumours were removed. Progressor tumours were allowed to grow for 3–4 weeks to ensure progression, while regressor tumours typically grew for 10–12 days before commencing spontaneous regression, at which point they were surgically removed. Progressor tumours were injected into mice 1–2 weeks prior to regressor tumours so that they could be surgically removed and analysed in parallel.
Flow cytometry
In vivo grown and excised tumours were cut into small pieces with a scalpel and incubated for 2–3 h at 37°C in DMEM containing 0.5 mg/ml collagenase II (Worthington Biochemical Corporation, Lakewood, N.J.), 0.5 mg/ml hyaluronidase (Sigma Chemical Company, St. Louis, Mo.) and 300 U/ml DNase (Sigma). Tumour debris was then further disaggregated by mashing with the end of a 5-ml syringe plunger. The suspension was strained through a 70-μm gauze to remove large debris, centrifuged through 1–2 ml FCS, washed with 10% FCS/DMEM and then placed into FACS tubes for labelling with the following antibodies; CD45 (30-F11), I-Ak (11-5.2), FasL (Kay-10), CD80 (16-10A1), CD86 (GL1), CD11c (HL3) and Fas (Jo2) (Pharmingen, Franklin Lakes, N.J.). Positive labelling for Annexin V (R&D Systems, Minneapolis, Minn.) was used to identify those cells undergoing apoptosis. Isotype control antibodies (Pharmingen) were used in parallel to ensure antibody specificity. Streptavidin allophycocyanate (Pharmingen) was routinely used to label biotinylated primary antibodies. Cells were incubated for 45 min at 4°C in primary or isotype control antibody at the same protein concentration (106 cells in 50 μl antibody, 1–2 μg antibody/106 cells). Cells were then washed by underlaying with 400 μl FCS and spun at 420 g before resuspending in 400 μl fresh DMEM/10% FCS for flow cytometry.
For acquisition of fluorescence intensity, a FACScalibur flow cytometer was used and CellQuest software (Becton Dickinson, Franklin Lakes, N.J.) was employed for analysis of acquired data. Routinely, 50,000 events were acquired. The CellQuest program was used to produce a gate around single viable cells based on forward/side scatter profiles. Tumour cells were identified as CD45− and a second antibody against one of the above markers was used to phenotype them. Isotype controls were subtracted from positively stained cells to determine the correct percentage of positive cells. A mean fluorescence index (MFI) was calculated by dividing the mean fluorescence of positively labelled cells by the mean fluorescence of isotype-matched negative control labelled cells. In this way, a MFI equal to 1 indicates that virtually no cells were positive for the particular label.
Immunohistochemistry to identify T cells in tumours
For the analysis of T-cell subsets infiltrating progressing and regressing skin tumours, 7 μm frozen vertical sections were prepared and immunohistochemically stained for CD4 (GK1.5) and CD8 (YTS-169) as we have previously described [15]. Briefly, groups of UV13-1 and LK-2 tumour-bearing mice were killed and their tumours surgically removed and intact solid tumours were snap-frozen in liquid nitrogen. Sections were fixed with acetone before labelling for CD4 and CD8, which enabled the quantification of the different T-cell subsets that infiltrated progressing and regressing skin tumours. Four random fields of the tumour sections, each 0.25 mm2 in area, were counted using a microscope at ×250 (Olympus BH-2). The area of the field was determined using a graticule.
Statistics
For the analysis of tumour percentages and the MFI, a paired Student's t-test was used. For analysis of T-cell numbers in tumours, an unpaired Student's t-test was used. In both cases, P values less than 0.05 were considered statistically significant.
