

Abstract
INTRODUCTION
Alzheimer's disease (AD) is the predominant dementia globally, with heterogeneous presentation and penetrance of clinical symptoms, variable presence of mixed pathologies, potential disease subtypes, and numerous associated endophenotypes. Beyond the difficulty of designing treatments that address the core pathological characteristics of the disease, therapeutic development is challenged by the uncertainty of which endophenotypic areas and specific targets implicated by those endophenotypes to prioritize for further translational research. However, publicly funded consortia driving large‐scale open science efforts have produced multiple omic analyses that address both disease risk relevance and biological process involvement of genes across the genome.
METHODS
Here we report the development of an informatic pipeline that draws from genetic association studies, predicted variant impact, and linkage with dementia associated phenotypes to create a genetic risk score. This is paired with a multi‐omic risk score utilizing extensive sets of both transcriptomic and proteomic studies to identify system‐level changes in expression associated with AD. These two elements combined constitute our target risk score that ranks AD risk genome‐wide. The ranked genes are organized into endophenotypic space through the development of 19 biological domains associated with AD in the described genetics and genomics studies and accompanying literature. The biological domains are constructed from exhaustive Gene Ontology (GO) term compilations, allowing automated assignment of genes into objectively defined disease‐associated biology. This rank‐and‐organize approach, performed genome‐wide, allows the characterization of aggregations of AD risk across biological domains.
RESULTS
The top AD‐risk‐associated biological domains are Synapse, Immune Response, Lipid Metabolism, Mitochondrial Metabolism, Structural Stabilization, and Proteostasis, with slightly lower levels of risk enrichment present within the other 13 biological domains.
DISCUSSION
This provides an objective methodology to localize risk within specific biological endophenotypes and drill down into the most significantly associated sets of GO terms and annotated genes for potential therapeutic targets.
Keywords: Alzheimer's disease, biological domain, endophenotype, genetics, proteomics, transcriptomics
1. BACKGROUND
Alzheimer's disease (AD) is a complex, heterogeneous, neurodegenerative disease defined by the extracellular aggregation of amyloid plaques and the intracellular accumulation of neurofibrillary tangles composed of paired helical filaments of hyperphosphorylated tau protein. 1 While amyloid and tau are hallmarks of the disease, recent large‐scale multi‐omic analyses emphasize the complexity of interwoven biological processes associated with AD pathogenesis. Over a decade ago the National Institute on Aging and the Alzheimer's Association began a joint initiative to capture this complexity within a disease ontology, the Common Alzheimer's and Related Dementias Research Ontology (CADRO). 2 The goal behind CADRO's development was to articulate the biological processes and cell types involved in AD pathology and progression and since its launch has been employed to characterize candidate AD therapeutics in clinical trials and track the diversification in investigational areas over time. 3 Establishing a diverse target portfolio enhances the potential for translational impact; the availability of therapeutic targets implicated in a variety of disease‐linked biology enables intervention through distinct mechanisms, which may be necessary to address the heterogeneous AD population and to be deployed in a coordinated manner. 4 While useful to classify the mechanism of action of trial therapeutics, the alignment between the gene target of a trial agent and its ontological classifier is performed manually based on the judgment of domain experts, which cannot be scaled genome‐wide without computationally amenable definitions.
A driving force behind the diversification of the AD target portfolio is an expanding view of AD biology due, in part, to recent efforts that have amassed a wealth of disease‐relevant molecular data from various patient cohorts. The Accelerating Medicines Partnership for Alzheimer's Disease (AMP‐AD) consortium, for example, has generated multiple omics datasets from post mortem brain samples (eg, including genomic, transcriptomic, proteomic, metabolomic) and made these data openly available on the AD Knowledge Portal. 5 These systems‐level investigations into AD are a rapidly increasing information domain, and each new study contributes large datasets that provide an objective view of disease processes across different biological vantage. However, each of these datasets can suggest hundreds of genes as potential new therapeutic targets without clear priority. Genome‐wide association studies (GWAS) alone have identified over 75 risk loci, 6 and analyses of transcriptomic 7 – 10 and proteomic 11 , 12 data have identified dozens of co‐expression modules that consist of hundreds to thousands of genes or proteins each. Currently over 600 targets have been nominated by AMP‐AD researchers for further therapeutic development (agora.adknowledgeportal.org). Furthermore, each of these studies implicates a diverse set of biological processes and endophenotypes that are altered in the genesis of, and response to, late‐onset progressive neurodegeneration in AD. The difficulty in performing a unified analysis of these divergent datasets is twofold: (1) there is no objective categorization of genes into disease‐relevant endophenotypes and (2) there is no genome‐wide methodology to integrate and assess AD‐associated risk measured in different studies.
In this paper, we describe data integration across modalities to score, rank, and organize potential AD therapeutic targets genome wide. First, we identified 19 biological domains that correspond to AD‐associated endophenotypes and define them using sets of Gene Ontology (GO) terms, with the intent to keep each domain siloed in a biologically coherent fashion. This provides an objective strategy to characterize gene targets into AD endophenotypes. Second, we developed a target risk score (TRS) that integrates signatures of risk from genetic association studies as well as signatures of differential expression in transcriptomic and proteomic data. We show that these tools can be applied to assess which specific genes within large datasets are elevated in disease risk and to group the most risk‐enriched genes within common biological domains, providing a framework for analysis that can be employed across research studies. While we observed that AD risk was distributed across all 19 biological domains, we found that the biological domains demonstrating the greatest AD risk association were Synapse, Immune Response, Lipid Metabolism, Mitochondrial Metabolism, Structural Stabilization, and Proteostasis. Each domain can be examined in more detail by elaborating specific elements of a biological process that are particularly risk‐enriched. For example, we identify electron transport chain complex I‐related factors within mitochondrial metabolism as one such focal point. The system described here represents the most comprehensive to date, providing genomic coverage of risk mapped onto known AD endophenotypes, spanning 27 genetic association studies, transcriptomic signatures from 1699 brains, proteomic signatures from 1188 brains, and 7127 GO terms structured within the 19 biological domain classifications. These tools are openly available to the research community as a part of the Target Enablement to Accelerate Therapy Development in AD (TREAT‐AD) efforts to facilitate the continued diversification of the AD drug development pipeline.
