

Abstract
Background
Alzheimer's disease (AD) is a neurodegenerative disorder distinguished by a swift cognitive deterioration accompanied by distinctive pathological hallmarks such as extracellular Aβ (β‐amyloid) peptides, neuronal neurofibrillary tangles (NFTs), sustained neuroinflammation, and synaptic degeneration. The elevated frequency of AD cases and its proclivity to manifest at a younger age present a pressing challenge in the quest for novel therapeutic interventions. Numerous investigations have substantiated the involvement of C/EBPβ in the progression of AD pathology, thus indicating its potential as a therapeutic target for AD treatment.
Aims
Several studies have demonstrated an elevation in the expression level of C/EBPβ among individuals afflicted with AD. Consequently, this review predominantly delves into the association between C/EBPβ expression and the pathological progression of Alzheimer's disease, elucidating its underlying molecular mechanism, and pointing out the possibility that C/EBPβ can be a new therapeutic target for AD.
Methods
A systematic literature search was performed across multiple databases, including PubMed, Google Scholar, and so on, utilizing predetermined keywords and MeSH terms, without temporal constraints. The inclusion criteria encompassed diverse study designs, such as experimental, case–control, and cohort studies, restricted to publications in the English language, while conference abstracts and unpublished sources were excluded.
Results
Overexpression of C/EBPβ exacerbates the pathological features of AD, primarily by promoting neuroinflammation and mediating the transcriptional regulation of key molecular pathways, including δ‐secretase, apolipoprotein E4 (APOE4), acidic leucine‐rich nuclear phosphoprotein‐32A (ANP32A), transient receptor potential channel 1 (TRPC1), and Forkhead BoxO (FOXO).
Discussion
The correlation between overexpression of C/EBPβ and the pathological development of AD, along with its molecular mechanisms, is evident. Investigating the pathways through which C/EBPβ regulates the development of AD reveals numerous multiple vicious cycle pathways exacerbating the pathological progression of the disease. Furthermore, the exacerbation of pathological progression due to C/EBPβ overexpression and its molecular mechanism is not limited to AD but also extends to other neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), Parkinson's disease (PD), and multiple sclerosis (MS).
Conclusion
The overexpression of C/EBPβ accelerates the irreversible progression of AD pathophysiology. Additionally, C/EBPβ plays a crucial role in mediating multiple pathways linked to AD pathology, some of which engender vicious cycles, leading to the establishment of feedback mechanisms. To sum up, targeting C/EBPβ could hold promise as a therapeutic strategy not only for AD but also for other degenerative diseases.
Keywords: Alzheimer's disease, C/EBPβ, neurodegenerative disease, therapy, transcription
C/EBPβ overexpression exacerbates the pathologic progression of AD, primarily by promoting neuroinflammation and mediating transcriptional regulation of several disease‐associated proteins, such as δ‐secretase and APOE4. Additionally, C/EBPβ stimulates the upregulation of ANP32A and TRPC1, leading to elevated levels of phosphorylated Tau, while inhibiting the expression of FOXO, which is associated with genes involved in longevity. The in‐depth research of the pathway through which C/EBPβ regulates the development of AD also reveals multiple vicious cycle pathways between C/EBPβ overexpression and the pathological progression of AD.

1. BACKGROUND
AD, a neurodegenerative condition marked by a gradual deterioration of cognitive abilities, stands as a prominent cause of mortality among individuals aged 65 and older. Its incidence escalates in tandem with the aging global population. 1 AD is typified by distinctive pathological hallmarks, encompassing extracellular Aβ (β‐amyloid) peptides, neuronal neurofibrillary tangles (NFTs), persistent neuroinflammation, and synaptic degeneration. 2 Aβ pathology arises from the aberrant sequential cleavage of amyloid precursor protein (APP) by β‐site APP cleaving enzyme 1 (BACE1) and γ‐secretase. This process leads to the accumulation of Aβ peptides, culminating in the formation of Aβ oligomers, fibrils, and plaques. NFTs primarily consist of hyperphosphorylated and truncated tau protein, which plays a crucial role in microtubule stabilization. 3 A majority of patients manifest late‐onset Alzheimer's disease, a non‐hereditary, multifactorial condition lacking a singular genetic etiology. However, prevailing evidence underscores the significant role of APOE4 in disease pathogenesis. 4 While age stands as the foremost risk factor for AD, several diseases and lifestyle factors elevate the susceptibility to this condition. These factors encompass traumatic brain injury, diabetes, hypertension, obesity, and various metabolic syndromes. 5 Individuals with AD exhibit detectable elevation of inflammation in the cerebrospinal fluid (CSF), reflecting immune activation in the early stages of AD pathology. 6 Research scholars have shown a significant interest in investigating the mechanisms underlying the onset and progression of AD. As our comprehension of AD has grown at the protein level, there has been a surge in studies investigating the generation of AD‐related pathologies through gene transcription processes in recent years.
The C/EBPs consist of a family comprising six transcription factor isotypes: C/EBPα, C/EBPβ, C/EBPδ, C/EBPε, C/EBPγ, and C/EBPζ. 7 C/EBPs act as pleiotropic transcription activators of various genes involved in energy metabolism, cell differentiation, and inflammation, often acting in synergy with other transcription factors. 8 , 9 C/EBPβ is evolutionarily conserved among species and expressed in multiple organ systems. 10 , 11 , 12 The gene is expressed as a single transcript that can be translated into three subtypes: the full‐length liver‐enriched activator protein (38‐kDa LAP1), the 34‐kDa liver‐enriched activator protein (LAP2), and the 21‐kDa liver‐enriched inhibitory protein (LIP). 13 The upregulation of C/EBPβ‐activated isotype full/LAP usually precedes the upregulation of inhibitory isoform LIP. 14 C/EBPβ mRNA is expressed in different regions of the adult rodent brain. 15 C/EBPβ promoted neurite regeneration and growth after nerve injury, 15 , 16 and promoted the expression of Aβ scavenger CD36 at the transcriptional level. 17 C/EBPβ has been shown to play a role in synaptic plasticity and memory formation, mainly in the hippocampus, neuronal differentiation, and hippocampal neurogenesis. 18
Recent research has demonstrated that there is an overexpression of C/EBPβ in patients with a variety of neurodegenerative diseases, and it is closely associated with the irreversible progression of AD. Acting as an enhancer‐binding protein, C/EBPβ facilitates gene expression by selectively binding to specific gene sequences. Genes linked to AD, including pro‐inflammatory, LMGN, and APOE genes, contain distinct sequences susceptible to C/EBPβ binding. 19 , 20 , 21 Furthermore, C/EBPβ mediates the transcription and expression of calcium‐related channel proteins, inducing endoplasmic retinal stress. This, in turn, leads to the dysregulation of phosphatase and protease activities, 22 FOXO inhibition, GABA neuron degeneration, and inactivation, 23 and even influences histone acetylation, 24 ultimately expediting AD progression and perpetuating an irreversible deleterious loop. C/EBPβ could emerge as a pivotal site and a promising therapeutic target in the pathogenesis of AD.
2. THE MOLECULAR PATHWAY MECHANISMS OF C/EBPβ REGULATING THE IRREVERSIBLE DEVELOPMENT OF AD
C/EBPβ exhibits elevated expression levels within the hippocampus, cortex, and cerebellum, correlating significantly with neuronal developmental processes. During central nervous system (CNS) development, in addition to playing multiple roles in diverting cortical precursors to neuronal fate and benefiting neurogenesis, the expression of C/EBPβ in the adult hippocampus has been shown to be primarily localized in the nucleus of granular neurons of the dentate gyrus and to play a key role in regulating the proliferation and differentiation of these cells in vitro and in vivo. 25 , 26 , 27 The comprehensive investigation of C/EBPβ's cellular and subcellular localization in the healthy CNS is yet to be undertaken. Notably, C/EBPβ has been directly associated with the progression of AD in humans. Protein levels are heightened in the brains of AD patients in comparison to those of healthy controls. Furthermore, the transcript of Cebpb (which encodes C/EBPβ) exhibits greater abundance in the brains of aged AD mouse models than in those of aged control mice. 19 The levels of C/EBPβ increase in neurons in an age‐dependent fashion. 28 Many pro‐inflammatory genes contain the underlying C/EBPβ consensus sequence, which plays a crucial role in central nervous inflammation. In AD, glial cells manifest heightened C/EBPβ levels due to sustained upregulation of pro‐inflammatory cytokines in the cortex, with a particularly notable increase observed in microglia. 29 , 30 Microglia exhibit heightened expression and nuclear localization of C/EBPβ. Furthermore, immunohistochemical studies have validated robust colocalization between C/EBPβ‐immunoreactive microglia and Aβ deposits and Aβ immunoreactive microvessels, along with pronounced immunoreactivity in pathologically vulnerable regions of the AD brain, contrasting with comparatively faint staining in corresponding areas of the non‐demented (ND) brain or pathologically spared regions of the AD brain (e.g., cerebellum). 31 Numerous studies have substantiated the pivotal role of C/EBPβ in the pathogenesis of AD, hastening its irreversible progression via diverse pathways (Table 1).
TABLE 1.
The molecular pathways of Alzheimer's disease progression are regulated by C/EBPβ.
| Molecular pathways | Mechanisms | Results |
|---|---|---|
| C/EBPβ‐inflammatory factors | Inflammatory factors activate glial cells, leading to an upregulation of C/EBPβ expression and nuclear translocation. C/EBPβ subsequently binds to the promoters of inflammatory factor genes, thereby promoting the expression of additional inflammatory factors |
inflammatory factors↑ Neuroinflammation↑ |
| C/EBPβ‐δ‐secretase | C/EBPβ binds to the promoter of the LMGN gene, thereby enhancing the expression of δ‐secretase, an enzyme responsible for cleaving both APP and Tau |
Aβ peptides↑ NFTs↑ |
| C/EBPβ‐APOE4 | C/EBPβ acts as a specific transcription factor for APOE to promote its gene transcription. ApoE4 exacerbates AD progression via both Aβ‐independent and Aβ‐dependent pathways |
Aβ peptides↑ synaptic function↓ Lipid droplet accumulation in neuron↑ C/EBPβ↑ |
| C/EBPβ‐TRPC1‐SOCE | C/EBPβ binds to the promoter sequence of TRPC1, thereby promoting the transcription of TRPC1, subsequently elevating the steady‐state calcium ion concentration in neurons ([Ca2+] I), augmenting ER stress, disrupting the balance of protein kinases and phosphatases, and worsening tauopathy |
hTau↑ p‐Tau↑ |
| C/EBPβ‐ANP32A | The activation of C/EBPβ prompts the transcription of the ANP32A gene, inhibits histone acetylation, and promotes the phosphorylation of Tau |
p‐Tau↑ autophagosome‐lysosome fusion↓ gene expression of long‐term memory↓ |
| C/EBPβ‐FOXO1 | C/EBPβ suppresses the promoter activity of REST and FOXO1, diminishes their transcription, promotes Aβ aggregation toxicity, and triggers the death of GABAergic neurons |
GABA neuron↓ Aβ↑ AEP↑ |
2.1. The role of C/EBPβ in neuroinflammation and its functional interaction with inflammatory mediators
Neuroinflammation plays a pivotal role in the propagation of several neurodegenerative disorders and stands as a significant contributing factor in the pathogenesis and advancement of AD. In the context of CNS inflammation, we primarily refer to the response of microglia and astrocytes to disruptions in homeostasis, which entail profound alterations in gene expression. These alterations frequently stem from cellular recognition of Aβ or other damage‐ or pathogen‐associated molecular patterns (collectively known as DAMPs or PAMPs). 32 , 33 Neuroglial cells assume a crucial role in various neurobiological processes, including neurogenesis, the preservation of neuronal well‐being, and the formation of neuronal circuits. They accomplish these functions through a spectrum of activities, including anti‐inflammatory responses, phagocytosis, steroid secretion, free radical scavenging, and cellular repair mechanisms. 34 , 35 Neuroinflammation manifests in a state of heightened activation among glial cells, characterized by the production of pro‐inflammatory cytokines. The excessive activation of inflammatory molecules intensifies neuroinflammation within the brain, precipitating the onset of AD and resulting in synaptic dysfunction, neuronal demise, and the impediment of neurogenesis, ultimately leading to irreversible cerebral damage. 36 Neuroinflammation in the context of neurodegenerative diseases frequently manifests as a persistent and unresolved chronic process, contributing significantly to disease progression. 37 Presently, a multitude of studies have underscored the involvement of C/EBPβ in the regulation of neuroinflammation, emphasizing its central role in the promotion of inflammatory gene expression. Notably, numerous pro‐inflammatory gene promoters harbor putative C/EBPβ consensus sequences. 38 Moreover, the expression and activation of C/EBPβ are subject to regulation by diverse extracellular signals, frequently involving intricate modulation by proinflammatory cues like IL‐6, IL‐43, and LPS. 39
2.1.1. C/EBPβ and astrocyte activation
The activation of astrocytes is influenced by various stimuli, including lipopolysaccharide (LPS), interleukin‐1β (IL‐1β), and tumor necrosis factor‐alpha (TNF‐α), which induce the expression of C/EBPβ and C/EBPδ genes in primary astrocytes, thereby promoting astrocyte activation. 40 , 41 Notably, LPS concentration dependently upregulates the expression of IL‐1β and TNF‐α in astrocytes. 42 The pro‐inflammatory cytokine interleukin‐1β (IL‐1β) is one of the most potent and characteristic signals triggering astrocyte activation in most neurodegenerative diseases, mediating neuroinflammation through activation of glial cells and subsequent changes in gene expression. 43 After IL‐1β‐mediated activation of glial cells, astrocyte C/EBPβ expression is induced by IL‐1β and localized to the nucleus, where it acts as a transcription factor to regulate gene expression in the nucleus. 40 , 44 Specifically, IL‐1β alters the mRNA expression of 32% (29/92) of inflammatory genes mediated by IL‐1β, with C/EBPβ regulating the expression of 52% (17/29) of these genes. In IL‐1β‐activated astrocytes, expression levels of nos‐2 and intercellular adhesion molecule‐1 (ICAM‐1) are elevated and decreased, respectively, compared to C/EBPβ‐deficient astrocytes. 45 , 46 Additionally, C/EBPβ inversely regulates the expression of cyclooxygenase‐2 (COX‐2) and bradykinin receptor B2 (BDKRB2). 47 Specifically, increased C/EBPβ expression enhances COX‐2 expression while diminishing BDKRB2 expression. IL‐1β signaling increases BDKRB2 and COX‐2 mRNA in astrocytes via extracellular regulated kinase (ERK) 1/2 and p38 kinase (p38K), respectively. 48 P38K inhibition blocked IL‐1β‐mediated C/EBPβ expression in astrocytes, whereas ERK1/2 inhibition enhanced expression. 49 This suggests that C/EBPβ may inhibit some ERK1/2‐regulated genes while activating p38K‐regulated genes, thereby influencing gene regulation. 48 Overexpression of C/EBPβ isoforms LAP1 and LAP2 also have profound effects on the induction of C3‐related gene promoter activity. 50 , 51 In response to nerve injury, astrocytes secrete tissue inhibitors of metalloproteinase‐1 (TIMP‐1). Altered TIMP‐1 levels have implications in various CNS diseases, including AD. Interestingly, overexpression of C/EBPβ in primary human astrocytes substantially enhances the activity of the −1718/+988 region of the TIMP‐1 promoter, leading to an increased TIMP‐1 expression. 44 Furthermore, C/EBPβ increases transforming growth factor‐β1 (TGF‐β1) promoter activity by binding to C/EBPβ binding sites in the −160 to −100 region of the TGF‐β1 promoter. This induction triggers the TGF‐β1/Smads signaling pathway, subsequently promoting astrocyte proliferation, which perpetuates the organism's state of disease. 52 In summary, C/EBPβ, through its functional interactions with inflammatory factors, exerts regulatory control over a multitude of human astrocyte‐related genes during neuroinflammation (Figure 1).
