

Abstract
ADAMTS13 consists of a reprolysin-type metallopro-tease domain followed by a disintegrin domain, a thrombospondin type 1 motif (TSP1), Cys-rich and spacer domains, seven more TSP1 motifs, and two CUB domains. ADAMTS13 limits platelet accumulation in microvascular thrombi by cleaving the Tyr1605-Met1606 bond in von Willebrand factor, and ADAMTS13 deficiency causes a lethal syndrome, thrombotic thrombocytopenic purpura. ADAMTS13 domains required for substrate recognition were localized by the characterization of recombinant deletion mutants. Constructs with C-terminal His6 and V5 epitopes were expressed by transient transfection of COS-7 cells or in a baculovirus system. No association with extracellular matrix or cell surface was detected for any ADAMTS13 variant by immunofluorescence microscopy or chemical modification. Both plasma and recombinant full-length ADAMTS13 cleaved von Willebrand factor subunits into two fragments of 176 kDa and 140 kDa. Recombinant ADAMTS13 was divalent metal ion-dependent and was inhibited by IgG from a patient with idiopathic thrombotic thrombocytopenic purpura. ADAMTS13 that was truncated after the metalloprotease domain, the disintegrin domain, the first TSP1 repeat, or the Cys-rich domain was not able to cleave von Willebrand factor, whereas addition of the spacer region restored protease activity. Therefore, the spacer region is necessary for normal ADAMTS13 activity toward von Willebrand factor, and the more C-terminal TSP1 and CUB domains are dispensable in vitro.
Thrombotic thrombocytopenic purpura (TTP)1 is a syndrome characterized by microangiopathic hemolytic anemia and thrombocytopenia, and it may be accompanied by neurological dysfunction, renal failure, and fever (1–3). If untreated, the mortality can exceed 90%, but plasma-exchange therapy has reduced the mortality to less than 20% (4). Although the patho-physiology of TTP is not fully understood, a plausible model has been proposed in which the proteolysis of von Willebrand factor (VWF) plays a central role (5). VWF is a multimeric protein that binds receptors on the surface of platelets and in connective tissue, thereby mediating the adhesion of platelets to sites of vascular injury. If unchecked, the process can lead to micro-vascular thrombosis. A plasma VWF-cleaving protease has been described that acts on the Tyr1605–Met1606 bond in the central A2 domain of the VWF subunit, and cleavage is stimulated by shear forces like those occurring at sites of thrombosis or by low concentrations of urea or guanidine (6, 7). This proteolytic reaction limits VWF-dependent platelet adhesion, and most adults with idiopathic TTP have an acquired autoantibody that inhibits the VWF-cleaving protease (8, 9). Therefore, therapeutic plasma exchange may ameliorate TTP by replacing the missing protease and removing the inhibitory antibody.
The VWF-cleaving protease was recently purified and identified as a new member of the ADAMTS family of metalloproteases (10, 11), so named for the combination of a disinte-grin-like and metalloprotease (reprolysin type), with thrombospondin type 1 motifs (12). The primary structure of the ADAMTS13 precursor was determined by cDNA cloning (13, 14) and by positional cloning in families with inherited ADAMTS13 deficiency (15). It consists of 1427 amino acid residues comprising a signal peptide, a short propeptide ending in the sequence RQRR, a reprolysin-like metalloprotease domain, a disintegrin domain, a thrombospondin-1 repeat (TSP1), a cysteine-rich domain and spacer characteristic of the ADAMTS family, seven additional TSP1 repeats, and two CUB domains (13–15). Several alternatively spliced mRNA species have been identified that could encode truncated forms of ADAMTS13 lacking various C-terminal domains (13–16).
The complex multidomain structure of ADAMTS13 is conserved across vertebrates as diverse as mammals, birds, and fish (17),2 suggesting that motifs outside the metalloprotease domain are required for its biological function. This concept is supported by the finding that missense mutations in domains far from the metalloprotease domain cause inherited ADAMTS13 deficiency and thrombotic microangiopathy (15, 18, 19). The specificity of ADAMTS13 for a single bond in VWF and the remarkable regulation of cleavage by tensile stress on the substrate suggest that accessory domains are critical for substrate recognition. However, the structural requirements for ADAMTS13 activity have not been determined. To address this question, ADAMTS13 constructs with nested C-terminal deletions were characterized. The results suggest that the spacer domain is required to recognize and cleave VWF. More distal domains may contribute to activity but do not appear to be necessary in vitro.
EXPERIMENTAL PROCEDURES
Plasmid Constructs—
A cDNA construct covering the ADAMTS13 coding sequence was assembled from partial cDNA clones (13) using a PCR-based strategy. The open reading frame of ADAMTS13 was amplified and cloned into pcDNA3.1/V5-His-TOPO (Invitrogen) to generate pcADAMTS13-V5-His. Constructs with C-terminal truncations were created similarly. This cloning strategy appends the following vector-encoded amino acid sequence onto the ADAMTS13 sequence: a linker region (KGNSADIQHSGGRSSLEGPRFE), the V5 epitope (GKPIPNPLLGLDST), a tripeptide (RTG), and a poly(His) tag (HHHHHH). All PCR amplifications were performed with high fidelity PfuTurbo Hotstart DNA polymerase (Stratagene, La Jolla, CA) in the presence of 7.5% Me2SO. The accuracy of all constructions was confirmed by DNA sequencing.
Using ADAMTS13 constructs in vector pcDNA3.1/V5-His-TOPO as the template, recombination sites attB1 and attB2 were introduced at the 5′- and 3′-ends of the insert by PCR with oligonucleotides 5′-ggg gac aag ttt gta caa aaa agc agg ctt cac cAT GCA CCA GCG TCA CCC CCG GGC AAG-3′ (attB1 site in lowercase) and 5′-ggg gac cac ttt gta caa gaa agc tgg gtc TCA ATG GTG ATG GTG ATG ATG ACC G-3′ (attB2 site in lowercase). PCR products were recombined with plasmid pDONR201 and BP Clonase to prepare entry clones according to the manufacturer’s instructions (Invitrogen). Entry clones were sequenced to verify the accuracy of the PCR and recombination reactions. Bacmid expression vectors were prepared by recombination of each entry clone with vector pDEST8 and LR Clonase (Invitrogen). Baculovirus stocks were prepared for each bacmid by transfection of Spodoptera frugiperda Sf9 cells and repeated amplification of the resultant virus.
Recombinant Protein Expression—
COS-1 or COS-7 cells (ATCC, Manassas, VA) were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, penicillin, and streptomycin. Cells were seeded on 6-well tissue culture plates at 45% confluence 12 h before transfection in serum-free Opti-MEM I (Invitrogen) containing 6 μl of LipofectAMINE 2000 (Invitrogen) per 1 μg of plasmid DNA. After 4–5 h, the transfection mixture was replaced with 2 ml of fresh serum-free Opti-MEM I. The medium was collected 48 h after transfection, and phenylmethylsulfonyl fluoride was added to a final concentration of 1 mM. Cell debris was removed by centrifugation, and the conditioned medium was stored at −70 °C.
Sf9 cells were cultured in 1-liter bottles at 27 °C with shaking in 100 ml of protein-free Sf-900 II SFM medium (Invitrogen) to a density of 2 × 106/ml. Baculovirus stocks were added at a multiplicity of infection of 0.1, and cultures were incubated for an additional 96 h. Media were dialyzed against phosphate-buffered saline (PBS), concentrated by ultrafiltration, and stored at −20 °C.
Recombinant proteins were visualized by SDS-PAGE and Western blotting with monoclonal anti-V5 antibody (Invitrogen), peroxidase-conjugated goat anti-mouse IgG (DAKO Corp., Carpinteria, CA), and the chemiluminescent ECL detection system (Amersham Biosciences) as described previously (20). The luminograms were scanned, and the relative amount of proteins detected was estimated by densitometry using NIH Image 1.62 (developed at the National Institutes of Health and available on the Internet at rsb.info.nih.gov/nih-image/). The absolute concentration of V5-tagged ADAMTS13 constructs was determined by standardization with the identically tagged Positope reference protein (Invitrogen).