RESEARCH IN CONTEXT
Systematic review: The data relevant to the review covered two areas: multi‐omic assessment of AD risk and disease‐linked endophenotypes. The authors reviewed the pertinent data in the literature in PubMed in addition to data within public repositories, such as AD Knowledge Portal, the GWAS Catalogue, and the Gene Ontology. All data sources employed are appropriately cited.
Interpretation: The analysis in this paper shows that endophenotypes identified in AD are enriched in multi‐omic association, supporting the linkage between AD pathogenesis and the mapped biological domains.
Future directions: The work in this paper broadly defines the endophenotypic areas involved in disease progression, develop and demonstrate a machine‐readable methodology for categorizing large datasets, and a scoring methodology for determining gene‐based risk genome wide. These methods will be employed in expanded multi‐omic data harmonization; further, each domain will be further subdivided by risk enrichment for greater biological process resolution.
2. METHODS
2.1. Alzheimer's disease biological domains
The development of the biological domains broadly encompasses two distinct processes: (1) the selection and (2) definition of each biological domain. The selection of the biological domains is guided by the attempt to exhaustively identify the endophenotypes and biological areas linked to AD pathogenesis. One of the most developed resources describing AD‐relevant endophenotypes is the Common Alzheimer's and Related Dementias Research Ontology (CADRO). 2 As CADRO is already in standard use for drug development classification, we leveraged this resource to help guide the initial stages of identification of relevant biological domains of AD (Table S1). The identification of biological domains was expanded beyond CADRO to be maximally inclusive of data derived from large‐scale consortia studies in different areas of disease relevance (Figure S1). Definition of the biological domains required a strategy that was (1) objective, (2) automatable, (3) easily intelligible, and (4) communally modifiable. Based on these criteria, we elected to use an exhaustive elaboration of GO terms associated with each biological domain as the core definition (Figure S2 and Table S2, see supplemental methods for more details).
2.2. TRS development and process
The goal behind the generation of the TREAT‐AD TRS is to develop a scoring infrastructure to assess AD risk association genome wide, leveraging and integrating all available data types. The contributing data types may evolve over time. Here we have initiated the process drawing from genetics, transcriptomics, and proteomics. The composite scoring method delineated below, and further elaborated in the supplemental methods, enables us to rank all genes for association with AD pathogenesis.
2.2.1. Genetic risk score component
The genetic component of AD risk, or genetic risk score, queries all ensemble gene loci for genetic associations with AD‐relevant traits across a range of studies (both GWAS/GWAX and quantitative trait locus [QTL]; see Table S3 for all studies queried). Variant‐level associations were assigned to genes, and summary metrics were calculated for each gene to capture pan‐study association strength and rank the predicted severity of both coding and non‐coding variants. Additionally, phenotype ontologies were queried to identify genes with phenotypic annotations that overlap with either AD [MONDO:0004975] or dementia [MONDO:0001627] (additional details in the supplementary methods). The genetic risk score for a target (Table S4) is calculated as the sum of the inverse rank for each of the following evidence categories, scaled to a total of three points: number of GWAS studies, minimum GWAS p value across studies, mean rank of minimum GWAS p value across studies, number of QTL studies, minimum QTL false discovery rate (FDR) across studies, mean rank of minimum QTL FDR across studies, coding variant summary, non‐coding variant summary, human phenotype score, model organism phenotype score, and MODEL‐AD strain and correlation.
2.2.2. Multi‐omic risk score component
A ratio of means meta‐analysis with a random effects model 13 was applied separately to both transcriptomics (Table S5) and proteomics (Table S6) datasets; modality‐specific weights were subsequently generated (see supplemental methods for details). To harness weights into a single value, a weighted adjustment by modality was applied (Table S7). Beyond collapsing the weight values into a single statistic, the omics harness is designed to weight proteomics more heavily than transcriptomics to account for the practicality of therapeutic intervention strategies at the protein level rather than the transcript level. Genes were ranked by their harness value and ranks were converted to 0–1 decimal. A binomial model was fit to predict the 0–1 adjusted rank from the second‐degree polynomial of the log harness values. This model was used to compute a multi‐omic weight from zero to one, where one corresponds to a greater harness value. For genes with no statistically significant RNA or protein values, this weight was set to zero and the weight was then multiplied by 2 to attain the points value of the multi‐omic risk score component (Table S8).
2.3. GSEA analysis using the biological domains
To assess the enrichment of GO terms subordinate to any of the biological domains, Gene Set Enrichment Analysis (GSEA) was performed using the gseGO function from the clusterProfiler R package, 14 and the results were then categorized into biological domains based on the GO ID of enriched terms. The input for each enrichment analysis were non‐zero target scores, in descending order. We performed this analysis separately for each component score: genetics, multi‐omics, and combined target risk. The displayed results include the normalized enrichment score (NES) as well as the Benjamini‐Hochberg corrected p value (p adj) for GO terms annotated to each biological domain. For clinical trial target enrichment analyses, the identities of genes and proteins in each set were used for GO term overrepresentation analysis using the “enrich GO” function in the clusterProfiler package, and the results were then categorized into biological domains based on the GO ID of enriched terms. A graphical model of the analysis pathway demonstrates how the pipeline processes are integrated into one workflow (Figure S3).
2.4. Other data
Several other datasets are used in this work. The list of AD GWAS hits is derived from integrating genes identified from three sources: the supplementary table that accompanies Neuner et al. (2020), 15 Table S5 from Bellenguez et al. (2022), which identifies all genome‐wide significant loci, 6 and the list of AD loci with genetic evidence compiled by the ADSP Gene Verification Committee (adsp.niagads.org/index.php/gvc‐top‐hits‐list/). The Open Targets 16 disease association scores for AD (https://platform.opentargets.org/disease/MONDO_0004975/associations), including data type scores, were accessed using the Open Targets application programming interface (API). For our investigation of clinical trial target genes, we obtained the list of “known drugs” from the Open Targets API (Table S9). Open Targets considers known drugs to be those drugs for which there is clinical precedence for investigation or approved for use in AD with a curated mechanism of action. The identities of currently nominated targets from the AMP‐AD consortium, listed on the Agora site (agora.adknowledgeportal.org/genes/nominated‐targets), were accessed using the synapseR R client. 17
3. RESULTS
3.1. AD biological domains
The primary goal of defining a structured set of biological domains is to standardize areas of disease‐associated biology to serve as a common reference point for the analysis of large datasets. Leveraging the CADRO ontology, with augmentation through literature curation and biological alignment with the employed multi‐omic datasets (see methods), we established 19 biological domains that covered the AD endophenotypic space (Table S1). We used 7127 unique GO terms (16.4% of all terms in the ontology) to annotate the biological domains. The number of terms annotated to each domain varies enormously (Figure 1A, Table S2); the Synapse domain requires 1379 terms to define, while Tau Homeostasis only requires 10 terms. The two smallest biological domains (Tau Homeostasis and APP Metabolism) focus on gene‐centric processes, requiring fewer terms to annotate than larger domains with broader biological focus, such as Lipid Metabolism or Proteostasis. Each biological domain was designed to be discrete, which is reflected in the sparse overlap of shared GO terms between domains (Figure 1A). The shared terms are truly inextricable; for example, the term “mitophagy,” defined as the “selective autophagy process in which a mitochondrion is degraded by macroautophagy,” legitimately resides in both the Mitochondrial Metabolism and Autophagy domains. In these cases, the repetition of a term between domains was allowed, as it represents a meaningful intersection of biological areas.