Results
A greater percentage of regressor tumour cells undergo apoptosis in vivo compared to progressor tumour cells
LK-2 regressor tumours injected subcutaneously into the flanks of C3H/HeN mice initially grew progressively, before undergoing spontaneous regression approximately 12 days after inoculation. In contrast, progressor tumours evade immunological destruction and continued to grow progressively in vivo. Skin tumours do not express the haemopoietic surface marker CD45, and it was on this basis that tumour cells were distinguished from inflammatory cells (R1; Fig. 1A, B). Therefore, monoclonal antibodies against CD45 together with annexin V labelling were used to detect apoptotic tumour cells by two-colour flow cytometry. Annexin V binds to phosphatidyl serine residues that appear on the surface of early apoptotic cells (Fig. 1C). Gating on these apoptotic CD45− tumour cells (M1; Fig. 1C), we found that a significantly greater percentage of regressor tumour cells were undergoing apoptosis in vivo (20% more tumour cells) compared to progressor tumour cells (Fig. 1D). This result confirmed the macroscopic phenotype of the two tumours and showed that skin tumour regression occurs via apoptosis.
Fig. 1A–D.
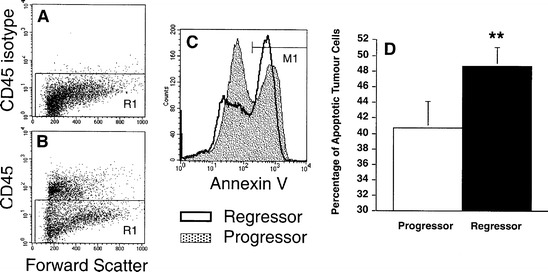
A greater proportion of CD45− regressor than progressor tumour cells undergo apoptosis. Single tumour cell suspensions were labelled for CD45-FITC and annexin V-biotin to determine the percentage of cells undergoing apoptosis in vivo. A Isotype control antibodies for CD45 show that nonspecific binding of antibodies was low. B Tumour cells were identified as CD45− (progressor tumour dotplot of forward scatter versus CD45 shown), with R1 being the region used to identify tumour cells. C Using a streptavidin allophycocyanate (APC) fluorochrome conjugate, annexin V identified those tumour cells undergoing early apoptosis (M1). D A paired Student's t-test revealed that a significantly greater percentage of regressor tumour cells (solid bar) were undergoing apoptosis in vivo than progressor tumour cells (open bar) (**P<0.005, n=4)
A greater percentage of progressor tumour cells express MHC class II and Fas compared to regressor tumour cells, while both tumours express low levels of costimulatory molecules
We were interested in the expression of the activation markers MHC II, CD80, CD86 as well as Fas on tumour cells to help explain why one tumour will be detected and destroyed by the immune system and the other will successfully evade this destruction. Therefore we labelled tumour cells (identified as CD45−; see Fig. 1A) for the surface antigens described above in a two-colour flow cytometry assay (Fig. 2A). We found that the vast majority of progressor tumour cells expressed high levels of both MHC II and Fas on their surface (Fig. 2B). In contrast, fewer than half of the regressor tumour cells growing in vivo expressed these molecules. Moreover, the MFI for MHC II and Fas expression on regressor tumours was 38% and 29% below that of progressor tumours, respectively. The high level of MHC II found on progressor tumours was similar to that found on CD45+CD11c+ professional APC infiltrating these same tumours (data not shown). This suggests that progressor tumours may have the ability to activate cells of the immune system in a similar way to APC. However, there was no difference in the percentage of tumour cells expressing either of the costimulatory molecules CD80 or CD86 (Fig. 2B), thus discounting the possibility that differences in the expression of these molecules determines whether skin tumours regress. Moreover, the low levels of CD80 expressed by both these tumour groups (Fig. 2A) was considerably lower (over 1000 times lower fluorescence intensity) than that expressed by tumour-infiltrating CD45+CD11c+ APC (as indicated by the thin solid line in Fig. 2A). Thus, progressor tumour expression of MHC II in the absence of costimulatory molecules may partially explain their ability to evade immune destruction.
Fig. 2A, B.