FIGURE 1.
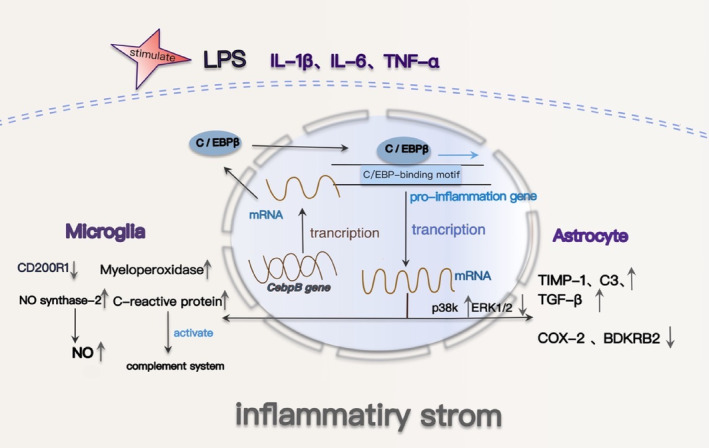
C/EBPβ regulates signaling pathways associated with brain inflammation in AD. C/EBPβ and astrocyte activation: After the activation of astrocytes, the expression of C/EBPβ increases and migrates to the nucleus to regulate gene expression in the nucleus as a transcription factor. C/EBPβ inhibited some ERK1/2 regulatory genes, activated P38K regulatory genes, downregulated ERK1/2 expression, and increased p38K expression, respectively, BDKRB2 expression retention and cox‐2 expression increase in astrocytes. C/EBPβ can also bind to C3, TIMP‐1, and TGF‐β1 promoters to promote their expression. C/EBPβ and microglia activation: After treatment with LPS, IL‐1β, IL‐6, or TNF‐α, C/EBPβ in microglia cells was significantly increased and its transfer to the nucleus was increased. C/EBPβ was bound to pro‐inflammatory gene promoters in a stimulatory and gene‐dependent manner to promote the expression of inflammatory genes in glia activation. iNOS, myeloperoxidase, C‐reactive protein, M‐CSF, and other promoters showed C/EBPβ‐binding sites. Inflammatory factors activate glial cells, induce the increase of C/EBPβ expression, and promote the increase of the expression of more inflammatory factors.
2.1.2. C/EBPβ and microglia activation
Elevated levels of C/EBPβ in activated microglia govern the expression of pivotal pro‐inflammatory genes, exerting a significant role in gene regulation within activated microglia. 53 Furthermore, the percentage of C/EBPβ‐immunopositive cells linked to Aβ peptides markedly exceeded those unrelated to Aβ plaques. Notably, strong co‐localization of C/EBPβ immunoreactivity occurred with Aβ deposits and Aβ‐immunoreactive microvessels, with concurrent co‐localization of C/EBPβ and microglia in the presence of Aβ peptides. 31 Microglial C/EBPβ also responds to the upregulation of C/EBP‐inducing cytokines or lipopolysaccharides, leading to upregulation and nuclear localization. Following treatment with C/EBP‐inducing pro‐inflammatory cytokines IL‐1β, IL‐6, or TNF‐α, C/EBPβ levels in microglia significantly increased, accompanied by enhanced nuclear translocation, akin to the effect of LPS treatment. 31 Treatment with lipopolysaccharide (LPS) or the combination of LPS and interferon γ (IFNγ) induced significant elevations in C/EBPβ mRNA and protein levels, along with enhanced DNA binding in glial cultures. C/EBPβ assumes a pivotal role as a component of the C/EBPs DNA‐binding complex during LPS and LPS+ IFNγ‐induced glial activation. During glial activation, C/EBPβ selectively binds to pro‐inflammatory gene promoters in a stimulatory and gene‐dependent fashion, thereby facilitating the expression of inflammatory genes. 54 Pro‐inflammatory factors promote the upregulation and translocation of C/EBPβ to the nucleus, subsequently fostering the production of inflammatory factors. Extensive evidence has demonstrated the neurocytotoxicity of the inducible nitric oxide synthase (iNOS) mechanism in the AD brain, with NO production assuming a prominent role in the neurotoxicity attributed to activated microglia. 55 Upregulation of C/EBPβ results in heightened iNOS expression via direct transcriptional mechanisms. 56 , 57 The absence of C/EBPβ in microglia led to diminished expression of IL‐1β and NO synthase‐2, resulting in reduced NO production. Additionally, it entirely abolished the neurotoxicity induced by co‐cultured microglia treated with LPS + IFNγ when in contact with neurons. 54 Moreover, C/EBPβ appears to downregulate CD200R1 receptor expression in microglia during inflammation, rendering them less responsive to inhibitory CD200 ligands on neurons. 58 The transcriptional regulation of macrophage colony‐stimulating factor (M‐CSF), a growth factor linked to inflammation and Aβ peptide formation in microglia, is orchestrated by C/EBPβ. 59 , 60 C‐reactive protein possesses the capability to activate the complement system and harbors a binding site for C/EBPβ within its promoter. 61 Myeloperoxidase similarly contains a promoter site for C/EBPβ. 62 The degradation of C/EBPβ in microglia resulted in the inhibition of neuroinflammation. 19 C/EBPβ plays an essential role in upregulating inflammatory gene expression and inducing neurotoxicity in microglia during neurodegenerative diseases (Figure 1). It serves as a pivotal transcription factor for reprogramming microglial activation, making it a potential therapeutic target to mitigate neuronal injury caused by neuroinflammation.
2.2. The C/EBPβ‐δ‐secretase axis is upregulated, resulting in increased levels of Aβ and NFT in AD
Asparagine endopeptidase (AEP), a cysteine protease, has been newly identified as δ‐secretase. It cleaves APP and Tau in an age‐dependent manner, leading to the formation of amyloid plaques and NFTs in AD. 63 δ‐secretase cleaves APP at residues N373 and N585 on the extracellular structural domain, enhancing Aβ production by BACE1. 64 It also cleaves Tau at sites N255 and N368, leading to accelerated Tau hyperphosphorylation and the subsequent accumulation of NFT. 65 Importantly, the tau (1–368) fragment is neurotoxic 66 and binds to, activating the transcription factor STAT1, thereby further up‐regulating BACE1 transcription and Aβ production. 67 Besides APP and tau, δ‐secretase also cleaves polyanionic serine‐arginine protein kinase 2 (SRPK2), a protein crucial in RNA splicing through the phosphorylation of SR splicing factors. 68 During tau mRNA splicing, SRPK undergoes δ‐secretase cleavage, nuclear translocation, and an increase in kinase activity. This results in enhanced exon 10 attachment and an imbalance in 4R‐tau and 3R‐tau expression, ultimately promoting tau aggregation in tauopathy. 69 Additionally, δ‐secretase cleaves SET, which functions as a DNase inhibitor, PP2A inhibitor, and a regulator of tau phosphorylation. 70 Fragments derived from δ‐secretase cleavage of SET lose their DNase inhibitor activity, induce genomic DNA incision, leading to neuronal cell death, 71 and inhibit PP2A activation, which triggers hyperphosphorylation and tau aggregation in AD 72 (Figure 2). Overexpression of δ‐secretase‐derived SET fragments in the brain similarly reproduces the key features of AD in rats. 73 In conclusion, it is widely validated that δ‐secretase plays an important role in the pathogenesis of AD.
FIGURE 2.
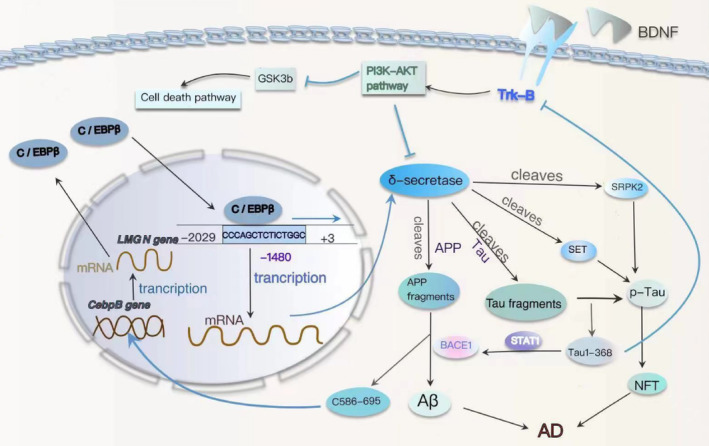
The molecular mechanisms underlying the C/EBPβ‐δ‐secretase signaling pathway expedite the progression of AD. The expression of δ‐secretase is regulated by the transcription factor C/EBPβ, which is regulated by the BDNF/TrkB signaling pathway. δ‐secretase is phosphorylated by Akt, which inhibits its activity, and SRPK2, which enhances its activity. The δ‐secretase cleaves APP and promotes the production of Aβ. The δ‐secretase also cleaves Tau, producing fragments that are prone to aggregation. The tau fragment derived from δ‐secretase‐enhanced BACE1 activity, which further promoted the production of Aβ. δ‐secretase‐derived SET fragments inhibit tau dephosphorylation, while δ secretase‐derived SRPK2 fragments affect tau selective splicing. Aβ and APP C586‐695 (C110) fragments ultimately stimulate the transcription of genes associated with AD by binding to and activating C/EBPβ. All of these pathways promote tau aggregation and the occurrence of AD.
Further investigation into the regulatory mechanism of δ‐secretase has demonstrated that complete depletion of C/EBPβ results in the total elimination of δ‐secretase expression. Within the −2029 to +3 base pair region of the LMGN (δ‐secretase gene) promoter, the highest transcriptional activity is observed. C/EBPβ substantially enhances LMGN promoter activity and associates with the GAATTCAGAGGC sequence located 1480 bases upstream of the LMGN promoter, facilitating LMGN mRNA transcription 74 (Figure 2). C/EBPβ serves as a crucial transcription factor that mediates the expression of δ‐secretase, thereby promoting the accelerated development of Alzheimer's disease. The levels of δ‐secretase protein in both the brain and spinal cord exhibit age‐dependent increases. 65 In the CNS, C/EBPβ modulates the expression of δ‐secretase in a time‐dependent fashion. 75 , 76 Simultaneously, both Aβ and the δ‐secretase‐truncated APP C586‐695 (C110) fragments stimulate the transcription of Alzheimer's disease‐related genes by binding to and activating C/EBPβ, which encompasses δ‐secretase. 77 , 78 C/EBPβ plays a pivotal role in regulating the transcription and activation of LMGN mRNA during aging, thereby mediating the initiation and progression of AD pathology. 79 In parallel, δ‐secretase engages in a feedback loop by cleaving APP and generating a protein‐hydrolyzed APP C586‐695 fragment, which selectively binds to and enhances the transcriptional activity of C/EBPβ. 77 C/EBPβ serves as the biological link in the detrimental cycle between δ‐secretase and Aβ in the context of AD.