FIGURE 1.
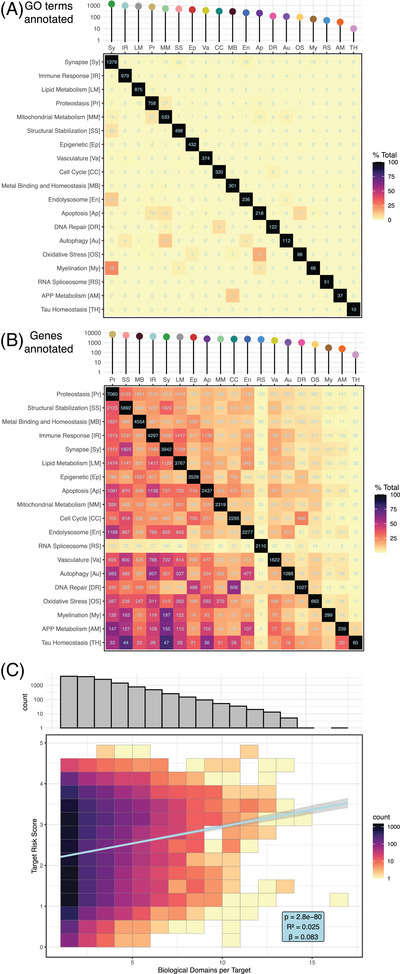
Biological domain demographics. (A) Number of GO terms employed to define each biological domain, shown as an interaction plot, with total number of terms per domain along diagonal and number of terms shared between domains arranged in external rows and columns. (B) Number of genes in each biological domain, organized as in (A), with total genes within each domain along diagonal and pairwise genes shared between domains heat mapped within rows and columns. (C) The top histogram shows the frequency of a gene annotation across the 19 biological domains for genes annotated to at least one biological domain. The histogram shows a decline in the number of genes annotated to multiple biological domains, with genes mapping to a single biological domain being the most numerous (4258). The lower plot shows the positive correlation between gene annotation in multiple biological domains and its target risk score, with higher scoring genes participating in multiple biological domains.
There is a greater overlap between genes annotated to different biological domains compared to the terms that overlap (Figure 1B). The number of genes annotated to each domain is roughly proportional to the number of GO terms per domain, with almost two orders of magnitude separating the largest domain (Proteostasis) and the smallest (Tau Homeostasis). Many genes are annotated to multiple GO terms subordinate to distinct biological domains, which may represent pleiotropic functions, a convergence of related processes, or both. While a plurality of annotated genes (30%) are annotated to a single biological domain, many participate in multiple domains each (Figure 1C), and genes annotated to more biological domains tend to have higher overall TRSs (see following discussion) (Pearson r = 0.158, p = 2.8 × 10−80).
3.2. TRS overview
The TREAT‐AD TRS is a metric designed to rank potential disease involvement of genes based on multiple independent lines of evidence to objectify the prioritization of potential targets and disease‐linked biology. The TRS has two components, genetic risk and multi‐omic risk, each derived from a meta‐analysis harmonizing multiple datasets. The genetic risk component is weighted more heavily, receiving up to three points, while multi‐omic risk has a maximum of two points. The rationale for providing more weight to the genetic component reflects the greater success in clinical trials for targets with genetic support. 18
3.2.1. Genetic risk score component
The genetic risk score component is a summary of genetic evidence supporting the target gene's association with late‐onset AD (LOAD), drawing from GWAS and quantitative trait locus (QTL) studies. 19 – 21 In total, 27 different association studies were queried (Table S3, Figure S4). We also assessed the predicted severity of identified variants (Figure S5) and phenotype overlap (Figure S6) between each gene and disease‐relevant terms. Gene‐level results for all evidence sources were summed to generate the genetic risk score (Figure 2A, Table S4). Genes contained within known AD GWAS loci are enriched among the top scores, as are gene targets identified by Open Targets (https://www.opentargets.org/), 16 , 22 a large‐scale effort to rank genes based on genetic support for translational relevance (Figure 2A). The TREAT‐AD genetics score has a weak positive correlation (Pearson r = 0.23, p = 8.4 × 10−6) with the Open Targets genetic association score for AD (Figure 2B), which is stronger when restricted to known AD GWAS genes (Pearson r = 0.29, p = 1.7 × 10−2).
FIGURE 2.
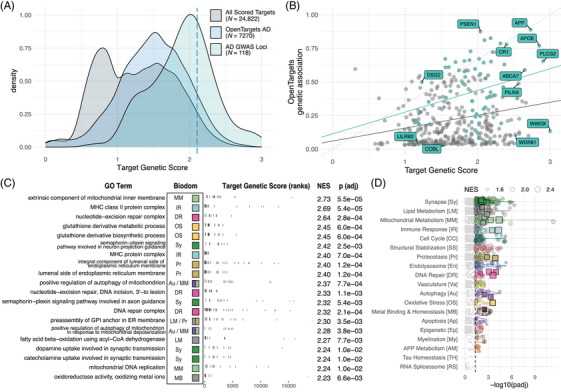
Genetic risk score. (A) Distribution of all genetic scores (gray), subset of targets evaluated by Open Targets platform (blue), and set of defined AD GWAS loci derived from various sources (green). Dashed blue line indicates the 95th percentile score. (B) Comparison of TREAT‐AD genetic risk score and Open Targets genetic association score. Plotted in green are the set of all genes within known AD GWAS loci scored by both metrics, several of which are labeled. The dashed line represents an equivalent score on each metric, and the solid color lines are the linear fit to each set. (C) Top GO terms significantly enriched using the TREAT‐AD target genetics score, arranged by normalized enrichment score (NES). For each GO term, the associated biological domains are indicated by the filled square and abbreviations (biological domain colors and abbreviations can be referenced from panel D). (D) Enrichment statistics for all biological domain terms. Each point is a GO term within the indicated biological domain and the size of the point is scaled by the GSEA normalized enrichment score (NES). The GO terms identified in panel (C) are indicated as square points with bold borders. The biological domains are ordered on the y‐axis by the number of significantly enriched GO terms identified from each domain.