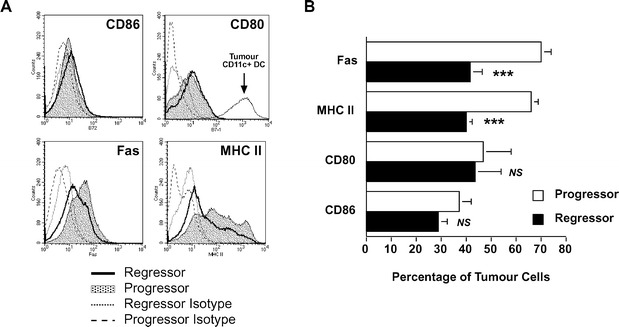
Progressor tumour cells express higher levels of MHC class II and Fas but negligible amounts of CD80 or CD86 costimulatory molecules. A Two-colour flow cytometry was used to identify tumour cells as CD45− along with a second marker against CD86, CD80, Fas or MHC II. Both progressor (shaded histogram) and regressor (thick-lined histogram) tumour expression of the markers are shown. Isotype-matched control antibodies for both progressor (dashed line) and regressor (dotted line) are indicated. The fluorescence intensity of CD80 on CD11c+CD45+ regressor infiltrating dendritic cells (as an example of APC expression of CD80) is shown by the thin solid line in the corresponding histogram. B Statistical analysis by a paired Student's t-test revealed that a significantly greater percentage of progressor tumour cells (open bars) expressed MHC II and Fas than regressor tumours (solid bars) (***P<0.001, n=7). There was no significant difference with respect to CD80 or CD86 expression
Progressor but not regressor skin tumour cells express surface FasL
Single tumour cell suspensions were made from in vivo grown tumours and then labelled for FasL. This enabled assessment of the surface expression of this molecule and to correlate this with tumour progression. We found that while regressor skin tumour cells did not express detectable levels of FasL (Fig. 3A), progressor skin tumours expressed significantly higher levels of this death molecule (Fig. 3B, D). To investigate whether UV13-1 progressor tumours constitutively express FasL, or whether growth in vivo is required for FasL expression, tumour cells grown in vitro were also labelled for FasL. However, in contrast to in vivo labelling, in vitro grown progressor tumour cells did not express detectable levels of FasL (Fig. 3C). In vitro grown regressor tumour cells also failed to express FasL (data not shown). Similar results were found when in vitro cultured progressor and regressor tumours were labelled for MHC class II molecules (data not shown). Attempts were made to induce expression of FasL and MHC II in vitro using a variety of different approaches including: (1) coculturing with phorbol-12-myristate 13-acetate (PMA) to induce activation, (2) using collagenase rather than trypsin to collect adherent tumour cells, (3) starving in vitro growing cells by using 0.5% rather than 10% FCS, (4) staining confluent and nonconfluent cells, and (5) LPS stimulation in vitro. All of these attempts failed to induce expression of either FasL or MHC II (data not shown). We conclude therefore that expression of FasL and MHC II on the surface of progressor tumours requires interactions with the surrounding stroma, immune cells and/or cytokines.
Fig. 3A–D.
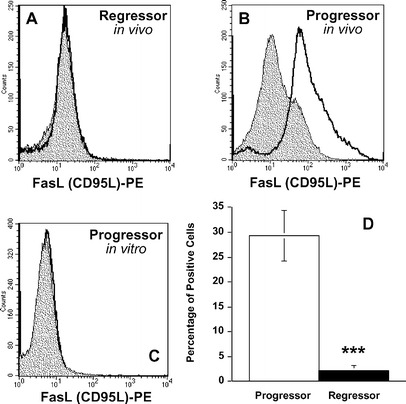
Progressor but not regressor tumour cells express FasL. Tumour cell suspensions were labelled for FasL by flow cytometry. A Regressor tumour cells from in vivo grown tumours did not express any detectable surface FasL (representative histogram of six independent experiments, n=6). B In contrast, progressor tumour cells from in vivo grown tumours expressed high levels of FasL (representative histogram of five independent experiments, n=5). C In vitro cultured tumour cells (progressor tumour shown) did not express FasL. D Because tumour cell suspensions were prepared and analysed on separate occasions, an unpaired Student's t-test was used which confirmed that a significantly greater percentage of progressor tumours (open bars) expressed FasL compared to regressor tumours (solid bars) (***P<0.001)
Regressor skin tumours are infiltrated by significantly greater numbers of CD4+ T cells than progressor tumours
In order to determine the number of CD4 and CD8 T cells infiltrating progressor and regressor skin tumours, frozen sections of tumours were stained and counted. We found that while both tumour groups were infiltrated by similar numbers of CD8+ T cells, regressor tumours were infiltrated by significantly greater numbers of CD4+ T cells (Fig. 4). There were almost 30% more CD4+ T cells found in regressor tumours compared to progressor tumours. These results suggest that CD4+ rather than CD8+ T cells play an important role in determining whether skin tumours regress.
