

Abstract
Background
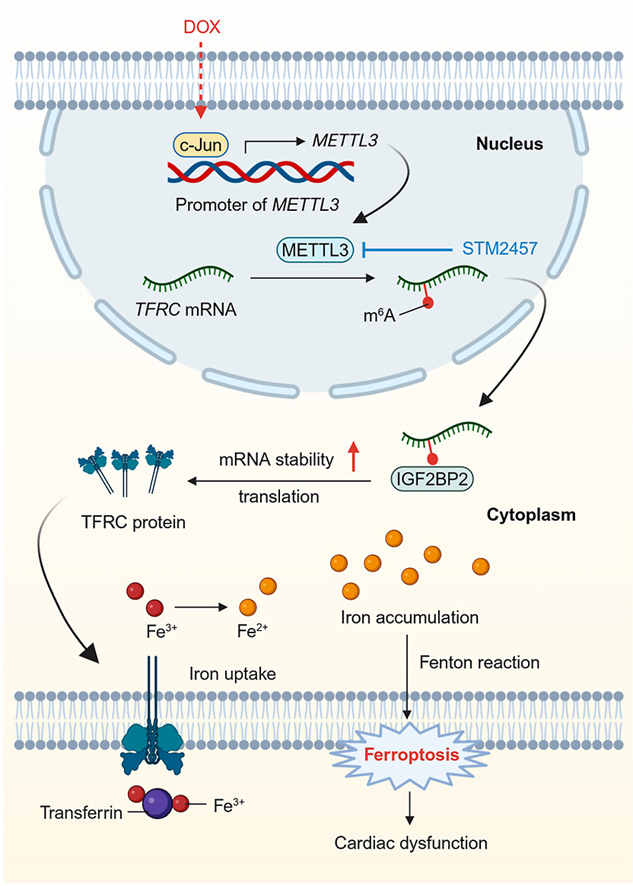
Doxorubicin (DOX) is a chemotherapeutic drug, while its clinical use is greatly limited by the life-threatening cardiotoxicity. N6-methyladenosine (m6A) RNA modification participates in varieties of cellular processes. Nonetheless, it remains elusive whether m6A modification and its methyltransferase METTL3 are involved in the progression of DOX-induced cardiotoxicity (DIC).
Methods
Mice were administrated with DOX (accumulative dosage of 20 mg/kg) repeatedly to establish a chronic DIC model. Cardiomyocyte-specific conditional METTL3 knockout mice were employed to evaluate the effects of altered m6A RNA modification on DIC. The effects of METTL3 on cardiomyocyte ferroptosis were also examined in response to DOX stimulation.
Results
DOX led to increased levels in m6A modification and METTL3 expression in cardiomyocytes in a c-Jun-dependent manner. METTL3-knockout mice exhibited improved cardiac function, remodeling and injury following DOX insult. Besides, inhibition of METTL3 alleviated DOX-induced iron accumulation and ferroptosis in cardiomyocytes, whereas METTL3 overexpression exerted the opposite effects. Mechanistically, METTL3 promoted m6A modification of TFRC mRNA, a critical gene governing iron uptake, and enhanced its stability through recognition of the m6A reader protein, IGF2BP2. Moreover, pharmacological administration of a highly selective METTL3 inhibitor STM2457 effectively ameliorated DIC in mice.
Conclusion
METTL3 plays a cardinal role in the etiology of DIC by regulating cardiac iron metabolism and ferroptosis through TFRC m6A modification. Inhibition of METTL3 might be a potential therapeutic avenue for DIC.
Keywords: METTT3, m6A modification, Doxorubicin-induced cardiotoxicity, Ferroptosis, TFRC
Graphical abstract
1. Introduction
Doxorubicin (DOX) is an anthracycline antineoplastic drug with a wide application in various types of hematopoietic malignancies and solid tumors [1,2]. Nonetheless, application of DOX is largely hampered due to development of severe cardiotoxicity, manifested as dilation of ventricles, progressive worsening of cardiac performance, and ultimately progressing to heart failure [1]. Importantly, the onset of DOX-induced cardiotoxicity (DIC) is closely associated with increased mortality and a poor prognosis in cancer survivors [3]. Therefore, it is imperative to discover the potential mechanisms and preventive strategies for DIC. Currently, several major pathological processes participate in the development of DIC, including cardiomyocyte cell death, disrupted autophagy/mitophagy, mitochondrial injury, and oxidative stress [1,[4], [5], [6], [7], [8], [9]]. Notably, recent mechanistic studies have indicated a critical role of ferroptosis in the onset and progression of DIC [[10], [11], [12], [13], [14], [15]].
Ferroptosis, an iron-dependent regulated cell death, is induced by iron overload, glutathione (GSH) deficiency, and excessive accumulation of phospholipid peroxides [16,17]. Emerging evidence has consolidated the involvement of iron accumulation and ferroptosis in multiple cardiac diseases, including DIC [[10], [11], [12], [13], [14], [15]]. DOX can provoke cardiac iron deposit, accumulation of lipid peroxides, and cardiomyocyte ferroptosis, resulting in cardiac injury and dysfunction [10]. Besides, DOX is shown to downregulate glutathione peroxidase 4 (GPX4), a critical enzyme detoxifying peroxidized lipids, leading to overwhelmed lipid peroxidation and ferroptosis [14]. Preclinical studies have also depicted that several ferroptotic inhibitors exhibit therapeutic efficacy in animal models of DIC [10,14,15,18,19]. These pieces of evidence suggest a crucial involvement of cardiomyocyte ferroptosis in the pathogenesis of DIC. However, the mechanism through which DOX triggers iron overload and subsequent ferroptosis in the heart is still not fully elucidated. Hence, identification of specific molecules and pathways responsible for DOX-induced ferroptosis is pertinent for the development of therapeutic targets to prevent DIC.
N6-methyladenosine (m6A) is the most abundant post-transcriptional modification in eukaryotic cell [20]. Recent findings have consolidated the nature of m6A modification as a reversible and dynamic event. The deposition and removal of such modification is regulated by methyltransferases (m6A writers), including methyltransferase-like 3 (METTL3), METTL14, and WTAP, and demethylases (erasers), encompassing FTO and ALKBH5 [20]. Notably, m6A modification is perceived to regulate a wide spectrum of RNA biology including splicing, nuclear export, translation, and mRNA stability and decoy, thereby influencing multiple biological processes, including stress responses, cell differentiation, and embryonic development [20,21]. Moreover, dysregulation of m6A has been reported in multiple types of cardiovascular diseases [22], including myocardial infarction [23], heart failure [24], cardiac hypertrophy [25,26], and atherosclerosis [27,28]. However, the etiological nature of m6A modification in DIC remains elusive.
Given the significance of the m6A modification in cardiovascular pathology [22], we aimed to explore its potential involvement in DIC. Here, we provided evidence for the first time that METTL3-mediated m6A modification is elevated in cardiomyocytes in the face of DIC challenge. Our results depicted that upregulated METTL3 catalyzes m6A modification of TFRC mRNA, thus contributing to DIC by prompting cardiac iron uptake and ferroptosis. We also revealed that pharmacological inhibition of METTL3 using a selective METTL3 inhibitor mitigated DIC. These findings suggest that targeting METTL3 alleviates DIC through inhibition of TFRC-mediated ferroptosis.
2. Materials and methods
2.1. Experimental animals and in vivo drug treatments
C57BL/6J mice, Mettl3flox/flox (Mettl3fl/fl) mice and Myh6-CreERT mice were purchased from Gempharmatech (Nanjing, China). Exon 2–4 of Mettl3 was employed as the knockout region. Conditional cardiomyocyte-specific Mettl3 deficiency mice (Mettl3CKO) were produced through breeding Mettl3fl/fl mice with Myh6-CreERT mice and intraperitoneally injected with tamoxifen (100 mg/kg) daily for 5 days. All mice were housed in a controlled environment with a 12/12 light-dark cycle at a constant temperature of 22 ± 2 °C with ad libitum availability to water and standard rodent chow. At the conclusion of each experiment, animals were humanely euthanized under general anesthesia using cervical dislocation under inhalation of 2.5% isoflurane anesthesia. All animal studies adhered to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals, and received approval from the Animal Care and Use Committee of Zhongshan Hospital Fudan University (approval No. 202209028Z).
To establish a chronic DIC model, eight-week-old male and female mice were given DOX (5 mg/kg, Cat No. D1515, Sigma-Aldrich) or saline once weekly for 4 weeks via intraperitoneal injection [29]. For experiments with STM2457 (Cat No. SML3360, Sigma-Aldrich), WT mice received STM2457 (50 mg/kg dissolved in 10% DMSO and 90% saline, i. p.) once daily [30]. Cardiac function was evaluated using echocardiography 1 week following the final DOX. Then, heart tissues were collected after echocardiography assessment for biochemical assays.
2.2. Echocardiography
Cardiac function was evaluated in anesthetized mice (2% isoflurane) by transthoracic echocardiography (VisualSonics VeVo 2100 Imaging System, Toronto, Canada). Left ventricular fractional shortening (LVFS) and left ventricular ejection fraction (LVEF) were recorded as previously described [31].
2.3. Histological assessment
Myocardial samples were incubated in paraformaldehyde (4%) prior to embedment in paraffin. Samples were sliced into sequential pieces (5-μm in thickness) and were stained with Hematoxylin and eosin (HE, Cat No. C0105 M, Beyotime Biotechnology) or Masson's Trichrome (Cat No. ab150686, Abcam). For immunohistochemical staining, sample slices were exposed overnight to anti-METTL3 (1:200, Cat No. 86132, Cell Signaling Technology) or anti- 4-hydroxynonenal (4-HNE, 1:100, Cat No. ab46545, Abcam) antibody, prior to nurturing with a secondary antibody. Images were captured through a digital microscope and were processed using Image J (v1.34S).