ADAMTS13 Assays—
ADAMTS13 protease activity was assayed by a collagen-binding method (21) with minor modifications (22). The multimer distribution of VWF after incubation with ADAMTS13 variants was assessed also by SDS-agarose gel electrophoresis and Western blotting with peroxidase-conjugated polyclonal anti-VWF antibody (catalog no. P0226, DAKO) as described previously (23).
The specific cleavage of VWF subunits by ADAMTS13 was confirmed by SDS-PAGE and Western blotting as described above, but samples were reduced with 2% β-mercaptoethanol, and proteins were detected with polyclonal anti-VWF antibody (23).
Immunofluorescence Microscopy—
COS-7 cells were grown to 80% confluence in 4-well Lab-Tek glass chamber slides (Nalge Nunc Int., Naperville, IL) in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum. Cells were washed once with PBS and transfected with plasmid DNA mixed with LipofectAMINE 2000 (1:6, w/v) in serum-free Opti-MEM I (Invitrogen). After 5 h, the medium was removed and replaced with fresh Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum. After 48 h, the cells were washed with ice-cold PBS and fixed for 10 min at −20 °C with ethanol:acetic acid (9:1) or at room temperature with 2% paraformaldehyde in PBS. The cells were washed three times with PBS, and nonspecific antibody binding sites were blocked with 1.5% bovine serum albumin in PBS for 30 min. The cells were incubated sequentially for 1 h each at 20 °C with anti-V5 IgG (1:1000) followed by Cy3-conjugated anti-mouse IgG (Jackson ImmunoResearch Laboratories, Inc.) in PBS containing 1.5% bovine serum albumin. The cells were washed three times with PBS between the incubations with antibodies. For nuclear staining, cells fixed with paraformaldehyde were permeabilized with 0.075% saponin for 3 min and stained with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA).
Extracellular Matrix Localization—
RFL-6 primary rat lung fibroblasts (CCL-192, ATCC) were maintained in Kaighn’s modification of Ham’s F-12 medium (ICN Biomedicals, Irvine, CA) with 20% fetal bovine serum, 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cells at 50% confluence in T25 flasks were transfected with 2 μg of pcADAMTS13-V5-His, 12 μl of LipofectAMINE, and 8 μl of Plus reagent (Invitrogen) in 2 ml of Opti-MEM I. At 48 h, medium was collected, and cells were washed extensively with ice-cold PBS. The cells were lysed on ice for 30 min in 2 ml of water containing 0.1% protease inhibitor mixture (Sigma) and 1 mM phenylmethylsulfonyl fluoride. The lysate was removed, and remaining cell membranes and matrix were washed extensively with ice-cold water. The extracellular matrix was solubilized by scraping into 100 μl of SDS-PAGE sample buffer (15 mM Tris-HCl, pH 6.8, 2.5% glycerol, 0.5% SDS, 178 mM β-mercaptoethanol, and 0.25% bromphenol blue). Equal percentages of lysate, medium, and matrix solutions were incubated at 100 °C for 5 min, then diluted 2.5-fold in 50 mM sodium citrate, pH 5.5, with or without 500 units of recombinant Streptomyces plicatus endoglycosidase H (New England Biolabs, Inc., Beverly, MA) and incubated at 37 °C for 2 h. Products were analyzed by SDS-PAGE and Western blotting with anti-V5 antibody or monoclonal anti-human fibronectin antibody (clone IST-3, catalog no. F0791, Sigma).
Cell Surface Biotinylation—
Transfected cells in 6-well plates were washed three times with ice-cold PBS and incubated at 4 °C for 30 min with 1 ml of PBS containing 1.5 mg/ml sulfosuccinimidyl-2-(biotinamido)ethyl-1,3-dithiopropionate (NHS-SS-Biotin, Pierce) (24). Reaction was stopped by washing with ice-cold PBS, and cells were lysed in 1.5 ml of PBS containing 1% Triton X-100 and 1% protease inhibitor mixture (Sigma). The lysate was clarified by centrifugation and split into two equal samples. ADAMTS13 proteins in one sample were concentrated by adsorption onto 100 μl of TALON metal affinity beads (BD Biosciences Clontech). Biotin-labeled proteins from the second sample were adsorbed onto 100 μl of streptavidin-agarose (Pierce) at room temperature for 2 h, and unbound ADAMTS13 in the flow-through fraction was concentrated by adsorption onto 100 μl of TALON beads. After washing four times with PBS, proteins were eluted from TALON or streptavidin beads with 40 μl of 0.4% SDS containing 5% β-mercaptoethanol. Samples (30 μl) were diluted with 30 μl of water and incubated at 37 °C for 16 h in a total volume of 50 μl containing 100 mM sodium citrate phosphate, pH 5.5, 2% Triton X-100, 4% β-mercaptoethanol, and 0.02% sodium azide without (control) or with 2 milliunits of recombinant S. plicatus endoglycosidase H (Oxford Glycosciences, Abingdon, UK) (24). Products were analyzed by SDS-PAGE and Western blotting with anti-V5 antibody.
RESULTS
Expression and Localization of Recombinant ADAMTS13 Variants—
Plasma ADAMTS13 consists of an N-terminal metalloprotease domain followed by several conserved structural motifs of unknown function. To assess the role of these various domains in substrate recognition, nested C-terminal deletions were constructed with a V5 epitope, and poly(His) tag was appended to facilitate detection of the recombinant proteins (Fig. 1). In a preliminary comparison of expression systems by Western blotting, stably transfected COS-7, CHO, or BHK cell lines expressed much less ADAMTS13 than did transiently transfected COS-7 cells, suggesting that high levels of ADAMTS13 confer a selective disadvantage.3 Therefore, ADAMTS13 variants were expressed in transiently transfected COS-7 cells. All constructs were synthesized with similar efficiency varying only ≈2-fold in the amount of secreted protein detected by Western blotting with anti-V5 antibody.
Fig. 1. ADAMTS13 constructs.
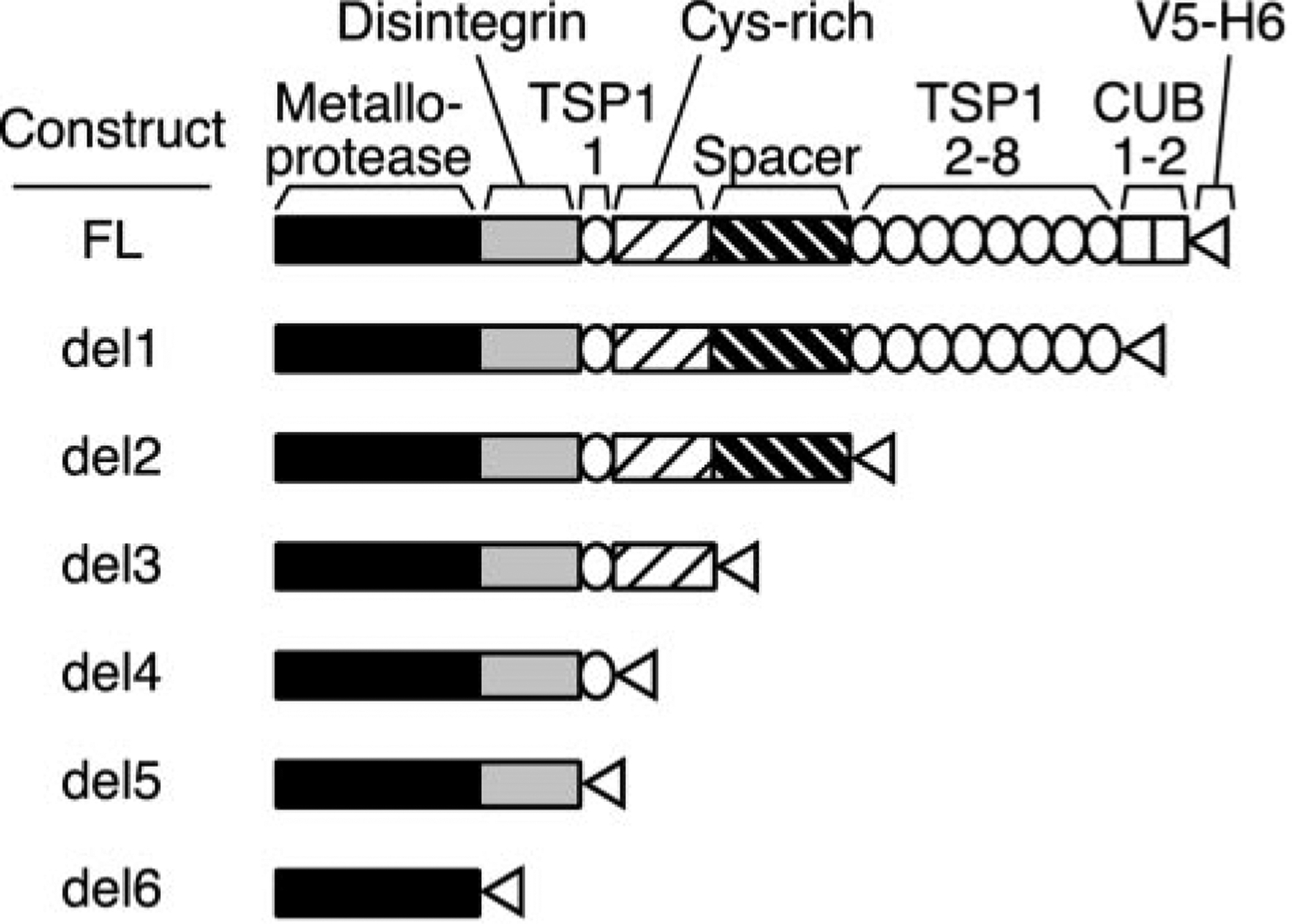
Secreted ADAMTS13 consists of a metalloprotease domain, a disintegrin domain, 8 thrombospondin-1 (TSP1) repeats, a Cys-rich and spacer domain, and two CUB domains. The signal peptide and propeptide that occur in the primary translation product (residues 1–74) are not part of mature ADAMTS13 and are not shown. Truncation mutants were constructed by inserting a V5 epitope, His6 tag, and termination codon after the following amino acid residues: full-length (FL) after Thr1427; del1 after Ala1191; del2 after Ala685; del3 after Cys555; del4 after Glu439; del5 after Gly385; and del6 after Gln289. The domains present in each construct are shown schematically.