Through the C/EBPβ‐δ‐secretase axis, C/EBPβ binds to enhancers in LMGN gene transcription and acts as a transcription factor, leading to an elevation in δ‐secretase expression, thus expediting the progression of AD. Furthermore, the C/EBPβ‐δ‐secretase axis mediates the interaction between numerous exogenous and endogenous risk factors and the development of AD. Intestinal inflammation or microbiota alterations activate the C/EBPβ‐δ‐secretase axis, initiating AD pathology in the gut, which subsequently ascends to the brain via the vagus nerve. 28 , 80 , 81 The concept of the gut‐brain axis has garnered increasing attention, and the C/EBPβ‐δ‐secretase axis further strengthens the connection between intestinal inflammation or microbiota and neurodegenerative diseases. Atherosclerosis (ATH) and AD are both age‐dependent inflammatory diseases associated with infiltrating macrophages, vascular pathology, and shared molecular factors. In recent experiments, it has been shown that C/EBPβ‐AEP signaling links atherosclerosis (ATH) to AD by mediating vascular pathology. 82 Inflammatory responses triggered by a high‐fat diet (HFD) activate neuronal C/EBPβ‐AEP signaling, resulting in the development of AD pathology and cognitive impairments. 83 Traumatic brain injury (TBI) induces the activation of the transcription factor C/EBPβ, leading to an upregulation of δ‐secretase expression. This, in turn, contributes to the pathogenesis of AD by fostering the production of Aβ, Tau hyperphosphorylation, and the induction of neuroinflammation and neurotoxicity. 84 The expression level of C/EBPβ exhibited a negative correlation with BDNF/pTrkB signaling. Decreased BDNF/TrkB signaling results in elevated inflammatory cytokines and the activation of the JAK2/STAT3 pathway, ultimately leading to an upregulation of C/EBPβ and δ‐secretase, consequently enhancing proteolytic APP and Tau fragmentation. 66 , 85 Over recent years, numerous studies have substantiated C/EBPβ and δ‐secretases as promising therapeutic targets for AD.
2.3. C/EBPβ mediates the transcription of APOE4, the key gene in AD
ApoE, a glycoprotein primarily expressed in astrocytes, microglia, neurons, and the choroid plexus, functions as a lipid transporter, particularly for cholesterol and cholesteryl esters. 86 In humans, the APOE gene exists in three polymorphic alleles (ε2, ε3, and ε4). APOE isoforms have been found to influence Aβ and Tau deposition in the brain, with APOE4 being the most potent genetic risk factor for AD. 87 It substantially elevates the risk of both early‐onset and late‐onset AD in a dose‐dependent manner. 88 ApoE4‐related pathogenesis can be categorized into Aβ‐related and Aβ‐independent mechanisms (Figure 3). ApoE plays a pivotal role in lipid transport and cholesterol homeostasis within the brain. Compared with other ApoE isoforms, ApoE4 isoforms show a low lipid state, are prone to form non‐lipid‐containing ApoE4 aggregates, and have poor lipid turnover between neurons and astrocytes, which leads to the accumulation of neuronal lipid droplets. 89 This promotes mitochondrial damage and reactive oxygen species (ROS) production. 90 In addition, ApoE4 also promotes the formation of endosomes and the degradation of synaptic receptors, including AMPA and NMDA, resulting in impaired synaptic function. 89 , 91 Peripheral apoE4 may also have potentially negative effects on the pathological processes in brain development. 92 ApoE can influence the neuroinflammatory process through various mechanisms; however, APOE4 astrocytes and microglia exhibit dysfunction in their response to inflammatory injury. 93 Experimental results demonstrate that mice expressing the human ApoE4 isoform exhibit a higher presence of Aβ deposits and neuritic amyloid plaques compared to mice expressing other human ApoE isoforms. 94 It has been reported that ApoE‐isoforms have an Aβ‐dependent effect on APP transcription and Aβ secretion. ApoE4 induces the strongest increase in APP transcription, 95 and ApoE4 also through mitogen‐activated protein kinase signaling 96 and Aβ seeding and Aβ aggregation into oligomers and fibrils, increasing Aβ production and aggregation, 97 while apoE4 reduces Aβ clearance. 88 Furthermore, ApoE4 exacerbates tau‐mediated pathogenesis, neurotoxic neuroinflammation, and neurodegeneration. 98 For instance, it increases tau neurotoxicity through its binding to the monoamine transporter 2 (VMAT2), inhibits the active transport of neurotransmitters into synaptic vesicles, and ultimately results in degeneration of the Locus Coeruleus. 99 In a word, the ApoE4 is a significant factor in the development of Alzheimer's disease occurred.
FIGURE 3.
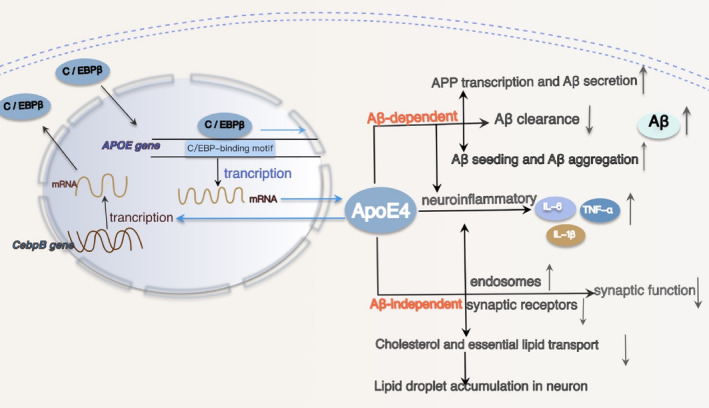
C/EBPβ mediates the expression of APOE4, leading to the accumulation of lipid droplets and Aβ. The transcription factor C/EBPβ binds and interacts with the C/EBP‐binding motifs within the APOE promoter −305 to +93 fragment, acting as a specific transcription factor for APOE and promoting its gene transcription. ApoE4 can influence lipid transport through an Aβ‐independent pathway, leading to mitochondrial damage and increased reactive oxygen species. Additionally, it can enhance Aβ production through an Aβ‐dependent pathway, concurrently amplifying neuroinflammatory responses. Conversely, ApoE4 strongly activates C/EBPβ, subsequently enhancing δ‐secretase cleavage of the increased APP.
ApoE production can be stimulated by a range of transcription factors, regulatory elements, hormones, cytokines, and lipids. 100 A positive correlation exists between C/EBPβ expression levels and ApoE levels, indicating a close involvement of C/EBPβ in regulating APOE transcription and protein expression. 101 The APOE promoter harbors multiple putative C/EBPβ binding motifs. Notably, the −305 to +93 fragment, which contains just one C/EBP‐binding motif, exhibits transcriptional activity akin to that of the full‐length promoter. This fragment encompasses the primary C/EBPβ‐binding site on the APOE promoter. 20 In the mouse model of AD, C/EBPβ governs ApoE expression across multiple cell types, encompassing neurons, astrocytes, and microglia. Additionally, it modulates Aβ‐induced ApoE transcription and protein expression, with a particular impact on ApoE4. 20 C/EBPβ functions as an APOE‐specific transcription factor by binding and interacting with the C/EBP‐binding motif within the −305 to +93 fragment of the APOE promoter, facilitating the transcription of APOE genes (Figure 3).
Neurons express ApoE, especially in stress response, particularly in the context of AD. 102 , 103 C/EBPβ is primarily responsible for the Aβ‐induced upregulation of ApoE expression, and Aβ42 oligomers produce C/EBPβ upregulation in a dose‐dependent manner, resulting in increased ApoE and AEP levels. 20 Remarkably, neuronal ApoE4 robustly triggers C/EBPβ activation, which, in turn, enhances δ‐secretase activity, leading to increased cleavage of APP and Tau in mice. This cascade promotes AD‐like pathology, with Thy1‐ApoE4/C/EBPβ mice exhibiting age‐dependent development of amyloid deposits, Tau aggregates, neurodegeneration, synaptic dysfunction, and cognitive impairment. 104 Conversely, ApoE4 exacerbates neuroinflammation and elevates Aβ levels, both of which can further stimulate C/EBPβ activation. 40 , 78 ApoE4, in conjunction with 27‐hydroxycholesterol (27‐OHC), collaboratively activates C/EBPβ/δ secretase signaling in neurons, mediating AD pathogenesis, which is dependent on the neuronal secretion of Aβ and inflammatory cytokines. 105 A feedback loop operates between C/EBPβ and ApoE4, wherein ApoE4 strongly activates C/EBPβ, contributing to pathological changes in AD. Conversely, overexpression of C/EBPβ preferentially mediates the effects of ApoE4. In conclusion, C/EBPβ serves as the principal transcription factor for APOE, notably APOE4, which represents a prominent genetic factor in AD and significantly contributes to the disease's development. C/EBPβ and ApoE reciprocally modulate and enhance their respective biological roles in the pathogenesis of AD. Breaking this detrimental cycle holds promise as an appealing strategy for intervening in the initiation and progression of AD.
2.4. Upregulation of C/EBPβ‐TRPC1‐SOCE signaling causes ER stress and dysregulation of protein enzymes and phosphatases, resulting in increased p‐Tau
Store‐operated calcium entry (SOCE) is of paramount importance in the CNS, and its dysregulation gives rise to neuroinflammation, synaptic dysfunction, and neuronal demise. 106 , 107 , 108 SOCE constitutes a ubiquitous Ca2+ entry pathway, activated following the depletion of endoplasmic reticulum (ER) calcium stores, initiated by the stimulation of plasma membrane receptors linked to phosphatidylinositol 4,5‐bisphosphate (PIP2) hydrolysis and inositol trisphosphate (IP3) production. 109 Key molecular constituents of the SOCE machinery encompass stromal interaction molecules (STIM1/2), Orai channels (ORAI2/3), and transient receptor potential channels (TRPC1‐7). Upon depletion of ER calcium stores, STIM1/2 detects reduced ER calcium levels, leading to oligomerization and subsequent translocation from ER‐like sites to the plasma membrane, where they interact with calcium‐conductive channels (ORAI and TRPC) to elicit calcium influx and replenish calcium stores. 110 , 111 TRPC1, the first identified mammalian TRPC protein, exhibits widespread expression in both neuronal and nonneuronal tissues and is currently the principal candidate component of the SOCE system, contributing significantly to SOCE in various cell types. 112 Cumulative evidence underscores the indispensable role of the TRPC1‐SOCE pathway in the initiation and progression of neurodegenerative disorders.
Overexpression of full‐length wild‐type human tau (known as hTau) is an early tau pathological hallmark observed in sporadic AD. The overexpression of intracellular tau can trigger TRPC1‐dependent SOCE signaling by elevating TRPC1 mRNA and protein levels. This elevation leads to an increase in the steady‐state intraneuronal calcium concentration ([Ca2+] I), heightened ER stress, an imbalance in protein kinase and phosphatase activity, and enhanced tau protein phosphorylation at AT8 and p‐S396, as previously documented. 22 ER stress leads to the activation of AKT/GSK3β signaling, resulting in robust tau phosphorylation. 113 Overexpression of hTau decreases the levels of PP2A‐C (the catalytic subunit of PP2A), while PP2A‐B (the regulatory subunit) remains unchanged, as demonstrated in previous research, 22 PP2A is known to be the primary tau phosphatase, regulating tau phosphorylation at various pathological sites, 114 This observation suggests that hTau overexpression diminishes PP2A activity, thereby promoting the accumulation of phosphorylated tau (p‐Tau) in the brain. The overexpression of TRPC1 leads to an elevation in phosphorylated CaMKII level, a major kinase that targets the phosphorylation of tau at Ser262, which can be activated by increased intracellular [Ca2+]. 115 In summary, overexpression of intracellular hTau can induce ER stress through a TRPC1‐dependent SOCE mechanism, resulting in the dysregulation of protein kinases and phosphatases, ultimately exacerbating tau pathology. Concurrently, TRPC1 overexpression leads to a significant reduction in dendritic spines within the hippocampal CA3 region, resulting in synaptic dysfunction and cognitive deficits. 22 SOCE‐mediated NFAT1‐NOX2‐NLRP1 inflammasome corpuscle in LPS‐induced neuronal damage and Aβ generation. 116 C/EBPβ, a transcription factor associated with aging and listed in the GenAge database, is abundantly expressed in the brain and can be activated by risk factors associated with AD. 24 C/EBPβ was verified as a transcription factor responsible for mediating TRPC1 gene expression (Figure 4). Overexpression of hTau upregulated C/EBPβ expression, subsequently enhancing TRPC1 mRNA transcription and protein expression. Conversely, the downregulation of C/EBPβ reduced hTau‐induced TRPC1 transcription and translation. 22 The overexpression of hTau leads to an elevation in TRPC1 transcription by activating the transcription factor C/EBPβ, which in turn this upregulation of hTau occurs via the C/EBPβ‐TRPC1‐SOCE signaling pathway, thereby forming another vicious cycle of AD development.
FIGURE 4.