GSEA using the genetics score to rank genes identifies significant GO terms from 17 biological domains; the biological domains with the largest number of enriched GO terms are Synapse, Lipid Metabolism, and Structural Stabilization (Figure 2C,D, Table S10). The Open Targets genetic association score enriches GO terms from 10 biological domains, with terms in the APP Metabolism domain by far the most significantly enriched and Synapse, Immune Response, and Lipid Metabolism being the domains with the most enriched terms (Figure S7C). The relative emphasis of APP Metabolism from the Open Targets score likely reflects the inclusion of evidence from early‐onset dominantly inherited AD, whereas the TREAT‐AD genetics score draws primarily from genetic associations of LOAD, the predominant and sporadic form of the disease. Notably, Synapse, Lipid Metabolism, Structural Stabilization, and Immune Response are among the biological domains with the largest number of enriched GO terms for both scores.
3.2.2. Multi‐omic risk score component
The multi‐omic risk score is a summary metric encapsulating available evidence supporting whether gene expression is altered in the brains of AD patients. The score makes use of proteomic and transcriptomic datasets generated as part of AMP‐AD consortium efforts (Tables S5 and S6). For each data modality (ie, transcriptomic or proteomic), a meta‐analysis of samples is used to generate weights for significantly differentially expressed genes based on observed fold changes (Figure 3A,B). Analyzing the directionality of expression change using GSEA, we find that, for both transcriptomics (Figure 3C, Figure S8C) and proteomics (Figure 3D, Figure S8D), Synapse and Mitochondrial Metabolism are the biological domains with the largest number of downregulated GO terms, and Immune Response and Structural Stabilization are the biological domains with the largest number of upregulated GO terms.
FIGURE 3.
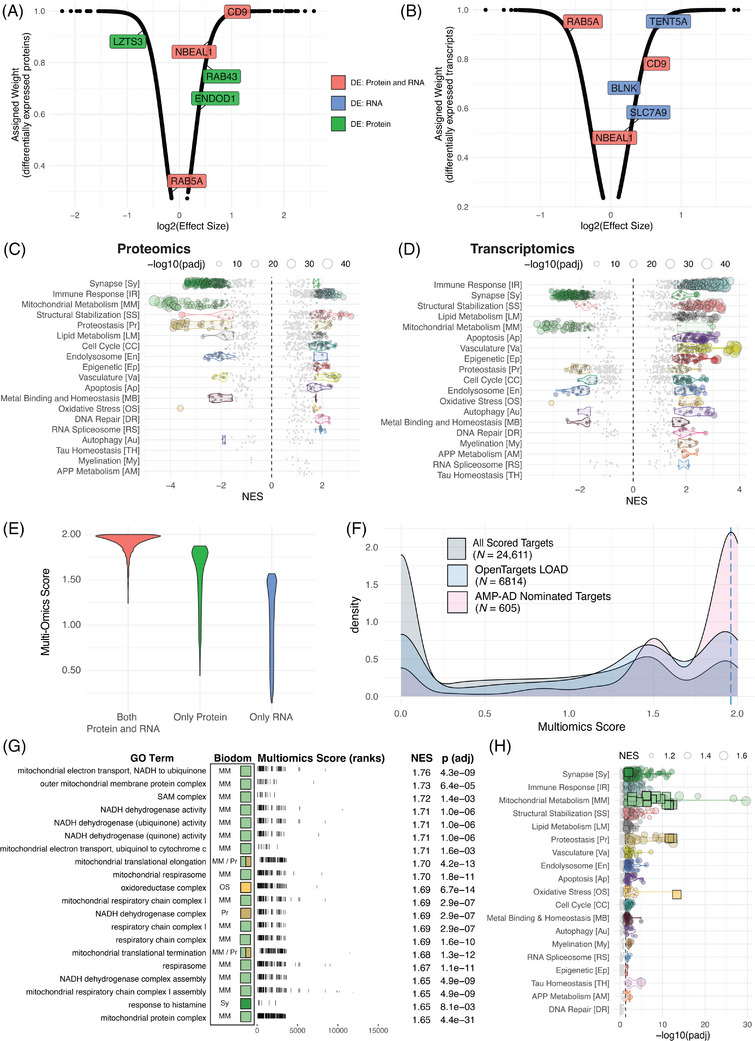
The multi‐omic score component of target risk score (TRS). Assigned weights as a function of log(effect size) of significantly differentially expressed (A) transcripts and (B) proteins. Labeled points in (A) and (B) represent example genes where the color indicates significantly differential expression in only RNA‐seq (blue), only proteomics (green), or both RNA‐seq and proteomics (red). Enrichment statistics for all biological domain GO terms based on proteomics (C) and transcriptomics (D) meta‐analysis treatment effect. Each point is a GO term within the indicated biological domain, the size of the point is scaled by the GSEA adjusted p value, and the NES indicates if the GO term is up‐ or downregulated. The biological domains are ordered on the y‐axis by the number of significantly enriched GO terms identified from each domain. (E) Assigned weights as a function of log(effect size) of significantly differentially expressed combined weight values as a function of log10 (harness adjusted scaled values), scaled to 2. (F) The distribution of multi‐omic scores for all scored genes (gray), those scored by the Open Targets platform (blue), and genes nominated by a team from the AMP‐AD consortium (pink). Dashed blue line indicates 95th percentile score. (G) Top GO terms significantly enriched using TREAT‐AD target multi‐omic score, arranged by normalized enrichment score (NES). For each GO term, the associated biological domains are indicated by the filled square and abbreviations (biological domain colors and abbreviations can be referenced from panel H). (H) Enrichment statistics for all biological domain terms. Each point is a GO term within the indicated biological domain, and the size of the point is scaled by the GSEA NES. The GO terms identified in panel (G) are indicated as square points with bold borders. The biological domains are ordered on the y‐axis by the number of significantly enriched GO terms identified from each domain.