Fig. 4.
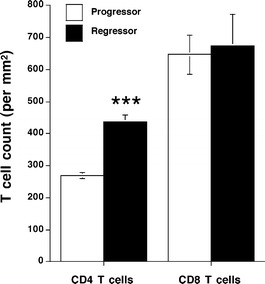
Regressor tumours were infiltrated by greater numbers of CD4+ but not CD8+ T cells than progressor tumours. Frozen progressor (open bars) and regressor (solid bars) tumours were stained for either CD4 or CD8 antigens to identify and determine the number of CD4 and CD8 T-cell subsets. While there was no significant difference between the tumours with regard to CD8 T cell numbers, regressor tumours were infiltrated by significantly greater numbers of CD4 T cells compared to progressor tumours (***P<0.001, n=4)
Discussion
We used a unique animal tumour model to show that one of the possible ways that skin tumours evade the immune system is by surface expression of molecules which influence immune responses. Progressor tumours express levels of MHC class II molecules on their surface comparable to dendritic cells, and significantly higher levels than regressor tumours. In isolation this seems paradoxical due to the role of class II MHC molecules in presenting peptides to CD4+ T cells and yet the progressor tumour evades immunological destruction. Whether expression of MHC class II by tumour cells is found appears to depend on the type of tumour being studied. We have previously shown that regression of human basal cell carcinoma is not dependent on MHC class II expression [16], while Glew et al. have demonstrated that cervical tumours express significantly high levels of this molecule [17]. HLA-DR expression increases as melanocytes progress to metastatic melanoma, indicating that HLA-DR expression may be a useful indicator of tumour grade [7, 8]. In contrast, Esteban et al. have shown that HLA-DR expression by human SCC in the larynx is associated with an excellent prognosis [18]. It is likely that discrepancies between these studies can be explained by a number of factors, including tumour type, stage of malignancy and coexpression of other molecules.
Our results showed that skin tumour regression in a murine model is associated with a significantly larger infiltration of CD4 but not CD8 T cells into the tumour mass. This agrees with our previous findings in humans [14, 19] and reports from others in mice showing that CD4+ T cells are required for regression [20, 21]. In a murine model similar to ours, Mumberg et al. used a UV-induced fibrosarcoma to show that CD4 but not CD8 T cells are responsible for the eradication of MHC class II-negative tumours, a process dependent on IFNγ [22]. Similarly, Baskar et al. have shown that while CD8 T cells are required for effective immunotherapy of mice with established tumours, it is the CD4 subset that is responsible for the rejection of tumours in naive mice [3]. Taken together, our results suggest that MHC II expression in the absence of costimulation by progressor tumours has the potential to limit activation of CD4 T cells to levels which, supported by macroscopic growth characteristics, are insufficient to support regression.