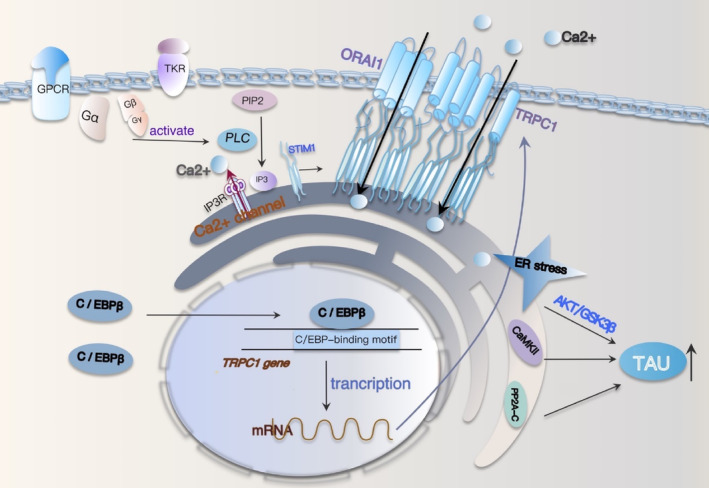
C/EBPβ‐TRPC1‐SOCE signaling causes ER stress and dysregulation of protein enzymes and phosphatases. After G protein‐coupled receptors and tyrosine receptors were activated, PLC was subsequently activated to generate IP3 from PIP2. As a calcium channel, IP3R binds to IP3 and is activated, resulting in calcium ion outflow. When Ca2+ storage in the ER is depleted, STIM1/2 senses a reduction in Ca2+ in the ER, which leads to oligomerization and subsequent transfer from ER‐like sites to the plasma membrane, where it interacts with calcium conduction channels (ORAIs and TRPCs) to induce Ca2+ influx and storage regeneration. In the context of hTau overexpression, activated C/EBPβ binds to the TRPC1 promoter sequence, promoting TRPC1 transcription, then increases neuronal steady‐state [Ca2+] I, enhances endoplasmic reticulum (ER) stress, imbalances protein kinases, and phosphatases, and exacerbates tauopathy. The C/EBPβ‐Trpc1‐soce‐signaling pathway in turn increases hTau, thus accelerating the progression of AD.
2.5. Activation of C/EBPβ induces transcription of the ANP32A gene, suppresses histone acetylation, and stimulates the formation of phosphorylated Tau
Synaptic damage and memory impairment are hallmark pathologies and symptoms associated with Alzheimer's disease. Epigenetic modifications, particularly protein acetylation within the nervous system, play a crucial role in the regulation of gene expression related to long‐term memory. 117 Acetylation of histones diminishes the electrostatic attraction between adjacent histones and DNA, thus fostering a more accessible chromatin configuration conducive to the transcription of genes associated with memory. 118 Histone acetylation, observed to be reduced in animal models of neurodegenerative diseases, including AD, is causally related to cognitive decline. 119 Histone acetylation and deacetylation processes are catalyzed by histone acetyltransferases (HATS) and histone deacetylases (HDACs), respectively. Additionally, cellular complex acetyltransferase inhibitor (INHAT) inhibits histone acetylation. 120 , 121 Two key components of INHAT, ANP32A (also known as I1 PP2A) and SET (also known as I2 PP2A), are selectively upregulated in brain regions affected by neurofibrillary pathology in AD. 122 , 123 , 124 , 125 ANP32A and SET together form an inhibitory complex, INHAT, which binds to histones and hinders their interaction with HAT. 126 , 127 The prevention of ANP32A elevation in the hippocampal CA3 region rescues learning and memory decline in htau transgenic mice, and downregulating ANP32A effectively ameliorates synaptic impairment in these mice. 24 Meanwhile, the phosphorylation and binding capacity of the microtubule‐associated protein tau are regulated by specific kinases and phosphatases. 128 The activation of tau kinases, particularly GSK3β, plays a pivotal role in tau hyperphosphorylation in AD and related tauopathies. 129 PP2A, the primary phosphatase controlling essential neuronal signaling pathways, mainly relies on the B55α subunit and is considered the principal tau phosphatase, accounting for 70% of total tau phosphatase activity. 130 , 131 , 132 The ANP32A and SET act as endogenous PP2A inhibitors, leading to elevated tau hyperphosphorylation levels. 123 , 124 Furthermore, the accumulation of AD‐like mitogen‐activated protein kinase (MAPT)/Tau disrupts autophagosome‐lysosome fusion by perturbing the ANP32A‐INHAT‐IST1‐ESCRT‐III pathway (Figure 5). This finding highlights the role of ANP32A in mediating a detrimental cycle of MAPT/Tau accumulation and autophagy deficiency during the chronic progression of AD neurodegeneration. 120
FIGURE 5.
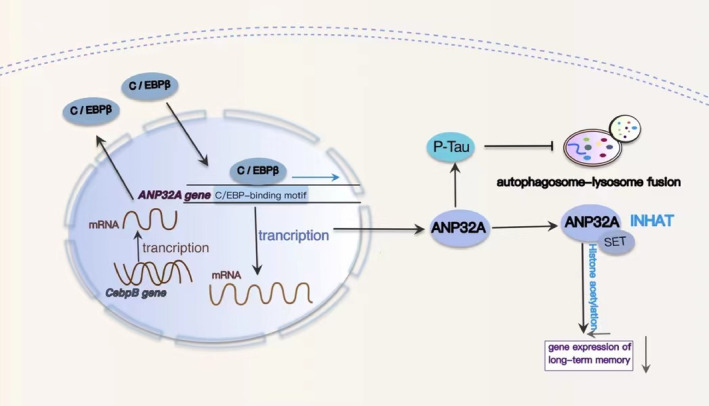
The molecular mechanism of C/EBPβ‐ANP32A‐INHAT/PP2A affecting cognitive function. AD‐related stressors increase total C/EBPβ levels and phosphorylated C/EBPβ levels. C/EBPβ induces ANP32 overexpression by binding to conserved recognition elements in the ANP32A proximal promoter region. ANP32A binds to SET to form an inhibitory complex (INHAT) that binds to histones and blocks their acetylation, ultimately resulting in reduced expression of life‐related genes. ANP32A, as an endogenous PP2A inhibitor, can increase the tau hyperphosphorylation level. AD‐like MAPT/Tau accumulation can inhibit autophagosome‐lysosome fusion by regulating the ANP32A‐INHAT‐IST1‐ESCRT‐III pathway, and overexpressed Tau can further promote Cebpb gene expression.
To further elucidate the underlying mechanisms contributing to the increased expression of ANP32A in AD, we discovered that ANP32A mRNA and protein levels were significantly elevated in response to hTau accumulation, Aβ1‐42 exposure, or H2O2 treatments. 24 To identify potential regulatory elements, we conducted a screening of ANP32A promoter binding sites using the transcription factor binding database, 133 and we identified a conserved C/EBPβ‐binding site in the proximal promoter region of ANP32A. 24 Notably, AD‐related stressors, such as tau overexpression, Aβ exposure, and oxidative stress, were found to enhance ANP32A gene transcription, with its expression under the regulation of C/EBPβ 24 (Figure 5). Stimulation of C/EBPβ led to the overexpression of ANP32A. 134 Additionally, AD‐related stressors were observed to elevate the levels of total and phosphorylated C/EBPβ, which were also increased in the brains of AD patients and transgenic animal models of AD. 31 , 78 The upregulation of ANP32A in AD may be a consequence of increased transcription factors C/EBPβ, suggesting that the upregulation of C/EBPβ may represent a key role leading to ANP32A overexpression, followed by histone acetylation and impaired cognition.
2.6. C/EBPβ selectively triggers inhibitory GABAergic neuronal degeneration by repressing FOXO1
FOXO, a member of the forkhead family of transcription factors that acts downstream of insulin/insulin‐like growth factor (IGF) signaling, plays conserved roles in longevity, cellular homeostasis, and cognitive performance. 135 It integrates signals from nutrient deprivation and stress stimuli to coordinate gene programs involved in cellular metabolism and protection against oxidative stress, thereby maintaining organelle and protein homeostasis. 136 , 137 , 138 FOXO is phosphorylated by Akt and Mst1 at residues T24 and S212, respectively. FOXO1 phosphorylated by Akt is sequestered in the cytoplasm, where it inhibits apoptotic genes, such as BIM (a mediator of Bcl‐2 cell death), promoting cell survival. 139 In contrast, MST1‐phosphorylated FOXO1 translocates to the nucleus, where it promotes apoptosis. 134 REST (RE1 silencing transcription factor, a transcription factor for FOXOs) and nerve activity are associated with IGF signaling, regulating the activity of the tripartite transcription factor. Nuclear REST and FOXO1 levels are strongly positively correlated with genes that repress excitation and synaptic functions, thereby delaying aging and extending lifespan. 140 The activity of FOXO/DAF‐16 (abnormal DAuer formation‐16) is required to reduce the toxicity of Aβ aggregates under conditions of low insulin signaling, 141 indicating a potential protective role in AD. C/EBPβ has also been implicated in mediating IGF‐1 and human insulin receptor (IR) expression by binding to its promoter. 16
The up‐regulation of neuronal C/EBPβ significantly affects insulin‐triggered P‐IR and p‐FOXO1 T24/FOXO1 activity, especially in GABAergic neurons, which renders GABAergic neurons more vulnerable to damage and variability during aging. 23 The loss of GABA and GABAergic interneurons in AD patients is a significant component of AD and may contribute to the network hyperactivity manifested as seizures. 142 The onset of seizures frequently precedes cognitive decline and may serve as a precursor to AD. 143 C/EBPβ and FOXO1 mutually repress each other. C/EBPβ overexpression inhibits FOXO1, FOXO3, and REST mRNA levels, leading to an increase in Bim and LGMN mRNA levels. Conversely, overexpression of FOXO1 reduces C/EBPβ mRNA levels, resulting in a decrease in both Bim and LGMN mRNAs (Figure 6). Additionally, it has been found that C/EBPβ directly binds to the REST and FOXO1 promoters, functioning as a transcriptional repressor. 23 C/EBPβ overexpression inhibited the transcription of FOXO and REST genes by binding to their promoters, which not only makes GABAergic neurons more susceptible to degeneration during aging but also selectively induces the increased expression of LGMN in GABAergic neurons and increases the production of AEP in GABAergic neurons, which together lead to the aggravation of cognitive dysfunction in AD.
FIGURE 6.
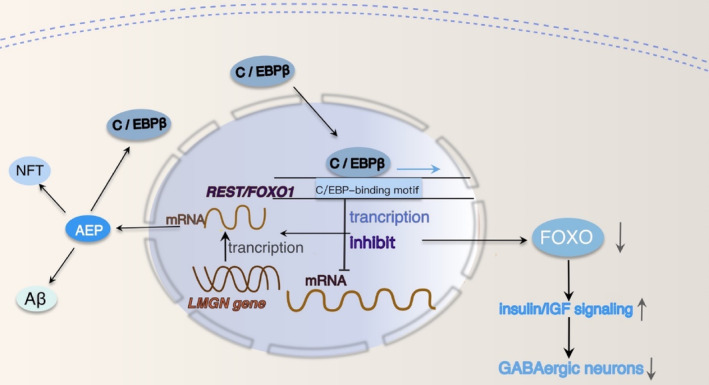
Molecular mechanisms of FOXO interact with C/EBPβ to accelerate AD progression. Overexpression of C/EBPβ inhibits REST and FOXO1 promoter activity and reduces REST and FOXO1 transcription. Decreased FOXO/DAF‐16 (abnormal DAuer formation‐16) activity increased insulin signaling, increased Aβ aggregation toxicity, and induced GABAergic neuronal degeneration and death. Inhibition of FOXO gene transcription can promote the transcription of LGMN, which leads to the increase of AEP and the accumulation of Aβ and Tau.