The cellular localization of ADAMTS13 proteins was assessed by immunofluorescence microscopy (Fig. 2). The variant containing only the metalloprotease domain (del6) exhibited a perinuclear distribution consistent with localization in the Golgi apparatus. Larger constructs exhibited a more extensively diffuse plus granular pattern consistent with transient localization in the endoplasmic reticulum during biosynthesis and secretion. Staining of nonpermeabilized cells, either in culture at 4 °C or after fixation with paraformaldehyde, did not identify any ADAMTS13 variant on the cell surface. The absence of cell-surface ADAMTS13 was confirmed for full-length ADAMTS13 by a chemical modification approach (Fig. 3). Cell surface proteins were biotinylated and recovered from cell ly-sates by affinity chromatography on avidin-agarose. Contamination of the biotinylated fraction by intracellular proteins was assessed by digestion with endoglycosidase H, which does not act on complex oligosaccharides of secreted ADAMTS13 in conditioned medium. Almost all of the ADAMTS13 was not biotinylated and had endoglycosidase H-sensitive oligosaccharides consistent with localization in the endoplasmic reticulum. Trace amounts of biotinylated ADAMTS13 had endoglycosidase H-sensitive oligosaccharides and probably represent intracellular protein derived from broken cells. Therefore, secreted ADAMTS13 does not bind detectably to COS-7 cells.
Fig. 2. Immunofluorescence localization of ADAMTS13.
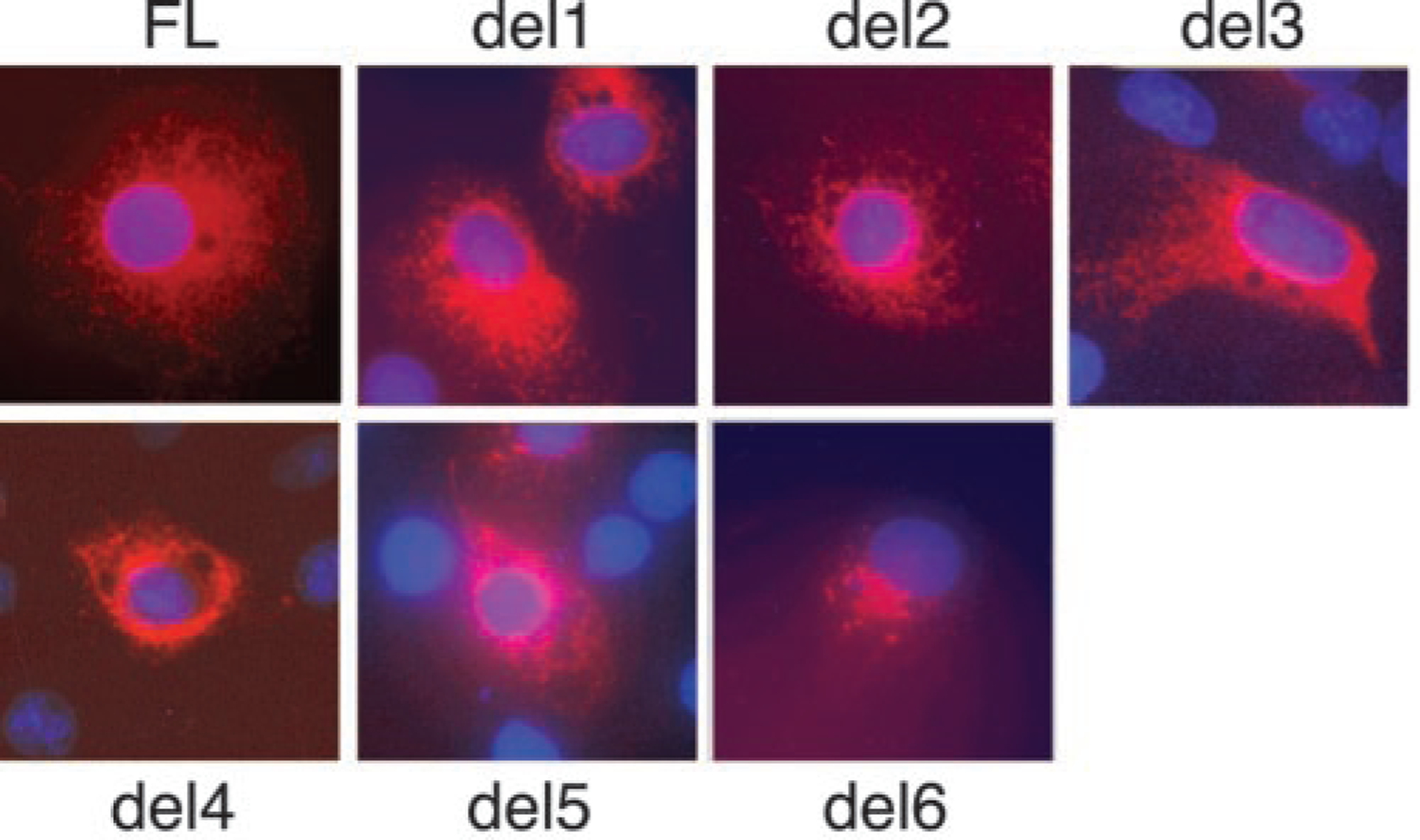
COS-7 cells expressing full-length ADAMTS13 (FL) or the indicated deletion mutants described in Fig. 1 were fixed with ethanol:acetic acid and incubated with anti-V5 IgG and Cy3-conjugated anti-mouse IgG (in red). Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (in blue).
Fig. 3. Surface biotinylation does not detect secreted cell-surface ADAMTS13.