The calculated weights for each modality are combined using a scoring harness (Table S7) that yields a higher score for targets with evidence for both, followed by targets with evidence for protein only, followed lastly by targets with evidence for RNA only (Figure 3E). The rationale for this harness is twofold: first, concordant evidence from multiple data modalities leads to higher confidence that a target gene's expression is altered in AD brains, and second, that protein levels more accurately reflect the biological state of an in vivo system. 11 , 23 The effect of the harness is tuned to impose this hierarchy without disregarding results specific to a single modality, striking a balance between the relevance of proteomic evidence to disease state versus the increased sensitivity of detection for transcriptomics.
We compare the distribution of all scored targets (Table S8) to those scored by the Open Targets platform 16 , 22 and those nominated for follow‐up by the AMP‐AD consortium. 5 , 24 The AMP‐AD nominated targets tend to have higher multi‐omic scores than the population of all targets (Figure 3F), and there is a very weak correlation between the multi‐omic score and the number of nominations received by a given target (r = 0.153; Figure S8E). Numerous targets receive only one nomination, yet are ranked among the highest multi‐omic scores, while there are many targets that received several nominations with a low multi‐omic score. AMP‐AD investigators used diverse methods beyond differential expression analysis to identify targets, and some modalities (eg, metabolomics) are not included within the current multi‐omic score. Future efforts will include work to integrate additional data modalities into the score.
Using the multi‐omic score to rank genes for GSEA results in the enrichment of GO terms from 18 biological domains. The domains with the largest number of enriched terms include Synapse, Immune Response, Structural Stabilization, and Mitochondrial Metabolism (Figure 3G,H, Table S11). The downregulated terms from the Mitochondrial Metabolism domain are the most significantly enriched (Figure 3H) and have the highest NES (Figure 3G). Interestingly, the top terms within Mitochondrial Metabolism focus upon mitochondrial translation and complex I of the electron transport chain (Figure 3G), showing these terms are the most significantly downregulated biological processes associated with AD.
3.2.3. Composite target risk score (TRS)
The TRS is derived by summing the component risk scores (ie, genetic and multi‐omic), and the highest ranked target scores 4.74 out of a maximum possible score of 5 (Table S12). As with the component scores, the top TRSs are enriched for GWAS loci, AMP‐AD nominated targets, and targets considered by the Open Targets platform (Figure 4A). Considering both the genetic and multi‐omic scores for each target (Figure 4B), the targets with the top TRSs tend to have relatively higher genetic scores. When we compare the TREAT‐AD TRS with the Open Targets target score (Figure S7A), we see that many targets receive a relatively higher score from the TRS, likely due to the unique inclusion of disease‐relevant omic datasets from the AMP‐AD consortium in the TRS. AD GWAS loci are generally scored highly by both the TREAT‐AD TRS and the Open Targets target score metrics.
FIGURE 4.
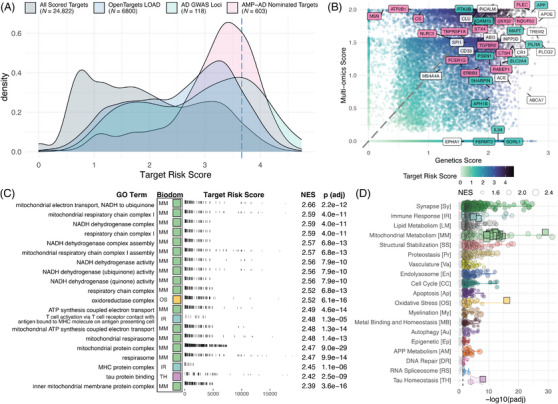
Composite TREAT‐AD target risk score (TRS). (A) Distribution of TRS for all scored targets (gray), those scored by the Open Targets platform (blue), those nominated by a team from the AMP‐AD consortium (pink), and the set of defined AD GWAS loci derived from various sources (green). Dashed blue line indicates the 95th percentile score. (B) Comparison of genetics score and multi‐omic score for all targets. Point color indicates the composite TRS for the target. Labeled genes represent a subset of either GWAS genes (green), AMP‐AD nominated genes (pink), or both (white). (C) Top GO terms significantly enriched using the TRS, arranged by normalized enrichment score (NES). For each term, the resident biological domains are indicated by the filled square (biological domain colors can be referenced from panel D). (D) Enrichment statistics for all biological domain GO terms. Each point is a GO term within the indicated biological domain, and the size of the point is scaled by the GSEA NES. The terms identified in panel (C) are indicated as square points with bold borders. The biological domain terms are ordered on the y‐axis by the number of significantly enriched terms identified from each domain.
GSEA using the overall TRS enriches 3142 GO terms, including 1358 (43.5%) annotated to at least one of the 19 biological domains (Table S13). The biological domains with the largest number of enriched GO terms are Synapse, Immune Response, and Lipid Metabolism (Figure 4D). In comparison, the Open Targets target score enriches terms from 16 biological domains, and the domains with the largest number of enriched terms are also Synapse, Lipid Metabolism, and Immune Response (Figure S7B).
GO terms enriched using the TRS have different enrichment strengths when using the component scores (Figure 5A, Tables S10, S11, and S13). For example, the Lipid Metabolism term “fatty acid metabolic process” is enriched using all scores, but the enrichment is more significant for the genetics (adjusted p value 5.9 × 10−6) than for the omics score (adjusted p value 7.9 × 10−3). The inverse is true for the “membrane raft” term, which is more significant using the multi‐omic (adjusted p value = 1.3 × 10−5) compared to the genetic scores (adjusted p value = 1.8 × 10−2). There is a similar pattern for terms related to developmental processes in the Immune Response domain. For instance, “myeloid leukocyte differentiation” shows a stronger enrichment from the genetic score, while terms related to mature structures and processes like “leukocyte degranulation” are more significantly enriched by the multi‐omic score (Figure 5B). Considering terms from the Synapse biological domain, more postsynaptic terms are enriched (40 terms) and are more significant relative to presynaptic terms (nine terms) with the cumulative TRS and the component scores (Figure 5C). The optimal targets should reflect a parity of ranking between score components, with evidence of both genetic and multi‐omic risk. Evidence of genetic risk without multi‐omic risk could indicate developmental processes that are less relevant in later stages, whereas enrichment of multi‐omic risk without genetic risk could implicate processes that change in response to disease pathology but are not causal in the underlying etiology. 18 , 25 , 26 , 27 , 28 , 29 Moreover, processes that are risk‐enriched across distinct measures increase the confidence of disease association. While the multi‐omic score enriches terms related to mitochondrial electron transport chain complexes I and III, the genetic score only enriches terms related to complex I (Figure 5D), yet both measures point to the centrality of electron transport chain relevant genes in our scored AD risk. GO terms close to the diagonal dashed line (Figure 5A) suggest that the risk associated with genes in these terms are embedded equivalently in each modality and merit consideration for further resource development. GO terms that are significantly enriched but do not fall into any biological domain (ie, Figure 5A, “none”) reflect either a categorization that is too general to be mapped into endophenotypic space, such as “ATP Metabolic Process,” or terms that map to high‐order positions within the ontology, such as “Cell Leading Edge” (Figure 5A).