In a tumour model similar to ours, Eberl et al. have found the expression of FasL by progressor but not regressor tumours [23], an observation that correlates with tumour growth characteristics. Whereas we made similar observations in our experiments, the previous study did not explore the possible role of other cell surface markers, nor did it correlate regression with T-cell infiltration. In the current study, we found the expression of FasL by progressor but not by regressor SCC grown in vivo but not in vitro. Regression was also accompanied by a marked increase in the percentage of apoptotic tumour cells but a decreased expression of Fas compared to progressor cells. These results show that (1) interactions between tumour cells and the surrounding stroma and/or immune cells is required for FasL expression by tumour cells, (2) destruction of SCC is an apoptotic rather than a necrotic event, (3) expression of FasL by progressor tumour cells correlates with their progressive growth characteristics, and (4) progression is not due to an absence of cell surface expressed Fas (i.e. regression cannot be due to one tumour cell killing another via a FasL-dependent mechanism).
Despite the evidence linking FasL expression with tumour progression, there are numerous reports in the literature refuting the idea that FasL is involved in tumour immune evasion. Indeed, recent reports present compelling evidence that FasL-expressing tumour cells may enhance antitumour immunity by facilitating antigen uptake by APC [13]. Research by Terheyden et al. has led to the suggestion that while FasL-expressing melanomas may not enable tumour immune escape, it does correlate with the stage of melanoma progression [24]. Later it was found that the discrepancies may be due to the in vitro cytotoxicity assays used to determine the functionality of FasL on tumour cells. In more recent studies, it has been shown that FasL on tumour cells cannot induce apoptosis in Fas-expressing target T cells [25]. However, these apparent contradictions have recently been clarified by Andreola et al. who demonstrated that lymphocyte apoptosis can be induced by tumour cells which secrete FasL-bearing microvesicles [26]. Thus it appears that (1) tumours do express FasL, (2) expression of FasL by some tumours can lead to the creation of an immune privileged status, and (3) expression of FasL is almost certainly linked to progression of some tumours. It is also likely that these apparently contradictory results have surfaced due to the study of different tumour models using different cytotoxicity assays.
CD80 and CD86 costimulation plays a pivotal role in the activation and differentiation of both CD4+ and CD8+ T cells [27]. However, antigens presented in association with MHC class II to CD4+ T cells without costimulation renders them anergic. While this is likely to be an important mechanism in immune regulation to self antigens, it appears that progressing skin tumours possess all the required molecules, such that they have the potential to abuse such a mechanism to their own immunological advantage. To that end, we have shown that when skin tumours express surface MHC II molecules in the absence of CD80 or CD86, they evade the immune system and progress. Because signalling through MHC II can prevent Fas-induced apoptosis [28], it is possible that the observed high expression of MHC II by progressor tumours is a counter-measure acquired by Fas+ tumour cells that enables them to survive a FasL-mediated T-cell attack. Therefore, progressor tumours may prevent T-cell activation by a coordinated mechanism that revolves around presentation of antigen via MHC II molecules without costimulation. This would render T cells anergic, or make them more susceptible to apoptosis via the Fas-FasL system. This hypothesis is supported by two additional key pieces of evidence: (1) the expression of FasL on the surface of progressor but not regressor tumour cells in vivo, and (2) a decreased CD4+ T cell count in progressor tumours, both of which appear to be key events in the progression of skin tumours.
In summary, we showed that progression of murine skin tumours in vivo was associated with expression of FasL and class II MHC, in the absence of costimulatory molecules (CD80 and CD86). It would be interesting to explore whether progression of human skin tumours is associated with a similar phenotype. Furthermore, in this model, regression is an apoptotic event which does not appear to be affected by the level of Fas expression on tumour cells. Finally, skin tumour progression is made possible by the lack of appropriate CD4+ T cell infiltration. This result corroborates our previous immunohistochemical findings on T-cell infiltrates in regressing human skin tumours. This accumulation of evidence leads us to hypothesize that these T cells could be rendered anergic or killed or both, by the CD45−FasL+MHC II+CD80−CD86− progressor tumour cells. We are currently investigating such possibilities. These results have highlighted some of the new and varied ways in which skin tumours may evade the immune system and therefore have important implications for the future design of immunological strategies aimed at promoting effective antitumour defences.