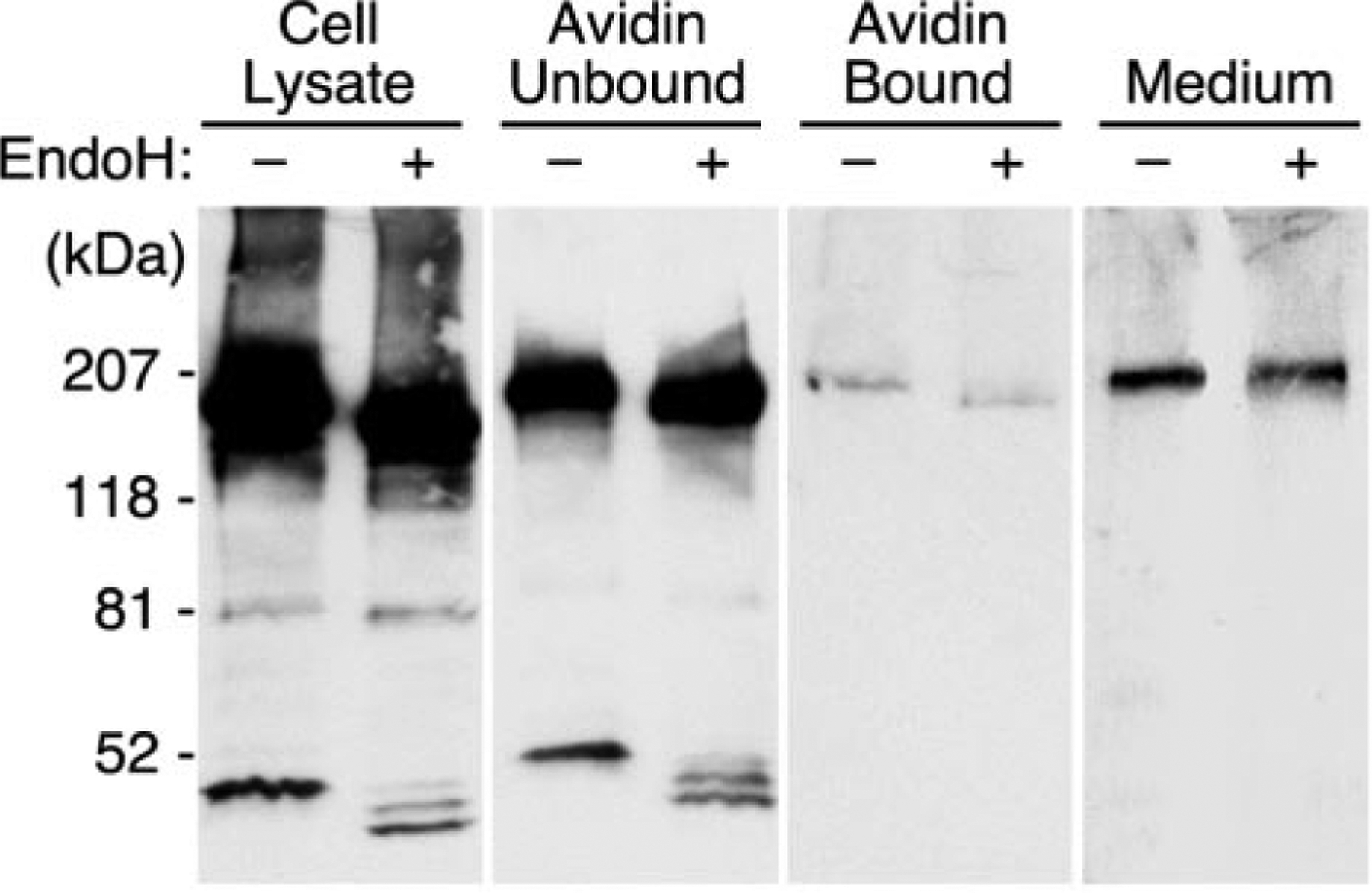
Full-length ADAMTS13 was expressed in COS-7 cells. Cell surface proteins were biotinylated and collected by adsorption on streptavidin-agarose. Samples of the initial cell lysate, the streptavidin flow-through fraction (Unbound), the streptavidin eluate (Bound), and conditioned medium (Medium) were digested with endoglycosidase H (EndoH). Products were separated by SDS-PAGE and visualized by Western blotting with anti-V5 antibody. The position of molecular mass standards is indicated at the left.
Because some ADAMTS proteases interact with extracellular matrix components, the association of ADAMTS13 with extracellular matrix was examined. Recombinant full-length ADAMTS13 was not detected in the extracellular matrix of transfected COS-1 cells by immunofluorescence or by Western blotting (data not shown), but COS-1 cells produce a relatively sparse matrix in culture. Therefore, similar experiments were performed in primary rat lung fibroblasts, which produce a more substantial matrix and have been used extensively in studies of extracellular matrix localization (25). In RFL-6 cells, the oligosaccharides of intracellular ADAMTS13 were endoglycosidase H-sensitive, whereas oligosaccharides of secreted ADAMTS13 were endoglycosidase H-resistant (Fig. 4). Trace amounts of endoglycosidase H-sensitive ADAMTS13 were recovered in solubilized extracellular matrix (Fig. 4), although endoglycosidase H-resistant fibronectin was abundant (data not shown). Overexposure did not demonstrate any ADAMTS13 in the matrix fraction that was resistant to endoglycosidase H. Similar results were obtained by immunofluorescence: fibronectin was detected readily in RFL-6 extracellular matrix, but ADAMTS13 was not. Thus, secreted ADAMTS13 does not appear to bind to the extracellular matrix of cultured RFL-6 or COS-1 cells.
Fig. 4. Secreted ADAMTS13 is undetectable in extracellular matrix.
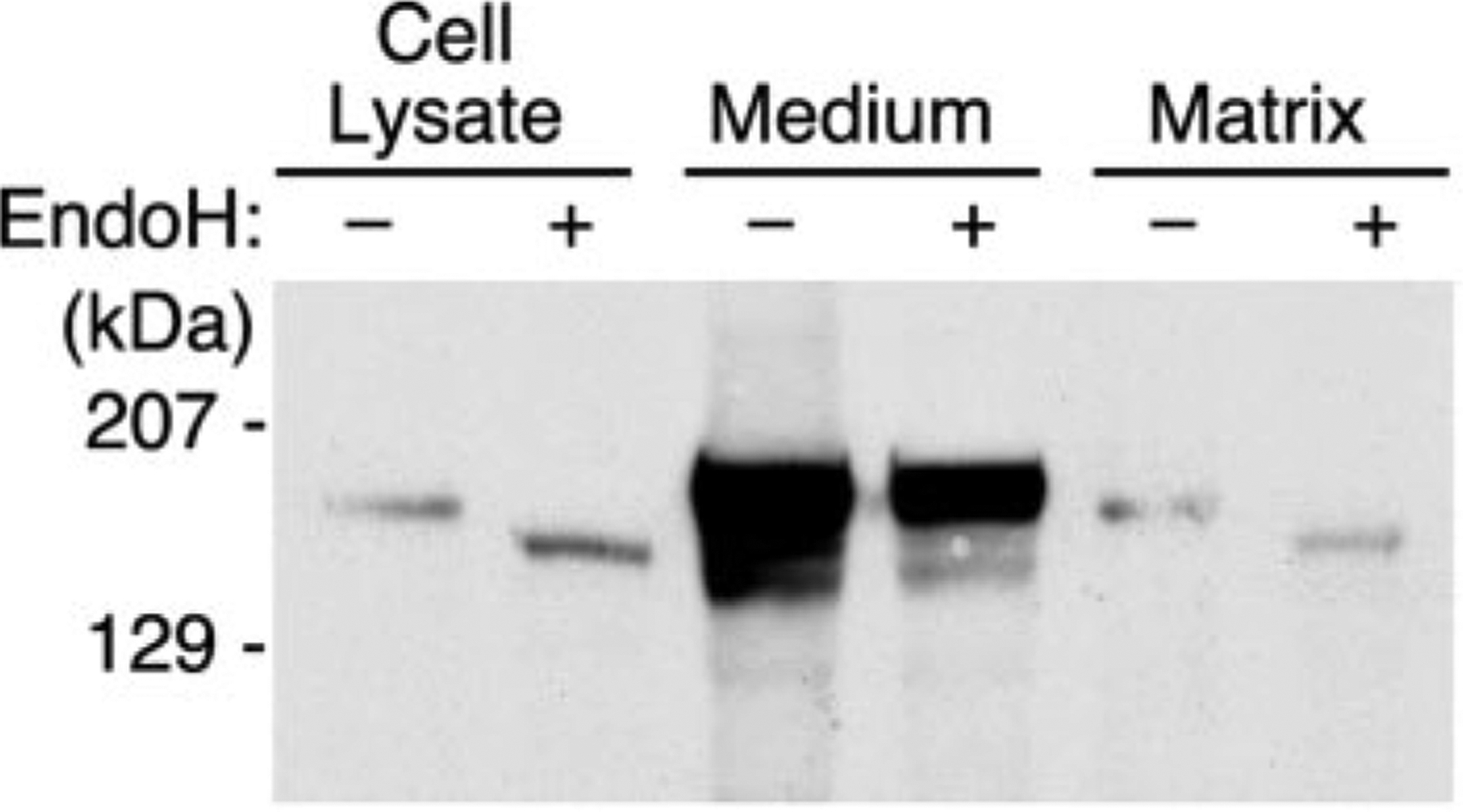
Full-length ADAMTS13 was expressed in RFL-6 cells. Cell lysate, conditioned medium, and extracellular matrix were prepared, and samples were digested with endoglycosidase H (EndoH) as described under “Experimental Procedures.” Products were separated by SDS-PAGE and visualized by Western blotting with anti-V5 antibody. The position of molecular mass standards is indicated at the left.