2.4. Biochemical detection
Mouse serum was collected and cardiac troponin T (cTnT) was measured with a cTnT test kit (Cat No. H149-4, Nanjing Jiancheng bioengineering Institute) per guideline from the manufacturer. GSH and GSSG levels were determined using a glutathione measurement package (Cat No. S0052, Beyotime Biotechnology). Malondialdehyde (MDA) was determined by a lipid peroxidation MDA analysis kit (Cat No. S0131, Beyotime Biotechnology). 4-HNE adduct was evaluated using a Lipid Peroxidation (4-HNE) package (Cat No. ab238538, Abcam). Lactate dehydrogenase (LDH) was assessed using a LDH kit (Cat No. ab102526, Abcam).
2.5. Transmission electron microscopy
The heart was harvested and was diced into little pieces (1 mm3) before being immersed in 2.5% glutaraldehyde (in 0.1 M sodium phosphate, pH 7.4) at 4 °C for a minimum of 24 h. Subsequently, tissues underwent dehydration using graded alcohols and were embedded in Epon Araldite before fixation in 1% OsO4 for 60 min. Ultrathin sections (75–80 nm) were obtained using a Leica ultramicrotome (Wetzlar, Germany) fitted with a Diatome diamond knife, and were then treated with uranyl acetate for 10 min and lead citrate for an additional 5 min. Samples were imaged through a Hitachi H600 transmission electron microscope (Hitachi, Japan) operated at 40–120 kV. Micrographs were derived with the Digital Micrograph software [32].
2.6. Isolation of adult mouse cardiomyocytes (AMCMs)
Hearts were swiftly exercised prior to perfusion with a modified Tyrode's solution. Subsequently, myocardium underwent digestion using a KHB buffer devoid of Ca2+ containing collagenase D for a duration of 20 min. Following this, tissues were sectioned and agitated before cell pellets were resuspended. Extracellular Ca2+ was gradually reintroduced to a concentration of 1.20 mM over half hour. Isolated ACMCs were then resuspended and subjected to primary culture on laminin-coated dishes or glass slides. An overall yield of 70% rod-shaped cells exhibiting clear sarcomere contour was considered a valid isolation. Those rod-shaped cardiomyocytes with distinct edges were chosen for mechanical recording [33].
2.7. Cell shortening/relengthening
Cardiomyocyte contractile capacity was evaluated with the assistance of a SoftEdge MyoCam system (IonOptix Corporation, Milton, MA, USA) coupled with an IX-70 Olympus microscope [32]. In brief, cells were paced at 0.5 Hz before mechanical properties were recorded including maximal velocities of shortening/relengthening (±dL/dt), peak shortening (PS), time-to-peak shortening (TPS), and time-to-90% relengthening (TR90).
2.8. Cell culture, transfection and in vitro treatment
Using 1- to 2-day-old Sprague-Dawley rat hearts, primary neonatal rat ventricular myocytes (NRCMs) were dispersed using collagenase (type II, Cat No. 9001-12-1, Worthington). Cell lysates were spined through a Percoll gradient. Both NRCMs and HL-1 cardiomyocytes were maintained in a Dulbecco's Modified Eagle Medium (DMEM, Gibco), supplemented with fetal bovine serum albumin (FBS, 10%, Gibco) under 5% CO2 at 37 °C. Plasmid or siRNA transfection was accomplished with Lipofectamine 3000 (Cat No. L3000008, Thermo Fisher) prior to biochemical evaluation 2–3 days later.
To discern the impact of STM2457 on DOX-evoked cardiotoxicity, HL-1 cardiomyocytes were challenged by DOX (1 μM, Cat No. D1515, Sigma-Aldrich) or STM2457 (5 μM, Cat No. SML3360, Sigma-Aldrich) in vitro for 24 h. To evaluate the involvement of ferroptosis, the ferroptosis inducers erastin (5 μM, Cat No. HY-15763, MedChemExpress), RSL3 (2.5 μM, Cat No. HY-100218A, MedChemExpress), ferroptosis inhibitor ferrostatin-1 (Fer-1, 5 μM, Cat No. HY-100579, MedChemExpress), the iron chelator deferoxamine (DFO, 5 μM, Cat No. HY-B1625, MedChemExpress), the antioxidant N-acetyl cysteine (NAC, 5 mM, Cat No. HY-B0215, MedChemExpress), the pyroptosis (caspase 1) inhibitor VX-765 (10 μM, Cat No. HY-13205, MedChemExpress), the apoptosis inhibitor Z-VAD-FMK (Z-V, 10 μM, Cat No. HY-16658B, MedChemExpress), or the necroptosis inhibitor Necrostatin-1 (Nec-1, 2 μM, Cat No. HY-15760, MedChemExpress) were supplemented to HL-1 cardiomyocytes for further experimentation.
2.9. Cell viability assay
Cell viability was evaluated in a 96-well plate using Cell Counting Kit-8 (Cat No. C0039, Beyotime). Cells were plated at 1 × 104 in each well. Absorbance was determined with a Multi-Plate Reader (Biotek Synergy) at 450-nm, prior to determination of percentage of cell viability.
2.10. Immunofluorescence staining
Cells were initially fixed with paraformaldehyde in phosphate buffered saline (4%) and were subsequently treated with Triton X-100 (0.2%). Following this, cardiomyocytes were exposed to anti-METTL3 antibody (1:200, Cat No. 86132, Cell Signaling), followed by incubation with secondary antibodies labeled with Alexa Fluor. Cells were visualized through a laser confocal microscope equipped with a 630 × oil immersion objective and were excited with 561-nm laser (Leica, Wetzlar, Germany).
2.11. Assessment of lipid peroxidation
Lipid peroxidation was assessed by Liperfluo (Cat No. L248, Dojindo). Following drug treatment, cells were exposed to 5 μM Liperfluo for 30 min. Then, cells were visualized through a ZEISS confocal laser scanning microscope (model LSM 800) at excitation wavelength of 488 nm and emission wavelength of 535 nm.
2.12. Real-time quantitative PCR (qPCR)
RNA was obtained from myocardial tissues or cells using a commercial kit (Cat No. 15596018, Invitrogen). RNA was utilized as the template to generate complementary DNA (cDNA) using a cDNA Synthesis Kit (Cat No. RR036A, Takara). qPCR was performed with the SYBR Premix Ex Taq II (Cat No. RR820A, Takara) and a CFX96 Real-Time System (Bio-Rad). Relative gene level was calculated with the assistance of 2−ΔΔCt method, with levels being normalized to GAPDH [34]. Primer information is shown in Table S1.
2.13. Immunoblotting
Samples (tissues and cells) were collected and lysed in a RIPA lysis buffer (Cat No. P0013B, Beyotime Biotechnology). Protein samples were isolated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) prior to transfer onto 0.22-μm PVDF membranes. Following 5% bovine serum albumin (BSA) blocking, PVDF membranes were cultivated with primary antibodies, including anti-METTL3 (1:1000, Cat No. 96391, Cell Signaling Technology), anti-METTL14 (1:1000, Cat No. 51104, Cell Signaling Technology), anti-WTAP (1:1000, Cat No. 56501, Cell Signaling Technology), anti-FTO (1:1000, Cat No. 51104, Cell Signaling Technology), anti-ALKBH5 (1:1000, Cat No. 80283, Cell Signaling Technology), anti-c-Jun (1:1000, Cat No. 9165, Cell Signaling Technology), anti-GPX4 (1:1000, ab125066, Abcam); anti-SLC7A11 (1:1000, ab307601, Abcam), anti-TFRC (1:1000, Cat No. ab109259, Abcam), anti-IGF2BP2 (1:1000, Cat No. 14672, Cell Signaling Technology), anti-GAPDH (1:2000, ab9485, Abcam), anti-Tubulin (1:1000, Cat No. 2144, Cell Signaling Technology).
2.14. m6A dot blot
Total RNA was purified using TRIzol (Cat No. 15596018, Invitrogen). RNAs (100 and 250 ng) were placed on a nylon membrane (Cat No. GERPN1210B, Sigma-Aldrich) prior to ultraviolet crosslinking and blocking (5% BSA) for 60 min. An anti-m6A antibody (1:1000, Cat No. 202003, Synaptic Systems) was cultivated overnight (4 °C) with the membranes. Following rinsing with 0.1% phosphate-buffered saline-Tween 20, samples were exposed to secondary antibody for 60 min at room temperature. Following rinsing, membranes were probed using a 3,3′-diaminobenzidine peroxidase substrate kit (Cat No. 36302ES01, Yeasen Biotechnology). Equal quantity of RNAs was added onto the membrane, before methylene blue staining to display total RNA level [35].
2.15. Quantification of m6A modifications
Total RNA was enriched by TRIzol (Cat No. 15596018, Invitrogen) prior to treatment of deoxyribonuclease I (Cat No. 04716728001, Sigma-Aldrich). RNA quality was assessed using NanoDrop. The alteration in global m6A methylation within mRNA was determined utilizing the EpiQuik m6A RNA Methylation Quantification Kit (Cat No. P-9005-48, Epigentek). Briefly, RNAs (200 ng) was applied to assay wells. Subsequently, capture antibody and detection antibody solutions were individually added to the assay wells at appropriate dilutions. m6A methylation levels were evaluated by measuring absorbance at a wavelength of 450 nm, and calculations were performed based on the standard curve.
2.16. Methylated RNA immunoprecipitation (MeRIP)-qPCR
The MeRIP protocol was previously outlined [28]. In sum, poly(A) RNA was initially isolated from 50 μg of total RNA using the Dynabeads mRNA Purification Kit (Cat No. 61006, Invitrogen), with 1/10 of the RNA reserved as the input control. Pierce Protein A/G Magnetic Beads (Cat No. 88803, Thermo Fisher) were prepared and incubated with anti-m6A antibody (5 μg, Cat No. 202003, Synaptic Systems) or rabbit IgG at 4 °C for 120 min. Following three rinses, the antibody-bound beads were combined with purified poly(A) RNA and 1 × IP buffer containing RNase inhibitors. Subsequently, methylated mRNAs were precipitated using glycogen and sodium acetate (3 M) at −80 °C overnight following proteinase K challenge. Further purification was evaluated through qPCR, with the corresponding m6A enrichment determined by normalization to the input.