FIGURE 5.

Comparing biological domain enrichments across risk modalities. (A) Relative significance of biological domain term enrichment using genetics score component (x‐axis), multi‐omic score component (y‐axis), and the overall TRS (point size). (B) Term enrichments from Immune Response biological domain, subset by immune cell development terms (blue), other immune cell terms (yellow), and other terms from the Immune Response biological domain (gray). (C) Term enrichments from Synapse biological domain, subset by postsynaptic terms (yellow), presynaptic terms (blue), and other terms from the Synapse biological domain (gray). (D) Term enrichments from Mitochondrial Metabolism biological domain, subset by mitochondrial electron transport chain complex I (yellow), complex III (blue), complex V (green), and other terms from the Mitochondrial Metabolism biological domain (gray). Complexes II and IV were not significantly enriched in these analyses.
3.3. Assessment of clinical trial targets
These resources are intended to facilitate target prioritization and therapeutic hypothesis development for AD. As an example use case, we characterize the gene targets of therapeutics under clinical investigation. We obtained data for 526 trials (1995 to 2025, Figure S9A), for 192 agents targeting 187 genes (Table S9). The 173 targets that are scored by our processes have a higher distribution of TRSs than do non‐targets (Figure 6A, Kolmogorov–Smirnov p = 3.7 × 10−8). Targets of agents that have entered phase 4 trials tend to have higher genetic risk scores than do targets in earlier phases (Figure 6B, Kolmogorov–Smirnov p values from .04 to .0066). This observation echoes previous reports regarding clinical trials in general, specifically that agents that progress to later phases are those that have stronger genetic evidence supporting disease involvement. 30 Overrepresentations tests using these targets show that terms from the Synapse, Vasculature, Tau Homeostasis, and APP Metabolism domains are the most significantly enriched (Figure 6C). Contrasting this to terms enriched from a similarly sized list of genes pulled from the top of the TRS distribution, we note larger p values overall but a relative emphasis on terms from the Mitochondrial Metabolism, Immune Response, Cell Cycle, and Proteostasis domains (Figure 6C). We also note the change in emphasis over time (Figure S9B), with a decreased representation of APP Metabolism and an increase in Autophagy and Immune Response, and that these more recent areas of emphasis are reflected in earlier phases of the development pipeline (Figure S9C).
FIGURE 6.
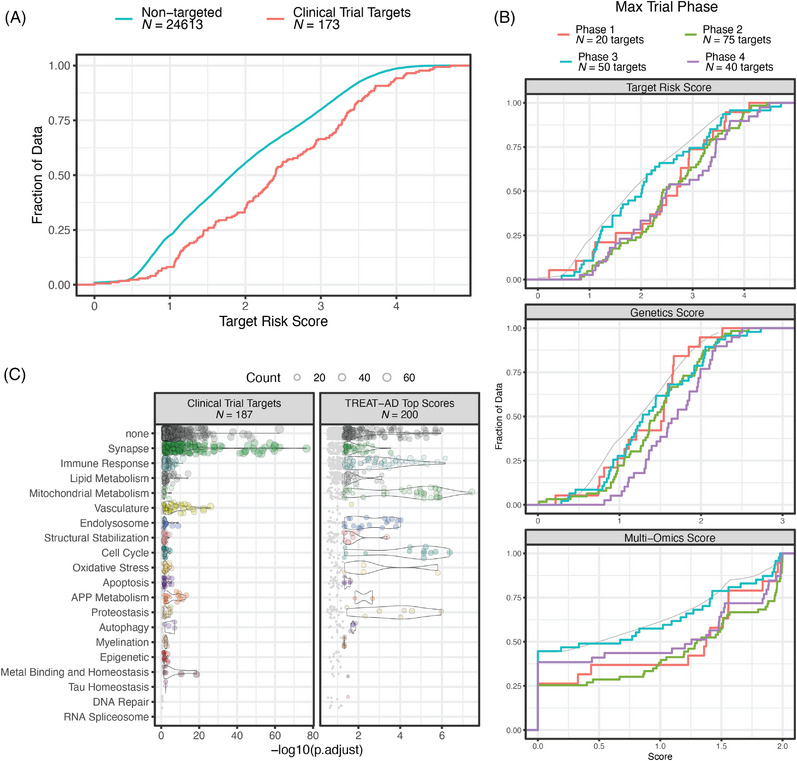
Evaluation of targets of agents in clinical trials for AD. (A) Empirical cumulative distribution functions (ECDFs) of overall target risk score (TRS) for genes identified as targets of agents in clinical trials (red line) and genes that are not targeted by any trial therapeutic (blue line). (B) ECDFs of the TRSs and component risk scores for targets of agents in clinical trial, broken out by maximum clinical trial phase for each agent. For each plot the gray line represents the distribution of non‐targeted genes. The distribution of genetics scores for targets of agents that have reached phase 4 trials is significantly higher than the distributions of targets of agents in other phases of development (Kolmogorov–Smirnov p values from .04 to .0066). (C) Significantly enriched GO terms for targets of agents in clinical trials and a comparably sized set of genes from the top of the TRS distribution. Each point is a term within the indicated biological domain, the position on the x‐axis shows the significance of the term enrichment, and the size of the point is scaled by the number of genes in that term in the lists. The name and size of each list is indicated above each plot.