3. MECHANISMS OF ACTION OF C/EBPβ IN OTHER NEURODEGENERATIVE DISEASES
C/EBPβ functions as a transcription factor, contributing to the progression of AD through diverse pathways. There is considerable experimental evidence that C/EBPβ can also contribute to the development of a variety of other degenerative diseases through several pathways. Parkinson's disease (PD) is characterized by the loss of dopaminergic neurons and the presence of intraneuronal Lewy body (LB) inclusions, of which aggregated α‐synuclein (α‐syn) is a major component. C/EBPβ exhibits age‐dependent regulation of MAOB (monoamine oxidase B) expression, a pivotal enzyme in dopamine metabolism, which can trigger oxidative stress in dopaminergic neurons and contribute to the aggregation of α‐syn, a hallmark protein associated with neurodegenerative disorders. 144 Additionally, C/EBPβ/AEP exhibits age‐dependent activation in Parkinson's disease, wherein it assumes a role in mediating the presence of α‐synuclein in both the gastrointestinal tract and the brain. 145 Deprivation of C/EBPβ provided substantial in vitro and in vivo neuroprotection to dopaminergic cells, concurrently reducing inflammatory responses and glial activation levels. 146 The inactivation of the mitochondrial electron transport chain (ETC) activates the C/EBPβ/AEP pathway, thereby triggering the pathogenesis of PD. Deficiencies in all three complexes of the mitochondrial ETC robustly hinder oxidative phosphorylation, leading to a significant increase in ROS levels and activation of the C/EBPβ/AEP pathway, resulting in dopaminergic neuron cell death. C/EBPβ can also affect mitochondrial autophagy by regulating mitochondrial transcription factor A (TFAM). 147 These deficiencies also contribute to constipation and dyskinesia, key factors in PD pathogenesis. 148 BDNF and Netrin‐1, which promote neuronal survival and regulate intestinal function, are strongly reduced in both the brain and gut of PD patients, and the deficiency of these nutritional factors in the gut causes dopaminergic neuron loss, constipation, and motor dysfunction. The transcription factor C/EBPβ, induced by inflammation and oxidative stress, acts as a potent repressor of BDNF and Netrin‐1, suppressing the expression of nutritional factors, and thereby triggering non‐motor and motor symptoms of PD. Its level exhibits a negative correlation with BDNF and Netrin‐1 in PD patients. 149 C/EBPβ preferentially mediates APOE4 expression, which would exacerbate α‐syn pathology. 150 In multiple sclerosis (MS), the expression level of C/EBPβ is increased, with LAP being the predominant isoform. Loss of C/EBPβ attenuates microglial cell viability and has a neuroprotective effect. 53 Interaction between myelin basic protein‐primed T cells and microglia activates NF‐κB and C/EBPβ in microglia, leading to the specific expression of proinflammatory cytokines IL‐1β, IL‐1α, TNF‐α, and IL‐1 in microglia. 151 In amyotrophic lateral sclerosis (ALS), C/EBPβ has also been shown to act as a transcription factor regulating the expression of proinflammatory genes and is a candidate for regulating the expression of potentially neurotoxic genes in ALS microglia. 152
4. C/EBPβ REPRESENTS A PROMISING POTENTIAL THERAPEUTIC TARGET FOR AD TREATMENT
C/EBPβ plays a pivotal role in the modulation of AD‐related protein expression via multiple pathways, making it a crucial target for AD therapy. At present, drugs directly targeting C/EBPβ are relatively scarce in neurodegenerative disease research. However, microglia‐targeting C/EBPβ inhibition has been shown to be a safe and effective method to reduce neurodegeneration driven by neuroinflammation. 53 There are indications that drug‐induced reduction of C/EBPβ levels may have neuroprotective effects. AD has a higher incidence in elderly women. 153 Elevated FSH levels following the perimenopausal period directly impact hippocampal and cortical neurons, triggering the expression of Cebpb, Lgmn, and App, accelerating the deposition of β‐amyloid and Tau, and impairing cognitive function. Blocking FSH eliminated FSH‐induced AD pathology by suppressing the neuronal C/EBPβ‐δ‐secretase pathway, potentially preventing and delaying the progression of AD pathology. 154 C/EBPβ in microglia is regulated post‐translationally by the ubiquitin ligase COP1 (also called RFWD2). Absent COP1, C/EBPβ rapidly accumulates and instigates a robust pro‐inflammatory and neurodegeneration‐associated gene program. Conversely, COP1 restoration inhibits microglial activation and alleviates neuroinflammation. 19 , 155 Although targeting C/EBPβ directly may pose challenges, chemical inducers of degradation could offer a potential approach for modulating C/EBPβ. In a germinal matrix hemorrhage model, Secukinumab mitigated reactive astrogliosis and diminished neurological deficits, in part, through the regulation of the IL‐17RA/C/EBPβ pathways. 156 Pruni Cortex, a medicinal herb, can suppress the iNOS gene by inhibiting PI3K/Akt signaling and mitigating C/EBPβ phosphorylation, attributed to its constituent, Sakura. 157 This has noteworthy therapeutic implications for neuroinflammation. Patchouli alcohol (PA) has also been demonstrated to significantly downregulate the protein expression of C/EBPβ and AEP in the hippocampus of Tg mice. This downregulation inhibits the deposition of Aβ plaques, excessive phosphorylation of tau proteins, neuroinflammation, and intestinal ecological imbalance, thereby improving cognitive deficits in the AD mouse model. These findings underscore the promise of PA as a naturally occurring compound deserving of further exploration for its potential in AD pharmacotherapy. 158 Indeed, in cancer treatment research, numerous natural and synthetic drugs have demonstrated the capacity to inhibit C/EBPβ production, diminish its activity, or facilitate its degradation through targeted approaches. Considering the overexpression of C/EBPβ in AD, it is plausible to anticipate that investigations into drug interventions targeting C/EBPβ hold significant potential for treating AD and other neurodegenerative conditions.
5. CONCLUSION
C/EBPβ, activated by inflammatory cytokines, is a pivotal transcription factor regulating key enzymes and proteins across pathways that drive AD progression. Neuroinflammation is a prevalent hallmark of neurodegenerative diseases. C/EBPβ regulates proinflammatory gene expression during glial activation and plays a pivotal role in microglial activation, inducing neurotoxicity. 54 The δ‐secretase, activated in the aging brain, is the key enzyme in the cleavage of APP and Tau, ultimately leading to cognitive impairment. 63 C/EBPβ primarily upregulates LMGN gene transcription by binding to the CCCAGCTCTGGC promoter sequence, promoting δ‐secretase expression. 79 APOE represents the most potent genetic risk factor for AD. C/EBPβ binds to the APOE promoter region (−305 to +93) and primarily mediates the expression of ApoE4 protein induced by Aβ. 20 C/EBPβ‐mediated vicious cycles occur in all three aforementioned pathways, reinforcing the irreversibility of AD progression. Furthermore, C/EBPβ functions as a transcription factor for the calcium channel protein TRPC1, enhancing C/EBPβ‐TRPC1‐SOCE signaling, leading to ER stress and dysregulation of proteases and phosphatases, resulting in increased phosphorylated hTau and exacerbating AD's pathological progression. 22 ANP32A, a critical component of INHAT, mediates the vicious cycle between Tau/MAPT accumulation and autophagy deficiency, with its expression primarily regulated by C/EBPβ at the transcriptional level. 24 Additionally, FOXO, a member of the FOXO transcription factor family, functions downstream of IGF‐like signaling and can be selectively inhibited by C/EBPβ in degenerating GABA neurons. 23 C/EBPβ promotes Aβ production and increases Tau phosphorylation through multiple pathways, resulting in elevated extracellular Aβ plaques and intraneuronal NFTs, thus mediating a vicious cycle of chronic neuroinflammation and neuronal loss.
The overexpression of C/EBPβ expedites the irreversible advancement of AD pathophysiology. Furthermore, C/EBPβ plays a pivotal role in mediating various pathways associated with AD pathology, some of which develop vicious loops, resulting in the formation of feedback mechanisms. The significant involvement of C/EBPβ in prevalent degenerative diseases such as PD, MS, and ALS has been established. Additionally, drugs that counteract C/EBPβ in cancer therapy have shown promising experimental outcomes. 159 For instance, Sesquiterpene lactone is frequently employed in cancer treatment research as a natural compound. Notably, Helenalin acetate can bind to the initial 21 amino acids of LAP*, disrupting its interaction with the coactivator p300 and consequently suppressing the transcription of the target gene. 160 Withaferin A and Celastrol have likewise been demonstrated to inhibit C/EBPβ activity through a mechanism similar to that of Helenalin acetate. 161 ST101, an innovative and selective peptide antagonist of C/EBPβ, binds to the leucine zipper domain of C/EBPβ, preventing its dimerization and augmenting the ubiquitin‐proteasome‐dependent degradation of C/EBPβ. 162 Research has demonstrated that LIP promotes cell survival during staurosporine‐ or taxol‐induced Hep3B cell death. 163 Targeting LAP*/LAP could potentially result in LIP overexpression. Consequently, this review aims to enhance our comprehension of the interplay between C/EBPβ signaling and the expression of pertinent target genes. It also underscores the potential of C/EBPβ as a therapeutic target for addressing AD and other degenerative conditions. Currently, our understanding of the regulatory mechanisms governing gene expression in pathogenic glial cells remains limited, and the effectiveness and potential side effects of C/EBPβ antagonists in neurodegenerative diseases remain unclear. Researchers can continue to explore C/EBPβ as a focal point for extensive and in‐depth investigations, with the aim of developing more efficacious treatment strategies and pharmaceutical interventions for AD, other neurodegenerative disorders, and even more challenging diseases.
AUTHOR CONTRIBUTIONS
QY and CBL drafted the manuscript, PCY, GYZ, WW, XQR, and JY contributed to the analysis of the results, and FZH and WDL reviewed and modified the manuscript. All authors read and approved the final manuscript.
FUNDING INFORMATION
This work was supported by grants from the National Natural Science Foundation of China (82271234; 82060219); Natural Science Foundation of Jiangxi Province (20212ACB216009; 20212BAB216048); Jiangxi Province “Double Thousand Plan” (jxsq2019201023); Youth Team Project of the Second Affiliated Hospital of Nanchang University (2019YNTD12003).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
CONSENT FOR PUBLICATION
All authors read and approved the final manuscript.
Yao Q, Long C, Yi P, et al. C/EBPβ: A transcription factor associated with the irreversible progression of Alzheimer's disease. CNS Neurosci Ther. 2024;30:e14721. doi: 10.1111/cns.14721
Contributor Information
Weidong Liang, Email: lwd0929@gmu.edu.cn.
Fuzhou Hua, Email: huafuzhou@126.com.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. 2023 Alzheimer's disease facts and figures. Alzheimers Dement. 2023;19(4):1598‐1695. doi: 10.1002/alz.13016 [DOI] [PubMed] [Google Scholar]
- 2. Jorfi M, Maaser‐Hecker A, Tanzi RE. The neuroimmune axis of Alzheimer's disease. Genome Med. 2023;15(1):6. doi: 10.1186/s13073-023-01155-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Holtzman DM, Morris JC, Goate AM. Alzheimer's disease: the challenge of the second century. Sci Transl Med. 2011;3(77):77sr1. doi: 10.1126/scitranslmed.3002369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rostagno AA. Pathogenesis of Alzheimer's disease. Int J Mol Sci. 2022;24(1):107. doi: 10.3390/ijms24010107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Larsson SC, Traylor M, Malik R, et al. Modifiable pathways in Alzheimer's disease: Mendelian randomisation analysis. BMJ. 2017;359:j5375. doi: 10.1136/bmj.j5375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gaetani L, Bellomo G, Parnetti L, Blennow K, Zetterberg H, di Filippo M. Neuroinflammation and Alzheimer's disease: a machine learning approach to CSF proteomics. Cells. 2021;10(8):1930. doi: 10.3390/cells10081930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hartl L, Roelofs JJTH, Dijk F, Bijlsma MF, Duitman JW, Spek CA. C/EBP‐family redundancy determines patient survival and lymph node involvement in PDAC. Int J Mol Sci. 2023;24(2):1537. doi: 10.3390/ijms24021537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ramji DP, Foka P. CCAAT/enhancer‐binding proteins: structure, function and regulation. Biochem J. 2002;365(Pt 3):561‐575. doi: 10.1042/bj20020508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu D, Zhang Y, Zhao C, et al. Disruption of C/EBPβ‐Clec7a axis exacerbates neuroinflammatory injury via NLRP3 inflammasome‐mediated pyroptosis in experimental neuropathic pain. J Transl Med. 2022;20(1):583. doi: 10.1186/s12967-022-03779-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Neufeld EJ, Skalnik DG, Lievens PMJ, Orkin SH. Human CCAAT displacement protein is homologous to the drosophila homeoprotein, cut. Nat Genet. 1992;1(1):50‐55. doi: 10.1038/ng0492-50 [DOI] [PubMed] [Google Scholar]