2.17. RNA stability
To determine TFRC stability, cells were incubated with 5 μg/ml actinomycin D (Cat No. HY-17559, MedChemExpress) to stop transcription [36]. RNAs were gathered at 0, 3, and 6 h following cessation of transcription. Total RNA was enriched, and TFRC level was evaluated using real-time qPCR.
2.18. RNA immunoprecipitation (RIP)
RIP was performed using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Cat No. 17–700, Sigma-Aldrich). In short, HL-1 cells were dispersed in a complete RIP lysis buffer. Magnetic beads conjugated with specific antibodies against IgG or IGF2BP2 (1:30, Cat No. ab188200, Abcam) were cultivated with samples for 4 h at room temperature. Next, RNA-protein complexes were cultivated with proteinase K to separate immunoprecipitated RNA [28]. The TFRC-IGF2BP2 interaction was evaluated using qPCR.
2.19. Chromatin immunoprecipitation (ChIP) assay
ChIP was conducted by the SimpleChIP Enzymatic Chromatin IP Kit (Magnetic Beads; Cat No. 9003, Cell Signaling Technology) [36]. Sequences for primer 1 (with a c-Jun binding site) denoted: 5′-CCAGAGAGGCAGAGAACATA-3’ (forward) and 5′-CATGATGGGAAGCTGGAGTA-3’ (reverse). Meanwhile, an anti-c-Jun (1:100; Cat No. 9165, Cell Signaling Technology) antibody was employed.
2.20. Iron measurement
Perl's staining was employed to evaluate cardiac iron accumulation. Deparaffinized paraffin sections (5 μm) were marked using the Prussian Blue staining kit (Cat No. ab150674, Abcam). In addition, cardiac iron (total, ferrous, and ferric) levels were determined using an Iron Assay Kit (Cat No. MAK025; Sigma). Samples were treated with the fluorescent probe FerroOrange (1 μM for 20 min, Cat No. F374, Dojindo) to enable live-cell imaging for intracellular Fe2+. Samples were visualized using a fluorescence microscope (excitation at 543-nm and emission at 580-nm). Levels of labile iron pool (LIP) were determined as previously described [37]. Cardiomyocytes were cultivated with calcein-AM (acetoxymethyl ester, 1 μM, Cat No. C3099, Thermo) for 15 min. The initial (F0) and chelated fluorescence intensity (FDFO, following100 μM iron chelator deferoxamine mesylate treatment, Cat No. D9533, Sigma-Aldrich) were recorded using a microplate reader (FLUOstar Optima, BMG LABTECH) to generate the relative quantity of labile iron pool (ΔF = FDFO − F0).
2.21. Statistical analysis
The quantitative data were presented as mean ± standard error of the mean (SEM). Statistical analysis was performed using Prism 8.0 software (GraphPad, San Diego, CA). For in vitro studies, since the experimental data set is an average of numerous cultured cells, the data was assumed normally distributed due to the central limit theorem. For in vivo studies, Shapiro-Wilk test was employed to test normality of data distribution. Comparison within two groups were conducted using the Student's t-test (two-tailed), or Welch's correction if equal standard deviations did not prevail through an F test. Comparison across multiple groups were determined using one-way ANOVA followed by a post hoc Tukey's analysis, and the Brown-Forsythe test was used to evaluate variance homogeneity. Mann-Whitney U test (two groups) or Kruskal-Wallis test with Dunn's multiple comparisons test (multiple groups) was applied to data failed normality test. A p value less than 0.05 was deemed being significant.
3. Results
3.1. DOX increases METTL3-dependent m6A modification in cardiomyocytes
To determine the possible involvement of m6A modification in DIC, HL-1 cells and neonatal rat cardiomyocytes (NRCMs) were challenged with DOX for 24 h, m6A levels were measured using m6A dot blot. The m6A levels were found significantly elevated in both HL-1 cells and NRCMs upon DOX insult (Fig. 1A and B). To further evaluate m6A modification in an in vivo setting, m6A levels were assesses in hearts from a mouse DIC model. Consistent with data from cardiomyocytes, m6A levels were significantly increased following DOX treatment (Fig. 1C). Besides, we also applied enzyme-linked immunosorbent assay (ELISA) to evaluate m6A levels. Consistently, DOX insult markedly upregulated m6A modification in HL-1 cells, NRCMs, and mouse hearts (Figs. S1A–C). These results collectively supported that m6A levels were increased in cardiomyocytes in the face of DOX insult.
Fig. 1.

DOX increases m6A modification and METTL3 expression in cardiomyocytes. (A)-(C) Representative m6A dot blot and statistical analysis of m6A level in DOX-treated HL-1 cells, neonatal rat ventricular cardiomyocytes (NRCMs), and mouse heart tissues. (D) and (E) Representative immunoblots and statistical analysis of protein expression of m6A methyltransferases and demethylases. (F) Representative immunohistochemical staining of METTL3 in hearts from a mouse model of DOX-induced cardiotoxicity. (G) Representative immunofluorescence staining of METTL3 in DOX-treated HL-1 cardiomyocytes. Mean ± SEM, n = 6 per group. P values were determined by unpaired two-tailed Student's t-test (A-C and E).*P < 0.05, and **P < 0.01.
Given that m6A modification is primarily orchestrated by methyltransferases and demethyltransferases [20]. Protein levels of these enzymes were examined. DOX treatment markedly upregulated METTL3 protein abundance in murine heart tissues, whereas protein abundance of METTL14, WTAP, FTO and ALKBH5 remained unaltered (Fig. 1D and E). Immunohistochemical analysis also showed increased METTL3 levels in hearts from DOX-treated mice (Fig. 1F). Likewise, METTL3 expression was increased in HL-1 cells with DOX treatment, with predominant nucleic localization (Fig. 1G). Taken together, our data have demonstrated that DOX led to increases in METTL3-dependent m6A RNA modification in cardiomyocytes.
3.2. METTL3 is induced through a c-Jun-dependent manner in response to DOX treatment
To investigate the mechanism of METTL3 upregulation in DIC, METTL3 mRNA level was examined in DOX-exposed cardiomyocytes. METTL3 mRNA expression was found significantly elevated in a time-dependent manner (Fig. S2A), denoting a possible transcriptional regulation of METTL3 in response to DOX stimuli. Next, the potential binding sites of METTL3 promoter for transcription factors were analyzed using online JASPAR database (http://jaspar.genereg.net/). The potential binding sites of c-Jun, with high predictive scores, were shown in the METTL3 promoter region (Fig. S2B and Table S2). Protein expressions of both c-Jun and METTL3 was also found elevated in DOX-treated cardiomyocytes in a time-dependent manner (Fig. 2A–C). We then silenced or overexpressed c-Jun in HL-1 cells, and found that c-Jun knockdown significantly reduced METTL3 levels (Fig. 2D and E, Figs. S2C and D), whereas c-Jun overexpression enhanced METTL3 mRNA and protein abundance (Fig. 2F and G, Figs. S2E and F). Furthermore, ChIP assay suggested that DOX insult promoted c-Jun binding to the METTL3 promoter (Fig. 2H and I). Collectively, these findings demonstrate that upregulated levels of c-Jun upon DOX challenge transcriptionally enhanced METTL3 expression in cardiomyocytes.
Fig. 2.

METTL3 is induced in DOX-treated cardiomyocytes in a c-Jun-dependent manner. (A)-(C) Representative Western blots and statistical analysis of METTL3 and c-Jun in DOX-challenged HL-1 cells (n = 4 per group). (D) Protein levels of c-Jun and METTL3 in DOX-treated HL-1 cells with c-Jun knockdown (n = 3 per group). (E) mRNA levels of METTL3 in DOX-treated HL-1 cells with c-Jun knockdown (n = 4 per group). (F) Protein levels of c-Jun and METTL3 in DOX-treated HL-1 cells with c-Jun overexpression (n = 3 per g mRNA levels of METTL3 in DOX-treated HL-1 cells with c-Jun expression (n = 4 per group)roup). (G). (H) and (I) ChIP-PCR assays for binding of c-Jun to the METTL3 promoter region in the absence or presence of DOX treatment (n = 4 per group). Mean ± SEM. P values were determined by one-way ANOVA with Tukey's post-hoc test (B, C, E, and G) or unpaired two-tailed Student's t-test (I).*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
3.3. Cardiomyocyte-specific deletion of METTL3 prevents DIC
To evaluate METTL3 loss-of-function response for DIC, Mettl3CKO mice were obtained by crossing Mettl3-Loxp (Mettl3fl/fl) mice with Myh6-Cre/ERT2 mice following tamoxifen stimulation (Fig. 3A). Western blots validated overtly reduced METTL3 protein abundance in hearts from Mettl3CKO mice (Figs. S3A and B). Following the experimental protocol indicated in Fig. 3B, DOX administration was applied in Mettl3fl/fl and Mettl3CKO mice for 4 weeks. A marked reduction in LVEF and LVFS was observed in Mettl3fl/fl mice 1 week following final dosage of DOX (Fig. 3C–E). In contrast, DOX-treated Mettl3CKO mice exhibited preserved cardiac function manifested by restored LVEF and LVFS (Fig. 3C–E). In addition, DOX treatment induced increased levels of cardiac fibrosis and injury markers (cTnT) in Mettl3fl/fl mice, which were mitigated in Mettl3CKO mice (Fig. 3F–H). DOX-treated Mettl3fl/fl mice exhibited drastic reduction in cardiomyocyte area and heart weight-to-tibial length (HW/TL) ratio, while these effects were essentially nullified in DOX-treated Mettl3CKO mice (Fig. 3I–K).