4. DISCUSSION
We have developed an integrated system‐level assessment of AD risk across data modalities along with a systematic enumeration and functionalization of AD endophenotypes. The risk scoring paradigm ranks genes genome‐wide drawing from multiple genetic association studies as well as large transcriptomic and proteomic datasets comprising data from thousands of post mortem samples drawn from multiple large brain bank studies. We have also developed 19 biological domains that align with known AD endophenotypes, defining each with sets of GO terms and annotated gene sets. The goal is to provide the scientific community with an evaluative resource that can be employed across studies to integrate knowledge about candidates for further investigation as well as provide a unified framework for defining AD endophenotypes. The biological domains described in this work have already been adopted by several groups as a common organizational reference. The expanding utilization of these biological domains will provide the opportunity for increased interoperability and harmonization across research efforts. Both methodologies are easily updated as new information becomes available as they draw from open‐source resources central to AD investigations.
There are several limitations to the methodology we employed. Our meta‐analysis approach prioritizes signatures of disease that span study populations and tissues of origin, and as such any pathological changes that are limited to specific tissues or study populations will be underemphasized. Furthermore, the metrics we developed to encapsulate risk rely on standard case‐control contrasts, while AD is heterogeneous with reported molecular sand pathological subtypes. 31 , 32 We have not addressed other contributors of risk, such as the impacts of age, sex, APOE allele or environmental exposures on pathogenesis. While these aspects of disease risk will be investigated independently in future work, their absence does not detract from the power of the integrative approach utilized here, creating an objective risk assessment and endophenotypic characterization strategy that can be employed to better illuminate the AD risk factors and subtypes that were not explored in the present work.
Gene set enrichment using the quantitative scores enables us to identify GO terms significantly enriched for specific biological areas of risk across and between data modalities. These analyses (Figures 2, 3, 4) demonstrate that different modalities emphasize independent aspects of disease risk but together are a powerful tool for automated mapping of risk into specific biological areas within disease‐associated endophenotypes (eg, Figure 5A, Tables S10, S11, and S13).
We investigated which biological domains were the most up‐ and downregulated based on post mortem differential expression analyses. The top two downregulated domains are Mitochondrial Metabolism and Synapse (Figure 3C,D). The downregulation of synaptic genes span both pre‐ and postsynaptic gene sets; however, there are three to four times as many postsynaptic GO terms enriched, and postsynaptic terms are more significantly enriched across all component risk scores (Figure 5C). This aligns with previous research into the role postsynaptic mechanisms play in the maintenance of dendrites and synaptic plasticity, lost during the cognitive decline of AD. 33 , 34 , 35 Likewise, numerous studies have demonstrated elements of mitochondrial dysfunction and hypometabolism in AD. 1 , 36 , 37 , 38 , 39 , 40 Furthermore, these two domains are associated with cognitive stability in aging 41 , 42 and may progress coordinately in AD. 37 , 42 While these analyses only identify the independent contributions of mitochondrial hypometabolism and synaptic dysfunction or loss, future work may be able to highlight aspects of biology at the intersection of these domains.
Multiple biological domains are upregulated in our analyses, most notably Immune Response, Structural Stabilization, Lipid Metabolism, and Proteostasis (Figure 3C,D). Immune involvement in the progression of AD pathology has attracted attention recently, particularly regarding the Janus faces of microglia: the homeostatic and degenerative roles. A meaningful discussion of these areas is beyond the scope of this work, but the topic has been extensively reviewed. 43 – 45 Structural stabilization has been highlighted by recent proteomics studies, suggesting roles for heparin binding, extracellular matrix, and cytoskeleton‐associated proteins in either AD resilience or progression. 11 , 46 Dysfunctions in lipid metabolism have been central to the findings of several recent AD metabolomics studies. 26 , 28 , 29 , 47 – 49 Proteostasis is also frequently implicated in AD, given the complex linkages between autophagy and proteome stability, changes in chaperone‐mediated protein processing, and endoplasmic reticulum stress responses, all of which may play a role. 50 – 52 The upregulated processes from these domains delineate areas of biology that merit further exploration for both mechanisms driving disease and compensatory processes that may facilitate future therapeutic development.
We assessed how the TREAT‐AD risk score compared with corresponding rankings from the Open Targets platform. 16 , 22 The two methodologies share several features: both integrate results from genetic association studies to implicate genes with variation that contribute to disease risk, both capture expression differences relevant to the disease, and both identify animal models with phenotypic relevance to disease. These similarities are reflected in the correlation observed between the two scores (eg, Figure S7A). The TRS is distinct in the inclusion of both omic datasets from numerous brain banks via AMP‐AD Consortium investigations and information from emerging AD mouse models via the MODEL‐AD Consortium. We benchmarked the scores from Open Targets and the TRS and observed very similar patterns of biological domain term enrichment (eg, Figure S7B), with the Synapse, Lipid Metabolism, and Immune Response domains among the strongest enrichments for both. However, the enrichments using the TRS also implicate Mitochondrial Metabolism (120 enriched terms) and Tau Homeostasis (two enriched terms) more strongly than the enrichments based on the Open Targets score (one enriched term and no enriched terms, respectively).
Finally, we used the TRS and biological domain framework to assess the targets of therapeutic agents under clinical investigation for AD. We find that while these targets tend to be higher scoring than non‐targets, they emphasize different biological domains than those implicated by the measures of risk utilized here. In particular, the clinical trial targets strongly enrich terms from the Synapse, Vasculature, and APP Metabolism domains. A comparably sized list of genes from the top of the TRS distribution has a relatively stronger emphasis on Mitochondrial Metabolism and Immune Response domains. These results suggest that the underwhelming performance of trial therapeutics may be due, in part, to targeting disease endophenotypes that are misaligned with risk.
5. CONCLUSION
This work is the largest integrated effort to combine genetic and multi‐omic AD risk scoring with an automatable system of endophenotypic genetic characterization. The dual processes of ranking genes by risk and assembling the risk areas into biological domains points to consistency between our analyses and observations made across the field, which further supports our approach to identifying focal areas of AD risk. The advantage of our system is that we can utilize the comprehensive representation of AD risk genes organized into specific areas of biology to expand the study of disease domain transitions by identifying potential points of convergence between interacting domains and examining the genetic entities at those crossroads. The Emory‐Sage‐SGC TREAT‐AD Center utilizes these foundational resources along with other analytical approaches to nominate specific dark targets for potential future therapeutic evaluation. These approaches will continue to be expanded and refined with newly emerging data, analytical approaches, and community feedback. Informatic and material resources developed for each nominated target, which will include endophenotype‐aligned cell‐based assays and validated experimental reagents, are openly available to the scientific community (treatad.org/data‐tools/target‐dashboard). We anticipate that these resources will help the field foster current efforts leading to the growth of novel translational approaches, to identify new targets and sets of targets, and additionally to the repurposing of therapeutics developed in divergent fields for use as standalone or combination therapies in the treatment of AD. The objective identification of the ranked areas of disease risk scored genome‐wide and organized into defined biological domains highlight the significance of multiple aspects of disease biology for translational development that current resources can help hone into specific subdomains – such as mitochondrial complex I related factors and postsynaptic targets and upregulated targets in the Immune Response and Structural Stabilization biological domains. Application to the development of therapeutic approaches in combination with recently approved drugs is an attractive concept based on these efforts, be they repurposed agents or novel therapeutic entities.