- 11. Lekstrom‐Himes J, Xanthopoulos KG. Biological role of the CCAAT/enhancer‐binding protein family of transcription factors. J Biol Chem. 1998;273(44):28545‐28548. doi: 10.1074/jbc.273.44.28545 [DOI] [PubMed] [Google Scholar]
- 12. Birkenmeier EH, Gwynn B, Howard S, et al. Tissue‐specific expression, developmental regulation, and genetic mapping of the gene encoding CCAAT/enhancer binding protein. Genes Dev. 1989;3(8):1146‐1156. doi: 10.1101/gad.3.8.1146 [DOI] [PubMed] [Google Scholar]
- 13. Huang B, Zhao W, Cai X, et al. Expression and activity of the transcription factor CCAAT/enhancer‐binding protein β (C/EBPβ) is regulated by specific pulse‐modulated radio frequencies in Oligodendroglial cells. Int J Mol Sci. 2023;24(13):11131. doi: 10.3390/ijms241311131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meir O, Dvash E, Werman A, Rubinstein M. C/EBP‐beta regulates endoplasmic reticulum stress‐triggered cell death in mouse and human models. PLoS One. 2010;5(3):e9516. doi: 10.1371/journal.pone.0009516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nadeau S, Hein P, Fernandes KJL, Peterson AC, Miller FD. A transcriptional role for C/EBP beta in the neuronal response to axonal injury. Mol Cell Neurosci. 2005;29(4):525‐535. doi: 10.1016/j.mcn.2005.04.004 [DOI] [PubMed] [Google Scholar]
- 16. Aghanoori MR, Agarwal P, Gauvin E, et al. CEBPβ regulation of endogenous IGF‐1 in adult sensory neurons can be mobilized to overcome diabetes‐induced deficits in bioenergetics and axonal outgrowth. Cell Mol Life Sci. 2022;79(4):193. doi: 10.1007/s00018-022-04201-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu J, Yu T, Pietronigro EC, et al. Peli1 impairs microglial Aβ phagocytosis through promoting C/EBPβ degradation. PLoS Biol. 2020;18(10):e3000837. doi: 10.1371/journal.pbio.3000837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen DY, Stern SA, Garcia‐Osta A, et al. A critical role for IGF‐II in memory consolidation and enhancement. Nature. 2011;469(7331):491‐497. doi: 10.1038/nature09667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ndoja A, Reja R, Lee SH, et al. Ubiquitin ligase COP1 suppresses Neuroinflammation by degrading c/EBPβ in microglia. Cell. 2020;182(5):1156‐1169.e12. doi: 10.1016/j.cell.2020.07.011 [DOI] [PubMed] [Google Scholar]
- 20. Xia Y, Wang ZH, Zhang J, et al. C/EBPβ is a key transcription factor for APOE and preferentially mediates ApoE4 expression in Alzheimer's disease. Mol Psychiatry. 2021;26(10):6002‐6022. doi: 10.1038/s41380-020-00956-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xiong J, Zhang Z, Ye K. C/EBPβ/AEP signaling drives Alzheimer's disease pathogenesis. Neurosci Bull. 2023;39(7):1173‐1185. doi: 10.1007/s12264-023-01025-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ye J, Yin Y, Yin Y, et al. Tau‐induced upregulation of C/EBPβ‐TRPC1‐SOCE signaling aggravates tauopathies: a vicious cycle in Alzheimer neurodegeneration. Aging Cell. 2020;19(9):e13209. doi: 10.1111/acel.13209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xia Y, Qadota H, Wang ZH, et al. Neuronal C/EBPβ/AEP pathway shortens life span via selective GABAnergic neuronal degeneration by FOXO repression. Sci Adv. 2022;8(13):eabj8658. doi: 10.1126/sciadv.abj8658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chai GS, Feng Q, Wang ZH, et al. Downregulating ANP32A rescues synapse and memory loss via chromatin remodeling in Alzheimer model. Mol Neurodegener. 2017;12(1):34. doi: 10.1186/s13024-017-0178-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cortes‐Canteli M, Aguilar‐Morante D, Sanz‐SanCristobal M, Megias D, Santos A, Perez‐Castillo A. Role of C/EBPβ transcription factor in adult hippocampal neurogenesis. PLoS One. 2011;6(10):e24842. doi: 10.1371/journal.pone.0024842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li Y, Schor J, Bartko J, Albert G, Halterman MW. The transcription factor C/EBPβ promotes vascular endothelial growth factor a expression and neural stem cell expansion. FEBS Lett. 2022;596(13):1661‐1671. doi: 10.1002/1873-3468.14405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pulido‐Salgado M, Vidal‐Taboada JM, Saura J. C/EBPβ and C/EBPδ transcription factors: basic biology and roles in the CNS. Prog Neurobiol. 2015;132:1‐33. doi: 10.1016/j.pneurobio.2015.06.003 [DOI] [PubMed] [Google Scholar]
- 28. Chen C, Liao J, Xia Y, et al. Gut microbiota regulate Alzheimer's disease pathologies and cognitive disorders via PUFA‐associated neuroinflammation. Gut. 2022;71(11):2233‐2252. doi: 10.1136/gutjnl-2021-326269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ejarque‐Ortiz A, Medina MG, Tusell JM, Pérez‐González AP, Serratosa J, Saura J. Upregulation of CCAAT/enhancer binding protein beta in activated astrocytes and microglia. Glia. 2007;55(2):178‐188. doi: 10.1002/glia.20446 [DOI] [PubMed] [Google Scholar]
- 30. Xiong J, Kang SS, Wang M, et al. FSH and ApoE4 contribute to Alzheimer's disease‐like pathogenesis via C/EBPβ/δ‐secretase in female mice. Nat Commun. 2023;14(1):6577. doi: 10.1038/s41467-023-42282-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Strohmeyer R, Shelton J, Lougheed C, Breitkopf T. CCAAT‐enhancer binding protein‐β expression and elevation in Alzheimer's disease and microglial cell cultures. PLoS One. 2014;9(1):e86617. doi: 10.1371/journal.pone.0086617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hegdekar N, Sarkar C, Bustos S, et al. Inhibition of autophagy in microglia and macrophages exacerbates innate immune responses and worsens brain injury outcomes. Autophagy. 2023;19(7):2026‐2044. doi: 10.1080/15548627.2023.2167689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rajesh Y, Kanneganti TD. Innate immune cell death in neuroinflammation and Alzheimer's disease. Cells. 2022;11(12):1885. doi: 10.3390/cells11121885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dhapola R, Hota SS, Sarma P, Bhattacharyya A, Medhi B, Reddy DHK. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer's disease. Inflammopharmacology. 2021;29(6):1669‐1681. doi: 10.1007/s10787-021-00889-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Singh D. Astrocytic and microglial cells as the modulators of neuroinflammation in Alzheimer's disease. J Neuroinflammation. 2022;19(1):206. doi: 10.1186/s12974-022-02565-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thakur S, Dhapola R, Sarma P, Medhi B, Reddy DHK. Neuroinflammation in Alzheimer's disease: current progress in molecular signaling and therapeutics. Inflammation. 2023;46(1):1‐17. doi: 10.1007/s10753-022-01721-1 [DOI] [PubMed] [Google Scholar]
- 37. Lecca D, Jung YJ, Scerba MT, et al. Role of chronic neuroinflammation in neuroplasticity and cognitive function: a hypothesis. Alzheimers Dement. 2022;18(11):2327‐2340. doi: 10.1002/alz.12610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ma J, Yang X, Chen X. C/EBPβ is a key transcription factor of ox‐LDL inducing THP‐1 cells to release multiple pro‐inflammatory cytokines. Inflamm Res. 2021;70(10–12):1191‐1199. doi: 10.1007/s00011-021-01509-3 [DOI] [PubMed] [Google Scholar]
- 39. Zgorzynska E, Dziedzic B, Markiewicz M, Walczewska A. Omega‐3 PUFAs suppress IL‐1β‐induced hyperactivity of immunoproteasomes in astrocytes. Int J Mol Sci. 2021;22(11):5410. doi: 10.3390/ijms22115410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cardinaux JR, Allaman I, Magistretti PJ. Pro‐inflammatory cytokines induce the transcription factors C/EBPbeta and C/EBPdelta in astrocytes. Glia. 2000;29(1):91‐97. [PubMed] [Google Scholar]
- 41. Hernandez VG, Lechtenberg KJ, Peterson TC, et al. Translatome analysis reveals microglia and astrocytes to be distinct regulators of inflammation in the hyperacute and acute phases after stroke. Glia. 2023;71(8):1960‐1984. doi: 10.1002/glia.24377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goshi N, Morgan RK, Lein PJ, Seker E. A primary neural cell culture model to study neuron, astrocyte, and microglia interactions in neuroinflammation. J Neuroinflammation. 2020;17(1):155. doi: 10.1186/s12974-020-01819-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Voet S, Srinivasan S, Lamkanfi M, van Loo G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med. 2019;11(6):e10248. doi: 10.15252/emmm.201810248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fields J, Gardner‐Mercer J, Borgmann K, Clark I, Ghorpade A. CCAAT/enhancer binding protein β expression is increased in the brain during HIV‐1‐infection and contributes to regulation of astrocyte tissue inhibitor of metalloproteinase‐1. J Neurochem. 2011;118(1):93‐104. doi: 10.1111/j.1471-4159.2011.07203.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pahan K, Jana M, Liu X, Taylor BS, Wood C, Fischer SM. Gemfibrozil, a lipid‐lowering drug, inhibits the induction of nitric‐oxide synthase in human astrocytes. J Biol Chem. 2002;277(48):45984‐45991. doi: 10.1074/jbc.M200250200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802(4):396‐405. doi: 10.1016/j.bbadis.2009.12.009 [DOI] [PubMed] [Google Scholar]
- 47. Hussien YA, Mansour DF, Nada SA, et al. Linagliptin attenuates thioacetamide‐induced hepatic encephalopathy in rats: modulation of C/EBP‐β and CX3CL1/Fractalkine, neuro‐inflammation, oxidative stress and behavioral defects. Life Sci. 2022;295:120378. doi: 10.1016/j.lfs.2022.120378 [DOI] [PubMed] [Google Scholar]
- 48. Fields J, Ghorpade A. C/EBPβ regulates multiple IL‐1β‐induced human astrocyte inflammatory genes. J Neuroinflammation. 2012;9:177. doi: 10.1186/1742-2094-9-177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fields J, Cisneros IE, Borgmann K, Ghorpade A. Extracellular regulated kinase 1/2 signaling is a critical regulator of interleukin‐1β‐mediated astrocyte tissue inhibitor of metalloproteinase‐1 expression. PLoS One. 2013;8(2):e56891. doi: 10.1371/journal.pone.0056891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fan B, Peng Q, Song S, et al. Nonstructural protein 1 of variant PEDV plays a key role in escaping replication restriction by complement C3. J Virol. 2022;96(18):e0102422. doi: 10.1128/jvi.01024-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Maranto J, Rappaport J, Datta PK. Role of C/EBP‐β, p38 MAPK, and MKK6 in IL‐1β‐mediated C3 gene regulation in astrocytes. J Cell Biochem. 2011;112(4):1168‐1175. doi: 10.1002/jcb.23032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Abraham S, Sweet T, Khalili K, Sawaya BE, Amini S. Evidence for activation of the TGF‐beta1 promoter by C/EBPbeta and its modulation by Smads. J Interf Cytokine Res. 2009;29(1):1‐7. doi: 10.1089/jir.2008.0036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pulido‐Salgado M, Vidal‐Taboada JM, Garcia Diaz‐Barriga G, et al. Myeloid C/EBPβ deficiency reshapes microglial gene expression and is protective in experimental autoimmune encephalomyelitis. J Neuroinflammation. 2017;14(1):54. doi: 10.1186/s12974-017-0834-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Straccia M, Gresa‐Arribas N, Dentesano G, et al. Pro‐inflammatory gene expression and neurotoxic effects of activated microglia are attenuated by absence of CCAAT/enhancer binding protein β. J Neuroinflammation. 2011;8:156. doi: 10.1186/1742-2094-8-156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ngu EL, Tan CY, Lai NJY, et al. Spirulina platensis suppressed iNOS and proinflammatory cytokines in lipopolysaccharide‐induced BV2 microglia. Metabolites. 2022;12(11):1147. doi: 10.3390/metabo12111147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jana M, Anderson JA, Saha RN, Liu X, Pahan K. Regulation of inducible nitric oxide synthase in proinflammatory cytokine‐stimulated human primary astrocytes. Free Radic Biol Med. 2005;38(5):655‐664. doi: 10.1016/j.freeradbiomed.2004.11.021 [DOI] [PubMed] [Google Scholar]
- 57. Trummer M, Galardon E, Mayer B, Steiner G, Stamm T, Kloesch B. Polysulfides derived from the hydrogen sulfide and persulfide donor P* inhibit IL‐1β‐mediated inducible nitric oxide synthase signaling in ATDC5 cells: are CCAAT/enhancer‐binding proteins β and δ involved in the anti‐inflammatory effects of hydrogen sulfide and polysulfides? Nitric Oxide. 2022;129:41‐52. doi: 10.1016/j.niox.2022.09.005 [DOI] [PubMed] [Google Scholar]