Proteolytic Activity of ADAMTS13 Variants—
Recombinant ADAMTS13 proteins were assayed under conditions previously established for plasma ADAMTS13. ADAMTS13 does not cleave VWF at a significant rate under conditions of physiological ionic strength and in the absence of fluid shear stress. However, reaction occurs readily in buffers of low ionic strength that are supplemented with low concentrations of urea or guanidine (6, 7). The relative concentration of recombinant ADAMTS13 variants was determined by Western blotting and densitometry (Fig. 5A), and equal amounts were incubated with purified plasma VWF in the presence of 1.5 m urea. The affinity of VWF for collagen varies directly with multimer length. Therefore, cleavage of VWF multimers can be assayed as a decrease in collagen binding activity (21) (Fig. 5B). At the concentration tested, the activity of full-length recombinant ADAMTS13 was similar to that in an equal volume of plasma. As described for plasma ADAMTS13, the activity of recombinant ADAMTS13 was inhibited by chelation of divalent metal ions or by autoantibodies to ADAMTS13 from a patient with idiopathic TTP (22) confirming the specificity of the reaction. Construct del1, which lacks both CUB domains, had normal activity toward VWF. Further deletion of TSP1 domains 2–8 had a variable effect. Among four experiments, the average activity of construct del2 was ≈70% relative to the full-length protein, but the standard error was large, and the activity of del2 was not significantly different from that of del1 or full-length ADAMTS13. Deletion of the spacer domain in construct del3 abolished activity, and the shorter constructs del4, del5, and del6 also were inactive. The results obtained in the collagen-binding assay were confirmed by direct visualization of VWF multimers. Full-length ADAMTS13 and truncation mutants del1 and del2 digested VWF rapidly into small multimers, whereas constructs del3, del4, del5, and del6 had no discernable effect on the VWF multimer distribution (data not shown).
Fig. 5. ADAMTS13 variants expressed in COS-7 cells.
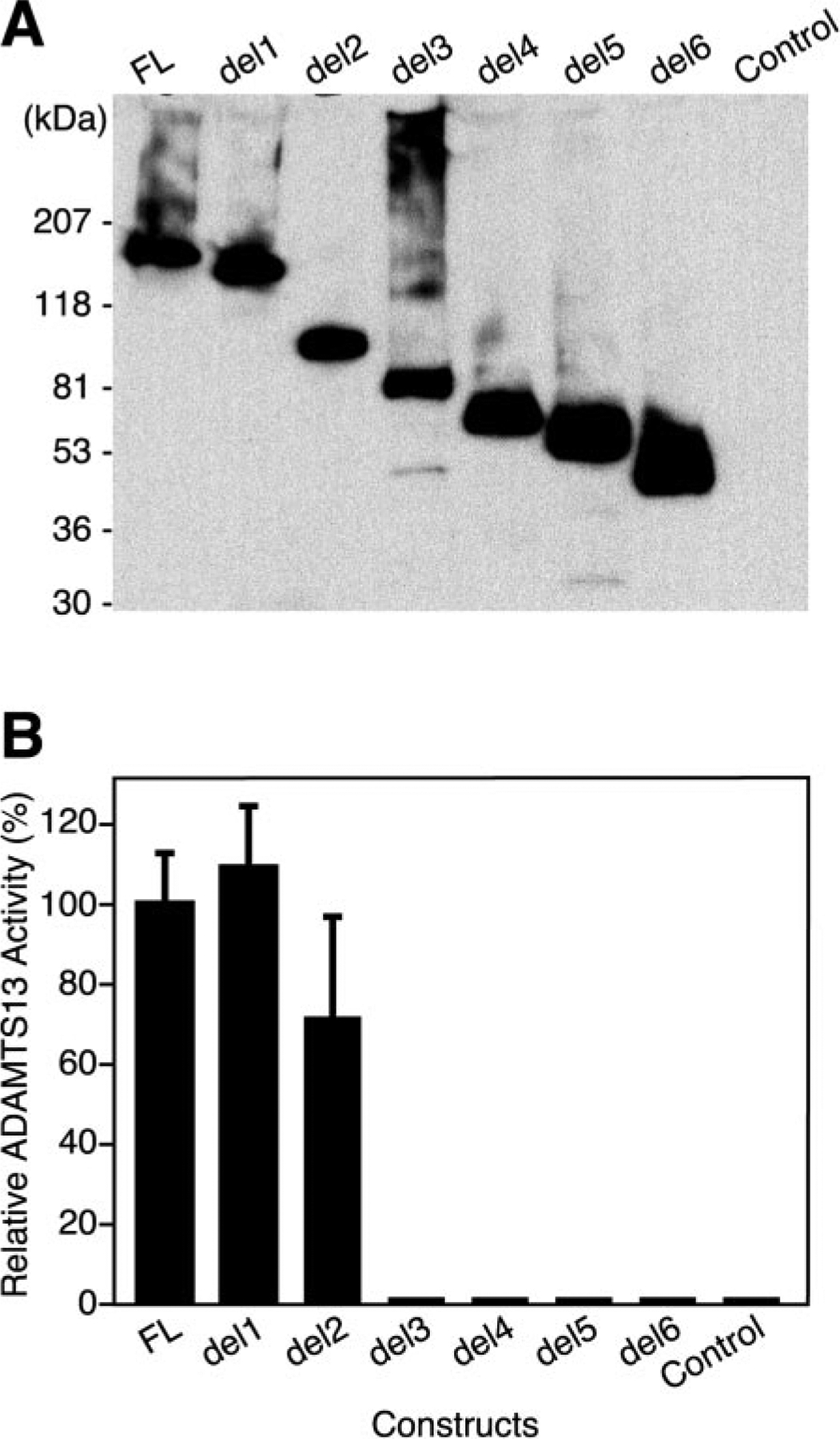
A, COS-7 cells were transiently transfected with plasmids encoding the indicated construct that is described under Fig. 1 or with vector only (Control). Conditioned medium was concentrated 5-fold, samples (50 μl) were analyzed by SDS-PAGE on a 5–15% gradient gel, and Western blotting was done with anti-V5 antibody. B, the ability of recombinant ADAMTS13 proteins to cleave plasma VWF was measured in a two-stage assay as described under “Experimental Procedures.” The relative concentration of recombinant ADAMTS13 proteins was determined by Western blotting with anti-V5 antibody and densitometry. Samples containing equivalent amounts of recombinant ADAMTS13 ~5 μl of concentrated conditioned medium) were incubated for 16 h with VWF substrate in buffer containing 5 mM Tris-HCl, pH 8.0, 3 mM BaCl2, and 1.5 m urea, and the integrity of the remaining VWF multimers was assessed in a collagen binding assay. The results were normalized to the values obtained with full-length ADAMTS13 (FL). The bars represent the S.E. of four independent assays.
Plasma ADAMTS13 (6, 7) and recombinant full-length ADAMTS13 (26) cleave the Tyr1605-Met1606 bond of the VWF subunit, generating characteristic fragments of 140 kDa and 176 kDa that also are produced during the normal catabolism of VWF in vivo (27). Active recombinant ADAMTS13 variants lacking the CUB domains (del1) or further lacking TSP1 repeats 2–8 (del2) produced VWF fragments similar to those generated by full-length recombinant or plasma ADAMTS13 (Fig. 6).