Fig. 3.

Cardiomyocyte-specific knockout of METTL3 protects against DOX-induced cardiotoxicity. (A) Illustrative experimental protocol for generation of METTL3CKO mice by crossing METTL3fl/fl mice with Myh6Cre/ERT2 mice followed by tamoxifen treatment. (B) Schematic timeline of tamoxifen treatment and DOX-induced cardiotoxicity model. (C)-(E) Representative M-mode echocardiographic image and analysis of left ventricular ejection fraction (LVEF) and fractional shortening (LVFS) in METTL3fl/fl and METTL3CKO mice in the presence or absence of DOX treatment. (F) and (G) Representative images of Masson trichrome staining and quantitative analysis of cardiac interstitial fibrosis. (H) Plasma cTnT levels in METTL3fl/fl and METTL3CKO mice. (I) and (J) Representative images of HE staining and quantitative analysis of cardiomyocyte areas. (K) Heart weight (HW)-to-tibial length (TL) ratio in METTL3fl/fl and METTL3CKO mice. Mean ± SEM. n = 6 per group. P values were determined by one-way ANOVA with Tukey's post-hoc test (D, E, G, H, J and K). ***P < 0.001.
To intuitively determine the role of METTL3 on cardiomyocyte contractile function, primary adult mouse cardiomyocytes from Mettl3fl/fl and Mettl3CKO mice were isolated to examine contractile parameters. DOX significantly reduced PS, ± dL/dt (Figs. S4A–E). DOX also prolonged TR90 without affecting TPS in cardiomyocytes from Mettl3fl/fl mice (Figs. S4F and G). In contrary, these cardiomyocyte mechanical defects were significantly attenuated or abolished in Mettl3CKO mice following DOX insult (Figs. S4A–G). Altogether, these findings highlight the protection of METTL3 ablation against DOX-induced cardiomyocyte anomalies.
3.4. METTL3 deficiency inhibits DOX-induced cardiac ferroptosis in vivo
A number studies have depicted an essential role for ferroptosis in the pathogenesis of DIC [[10], [11], [12], [13], [14], [15]]. We examined the possible involvement of METTL3 in the regulation of ferroptosis in response to DOX treatment. Intriguingly, DOX caused a significant increase of cardiac Ptgs2 mRNA level (a biochemical hallmark for ferroptosis) in Mettl3fl/fl mice, which was abolished in Mettl3CKO mice (Fig. 4A). Besides, levels of 4-HNE, a key biomarker of lipid peroxidation, were significantly elevated in cardiac tissues of Mettl3fl/fl mice following DOX treatment (Fig. 4B and C). In contrast, DOX-treated Mettl3CKO mice exhibited markedly reduced cardiac 4-HNE abundance (Fig. 4B and C). Similarly, DOX insult led to increased levels of cardiac MDA, another end-product of lipid peroxidation, in Mettl3fl/fl mice, which was mitigated in Mettl3CKO mice (Fig. 4D). DOX also significantly lowered GSH/GSSG ratio in heart tissues from Mettl3fl/fl mice, where cardiomyocyte-specific knockout of METTL3 markedly attenuated GSH decline (Fig. 4E). In addition, increases in protein abundance of GPX4 and SLC7A11, two crucial biomarkers resistant to ferroptosis, were observed in Mettl3CKO mice compared with Mettl3fl/fl mice after DOX treatment (Fig. 4F–H). Finally, we examined mitochondrial morphological features of ferroptosis, and noted ruptured mitochondria in conjunction with reduced cristae density and distorted cristae in DOX-treated Mettl3fl/fl mice (Fig. 4I). However, METTL3 knockout essentially preserved mitochondrial integrity in response to DOX treatment (Fig. 4I). Taken together, these findings support the protective property of METTL3 ablation in DOX-induced cardiac ferroptosis.
Fig. 4.

Cardiomyocyte-specific knockout of METTL3 attenuates DOX-induced cardiac ferroptosis. (A) Relative mRNA levels of Ptgs2 in heart tissues of METTL3fl/fl and METTL3CKO mice (n = 6 per group). (B) Representative immunohistochemical images stained with 4-hydroxynonenal (4-HNE), and quantitative analysis of 4-HNE-positive area (n = 5 per group). (C) ELISA quantification of 4-HNE adducts in heart tissues of METTL3fl/fl and METTL3CKO mice (n = 6 per group). (D) Malondialdehyde (MDA) levels in heart tissues (n = 5 per group). (E) GSH/GSSG levels in heart tissues (n = 5 per group). (F)-(H) Protein levels of GPX4 and SLC7A11 in heart tissues (n = 6 per group); (I) Representative electron microscopy images of mitochondria in heart tissues. Mean ± SEM. P values were determined by one-way ANOVA with Tukey's post-hoc test (A-E, G and H).*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
3.5. Inhibition of METTL3 attenuates DOX-induced cardiomyocyte ferroptosis in vitro
To further determine the regulatory role of METTL3 in cardiomyocyte ferroptosis upon DOX challenge, ferroptosis markers were also examined in METTL3-knockdown or METTL3-overexpressed HL-1 cardiomyocytes in vitro. Knockdown of METTL3 dramatically inhibited cell death and LDH release triggered by the ferroptosis inducer erastin or RSL3 (Figs. S5A and B). In contrast, overexpression of METTL3 reduced cell viability and enhanced cell susceptibility to erastin and RSL3, accompanied by elevated LDH release (Figs. S5C and D). However, METTL3 overexpression-evoked anomalies were significantly reversed by the ferroptosis inhibitors Fer-1, DFO or NAC, but not by apoptosis inhibitor Z-V, or necroptosis inhibitor Nec-1, or pyroptosis inhibitor VX-765 (Figs. S5C and D). We further evaluated the anti-ferroptotic effects of METTL3 deficiency in response to DOX insult. DOX evoked a robust lipid peroxidation, increased MDA levels, and dropped GSH/GSSG ratio in HL-1 cardiomyocytes, which were abolished by METTL3 knockdown (Figs. S5E–G). Not surprisingly, overexpression of METTL3 significantly aggravated DOX-induced ferroptosis, indicated by overwhelmed lipid peroxidation, elevated MDA level, and reduced GSH/GSSG ratio (Figs. S5H–J).
Next, we tested the efficacy of STM2457, a highly selective inhibitor of METTL3 [30], on DOX-induced cardiomyocyte ferroptosis. STM2457 significantly inhibited DOX-evoked cell death and LDH release, which were mimicked by Fer-1, DFO, or NAC (Figs. S6A and B). Furthermore, STM2457 significantly attenuated DOX-induced cardiomyocyte ferroptosis, as manifested by decreased lipid peroxidation and MDA production (Figs. S6C and D), improved GSH/GSSG ratio (Fig. S6E), as well as upregulated GPX4 and SLC7A11 (Figs. S6F–H). Taken together, these results indicate an obligatory role for METTL3 In DOX-evoked ferroptosis and cell injury.
3.6. Inhibition of METTL3 restores iron homeostasis by regulating TFRC-mediated iron uptake
Since iron is critical for execution of ferroptosis by facilitating the generation of lipid peroxides through the Fenton reaction [38], we investigated whether METTL3 altered cardiac iron metabolism in vivo and in vitro. DOX exposure led to pronounced iron deposition in myocardium from Mettl3fl/fl mice as detected using Prussian blue staining, which was mitigated in Mettl3CKO mice (Fig. 5A). Specifically, total, ferrous, and ferric iron were all increased in DOX-treated Mettl3fl/fl hearts, the effects of which were diminished in Mettl3CKO mice (Fig. 5B). Consistently, in vitro analysis showed that METTL3 knockdown reduced cellular ferrous iron in DOX-cardiomyocytes, as indicated by FerroOrange staining and LIP (Fig. 5C and D). These effects were also mimicked by the METTL3 inhibitor STM2457 (Figs. S7A and B). In contrast, METTL3 overexpression evoked a higher level of ferrous iron in DOX-treated HL-1 cells (Fig. 5E and F). Considering the critical role of iron in ferroptosis, inhibition of METTL3 likely attenuates DOX-induced ferroptosis through reducing cardiomyocyte iron accumulation.
Fig. 5.

METTL3 regulates TFRC-mediated iron uptake in vivo and in vitro. (A) Representative images of Prussian blue staining of iron deposition in mouse heart tissues. (B) Total, ferrous, and ferric iron levels in mouse heart tissues (n = 5 per group). (C) and (D) Representative images of cytoplasmic ferrous iron stained using FerroOrange and labile iron pool (LIP) levels in HL-1 cardiomyocytes with or without METTL3 knockdown (n = 3 per group). (E) and (F) Representative images of FerroOrange staining and LIP levels in HL-1 cardiomyocytes with or without METTL3 overexpression (n = 3 per group). (G) Relative mRNA levels of TFRC, DMT1, FPN, FTH1, FTL, FPN, and NCOA4 in HL-1 cells treated with DOX in the absence or presence of METTL3 knockdown (n = 3 per group). (H) and (I) Protein expression of TFRC in HL-1 cardiomyocytes with or without METTL3 knockdown (n = 6 per group). (J) and (K) Representative and pooled protein expression of TFRC in HL-1 cardiomyocytes with or without METTL3 overexpression (n = 6 per group). Mean ± SEM. P values were determined by one-way ANOVA with Tukey's post-hoc test (B, D, F, I, and K) or unpaired two-tailed Student's t-test (G).**P < 0.01, and ***P < 0.001.