Acknowledgements
The authors would like to thank Dr. Lily Zhuang for assisting with the frozen tumour sections. This work was supported by funds provided by the National Health and Medical Research Council of Australia and The University of Sydney Cancer Research Foundation.
References
- 1.Tan J Exp Med. 1993;177:165. doi: 10.1084/jem.177.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baskar Proc Natl Acad Sci U S A. 1993;90:5687. [Google Scholar]
- 3.Baskar J Exp Med. 1995;181:619. doi: 10.1084/jem.181.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Cancer Res. 1994;54:5420. [PubMed] [Google Scholar]
- 5.Yang J Immunol. 1995;154:2794. [PubMed] [Google Scholar]
- 6.Yang Gene Ther. 1999;6:253. doi: 10.1038/sj.gt.3300820. [DOI] [PubMed] [Google Scholar]
- 7.Holzmann Int J Cancer. 1987;39:466. doi: 10.1002/ijc.2910390410. [DOI] [PubMed] [Google Scholar]
- 8.Brocker Int J Cancer. 1985;36:29. doi: 10.1002/ijc.2910360106. [DOI] [PubMed] [Google Scholar]
- 9.Hahne Science. 1996;274:1363. [Google Scholar]
- 10.Nishimatsu Cancer Immunol Immunother. 1999;48:56. doi: 10.1007/s002620050548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chappell Cancer Res. 1999;59:59. [PMC free article] [PubMed] [Google Scholar]
- 12.Restifo Nat Med. 2000;6:493. doi: 10.1038/74955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tada J Immunol. 2002;169:2241. doi: 10.4049/jimmunol.169.5.2241. [DOI] [PubMed] [Google Scholar]
- 14.Cavanagh Cancer Res. 1996;56:2607. [PubMed] [Google Scholar]
- 15.Patel Br J Dermatol. 1994;131:789. doi: 10.1111/j.1365-2133.1994.tb08580.x. [DOI] [PubMed] [Google Scholar]
- 16.Hunt Br J Dermatol. 1994;130:1. doi: 10.1111/j.1365-2133.1994.tb06873.x. [DOI] [PubMed] [Google Scholar]
- 17.Glew Cancer Res. 1992;52:4009. [PubMed] [Google Scholar]
- 18.Esteban Clin Exp Metastasis. 1990;8:319. doi: 10.1007/BF01810678. [DOI] [PubMed] [Google Scholar]
- 19.Halliday World J Surg. 1995;19:352. doi: 10.1007/BF00299157. [DOI] [PubMed] [Google Scholar]
- 20.Hung J Exp Med. 1998;188:2357. doi: 10.1084/jem.188.12.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aruga J Immunother. 2000;23:225. doi: 10.1097/00002371-200003000-00007. [DOI] [PubMed] [Google Scholar]
- 22.Mumberg Proc Natl Acad Sci U S A. 1999;96:8633. doi: 10.1073/pnas.96.15.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eberl Int J Cancer. 1999;81:772. doi: 10.1002/(sici)1097-0215(19990531)81:5<772::aid-ijc18>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 24.Terheyden J Invest Dermatol. 1999;112:899. doi: 10.1046/j.1523-1747.1999.00607.x. [DOI] [PubMed] [Google Scholar]
- 25.Favre-Felix J Immunol. 2000;164:5023. doi: 10.4049/jimmunol.164.10.5023. [DOI] [PubMed] [Google Scholar]
- 26.Andreola J Exp Med. 2002;195:1303. doi: 10.1084/jem.20011624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McAdam Immunol Rev. 1998;165:231. doi: 10.1111/j.1600-065x.1998.tb01242.x. [DOI] [PubMed] [Google Scholar]
- 28.Catlett J Immunol. 2001;166:6019. doi: 10.4049/jimmunol.166.10.6019. [DOI] [PubMed] [Google Scholar]