Fig. 6. Cleavage of VWF subunits by ADAMTS13 variants.
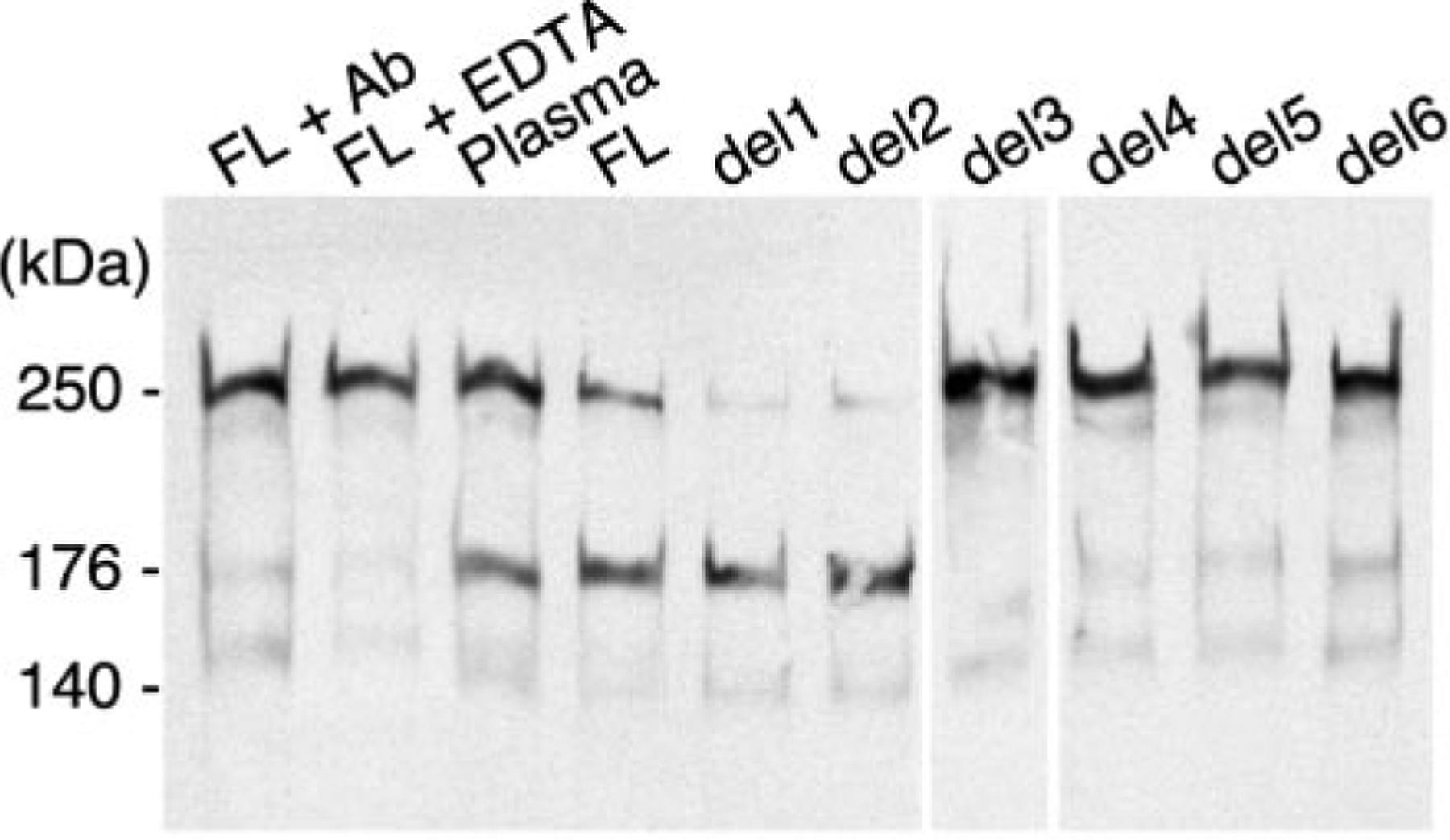
VWF (10 μg/ml) was incubated with the indicated recombinant ADAMTS13 protein expressed in COS-7 cells or with normal human plasma (Plasma) in buffer containing 5 mM Tris-HCl, pH 8.0, 3 mM BaCl2 and 1.5 m urea as described under “Experimental Procedures.” Control reactions containing full-length recombinant ADAMTS13 (FL) were performed in the presence of IgG (2.4 mg/ml) from a patient with autoimmune TTP (lane 1) or 10 mM EDTA (lane 2). Products were analyzed by SDS-PAGE under reducing conditions on a 5% gel and Western blotting with polyclonal anti-VWF antibody. The mass of the intact VWF subunit and the cleavage products generated by plasma ADAMTS13 are indicated at the left.
The results obtained with recombinant ADAMTS13 expressed in mammalian COS-7 cells were confirmed with selected variants expressed in baculovirus-infected Sf9 cells (Fig. 7). Proteins expressed in Sf9 cells had slightly lower apparent mass than their counterparts expressed in COS-7 cells, suggesting that Sf9 cells may add smaller oligosaccharides. Full-length ADAMTS13 expressed in both cell types gave rise to a similar C-terminal degradation product of ≈45 kDa (Fig. 7), suggesting that a site between TSP1 repeats 6 and 8 may be particularly susceptible to proteolysis.
Fig. 7. Comparison of ADAMTS13 variants expressed in COS-7 and Sf9 cells.
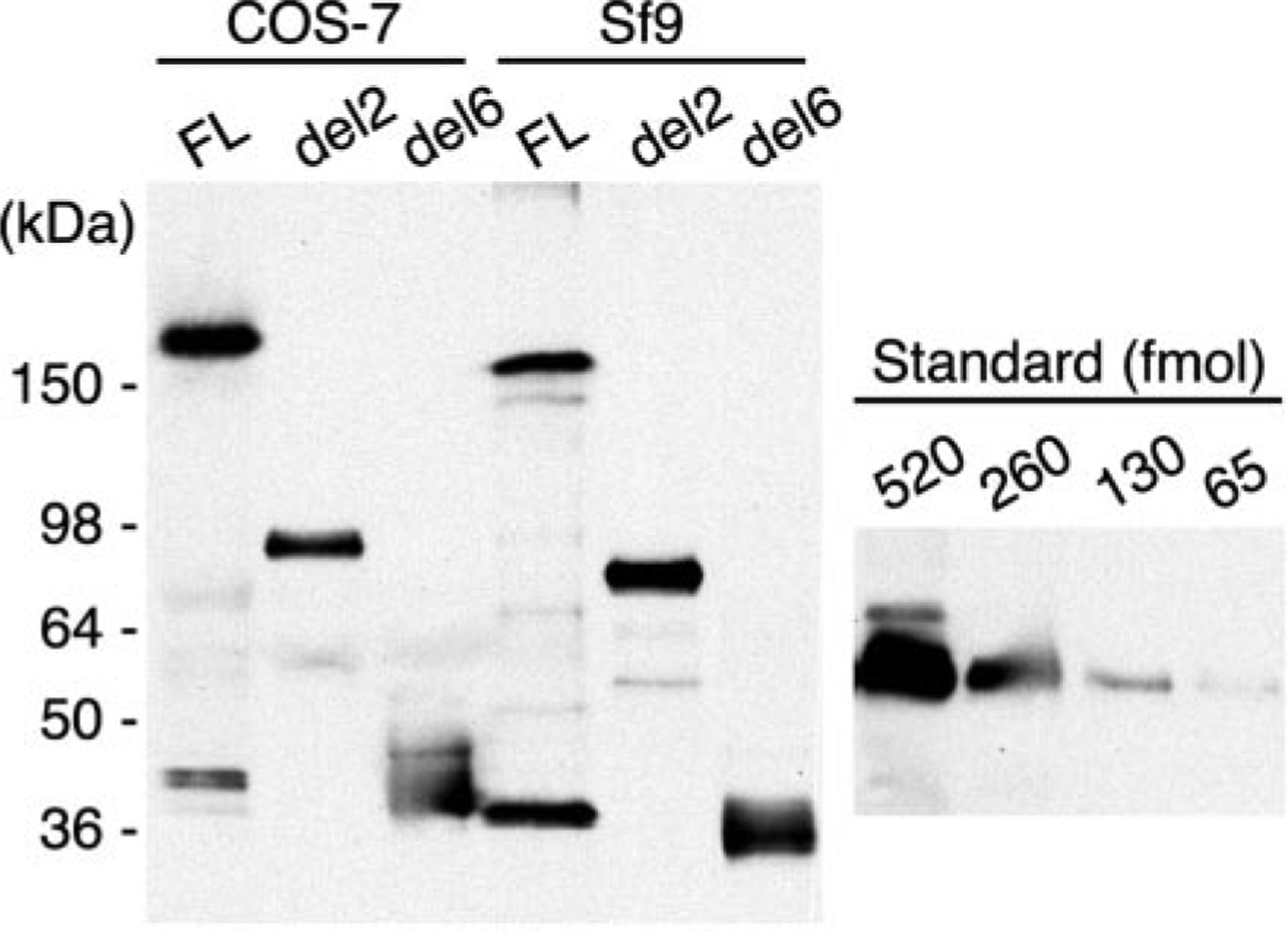
The indicated ADAMTS13 constructs were expressed by transient transfection of COS-7 cells or by baculovirus infection of Sf9 cells. Samples of concentrated conditioned medium were assayed for VWF cleaving activity in a collagen-binding assay. COS-7 cells: full-length (FL), 1.4 units/ml; del2, 0.73 units/ml, del6, 0 units/ml. Sf9 cells: full-length, 0.67 units/ml; del2, 1.42 units/ml; del6, 0 units/ml. The amount of recombinant protein was determined by SDS-PAGE, Western blotting with anti-V5 antibody and densitometry as described under “Experimental Procedures.” COS-7 cells: full-length, 12.9 nmol/liter; del2, 9.0 nmol/liter; del6, 12.9 nmol/liter. Sf9 cells: full-length, 6.4 nmol/liter; del2, 13.1 nmol/liter; del6, 13.4 nmol/liter. The signals obtained for the standard protein (Positope, Invitrogen) are shown at the right. Its apparent mass is 53 kDa, and its mass calculated from amino acid sequence is 47.7 kDa.