To investigate how METTL3 regulated cardiomyocyte iron homeostasis, cardiac expression of key iron metabolism-related genes was evaluated, including genes participating in iron uptake (TFRC and DMT1), iron export (FPN), iron storage (FTH1 and FTL), and ferritinophagy (NCOA4). Our results revealed that METTL3 knockdown overtly downregulated TFRC mRNA level in DOX-treated cardiomyocytes, without affecting other iron-regulatory gene levels (Fig. 5G). Furthermore, DOX induced a marked upregulation in TFRC protein levels, which was abolished and aggravated by METTL3 knockdown and overexpression, respectively (Fig. 5H–K). Consistently, the METTL3 inhibitor STM2457 significantly attenuated DOX-induced upregulation of TFRC mRNA and protein levels (Figs. S7C–E). These findings favor that DOX upregulates METTL3 to disturb cardiomyocyte iron homeostasis by promoting TFRC-mediated iron uptake.
3.7. METTL3-regulated ferroptosis is TFRC-dependent in DOX-treated cardiomyocytes
To further elucidate whether TFRC is required in METTL3-regulated iron metabolism and ferroptosis, we overexpressed TFRC in the METTL3 knocked down HL-1 cardiomyocytes and subjected cells to DOX treatment. Overexpression of TFRC significantly diminished shMettl3-induced reduction of cellular ferrous iron level (Fig. 6A and B). In addition, knockdown of METTL3 alleviated DOX-induced ferroptosis, indicated by increased cell viability (Fig. 6C), reduced LDH release (Fig. 6D), upregulated GPX4 expression (Fig. 6E and F), decreased lipid peroxidation and MDA levels (Fig. 6G and H), and increased GSH/GSSG (Fig. 6I). However, the anti-ferroptotic effects shMettl3 were all diminished by TFRC overexpression in DOX-treated cardiomyocytes (Fig. 6C–I). Collectively, our findings suggest that anti-ferroptotic effects induced by METTL3 deficiency is TFRC-dependent.
Fig. 6.

METTL3-regulated ferroptosis is TFRC-dependent in DOX-treated cardiomyocytes. HL-1 were treated with DOX (1 μM) with or without METTL3 knockdown or TFRC overexpression. (A) Representative FerroOrange staining images in HL-1 cells with indicated treatments. (B) Labile iron pool (LIP) levels in HL-1 cells with indicated treatments (n = 3 per group). (C) Cell viability in HL-1 cells with indicated treatments (n = 3 per group). (D) LDH release in HL-1 cells with indicated treatments (n = 4 per group). (E) and (F) Protein levels of GPX4 in HL-1 cells with indicated treatments (n = 3 per group). (G) Representative Liperfluo staining images in HL-1 cells with indicated treatments. (H) Malondialdehyde (MDA) levels in HL-1 cells with indicated treatments (n = 5 per group). (I) GSH/GSSG ratios in HL-1 cells with indicated treatments (n = 5 per group). Mean ± SEM. P values were determined by one-way ANOVA with Tukey's post-hoc test (B-D, F, H, and I). *P < 0.05, **P < 0.01, ***P < 0.001and ****P < 0.0001.
3.8. METTL3 catalyzes TFRC mRNA m6A modification and enhances its stability through IGF2BP2
Next, we questioned how METTL3 maneuvered TFRC expression in DOX-treated cardiomyocytes. Given that METTL3 is a m6A writer to regulate multiple aspects of RNA biology, including stability, translation efficiency, and degradation [20], we wondered whether altered TFRC expression is attributable to m6A modification. First, we found that knockdown of METTL3 significantly reduced m6A modification in HL-1 cardiomyocytes (Fig. 7A). Next, potential m6A binding sites of TFRC mRNA were predicted with the SRAMP database (http://www.cuilab.cn/sramp). We verified 12 potential m6A sites located on the TFRC mRNA sequence (Fig. S8A and Table S3). Among these sites, 3 were predicted as very high confidence (Fig. 7B). The MeRIP-qPCR assay demonstrated that DOX induced a substantial hypermethylation of TFRC mRNA at the site 2488, which was abolished by METTL3 knockdown (Fig. 7B). Consistently, overexpression of METTL3 significantly increased m6A level of TFRC, whereas STM2457 dramatically reduced m6A level of TFRC (Figs. S8B and C). In addition, inhibition of METTL3 through shRNA or STM2457 caused a shorten mRNA half-life of TFRC (Fig. 7C and S8D). These data suggest that METTL3-mediated TFRC m6A modification sustains the mRNA stability in DOX-treated cardiomyocytes.
Fig. 7.

TFRC serves as a target for METTL3-mediated m6A modification via an IGF2BP2-dependent manner. (A) Representative m6A dot blot and statistical analysis of m6A with or without METTL3 knockdown (n = 3 per group). (B) MeRIP-qPCR analysis showing location of given m6A methylation sites in TFRC (n = 4 per group). (C) Decay rate of TFRC mRNA after treatment of actinomycin D (ActD, 5 μg/ml) in METTL3 knockdown HL-1 cells (n = 3 per group). (D) Relative mRNA levels of TFRC in HL-1 cells knocked down with YTH and IGF2BP reader proteins (n = 4 per group). (E) and (F) Relative protein level of TFRC in the absence or presence of IGF2BP2 knockdown (n = 3 per group). (G) Sequences of TFRC RNA fragments with either WT or mutated (MUT) m6A sites. (H) RIP assay showing the binding affinity between TFRC mRNA and IGF2BP2 with or without METTL3 knockdown (n = 4 per group). (I) Decay rate of TFRC mRNA following ActD administration in IGF2BP2 knockdown HL-1 cells (n = 3 per group). Mean ± SEM. P values were determined by one-way ANOVA with Tukey's post-hoc test (F, and H) or unpaired two-tailed Student's t-test (A, C, D, and I). P < 0.05, **P < 0.01, and ***P < 0.001.
It is well perceived that m6A modification exerts various functional consequences on mRNAs driven through binding to m6A reader proteins [20]. To identify the m6A reader proteins responsible for recognition of METTL3-mediated m6A on TFRC mRNA, key reader genes including YTHDF1, YTHDF2, YTHDF3, IGF2BP1, IGF2BP2, and IGF2BP3, were knocked down in cardiomyocytes prior to DOX treatment (Fig. S9A). Knockdown of IGF2BP2, but not any other readers, led to significant downregulation of TFRC mRNA (Fig. 7D). Besides, IGF2BP2 knockdown also reduced TFRC protein abundance (Fig. 7E and F). In addition, IGF2BP2 knockdown significantly inhibited DOX-induced iron accumulation, reduced cell viability, increased LDH release, lipid peroxidation, and reduced GSH/GSSG ratio in cardiomyocytes (Figs. S9B–H). To further validate the binding of IGF2BP2 to TFRC mRNA, RNA fragments of TFRC were constructed with either WT or mutated m6A site (Fig. 7G). RNA immunoprecipitation (RIP) assay showed that both mutated m6A site and METTL3 knockdown abrogated the perceived interactions between IGF2BP2 and TFRC mRNA (Fig. 7H). Furthermore, the RNA stability assay showed that IGF2BP2 knockdown also compromised the stability of TFRC mRNA (Fig. 7I). Altogether, METTL3-mediated m6A modification promotes stabilization of TFRC mRNA under DOX insult, through recognition of m6A by IGF2BP2 reader protein.
3.9. Pharmacological inhibition of METTL3 prevents DIC and cardiomyocyte ferroptosis
Having established the essence of METTL3 in DIC, we finally investigated whether DIC is amendable by pharmacological inhibition of METTL3 in mice, which was achieved by a highly selective METTL3 inhibitor, STM2457 [30]. Following the study protocol illustrated in Fig. 8A, DOX was administered in C57BL/6J mice for 4 weeks, concurrent with daily treatment of STM2457 (50 mg/kg, i. p.). Our results indicated that STM2457 significantly alleviated DOX-evoked unfavorable changes in LVEF, LVSF, interstitial fibrosis, plasma cTnT level, heart size and weight in mice (Fig. 8B–H). Meanwhile, STM2457 also lowered 4-HNE and MDA levels, elevated GSH/GSSG ratio, and reduced cardiac iron content in DOX-challenged mice (Fig. 8I-L). These data clearly indicate a protective role of METTL3 inhibition against DOX-induced iron accumulation, ferroptosis, and cardiotoxicity.
Fig. 8.

Pharmacological inhibition of METTL3 protects against DOX-induced ferroptosis and cardiotoxicity. (A) Schematic timeline of DOX and STM2457 treatments. (B) Representative M-mode echocardiographic, Masson trichrome staining and HE staining images. (C) and (D) Statistical analysis of left ventricular ejection fraction (LVEF) and fractional shortening (LVFS). (E) Quantitative analysis of cardiac interstitial fibrosis. (F) Plasma cTnT levels in mice treated with DOX or STM2457. (G) and (H) Quantitative analysis of cardiomyocyte areas and heart weight (HW)-to-tibial length (TL) ratio in mice treated with DOX or STM2457. (I) Representative immunoblotting of 4-HNE, and quantitative analysis of 4-HNE levels in heart tissues of mice with indicated treatments. (J) Malondialdehyde (MDA) levels in heart tissues. (K) GSH/GSSG levels in heart tissues. (K) Total, ferrous, and ferric iron levels in mouse heart tissues with indicated treatments. Mean ± SEM. n = 6 per group. P values were determined by one-way ANOVA with Tukey's post-hoc test (C, D, E, F, and G-L). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
4. Discussion
m6A modification, the most prevalent RNA modification, is known to participate in the pathogenesis of cardiovascular diseases [22]. Here, we connected m6A modification with DIC, a major cause for discontinuation of DOX chemotherapy in cancer patients [1]. Our results suggested that DOX insult elevated METTL3-mediated m6A modification in cardiomyocytes in a c-Jun-dependent manner. Moreover, inhibition of METTL3 was capable of suppressing DOX-induced ferroptosis and cardiotoxicity. Mechanistical studies further revealed that METTL3 promoted TFRC m6A modification to sustain the RNA stability in an IGF2BP2-dependent fashion. In this context, METTL3 enhanced TFRC expression, promoting increased iron uptake and subsequent ferroptosis in cardiomyocytes. Treatment of a novel selective METTL3 inhibitor, STM2457, protected DOX-induced ferroptosis and cardiotoxicity. Collectively, these findings display the promises of the METTL3/TFRC axis as a therapeutic target for DIC (Fig. S10).