- 58. Dentesano G, Straccia M, Ejarque‐Ortiz A, et al. Inhibition of CD200R1 expression by C/EBP β in reactive microglial cells. J Neuroinflammation. 2012;9:165. doi: 10.1186/1742-2094-9-165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li W, Tanikawa T, Kryczek I, et al. Aerobic glycolysis controls myeloid‐derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple‐negative breast cancer. Cell Metab. 2018;28(1):87‐103.e6. doi: 10.1016/j.cmet.2018.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Spangenberg E, Severson PL, Hohsfield LA, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer's disease model. Nat Commun. 2019;10(1):3758. doi: 10.1038/s41467-019-11674-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang MY, Zhang CM, Zhou HH, et al. Identification of a distal enhancer that determines the expression pattern of acute phase marker C‐reactive protein. J Biol Chem. 2022;298(8):102160. doi: 10.1016/j.jbc.2022.102160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ford AM, Bennett CA, Healy LE, Towatari M, Greaves MF, Enver T. Regulation of the myeloperoxidase enhancer binding proteins Pu1, C‐EBP alpha, ‐beta, and ‐delta during granulocyte‐lineage specification. Proc Natl Acad Sci USA. 1996;93(20):10838‐10843. doi: 10.1073/pnas.93.20.10838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wu Z, Zhu R, Yu Y, et al. Spinal cord injury‐activated C/EBPβ‐AEP axis mediates cognitive impairment through APP C586/tau N368 fragments spreading. Prog Neurobiol. 2023;227:102467. doi: 10.1016/j.pneurobio.2023.102467 [DOI] [PubMed] [Google Scholar]
- 64. Zhang Z, Song M, Liu X, et al. Delta‐secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer's disease. Nat Commun. 2015;6:8762. doi: 10.1038/ncomms9762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang Z, Song M, Liu X, et al. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer's disease. Nat Med. 2014;20(11):1254‐1262. doi: 10.1038/nm.3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wu Z, Chen C, Kang SS, et al. Neurotrophic signaling deficiency exacerbates environmental risks for Alzheimer's disease pathogenesis. Proc Natl Acad Sci USA. 2021;118(25):e2100986118. doi: 10.1073/pnas.2100986118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang Z, Li XG, Wang ZH, et al. δ‐Secretase‐cleaved tau stimulates Aβ production via upregulating STAT1‐BACE1 signaling in Alzheimer's disease. Mol Psychiatry. 2021;26(2):586‐603. doi: 10.1038/s41380-018-0286-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hong Y, Chan CB, Kwon IS, et al. SRPK2 phosphorylates tau and mediates the cognitive defects in Alzheimer's disease. J Neurosci. 2012;32(48):17262‐17272. doi: 10.1523/jneurosci.3300-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang ZH, Liu P, Liu X, Yu SP, Wang JZ, Ye K. Delta‐secretase (AEP) mediates tau‐splicing imbalance and accelerates cognitive decline in tauopathies. J Exp Med. 2018;215(12):3038‐3056. doi: 10.1084/jem.20180539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liu C, Götz J. How it all started: tau and protein phosphatase 2A. J Alzheimers Dis. 2013;37(3):483‐494. doi: 10.3233/jad-130503 [DOI] [PubMed] [Google Scholar]
- 71. Liu Z, Jang SW, Liu X, et al. Neuroprotective actions of PIKE‐L by inhibition of SET proteolytic degradation by asparagine endopeptidase. Mol Cell. 2008;29(6):665‐678. doi: 10.1016/j.molcel.2008.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Basurto‐Islas G, Grundke‐Iqbal I, Tung YC, Liu F, Iqbal K. Activation of asparaginyl endopeptidase leads to tau hyperphosphorylation in Alzheimer disease. J Biol Chem. 2013;288(24):17495‐17507. doi: 10.1074/jbc.M112.446070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bolognin S, Blanchard J, Wang X, et al. An experimental rat model of sporadic Alzheimer's disease and rescue of cognitive impairment with a neurotrophic peptide. Acta Neuropathol. 2012;123(1):133‐151. doi: 10.1007/s00401-011-0908-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang ZH, Gong K, Liu X, et al. Author correction: C/EBPβ regulates delta‐secretase expression and mediates pathogenesis in mouse models of Alzheimer's disease. Nat Commun. 2019;10(1):5452. doi: 10.1038/s41467-019-13553-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang H, Liu X, Chen S, Ye K. Spatiotemporal activation of the C/EBPβ/δ‐secretase axis regulates the pathogenesis of Alzheimer's disease. Proc Natl Acad Sci USA. 2018;115(52):e12427‐e12434. doi: 10.1073/pnas.1815915115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Correction for Wang et al., Spatiotemporal activation of the C/EBPβ/δ‐secretase axis regulates the pathogenesis of Alzheimer's disease. Proc Natl Acad Sci USA. 2019;116(51):26090. doi: 10.1073/pnas.1920626117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yao Y, Kang SS, Xia Y, et al. A delta‐secretase‐truncated APP fragment activates CEBPB, mediating Alzheimer's disease pathologies. Brain. 2021;144(6):1833‐1852. doi: 10.1093/brain/awab062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ramberg V, Tracy LM, Samuelsson M, Nilsson LNG, Iverfeldt K. The CCAAT/enhancer binding protein (C/EBP) δ is differently regulated by fibrillar and oligomeric forms of the Alzheimer amyloid‐β peptide. J Neuroinflammation. 2011;8:34. doi: 10.1186/1742-2094-8-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang ZH, Gong K, Liu X, et al. C/EBPβ regulates delta‐secretase expression and mediates pathogenesis in mouse models of Alzheimer's disease. Nat Commun. 2018;9(1):1784. doi: 10.1038/s41467-018-04120-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chen C, Zhou Y, Wang H, et al. Gut inflammation triggers C/EBPβ/δ‐secretase‐dependent gut‐to‐brain propagation of Aβ and tau fibrils in Alzheimer's disease. EMBO J. 2021;40(17):e106320. doi: 10.15252/embj.2020106320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Xia Y, Xiao Y, Wang ZH, et al. Bacteroides Fragilis in the gut microbiomes of Alzheimer's disease activates microglia and triggers pathogenesis in neuronal C/EBPβ transgenic mice. Nat Commun. 2023;14(1):5471. doi: 10.1038/s41467-023-41283-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Liao J, Chen G, Liu X, et al. C/EBPβ/AEP signaling couples atherosclerosis to the pathogenesis of Alzheimer's disease. Mol Psychiatry. 2022;27(7):3034‐3046. doi: 10.1038/s41380-022-01556-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Liu P, Wang ZH, Kang SS, et al. High‐fat diet‐induced diabetes couples to Alzheimer's disease through inflammation‐activated C/EBPβ/AEP pathway. Mol Psychiatry. 2022;27:3396‐3409. doi: 10.1038/s41380-022-01600-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wu Z, Wang ZH, Liu X, et al. Traumatic brain injury triggers APP and tau cleavage by delta‐secretase, mediating Alzheimer's disease pathology. Prog Neurobiol. 2020;185:101730. doi: 10.1016/j.pneurobio.2019.101730 [DOI] [PubMed] [Google Scholar]
- 85. Wang ZH, Xiang J, Liu X, et al. Deficiency in BDNF/TrkB neurotrophic activity stimulates δ‐secretase by upregulating C/EBPβ in Alzheimer's disease. Cell Rep. 2019;28(3):655‐669.e5. doi: 10.1016/j.celrep.2019.06.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Blanchard JW, Akay LA, Davila‐Velderrain J, et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature. 2022;611(7937):769‐779. doi: 10.1038/s41586-022-05439-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Koutsodendris N, Nelson MR, Rao A, Huang Y. Apolipoprotein E and Alzheimer's disease: findings, hypotheses, and potential mechanisms. Annu Rev Pathol. 2022;17:73‐99. doi: 10.1146/annurev-pathmechdis-030421-112756 [DOI] [PubMed] [Google Scholar]
- 88. Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15(9):501‐518. doi: 10.1038/s41582-019-0228-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fernández‐Calle R, Konings SC, Frontiñán‐Rubio J, et al. APOE in the bullseye of neurodegenerative diseases: impact of the APOE genotype in Alzheimer's disease pathology and brain diseases. Mol Neurodegener. 2022;17(1):62. doi: 10.1186/s13024-022-00566-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Pires M, Rego AC. Apoe4 and Alzheimer's disease pathogenesis‐mitochondrial deregulation and targeted therapeutic strategies. Int J Mol Sci. 2023;24(1):778. doi: 10.3390/ijms24010778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Huang YA, Zhou B, Nabet AM, Wernig M, Südhof TC. Differential signaling mediated by ApoE2, ApoE3, and ApoE4 in human neurons parallels Alzheimer's disease risk. J Neurosci. 2019;39(37):7408‐7427. doi: 10.1523/jneurosci.2994-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Giannisis A, Patra K, Edlund AK, et al. Brain integrity is altered by hepatic APOE ε4 in humanized‐liver mice. Mol Psychiatry. 2022;27(8):3533‐3543. doi: 10.1038/s41380-022-01548-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lanfranco MF, Sepulveda J, Kopetsky G, Rebeck GW. Expression and secretion of apoE isoforms in astrocytes and microglia during inflammation. Glia. 2021;69(6):1478‐1493. doi: 10.1002/glia.23974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Holtzman DM, Bales KR, Tenkova T, et al. Apolipoprotein E isoform‐dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2000;97(6):2892‐2897. doi: 10.1073/pnas.050004797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Huang YA, Zhou B, Wernig M, Südhof TC. ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell. 2017;168(3):427‐441.e21. doi: 10.1016/j.cell.2016.12.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kloske CM, Wilcock DM. The important Interface between Apolipoprotein E and Neuroinflammation in Alzheimer's disease. Front Immunol. 2020;11:754. doi: 10.3389/fimmu.2020.00754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Liu CC, Zhao N, Fu Y, et al. ApoE4 accelerates early seeding of amyloid pathology. Neuron. 2017;96(5):1024‐1032.e3. doi: 10.1016/j.neuron.2017.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Baek MS, Cho H, Lee HS, Lee JH, Ryu YH, Lyoo CH. Effect of APOE ε4 genotype on amyloid‐β and tau accumulation in Alzheimer's disease. Alzheimers Res Ther. 2020;12(1):140. doi: 10.1186/s13195-020-00710-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kang SS, Ahn EH, Liu X, et al. ApoE4 inhibition of VMAT2 in the locus coeruleus exacerbates tau pathology in Alzheimer's disease. Acta Neuropathol. 2021;142(1):139‐158. doi: 10.1007/s00401-021-02315-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kockx M, Traini M, Kritharides L. Cell‐specific production, secretion, and function of apolipoprotein E. J Mol Med. 2018;96(5):361‐371. doi: 10.1007/s00109-018-1632-y [DOI] [PubMed] [Google Scholar]
- 101. Xu M, Zhang DF, Luo R, et al. A systematic integrated analysis of brain expression profiles reveals YAP1 and other prioritized hub genes as important upstream regulators in Alzheimer's disease. Alzheimers Dement. 2018;14(2):215‐229. doi: 10.1016/j.jalz.2017.08.012 [DOI] [PubMed] [Google Scholar]
- 102. Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26(19):4985‐4994. doi: 10.1523/jneurosci.5476-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer's diseases. Neurobiol Dis. 2014;72 Pt A:3‐12. doi: 10.1016/j.nbd.2014.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wang ZH, Xia Y, Wu Z, et al. Neuronal ApoE4 stimulates C/EBPβ activation, promoting Alzheimer's disease pathology in a mouse model. Prog Neurobiol. 2022;209:102212. doi: 10.1016/j.pneurobio.2021.102212 [DOI] [PubMed] [Google Scholar]
- 105. Wang ZH, Xia Y, Liu P, et al. ApoE4 activates C/EBPβ/δ‐secretase with 27‐hydroxycholesterol, driving the pathogenesis of Alzheimer's disease. Prog Neurobiol. 2021;202:102032. doi: 10.1016/j.pneurobio.2021.102032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Szteyn K, Gomez R, Berg KA, Jeske NA. Divergence in endothelin‐1‐ and bradykinin‐activated store‐operated calcium entry in afferent sensory neurons. ASN Neuro. 2015;7(2):175909141557871. doi: 10.1177/1759091415578714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Sun S, Zhang H, Liu J, et al. Reduced synaptic STIM2 expression and impaired store‐operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron. 2014;82(1):79‐93. doi: 10.1016/j.neuron.2014.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hao B, Lu Y, Wang Q, Guo W, Cheung KH, Yue J. Role of STIM1 in survival and neural differentiation of mouse embryonic stem cells independent of Orai1‐mediated Ca2+ entry. Stem Cell Res. 2014;12(2):452‐466. doi: 10.1016/j.scr.2013.12.005 [DOI] [PubMed] [Google Scholar]
- 109. Cheng KT et al. Contribution and regulation of TRPC channels in store‐operated Ca2+ entry. Curr Top Membr. 2013;71:149‐179. doi: 10.1016/b978-0-12-407870-3.00007-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zhang SL, Yu Y, Roos J, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437(7060):902‐905. doi: 10.1038/nature04147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhang H, Sun S, Wu L, et al. Store‐operated Calcium Channel complex in postsynaptic spines: a new therapeutic target for Alzheimer's disease treatment. J Neurosci. 2016;36(47):11837‐11850. doi: 10.1523/jneurosci.1188-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ambudkar IS, Ong HL, Liu X, Bandyopadhyay B, Cheng KT. TRPC1: the link between functionally distinct store‐operated calcium channels. Cell Calcium. 2007;42(2):213‐223. doi: 10.1016/j.ceca.2007.01.013 [DOI] [PubMed] [Google Scholar]