In COS-7 cells, all variants were expressed at a level of ≈9–13 nmol/liter conditioned medium. Surprisingly, expression in Sf9 cells was not markedly better. All species were expressed at ≈6–13 nmol/liter conditioned medium. Optimal expression required a low multiplicity of infection of 0.1, and almost no ADAMTS13 protein was recovered at a more typical multiplicity of infection of 1 or 5, suggesting that active ADAMTS13 may be toxic to insect cells.
Whether expressed in COS-7 cells or Sf9 cells, full-length ADAMTS13 and the variant truncated after the spacer domain (del2) had similar specific activities of ≈80–110 units/nmol (Fig. 7), confirming that domains C-terminal to the spacer domain are not required for proteolytic activity in vitro. If full-length recombinant and plasma ADAMTS13 (≈170 kDa) have similar specific activity, the results suggest that the plasma concentration of active ADAMTS13 is ≈1.6 μg/ml, which is similar to the value of 1 μg/ml based upon the recovery of ADAMTS13 during purification from plasma (11).
DISCUSSION
The complex modular structure of many ADAMTS proteases is highly conserved among vertebrates, suggesting that their noncatalytic domains perform specific selectable functions. The properties of several ADAMTS proteases supports this conclusion. For example, the Cys-rich and spacer domains and three TSP1 repeats of ADAMTS1 are necessary to bind heparin or extracellular matrix (28, 29). The C-terminal TSP1 domains of ADAMTS1 are reported to bind endothelial cells (29) and to have anti-angiogenic activity, which is attributed to binding and sequestration of vascular endothelial growth factor (30). The TSP1 motif of ADAMTS4 is required for aggrecan cleavage (31), and its Cys-rich and spacer domains bind sulfated glycosaminoglycans (32). ADAMTS12 also employs C-terminal TSP1 domains to bind extracellular matrix (33). Therefore, ADAMTS structures remote from the metalloprotease domain can participate in tissue localization, substrate recognition, and possibly other activities unrelated to proteolysis.
Although several ADAMTS proteases associate with extra-cellular matrix or cell surfaces, ADAMTS13 does not appear to fit this pattern. Full-length ADAMTS13 did not bind detectably to the surface of COS-7 cells (Fig. 3) and was not incorporated into the extracellular matrix of COS-1 or RFL-6 cells (Fig. 4). If ADAMTS13 binds cell surfaces or matrix in vivo, such interactions could depend on binding partners that are not expressed in these cultured cell lines.
The properties of truncated ADAMTS13 proteins suggest that substrate recognition requires the participation of several domains. Full-length recombinant ADAMTS13 was able to cleave VWF with apparently normal specificity, but truncation mutants lacking the spacer domain could not (Figs. 5–7). Therefore, TSP1 repeats 2–8 and the CUB domains are not required for VWF cleavage in vitro. Whether they are important under fluid shear stress or under more physiological conditions in vivo requires further study. If these C-terminal domains were necessary in vivo, one might predict the occurrence of patients with inherited TTP caused by mutations in the ADAMTS13 gene but with normal plasma ADAMTS13 activity upon laboratory testing. To date, however, all reported mutations distal to the spacer domain have been associated with severe ADAMTS13 deficiency (15, 19, 34).
Deletion of the ADAMTS13 spacer domain abolished the activity of construct del3 toward VWF (Figs. 5–7), suggesting that the spacer domain participates in substrate recognition. This conclusion would be strengthened if construct del3 and smaller variants were shown to have catalytic activity toward another substrate, to react with an active site inhibitor, or to have native conformation by another method. Unfortunately, no ADAMTS13 substrates other than VWF and certain fragments thereof have been reported. A variety of other proteins and small synthetic substrates have been tested, but none were cleaved detectably; also, with the exception of metal-ion chelators (6, 7), no synthetic or natural active site inhibitor including α2-macroglobulin4 has been shown to react with ADAMTS13. The efficient synthesis and secretion of small inactive ADAMTS13 constructs (Figs. 2 and 7) suggest they are folded properly, but direct evidence for structural integrity will require a different experimental approach.
Although not addressed in this study, the more proximal disintegrin and Cys-rich domains and the first TSP1 repeat may also participate in substrate recognition. For example, the mutation P475S in the ADAMTS13 Cys-rich domain was identified in a Japanese patient with inherited TTP; the corresponding recombinant mutant ADAMTS13 was secreted efficiently but had extremely low activity toward VWF (18). Other missense mutations that cause inherited ADAMTS13 deficiency have been found in the disintegrin, Cys-rich, and first TSP1 domains (15, 19), but protein expression studies have not yet been reported for them.
Proteolytic processing can generate distinct forms of several ADAMTS proteases, and this variation may control tissue localization or substrate specificity. For example, ADAMTS4 is autoproteolytically cleaved into products that lack variable portions of the spacer and Cys-rich domains, and the truncated forms have reduced affinity for sulfated glycosaminoglycans (32). Some cells remove the C-terminal TSP1 repeats from ADAMTS1, which reduces its affinity for heparin and endothelial cells (29). At least one site within the C-terminal TSP1 repeats of recombinant full-length ADAMTS13 appears to be sensitive to cleavage (Fig. 7), suggesting that it could be subject to proteolytic processing in vivo. Such processing could occur intracellularly as indicated by the sensitivity of the C-terminal fragment to digestion with endoglycosidase H (Fig. 3). Additional structural heterogeneity for several ADAMTS proteases may be produced by alternative mRNA splicing (16, 35, 36), although the biological significance of the predicted products has not been established. In the case of ADAMTS13, alternatively spliced forms have been cloned that encode variants truncated after the metalloprotease domain, the spacer domain, the second TSP1 repeat, or the first CUB domain (13–15). Similar deletion mutants (Fig. 1) were secreted efficiently by COS-7 cells and Sf9 cells, and ADAMTS13 purified from plasma was shown to contain species of 110, 130, 140, and 150 kDa that had the same N-terminal amino acid sequence (11). Thus, truncated forms of ADAMTS13 may circulate in vivo and could potentially be derived from alternatively spliced transcripts. Based on the properties of recombinant ADAMTS13 mutants (Figs. 5–7), such variation in the complement of C-terminal domains would dramatically affect ADAMTS13 substrate specificity.