Recent studies have suggested that m6A modification participates in important pathophysiological processes in multiple cardiovascular diseases [22]. For example, METTL3-mediated m6A modification was increased in cardiomyocytes underwent ischemia/reperfusion injury [35]. Inhibition of METTL3 was also shown to promote autophagic flux, and reduce injury in hypoxia/reoxygenation-treated cardiomyocytes [35]. Paradoxically, Dorn and colleagues revealed that METTL3 knockout in cardiomyocytes promoted maladaptive cardiac remodeling and dysfunction following pressure overload stimulation [26]. Our findings suggested that cardiomyocyte-specific knockout of METTL3 protected against DIC and cardiomyocyte ferroptosis. The discrepancy with regards to conflicting roles of METTL3 in various pathological settings may be attributable to differences between DOX insult and pressure overload. Besides, Dorn's study employed a transgenic mouse model where METTL3 was knocked out during embryonic development, while we hereby utilized tamoxifen-induced knockout mouse model. It is noteworthy that METTL3 might be involved in the regulation of heart development.
Although METTL3 and associated m6A modification have been demonstrated in the regulation of various pathologies [20], the upstream regulators or transcription factors of METTL3 remain largely unclear. An earlier report demonstrated that P300-induced H3K27 acetylation in METTL3 promoter region activated METTL3 transcription in gastric cancer [39]. In another independent study, the TATA box-binding protein (TBP) was shown to transcriptionally upregulate METTL3 expression in cervical cancer cells [40]. c-Jun is a transcription factor that binds to DNA and participates in the regulation of gene transcription [41]. Earlier studies have noted the involvement of c-Jun in the regulation of cardiac remodeling and injury [42,43]. Here, we found upregulated c-Jun level upon DOX challenge, which is capable of transcriptionally turning on METTL3 possibly through binding to the METTL3 promoter region.
During the past 5 years, multiple investigations have validated that ferroptosis serves as a crucial pathogenesis for heart diseases including DIC [[10], [11], [12], [13], [14], [15]]. A previous study published in 2019 first demonstrated that inhibition of ferroptosis, instead of other forms of cell death, overtly improved DOX-induced mouse mortality [10]. Besides, DOX was shown to decrease cardiac GPX4 expression, which resulted in defects in the resistance to lipid peroxidation and onset of ferroptosis [14]. In this context, GPX4 overexpression significantly attenuated DIC through inhibition of ferroptosis [14]. Moreover, various medications capable of inhibiting ferroptosis exhibited therapeutic promises in various preclinical models of DIC, including dexrazoxane [10] and Fer-1 [10,14]. Therefore, inhibition of ferroptosis might be a therapeutic strategy against DIC. Interestingly, ferroptosis-associated pathways are regulated by post-translational and epigenetic modifications, including m6A modification. For example, a recent study revealed that METTL3 promoted ferroptosis via m6A modification of HIF-1α (hypoxia-inducible factor-1α) signaling pathway in sepsis-induced acute lung injury [44]. Here, our findings suggested that genetic ablation and pharmacological inhibition of METTL3 overtly suppressed cardiomyocyte ferroptosis in response to DOX treatment.
Next, possible molecular mechanisms behind METTL3-regulated ferroptosis were unveiled in our present study. Cellular iron overload is indispensable for the execution of ferroptosis by generating lipid peroxides through the Fenton reaction [16]. Notably, excessive iron deposit has been observed in the mitochondria of hearts from patients with end-stage DIC [45]. Besides, previous studies also suggested that systemic iron accumulation caused by genetic modification or an iron-enriched diet, increased the susceptibility to DIC in mice [46,47]. Moreover, iron chelators including dexrazoxane and DFO, were able to protect against DIC in preclinical models [10,48]. Notably, dexrazoxane is a commonly employed drug for clinical treatment of DIC, exerting its cardioprotective effects through its anti-oxidant, iron-chelating, and anti-DNA damage properties [49,50]. Therefore, we speculated that METTL3 might regulate cardiac iron level and iron metabolism to promote ferroptosis following DOX exposure. Our results showed inhibition and overexpression of METTL3 significantly reduced and increased, respectively, iron levels in the heart. Notably, intracellular iron homeostasis is orchestrated by multiple iron metabolism-related genes governing iron uptake, export, and release [16]. Among these genes, TFRC, which encodes transferrin receptor 1, drives iron uptake through the binding of iron-bound transferrin [51]. Previous studies also showed that hearts or cardiomyocytes from DOX-treated mice exhibited an increased TFRC expression [48,52,53]. Besides, inhibition of TFRC in bovine aortic endothelial cells markedly attenuated DOX-induced iron overload, oxidative stress, and cell death [52]. Moreover, a recent study also demonstrated that TFRC knockdown attenuated DIC and cardiomyocyte ferroptosis, whereas overexpression of TFRC mimicked the ferroptotic cell death and cardiotoxicity induced by DOX [54]. Our present data found that METTL3 was responsible for DOX-induced increase in TFRC level and iron uptake in cardiomyocytes. These findings suggest that DOX induces METTL3 to promote TFRC-mediated iron uptake in cardiomyocytes, ultimately leading to iron overload and ferroptosis.
Given the biological feature of METTL3 as a m6A writer in the regulation of RNA biology [20], METTL3 may catalyze m6A modification of TFRC mRNA to foster its abundance. In this context, we verified METTL3-regulated TFRC m6A modification using MeRIP-qPCR assay. Normally, the fate of m6A-modificated mRNA depends on the binding and recognition of various m6A reader proteins, including YTH and IGF2BP family members [20]. Among these readers, IGF2BP2 serves as a predominant m6A readers to bind to m6A modifications and therefore enhance RNA stability, ultimately upregulating gene expression [55]. For instance, IGF2BP2-mediated recognition in TAB3 m6A modification promotes its mRNA stability, contributing to renal inflammation in acute kidney injury [36]. Here in our study, IGF2BP2, as opposed to any other readers, participates in the maintenance of TFRC mRNA stability via recognition of METTL3-mediated m6A modification.
Our study also supported the translational potential of METTL3-targeted therapy in DIC with STM2457. STM2457 has displayed proven therapeutic benefits in myeloid leukemia by inhibiting cancer cell growth and proliferation [30], indicating its promises in disease management. Besides, STM2457 also exhibited therapeutic effects in other malignancies (e.g., small cell lung cancer, hepatocellular carcinoma, pancreatic cancer) [[56], [57], [58]] and non-neoplastic diseases (e.g., kidney fibrosis, septic lung injury) [59,60]. Using a chronic DIC mouse model, STM2457 was found to significantly attenuate DOX-induced iron accumulation, ferroptosis and cardiac dysfunction. As such, STM2457 should be considered as a promising METTL3 inhibitor for future clinical prevention and treatment of DIC.
One experimental limitation prevails in our current study in that m6A modification and METTL3 levels were unable to be validated in human heart samples due to apparent difficulties to access human samples. Moreover, the effects of METTL3 were not assessed in tumor-bearing mouse model of DIC. Thus, whether inhibition of METTL3 will alter the efficacy of chemotherapeutic effects of DOX remains uncertain.
In conclusion, our findings have demonstrated that METTL3 abundance is highly upregulated through transcriptional activation of c-Jun in the heart upon DOX insult. METTL3 fosters cardiomyocyte ferroptosis and DIC by promoting TFRC mRNA m6A modification to sustain TFRC stability through an IGF2BP2-dependent mechanism. Therefore, targeting the METTL3/TFRC axis may become a potential therapeutic option in the treatment of DIC.
Funding
This work was supported in part by the National Natural Science Foundation of China (82130011, 92249301, RFIS82250710173).
CRediT authorship contribution statement
Lin Wu: Writing – review & editing, Writing – original draft, Methodology, Investigation, Conceptualization. Yuxin Du: Writing – review & editing, Methodology, Investigation. Litao Wang: Writing – review & editing, Methodology, Investigation. Yingmei Zhang: Writing – review & editing, Supervision, Methodology, Investigation. Jun Ren: Writing – review & editing, Supervision, Funding acquisition, Conceptualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
None.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2024.103157.
Contributor Information
Yingmei Zhang, Email: zhang.yingmei@zs-hospital.sh.cn.