- 113. Yuan YH, Yan WF, Sun JD, Huang JY, Mu Z, Chen NH. The molecular mechanism of rotenone‐induced α‐synuclein aggregation: emphasizing the role of the calcium/GSK3β pathway. Toxicol Lett. 2015;233(2):163‐171. doi: 10.1016/j.toxlet.2014.11.029 [DOI] [PubMed] [Google Scholar]
- 114. Liu F, Grundke‐Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22(8):1942‐1950. doi: 10.1111/j.1460-9568.2005.04391.x [DOI] [PubMed] [Google Scholar]
- 115. Sironi JJ, Yen SH, Gondal JA, Wu Q, Grundke‐Iqbal I, Iqbal K. Ser‐262 in human recombinant tau protein is a markedly more favorable site for phosphorylation by CaMKII than PKA or PhK. FEBS Lett. 1998;436(3):471‐475. doi: 10.1016/s0014-5793(98)01185-5 [DOI] [PubMed] [Google Scholar]
- 116. Sun Z, Li X, Yang L, et al. SOCE‐mediated NFAT1‐NOX2‐NLRP1 inflammasome involves in lipopolysaccharide‐induced neuronal damage and Aβ generation. Mol Neurobiol. 2022;59(5):3183‐3205. doi: 10.1007/s12035-021-02717-y [DOI] [PubMed] [Google Scholar]
- 117. Burns AM, Gräff J. Cognitive epigenetic priming: leveraging histone acetylation for memory amelioration. Curr Opin Neurobiol. 2021;67:75‐84. doi: 10.1016/j.conb.2020.08.011 [DOI] [PubMed] [Google Scholar]
- 118. Brownell JE, Allis CD. Special HATs for special occasions: linking histone acetylation to chromatin assembly and gene activation. Curr Opin Genet Dev. 1996;6(2):176‐184. doi: 10.1016/s0959-437x(96)80048-7 [DOI] [PubMed] [Google Scholar]
- 119. Gräff J, Kim D, Dobbin MM, Tsai LH. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol Rev. 2011;91(2):603‐649. doi: 10.1152/physrev.00012.2010 [DOI] [PubMed] [Google Scholar]
- 120. Feng Q, Luo Y, Zhang XN, et al. MAPT/tau accumulation represses autophagy flux by disrupting IST1‐regulated ESCRT‐III complex formation: a vicious cycle in Alzheimer neurodegeneration. Autophagy. 2020;16(4):641‐658. doi: 10.1080/15548627.2019.1633862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wang J, Yun F, Sui J, Liang W, Shen D, Zhang Q. HAT‐ and HDAC‐targeted protein acetylation in the occurrence and treatment of epilepsy. Biomedicine. 2022;11(1):88. doi: 10.3390/biomedicines11010088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Zhang Y, Ma RH, Li XC, et al. Silencing [formula: see text] rescues tau pathologies and memory deficits through rescuing PP2A and inhibiting GSK‐3β signaling in human tau transgenic mice. Front Aging Neurosci. 2014;6:123. doi: 10.3389/fnagi.2014.00123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Tsujio I, Zaidi T, Xu J, Kotula L, Grundke‐Iqbal I, Iqbal K. Inhibitors of protein phosphatase‐2A from human brain structures, immunocytological localization and activities towards dephosphorylation of the Alzheimer type hyperphosphorylated tau. FEBS Lett. 2005;579(2):363‐372. doi: 10.1016/j.febslet.2004.11.097 [DOI] [PubMed] [Google Scholar]
- 124. Tanimukai H, Grundke‐Iqbal I, Iqbal K. Up‐regulation of inhibitors of protein phosphatase‐2A in Alzheimer's disease. Am J Pathol. 2005;166(6):1761‐1771. doi: 10.1016/s0002-9440(10)62486-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Liu GP, Wei W, Zhou X, et al. Silencing PP2A inhibitor by lenti‐shRNA interference ameliorates neuropathologies and memory deficits in tg2576 mice. Mol Ther. 2013;21(12):2247‐2257. doi: 10.1038/mt.2013.189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Seo SB, McNamara P, Heo S, Turner A, Lane WS, Chakravarti D. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell. 2001;104(1):119‐130. doi: 10.1016/s0092-8674(01)00196-9 [DOI] [PubMed] [Google Scholar]
- 127. Seo SB, Macfarlan T, McNamara P, et al. Regulation of histone acetylation and transcription by nuclear protein pp32, a subunit of the INHAT complex. J Biol Chem. 2002;277(16):14005‐14010. doi: 10.1074/jbc.M112455200 [DOI] [PubMed] [Google Scholar]
- 128. Zhang H, Wang X, Xu P, et al. Tolfenamic acid inhibits GSK‐3β and PP2A mediated tau hyperphosphorylation in Alzheimer's disease models. J Physiol Sci. 2020;70(1):29. doi: 10.1186/s12576-020-00757-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kaur P, Khera A, Alajangi HK, et al. Role of tau in various Tauopathies, treatment approaches, and emerging role of nanotechnology in neurodegenerative disorders. Mol Neurobiol. 2023;60(3):1690‐1720. doi: 10.1007/s12035-022-03164-z [DOI] [PubMed] [Google Scholar]
- 130. Park HJ, Lee KW, Oh S, et al. Protein phosphatase 2A and its methylation modulating enzymes LCMT‐1 and PME‐1 are dysregulated in Tauopathies of progressive Supranuclear palsy and Alzheimer disease. J Neuropathol Exp Neurol. 2018;77(2):139‐148. doi: 10.1093/jnen/nlx110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Nunbhakdi‐Craig V, Schuechner S, Sontag JM, et al. Expression of protein phosphatase 2A mutants and silencing of the regulatory B alpha subunit induce a selective loss of acetylated and detyrosinated microtubules. J Neurochem. 2007;101(4):959‐971. doi: 10.1111/j.1471-4159.2007.04503.x [DOI] [PubMed] [Google Scholar]
- 132. Hu W, Wang Z, Zhang H, et al. Chk1 inhibition ameliorates Alzheimer's disease pathogenesis and cognitive dysfunction through CIP2A/PP2A signaling. Neurotherapeutics. 2022;19(2):570‐591. doi: 10.1007/s13311-022-01204-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Sandelin A, Alkema W, Engström P, Wasserman WW, Lenhard B. JASPAR: an open‐access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91‐D94. doi: 10.1093/nar/gkh012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lehtinen MK, Yuan Z, Boag PR, et al. A conserved MST‐FOXO signaling pathway mediates oxidative‐stress responses and extends life span. Cell. 2006;125(5):987‐1001. doi: 10.1016/j.cell.2006.03.046 [DOI] [PubMed] [Google Scholar]
- 135. Kim SY, Webb AE. Neuronal functions of FOXO/DAF‐16. Nutr Healthy Aging. 2017;4(2):113‐126. doi: 10.3233/nha-160009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014;39(4):159‐169. doi: 10.1016/j.tibs.2014.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Kikis EA, Gidalevitz T, Morimoto RI. Protein homeostasis in models of aging and age‐related conformational disease. Adv Exp Med Biol. 2010;694:138‐159. doi: 10.1007/978-1-4419-7002-2_11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40(2):427‐446. doi: 10.1016/s0896-6273(03)00606-8 [DOI] [PubMed] [Google Scholar]
- 139. Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96(6):857‐868. doi: 10.1016/s0092-8674(00)80595-4 [DOI] [PubMed] [Google Scholar]
- 140. Zullo JM, Drake D, Aron L, et al. Regulation of lifespan by neural excitation and REST. Nature. 2019;574(7778):359‐364. doi: 10.1038/s41586-019-1647-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age‐onset proteotoxicity. Science. 2006;313(5793):1604‐1610. doi: 10.1126/science.1124646 [DOI] [PubMed] [Google Scholar]
- 142. Najm R, Jones EA, Huang Y. Apolipoprotein E4, inhibitory network dysfunction, and Alzheimer's disease. Mol Neurodegener. 2019;14(1):24. doi: 10.1186/s13024-019-0324-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. DiFrancesco JC, Tremolizzo L, Polonia V, et al. Adult‐onset epilepsy in Presymptomatic Alzheimer's disease: a retrospective study. J Alzheimers Dis. 2017;60(4):1267‐1274. doi: 10.3233/jad-170392 [DOI] [PubMed] [Google Scholar]
- 144. Wu Z, Xia Y, Wang Z, et al. C/EBPβ/δ‐secretase signaling mediates Parkinson's disease pathogenesis via regulating transcription and proteolytic cleavage of α‐synuclein and MAOB. Mol Psychiatry. 2021;26(2):568‐585. doi: 10.1038/s41380-020-0687-7 [DOI] [PubMed] [Google Scholar]
- 145. Wang H, Chen G, Ahn EH, et al. C/EBPβ/AEP is age‐dependently activated in Parkinson's disease and mediates α‐synuclein in the gut and brain. NPJ Parkinsons Dis. 2023;9(1):1. doi: 10.1038/s41531-022-00430-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Morales‐Garcia JA, Gine E, Hernandez‐Encinas E, et al. CCAAT/enhancer binding protein β silencing mitigates glial activation and neurodegeneration in a rat model of Parkinson's disease. Sci Rep. 2017;7(1):13526. doi: 10.1038/s41598-017-13269-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Sierra‐Magro A, Bartolome F, Lozano‐Muñoz D, et al. C/EBPβ regulates TFAM expression, mitochondrial function and autophagy in cellular models of Parkinson's disease. Int J Mol Sci. 2023;24(2):1459. doi: 10.3390/ijms24021459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Ahn EH, Lei K, Kang SS, et al. Mitochondrial dysfunction triggers the pathogenesis of Parkinson's disease in neuronal C/EBPβ transgenic mice. Mol Psychiatry. 2021;26(12):7838‐7850. doi: 10.1038/s41380-021-01284-x [DOI] [PubMed] [Google Scholar]
- 149. Ahn EH, Kang SS, Liu X, et al. BDNF and Netrin‐1 repression by C/EBPβ in the gut triggers Parkinson's disease pathologies, associated with constipation and motor dysfunctions. Prog Neurobiol. 2021;198:101905. doi: 10.1016/j.pneurobio.2020.101905 [DOI] [PubMed] [Google Scholar]
- 150. Davis AA, Inman CE, Wargel ZM, et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci Transl Med. 2020;12(529):eaay3069. doi: 10.1126/scitranslmed.aay3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Dasgupta S, Jana M, Liu X, Pahan K. Role of very‐late antigen‐4 (VLA‐4) in myelin basic protein‐primed T cell contact‐induced expression of proinflammatory cytokines in microglial cells. J Biol Chem. 2003;278(25):22424‐22431. doi: 10.1074/jbc.M301789200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Valente T, Mancera P, Tusell JM, Serratosa J, Saura J. C/EBPβ expression in activated microglia in amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33(9):2186‐2199. doi: 10.1016/j.neurobiolaging.2011.09.019 [DOI] [PubMed] [Google Scholar]
- 153. Yeung CHC, Au Yeung SL, Kwok MK, Zhao JV, Schooling CM. The influence of growth and sex hormones on risk of Alzheimer's disease: a Mendelian randomization study. Eur J Epidemiol. 2023;38(7):745‐755. doi: 10.1007/s10654-023-01015-2 [DOI] [PubMed] [Google Scholar]
- 154. Xiong J, Kang SS, Wang Z, et al. FSH blockade improves cognition in mice with Alzheimer's disease. Nature. 2022;603(7901):470‐476. doi: 10.1038/s41586-022-04463-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. He C, Wang T, Han Y, Zuo C, Wang G. E3 ubiquitin ligase COP1 confers neuroprotection in cerebral ischemia/reperfusion injury via regulation of transcription factor C/EBPβ in microglia. Int J Biol Macromol. 2022;222(Pt B):1789‐1800. doi: 10.1016/j.ijbiomac.2022.09.264 [DOI] [PubMed] [Google Scholar]
- 156. Liu SP, Huang L, Flores J, et al. Secukinumab attenuates reactive astrogliosis via IL‐17RA/(C/EBPβ)/SIRT1 pathway in a rat model of germinal matrix hemorrhage. CNS Neurosci Ther. 2019;25(10):1151‐1161. doi: 10.1111/cns.13144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Yamauchi Y, Okuyama T, Ishii T, Okumura T, Ikeya Y, Nishizawa M. Sakuranetin downregulates inducible nitric oxide synthase expression by affecting interleukin‐1 receptor and CCAAT/enhancer‐binding protein β. J Nat Med. 2019;73(2):353‐368. doi: 10.1007/s11418-018-1267-x [DOI] [PubMed] [Google Scholar]
- 158. Xu QQ, Su ZR, Yang W, Zhong M, Xian YF, Lin ZX. Patchouli alcohol attenuates the cognitive deficits in a transgenic mouse model of Alzheimer's disease via modulating neuropathology and gut microbiota through suppressing C/EBPβ/AEP pathway. J Neuroinflammation. 2023;20(1):19. doi: 10.1186/s12974-023-02704-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Falkenberg KD, Jakobs A, Matern JC, et al. Withaferin a, a natural compound with anti‐tumor activity, is a potent inhibitor of transcription factor C/EBPβ. Biochim Biophys Acta, Mol Cell Res. 2017;1864(7):1349‐1358. doi: 10.1016/j.bbamcr.2017.05.003 [DOI] [PubMed] [Google Scholar]
- 160. Jakobs A, Steinmann S, Henrich SM, Schmidt TJ, Klempnauer KH. Helenalin acetate, a natural Sesquiterpene lactone with anti‐inflammatory and anti‐cancer activity, disrupts the cooperation of CCAAT box/enhancer‐binding protein β (C/EBPβ) and Co‐activator p300. J Biol Chem. 2016;291(50):26098‐26108. doi: 10.1074/jbc.M116.748129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Schmidt TJ, Klempnauer KH. Natural products with antitumor potential targeting the MYB‐C/EBPβ‐p300 transcription module. Molecules. 2022;27(7):2077. doi: 10.3390/molecules27072077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Darvishi E, Ghamsari L, Leong SF, et al. Anticancer activity of ST101, a novel antagonist of CCAAT/enhancer binding protein β. Mol Cancer Ther. 2022;21(11):1632‐1644. doi: 10.1158/1535-7163.Mct-21-0962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Saint‐Auret G, Danan JL, Hiron M, et al. Characterization of the transcriptional signature of C/EBPbeta isoforms (LAP/LIP) in Hep3B cells: implication of LIP in pro‐survival functions. J Hepatol. 2011;54(6):1185‐1194. doi: 10.1016/j.jhep.2010.09.021 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.