Acknowledgments—
We thank Claudine Mazurier (CRTS, Lille, France) for providing purified human plasma VWF and Robert Mecham (Washington University, St. Louis, MO) for providing RFL-6 cells and for advice on assessing the extracellular matrix localization of proteins.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: TTP, thrombotic thrombocytopenic purpura; PBS, phosphate-buffered saline; TSP1, thrombospondin type 1; VWF, von Willebrand factor.
J. E. Sadler, unpublished observations.
X. Zheng, K. Nishio, E. M. Majerus, and J. E. Sadler, unpublished observations.
X. Zheng and J. E. Sadler, unpublished data.
REFERENCES
- 1.George JN (2000) Blood 96, 1223–1229 [PubMed] [Google Scholar]
- 2.Furlan M, and Lämmle B (2001) Best Pract. Res. Clin. Haematol 14, 437–454 [DOI] [PubMed] [Google Scholar]
- 3.Moake JL (2002) Annu. Rev. Med 53, 75–88 [DOI] [PubMed] [Google Scholar]
- 4.Rock GA, Shumak KH, Buskard NA, Blanchette VS, Kelton JG, Nair RC, and Spasoff RA (1991) N. Engl. J. Med 325, 393–397 [DOI] [PubMed] [Google Scholar]
- 5.Moake JL, Rudy CK, Troll JH, Weinstein MJ, Colannino NM, Azocar J, Seder RH, Hong SL, and Deykin D (1982) N. Engl. J. Med 307, 1432–1435 [DOI] [PubMed] [Google Scholar]
- 6.Furlan M, Robles R, and Lämmle B (1996) Blood 87, 4223–4234 [PubMed] [Google Scholar]
- 7.Tsai H-M (1996) Blood 87, 4235–4244 [PubMed] [Google Scholar]
- 8.Furlan M, Robles R, Galbusera M, Remuzzi G, Kyrle PA, Brenner B, Krause M, Scharrer I, Aumann V, Mittler U, Solenthaler M, and Lämmle B (1998) N. Engl. J. Med 339, 1578–1584 [DOI] [PubMed] [Google Scholar]
- 9.Tsai HM, and Lian EC (1998) N. Engl. J. Med 339, 1585–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujikawa K, Suzuki H, McMullen B, and Chung D (2001) Blood 98, 1662–1666 [DOI] [PubMed] [Google Scholar]
- 11.Gerritsen HE, Robles R, Lämmle B, and Furlan M (2001) Blood 98, 1654–1661 [DOI] [PubMed] [Google Scholar]
- 12.Hurskainen TL, Hirohata S, Seldin MF, and Apte SS (1999) J. Biol. Chem 274, 25555–25563 [DOI] [PubMed] [Google Scholar]
- 13.Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, and Fujikawa K (2001) J. Biol. Chem 276, 41059–41063 [DOI] [PubMed] [Google Scholar]
- 14.Soejima K, Mimura N, Hirashima M, Maeda H, Hamamoto T, Nakagaki T, and Nozaki C (2001) J. Biochem. (Tokyo) 130, 475–480 [DOI] [PubMed] [Google Scholar]
- 15.Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, Yang AY, Siemieniak DR, Stark KR, Gruppo R, Sarode R, Shurin SB, Chandrasekaran V, Stabler SP, Sabio H, Bouhassira EE, Upshaw JD Jr., Ginsburg D, and Tsai HM (2001) Nature 413, 488–494 [DOI] [PubMed] [Google Scholar]
- 16.Cal S, Obaya AJ, Llamazares M, Garabaya C, Quesada V, and Lopez-Otin C (2002) Gene (Amst.) 283, 49–62 [DOI] [PubMed] [Google Scholar]
- 17.Sadler JE (2002) Proc. Natl. Acad. Sci. U. S. A 99, 11552–11554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kokame K, Matsumoto M, Soejima K, Yagi H, Ishizashi H, Funato M, Tamai H, Konno M, Kamide K, Kawano Y, Miyata T, and Fujimura Y (2002) Proc. Natl. Acad. Sci. U. S. A 99, 11902–11907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schneppenheim R, Budde U, Oyen F, Angerhaus D, Aumann V, Drewke E, Hassenpflug W, Haberle J, Kentouche K, Kohne E, Kurnik K, Mueller-Wiefel D, Obser T, Santer R, and Sykora KW (2003) Blood 101, 1845–1850 [DOI] [PubMed] [Google Scholar]
- 20.Zheng X, and Sadler JE (2002) J. Biol. Chem 277, 6858–6863 [DOI] [PubMed] [Google Scholar]
- 21.Gerritsen HE, Turecek PL, Schwarz HP, Lämmle B, and Furlan M (1999) Thromb. Haemostasis 82, 1386–1389 [PubMed] [Google Scholar]
- 22.Zheng X, Pallera AM, Goodnough LT, Sadler JE, and Blinder MA (2003) Ann. Intern. Med 138, 105–108 [DOI] [PubMed] [Google Scholar]
- 23.Raines G, Aumann H, Sykes S, and Street A (1990) Thromb. Res 60, 201–212 [DOI] [PubMed] [Google Scholar]
- 24.Zheng X, Lu D, and Sadler JE (1999) J. Biol. Chem 274, 1596–1606 [DOI] [PubMed] [Google Scholar]
- 25.Segade F, Trask BC, Broekelmann TJ, Pierce RA, and Mecham RP (2002) J. Biol. Chem 277, 11050–11057 [DOI] [PubMed] [Google Scholar]
- 26.Plaimauer B, Zimmermann K, Volkel D, Antoine G, Kerschbaumer R, Jenab P, Furlan M, Gerritsen H, Lämmle B, Schwarz HP, and Scheiflinger F (2002) Blood 100, 3626–3632 [DOI] [PubMed] [Google Scholar]
- 27.Dent JA, Berkowitz SD, Ware J, Kasper CK, and Ruggeri ZM (1990) Proc. Natl. Acad. Sci. U. S. A 87, 6306–6310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuno K, and Matsushima K (1998) J. Biol. Chem 273, 13912–13917 [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez-Manzaneque JC, Milchanowski AB, Dufour EK, Leduc R, and Iruela-Arispe ML (2000) J. Biol. Chem 275, 33471–33479 [DOI] [PubMed] [Google Scholar]
- 30.Luque A, Carpizo DR, and Iruela-Arispe ML (2003) J. Biol. Chem, 278, 23656–23665 [DOI] [PubMed] [Google Scholar]
- 31.Tortorella M, Pratta M, Liu RQ, Abbaszade I, Ross H, Burn T, and Arner E (2000) J. Biol. Chem 275, 25791–25797 [DOI] [PubMed] [Google Scholar]
- 32.Flannery CR, Zeng W, Corcoran C, Collins-Racie LA, Chockalingam PS, Hebert T, Mackie SA, McDonagh T, Crawford TK, Tomkinson KN, LaVallie ER, and Morris EA (2002) J. Biol. Chem 277, 42775–42780 [DOI] [PubMed] [Google Scholar]
- 33.Cal S, Arguelles JM, Fernandez PL, and Lopez-Otin C (2001) J. Biol. Chem 276, 17932–17940 [DOI] [PubMed] [Google Scholar]
- 34.Antoine G, Zimmermann K, Plaimauer B, Grillowitzer M, Studt JD, Lämmle B, and Scheiflinger F (2003) Br. J. Haematol 120, 821–824 [DOI] [PubMed] [Google Scholar]
- 35.Colige A, Sieron AL, Li SW, Schwarze U, Petty E, Wertelecki W, Wilcox W, Krakow D, Cohn DH, Reardon W, Byers PH, Lapiere CM, Prockop DJ, and Nusgens BV (1999) Am. J. Hum. Genet 65, 308–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernandes RJ, Hirohata S, Engle JM, Colige A, Cohn DH, Eyre DR, and Apte SS (2001) J. Biol. Chem 276, 31502–31509 [DOI] [PubMed] [Google Scholar]