Jun Ren, Email: ren.jun@zs-hospital.sh.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Wu L., Wang L., Du Y., Zhang Y., Ren J. Mitochondrial quality control mechanisms as therapeutic targets in doxorubicin-induced cardiotoxicity. Trends Pharmacol. Sci. 2023;44:34–49. doi: 10.1016/j.tips.2022.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Lyon A.R., Lopez-Fernandez T., Couch L.S., Asteggiano R., Aznar M.C., Bergler-Klein J., Boriani G., Cardinale D., Cordoba R., Cosyns B., Cutter D.J., de Azambuja E., de Boer R.A., Dent S.F., Farmakis D., Gevaert S.A., Gorog D.A., Herrmann J., Lenihan D., Moslehi J., Moura B., Salinger S.S., Stephens R., Suter T.M., Szmit S., Tamargo J., Thavendiranathan P., Tocchetti C.G., van der Meer P., van der Pal H.J.H., Group ESCSD 2022 ESC guidelines on cardio-oncology developed in collaboration with the European hematology association (EHA), the European society for therapeutic radiology and oncology (ESTRO) and the international cardio-oncology society (IC-OS) Eur Heart J Cardiovasc Imaging. 2022;23:e333–e465. doi: 10.1093/ehjci/jeac106. [DOI] [PubMed] [Google Scholar]
- 3.Swain S.M., Whaley F.S., Ewer M.S. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97:2869–2879. doi: 10.1002/cncr.11407. [DOI] [PubMed] [Google Scholar]
- 4.Wu L., Sowers J.R., Zhang Y., Ren J. Targeting DNA damage response in cardiovascular diseases: from pathophysiology to therapeutic implications. Cardiovasc. Res. 2023;119:691–709. doi: 10.1093/cvr/cvac080. [DOI] [PubMed] [Google Scholar]
- 5.Yu W., Xu H., Sun Z., Du Y., Sun S., Abudureyimu M., Zhang M., Tao J., Ge J., Ren J., Zhang Y. TBC1D15 deficiency protects against doxorubicin cardiotoxicity via inhibiting DNA-PKcs cytosolic retention and DNA damage. Acta Pharm. Sin. B. 2023;13:4823–4839. doi: 10.1016/j.apsb.2023.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma Y., Ma J., Lu L., Xiong X., Shao Y., Ren J., Yang J., Liu J. Melatonin restores autophagic flux by activating the Sirt3/TFEB signaling pathway to attenuate doxorubicin-induced cardiomyopathy. Antioxidants. 2023;12 doi: 10.3390/antiox12091716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang M., Abudureyimu M., Wang X., Zhou Y., Zhang Y., Ren J. PHB2 ameliorates Doxorubicin-induced cardiomyopathy through interaction with NDUFV2 and restoration of mitochondrial complex I function. Redox Biol. 2023;65 doi: 10.1016/j.redox.2023.102812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu W., Qin X., Zhang Y., Qiu P., Wang L., Zha W., Ren J. Curcumin suppresses doxorubicin-induced cardiomyocyte pyroptosis via a PI3K/Akt/mTOR-dependent manner. Cardiovasc. Diagn. Ther. 2020;10:752–769. doi: 10.21037/cdt-19-707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang X., Wang S., Wang L., Ceylan A.F., Ren J., Zhang Y. Mitophagy inhibitor liensinine suppresses doxorubicin-induced cardiotoxicity through inhibition of Drp1-mediated maladaptive mitochondrial fission. Pharmacol. Res. 2020;157 doi: 10.1016/j.phrs.2020.104846. [DOI] [PubMed] [Google Scholar]
- 10.Fang X., Wang H., Han D., Xie E., Yang X., Wei J., Gu S., Gao F., Zhu N., Yin X., Cheng Q., Zhang P., Dai W., Chen J., Yang F., Yang H.T., Linkermann A., Gu W., Min J., Wang F. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116:2672–2680. doi: 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He H., Wang L., Qiao Y., Yang B., Yin D., He M. Epigallocatechin-3-gallate pretreatment alleviates doxorubicin-induced ferroptosis and cardiotoxicity by upregulating AMPKalpha2 and activating adaptive autophagy. Redox Biol. 2021;48 doi: 10.1016/j.redox.2021.102185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou K., Shen J., Yan J., Zhai C., Zhang J., Pan J.A., Zhang Y., Jiang Y., Wang Y., Lin R.Z., Cong H., Gao S., Zong W.X. Loss of TRIM21 alleviates cardiotoxicity by suppressing ferroptosis induced by the chemotherapeutic agent doxorubicin. EBioMedicine. 2021;69 doi: 10.1016/j.ebiom.2021.103456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ta N., Qu C., Wu H., Zhang D., Sun T., Li Y., Wang J., Wang X., Tang T., Chen Q., Liu L. Mitochondrial outer membrane protein FUNDC2 promotes ferroptosis and contributes to doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci U S A. 2022;119 doi: 10.1073/pnas.2117396119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tadokoro T., Ikeda M., Ide T., Deguchi H., Ikeda S., Okabe K., Ishikita A., Matsushima S., Koumura T., Yamada K.I., Imai H., Tsutsui H. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. 2020;5 doi: 10.1172/jci.insight.132747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y., Yan S., Liu X., Deng F., Wang P., Yang L., Hu L., Huang K., He J. PRMT4 promotes ferroptosis to aggravate doxorubicin-induced cardiomyopathy via inhibition of the Nrf2/GPX4 pathway. Cell Death Differ. 2022;29:1982–1995. doi: 10.1038/s41418-022-00990-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang X., Stockwell B.R., Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021;22:266–282. doi: 10.1038/s41580-020-00324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang X., Ardehali H., Min J., Wang F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 2023;20:7–23. doi: 10.1038/s41569-022-00735-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X., Liang J., Qu L., Liu S., Qin A., Liu H., Wang T., Li W., Zou W. Exploring the role of ferroptosis in the doxorubicin-induced chronic cardiotoxicity using a murine model. Chem. Biol. Interact. 2022;363 doi: 10.1016/j.cbi.2022.110008. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H., Wang Z., Liu Z., Du K., Lu X. Protective effects of dexazoxane on rat ferroptosis in doxorubicin-induced cardiomyopathy through regulating HMGB1. Front Cardiovasc Med. 2021;8 doi: 10.3389/fcvm.2021.685434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boulias K., Greer E.L. Biological roles of adenine methylation in RNA. Nat. Rev. Genet. 2023;24:143–160. doi: 10.1038/s41576-022-00534-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu L., Zhang Y., Ren J. Targeting non-coding RNAs and N(6)-methyladenosine modification in hepatocellular carcinoma. Biochem. Pharmacol. 2024;223 doi: 10.1016/j.bcp.2024.116153. [DOI] [PubMed] [Google Scholar]
- 22.Kumari R., Ranjan P., Suleiman Z.G., Goswami S.K., Li J., Prasad R., Verma S.K. mRNA modifications in cardiovascular biology and disease: with a focus on m6A modification. Cardiovasc. Res. 2022;118:1680–1692. doi: 10.1093/cvr/cvab160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li S., Shen S., Xu H., Cai S., Yuan X., Wang C., Zhang X., Chen S., Chen J., Shi D.L., Zhang L. IGF2BP3 promotes adult myocardial regeneration by stabilizing MMP3 mRNA through interaction with m6A modification. Cell Death Dis. 2023;9:164. doi: 10.1038/s41420-023-01457-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berulava T., Buchholz E., Elerdashvili V., Pena T., Islam M.R., Lbik D., Mohamed B.A., Renner A., von Lewinski D., Sacherer M., Bohnsack K.E., Bohnsack M.T., Jain G., Capece V., Cleve N., Burkhardt S., Hasenfuss G., Fischer A., Toischer K. Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur. J. Heart Fail. 2020;22:54–66. doi: 10.1002/ejhf.1672. [DOI] [PubMed] [Google Scholar]
- 25.Zhang B., Jiang H., Wu J., Cai Y., Dong Z., Zhao Y., Hu Q., Hu K., Sun A., Ge J. m6A demethylase FTO attenuates cardiac dysfunction by regulating glucose uptake and glycolysis in mice with pressure overload-induced heart failure. Signal Transduct. Targeted Ther. 2021;6:377. doi: 10.1038/s41392-021-00699-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dorn L.E., Lasman L., Chen J., Xu X., Hund T.J., Medvedovic M., Hanna J.H., van Berlo J.H., Accornero F. The N(6)-methyladenosine mRNA methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation. 2019;139:533–545. doi: 10.1161/CIRCULATIONAHA.118.036146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang Q., Chen S., Wang X., Yang X., Chen L., Huang T., Zheng Y., Zheng X., Wu X., Sun Y., Wu J. Exercise mitigates endothelial pyroptosis and atherosclerosis by downregulating NEAT1 through N6-methyladenosine modifications. Arterioscler. Thromb. Vasc. Biol. 2023;43:910–926. doi: 10.1161/ATVBAHA.123.319251. [DOI] [PubMed] [Google Scholar]
- 28.Chien C.S., Li J.Y., Chien Y., Wang M.L., Yarmishyn A.A., Tsai P.H., Juan C.C., Nguyen P., Cheng H.M., Huo T.I., Chiou S.H., Chien S. METTL3-dependent N(6)-methyladenosine RNA modification mediates the atherogenic inflammatory cascades in vascular endothelium. Proc Natl Acad Sci U S A. 2021;118 doi: 10.1073/pnas.2025070118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li D.L., Wang Z.V., Ding G., Tan W., Luo X., Criollo A., Xie M., Jiang N., May H., Kyrychenko V., Schneider J.W., Gillette T.G., Hill J.A. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation. 2016;133:1668–1687. doi: 10.1161/CIRCULATIONAHA.115.017443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yankova E., Blackaby W., Albertella M., Rak J., De Braekeleer E., Tsagkogeorga G., Pilka E.S., Aspris D., Leggate D., Hendrick A.G., Webster N.A., Andrews B., Fosbeary R., Guest P., Irigoyen N., Eleftheriou M., Gozdecka M., Dias J.M.L., Bannister A.J., Vick B., Jeremias I., Vassiliou G.S., Rausch O., Tzelepis K., Kouzarides T. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593:597–601. doi: 10.1038/s41586-021-03536-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi H., Gao Y., Dong Z., Yang J., Gao R., Li X., Zhang S., Ma L., Sun X., Wang Z., Zhang F., Hu K., Sun A., Ge J. GSDMD-mediated cardiomyocyte pyroptosis promotes myocardial I/R injury. Circ. Res. 2021;129:383–396. doi: 10.1161/CIRCRESAHA.120.318629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Min J., Wu L., Liu Y., Song G., Deng Q., Jin W., Yu W., Abudureyimu M., Pei Z., Ren J. Empagliflozin attenuates trastuzumab-induced cardiotoxicity through suppression of DNA damage and ferroptosis. Life Sci. 2023;312 doi: 10.1016/j.lfs.2022.121207. [DOI] [PubMed] [Google Scholar]
- 33.Pei Z., Liu Y., Liu S., Jin W., Luo Y., Sun M., Duan Y., Ajoolabady A., Sowers J.R., Fang Y., Cao F., Xu H., Bi Y., Wang S., Ren J. FUNDC1 insufficiency sensitizes high fat diet intake-induced cardiac remodeling and contractile anomaly through ACSL4-mediated ferroptosis. Metabolism. 2021;122 doi: 10.1016/j.metabol.2021.154840. [DOI] [PubMed] [Google Scholar]
- 34.Bi Y., Liu S., Qin X., Abudureyimu M., Wang L., Zou R., Ajoolabady A., Zhang W., Peng H., Ren J., Zhang Y. FUNDC1 interacts with GPx4 to govern hepatic ferroptosis and fibrotic injury through a mitophagy-dependent manner. J. Adv. Res. 2024;55:45–60. doi: 10.1016/j.jare.2023.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song H., Feng X., Zhang H., Luo Y., Huang J., Lin M., Jin J., Ding X., Wu S., Huang H., Yu T., Zhang M., Hong H., Yao S., Zhao Y., Zhang Z. METTL3 and ALKBH5 oppositely regulate m(6)A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy. 2019;15:1419–1437. doi: 10.1080/15548627.2019.1586246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J.N., Wang F., Ke J., Li Z., Xu C.H., Yang Q., Chen X., He X.Y., He Y., Suo X.G., Li C., Yu J.T., Jiang L., Ni W.J., Jin J., Liu M.M., Shao W., Yang C., Gong Q., Chen H.Y., Li J., Wu Y.G., Meng X.M. Inhibition of METTL3 attenuates renal injury and inflammation by alleviating TAB3 m6A modifications via IGF2BP2-dependent mechanisms. Sci. Transl. Med. 2022;14 doi: 10.1126/scitranslmed.abk2709. [DOI] [PubMed] [Google Scholar]
- 37.Chen P.H., Wu J., Ding C.C., Lin C.C., Pan S., Bossa N., Xu Y., Yang W.H., Mathey-Prevot B., Chi J.T. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ. 2020;27:1008–1022. doi: 10.1038/s41418-019-0393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ajoolabady A., Aslkhodapasandhokmabad H., Libby P., Tuomilehto J., Lip G.Y.H., Penninger J.M., Richardson D.R., Tang D., Zhou H., Wang S., Klionsky D.J., Kroemer G., Ren J. Ferritinophagy and ferroptosis in the management of metabolic diseases. Trends Endocrinol Metab. 2021;32:444–462. doi: 10.1016/j.tem.2021.04.010. [DOI] [PubMed] [Google Scholar]
- 39.Wang Q., Chen C., Ding Q., Zhao Y., Wang Z., Chen J., Jiang Z., Zhang Y., Xu G., Zhang J., Zhou J., Sun B., Zou X., Wang S. METTL3-mediated m(6)A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2020;69:1193–1205. doi: 10.1136/gutjnl-2019-319639. [DOI] [PubMed] [Google Scholar]
- 40.Li Z., Peng Y., Li J., Chen Z., Chen F., Tu J., Lin S., Wang H. N(6)-methyladenosine regulates glycolysis of cancer cells through PDK4. Nat. Commun. 2020;11:2578. doi: 10.1038/s41467-020-16306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meng Q., Xia Y. c-Jun, at the crossroad of the signaling network. Protein Cell. 2011;2:889–898. doi: 10.1007/s13238-011-1113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Windak R., Muller J., Felley A., Akhmedov A., Wagner E.F., Pedrazzini T., Sumara G., Ricci R. The AP-1 transcription factor c-Jun prevents stress-imposed maladaptive remodeling of the heart. PLoS One. 2013;8 doi: 10.1371/journal.pone.0073294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nadruz W., Jr., Kobarg C.B., Kobarg J., Franchini K.G. c-Jun is regulated by combination of enhanced expression and phosphorylation in acute-overloaded rat heart. Am. J. Physiol. Heart Circ. Physiol. 2004;286:H760–H767. doi: 10.1152/ajpheart.00430.2003. [DOI] [PubMed] [Google Scholar]
- 44.Zhang H., Wu D., Wang Y., Guo K., Spencer C.B., Ortoga L., Qu M., Shi Y., Shao Y., Wang Z., Cata J.P., Miao C. METTL3-mediated N6-methyladenosine exacerbates ferroptosis via m6A-IGF2BP2-dependent mitochondrial metabolic reprogramming in sepsis-induced acute lung injury. Clin. Transl. Med. 2023;13 doi: 10.1002/ctm2.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ichikawa Y., Ghanefar M., Bayeva M., Wu R., Khechaduri A., Naga Prasad S.V., Mutharasan R.K., Naik T.J., Ardehali H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Invest. 2014;124:617–630. doi: 10.1172/JCI72931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miranda C.J., Makui H., Soares R.J., Bilodeau M., Mui J., Vali H., Bertrand R., Andrews N.C., Santos M.M. Hfe deficiency increases susceptibility to cardiotoxicity and exacerbates changes in iron metabolism induced by doxorubicin. Blood. 2003;102:2574–2580. doi: 10.1182/blood-2003-03-0869. [DOI] [PubMed] [Google Scholar]
- 47.Panjrath G.S., Patel V., Valdiviezo C.I., Narula N., Narula J., Jain D. Potentiation of Doxorubicin cardiotoxicity by iron loading in a rodent model. J. Am. Coll. Cardiol. 2007;49:2457–2464. doi: 10.1016/j.jacc.2007.02.060. [DOI] [PubMed] [Google Scholar]
- 48.Pan J., Xiong W., Zhang A., Zhang H., Lin H., Gao L., Ke J., Huang S., Zhang J., Gu J., Chang A.C.Y., Wang C. The imbalance of p53-park7 signaling Axis induces iron homeostasis dysfunction in doxorubicin-challenged cardiomyocytes. Adv. Sci. 2023 doi: 10.1002/advs.202206007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deng S., Yan T., Jendrny C., Nemecek A., Vincetic M., Godtel-Armbrust U., Wojnowski L. Dexrazoxane may prevent doxorubicin-induced DNA damage via depleting both topoisomerase II isoforms. BMC Cancer. 2014;14:842. doi: 10.1186/1471-2407-14-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wallace K.B., Sardao V.A., Oliveira P.J. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ. Res. 2020;126:926–941. doi: 10.1161/CIRCRESAHA.119.314681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koleini N., Shapiro J.S., Geier J., Ardehali H. Ironing out mechanisms of iron homeostasis and disorders of iron deficiency. J. Clin. Invest. 2021;131 doi: 10.1172/JCI148671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kotamraju S., Chitambar C.R., Kalivendi S.V., Joseph J., Kalyanaraman B. Transferrin receptor-dependent iron uptake is responsible for doxorubicin-mediated apoptosis in endothelial cells: role of oxidant-induced iron signaling in apoptosis. J. Biol. Chem. 2002;277:17179–17187. doi: 10.1074/jbc.M111604200. [DOI] [PubMed] [Google Scholar]
- 53.Zhuang S., Ma Y., Zeng Y., Lu C., Yang F., Jiang N., Ge J., Ju H., Zhong C., Wang J., Zhang J., Jiang S. METTL14 promotes doxorubicin-induced cardiomyocyte ferroptosis by regulating the KCNQ1OT1-miR-7-5p-TFRC axis. Cell Biol. Toxicol. 2021 doi: 10.1007/s10565-021-09660-7. [DOI] [PubMed] [Google Scholar]
- 54.Yu W., Hu Y., Liu Z., Guo K., Ma D., Peng M., Wang Y., Zhang J., Zhang X., Wang P., Zhang J., Liu P., Lu J. Sorting nexin 3 exacerbates doxorubicin-induced cardiomyopathy via regulation of TFRC-dependent ferroptosis. Acta Pharm. Sin. B. 2023;13:4875–4892. doi: 10.1016/j.apsb.2023.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dai N. The diverse functions of IMP2/IGF2BP2 in metabolism. Trends Endocrinol Metab. 2020;31:670–679. doi: 10.1016/j.tem.2020.05.007. [DOI] [PubMed] [Google Scholar]
- 56.Sun Y., Shen W., Hu S., Lyu Q., Wang Q., Wei T., Zhu W., Zhang J. METTL3 promotes chemoresistance in small cell lung cancer by inducing mitophagy. J. Exp. Clin. Cancer Res. 2023;42:65. doi: 10.1186/s13046-023-02638-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang L., Yang Q., Zhou Q., Fang F., Lei K., Liu Z., Zheng G., Zhu L., Huo J., Li X., Peng S., Kuang M., Lin S., Huang M., Xu L. METTL3-m(6)A-EGFR-axis drives lenvatinib resistance in hepatocellular carcinoma. Cancer Lett. 2023;559 doi: 10.1016/j.canlet.2023.216122. [DOI] [PubMed] [Google Scholar]
- 58.Hao S., Sun H., Sun H., Zhang B., Ji K., Liu P., Nie F., Han W. STM2457 inhibits the invasion and metastasis of pancreatic cancer by down-regulating BRAF-activated noncoding RNA N6-methyladenosine modification. Curr. Issues Mol. Biol. 2023;45:8852–8863. doi: 10.3390/cimb45110555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jung H.R., Lee J., Hong S.P., Shin N., Cho A., Shin D.J., Choi J.W., Kim J.I., Lee J.P., Cho S.Y. Targeting the m(6)A RNA methyltransferase METTL3 attenuates the development of kidney fibrosis. Exp. Mol. Med. 2024;56:355–369. doi: 10.1038/s12276-024-01159-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jia J., Yuan Y., He Y., Wasti B., Duan W., Chen Z., Li D., Sun W., Zeng Q., Ma L., Zhang X., Liu S., Zhang D., Liu L., Liu Q., Liang H., Wang G., Xiang X., Xiao B. Inhibition of METTL3 alleviated LPS-induced alveolar epithelial cell apoptosis and acute lung injury via restoring neprilysin expression. Life Sci. 2023;333 doi: 10.1016/j.lfs.2023.122148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.