CONFLICT OF INTEREST STATEMENT
G.A. Cary, J.C. Wiley, S. Keegan, L. Heath, R.R. Butler III, L.M. Mangravite, and A.K. Greenwood: No conflicts of interest. J. Gockley: Currently a full‐time employee of Cajal Neuroscience. B.A. Logsdon: Currently a full‐time employee of Cajal Neuroscience. F.M. Longo: Is a founder of, has equity interest in, and serves as a paid consultant for PharmatrophiX, a company engaged in the development of small‐molecule treatments for neurodegenerative and other disorders. F.L. serves as a paid consultant for Red Tree Venture Capital, a firm investing in biotechnology companies developing treatments for neurological and other disorders. A. Levey: A.L. is a founder, has equity interest in, and serves as a paid consultant for EmTheraPro, a company developing novel biomarkers and treatments for AD and related disorders. A.L. serves as a paid consultant for Karuna Pharmaceuticals, Cognito Therapeutics, and MEPSGEN and receives royalties from Linus Health. G.W. Carter: G.W.C. has served as paid consultant for Astrex Therapeutics. Author disclosures are available in the supporting information.
CONSENT STATEMENT
No human participants were recruited for this work. The details of the Institutional Review Board (IRB)/oversight body that provided approval or exemption for the research described are given as follows: Western Institutional Review Board—Copernicus Group (WCG) IRB of Sage Bionetworks gave ethical approval for this work.
Supporting information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors would like to acknowledge the support of Drs. Lea T. Grinberg, Joshua M. Shulman, David Li‐Kroeger, Jessica E. Young, Suman Jayadev, Ranjita Betarbet, and Benoit Lehallier for insightful discussions and suggestions during the development of these resources. The Target Enablement to Accelerate Therapy Development for Alzheimer's Disease (TREAT‐AD) Consortium was established by the NIA. The research reported in this manuscript was led by the Emory‐Sage‐SGC TREAT‐AD Center and supported by grant U54AG065187 from the NIA. Certain data used in this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer's Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24AG041689), funded by the NIA; detailed citations and accessions can be found in Table S3. Data used in this study were obtained from the Accelerating Medicines Partnership Program for Alzheimer's Disease (AMP‐AD) Consortium members below: Mayo RNAseq Study: Study data were provided by the following sources: The Mayo Clinic Alzheimer's Disease Genetic Studies, led by Dr. Nilufer Ertekin‐Taner and Dr. Steven G. Younkin, Mayo Clinic, Jacksonville, FL, using samples from the Mayo Clinic Study of Aging, the Mayo Clinic Alzheimer's Disease Research Center, and the Mayo Clinic Brain Bank. Data collection was supported through funding by NIA grants P50 AG016574, R01 AG032990, U01 AG046139, R01 AG018023, U01 AG006576, U01 AG006786, R01 AG025711, R01 AG017216, R01 AG003949, NINDS grant R01 NS080820, CurePSP Foundation, and support from Mayo Foundation. Study data include samples collected through the Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson's Disease and Related Disorders), the NIA (P30 AG19610 Arizona Alzheimer's Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer's Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05‐901, and 1001 to the Arizona Parkinson's Disease Consortium), and the Michael J. Fox Foundation for Parkinson's Research. Religious Orders Study/Memory and Aging Project (ROSMAP): We are grateful to the participants in the Religious Order Study and the Memory and Aging Project. This work was supported by the US National Institutes of Health (U01 AG046152, R01 AG043617, R01 AG042210, R01 AG036042, R01 AG036836, R01 AG032990, R01 AG18023, RC2 AG036547, P50 AG016574, U01 ES017155, KL2 RR024151, K25 AG041906‐01, R01 AG30146, P30 AG10161, R01 AG17917, R01 AG15819, K08 AG034290, P30 AG10161, and R01 AG11101). Mount Sinai Brain Bank (MSBB): This work was supported by grants R01AG046170, RF1AG054014, RF1AG057440, and R01AG057907 from the NIH/NIA. R01AG046170 is a component of the AMP‐AD Target Discovery and Preclinical Validation Project. Brain tissue collection and characterization was supported by NIH HHSN271201300031C.
Cary GA, Wiley JC, Gockley J, et al. Genetic and multi‐omic risk assessment of Alzheimer's disease implicates core associated biological domains. Alzheimer's Dement. 2024;10:e12461. 10.1002/trc2.12461
Gregory A. Cary and Jesse C. Wiley participated equally towards the completion of this work.
REFERENCES
- 1. Knopman DS, Amieva H, Petersen RC, et al. Alzheimer disease. Nat Rev Dis Primers. 2021;7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Refolo LM, Snyder H, Liggins C, et al. Common Alzheimer's disease research ontology: National Institute on Aging and Alzheimer's Association collaborative project. Alzheimers Dement. 2012;8:372‐375. [DOI] [PubMed] [Google Scholar]
- 3. Cummings J, Lee G, Nahed P, Kambar MEZN, Zhong K, Fonseca J. Alzheimer's disease drug development pipeline: 2022. Alzheimers Dement (N Y). 2022;8:e12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gauthier S, Alam J, Fillit H, et al. Combination Therapy for Alzheimer's Disease: perspectives of the EU/US CTAD Task Force. J Prev Alzheimers Dis. 2019;6:164‐168. [DOI] [PubMed] [Google Scholar]
- 5. Greenwood AK, Montgomery KS, Kauer N, et al. The AD knowledge portal: a repository for multi‐omic data on Alzheimer's disease and aging. Curr Protoc Hum Genet. 2020;108:e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bellenguez C, Küçükali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54:412‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li J‐W, Wang R‐L, Xu J, et al. Methylene blue prevents osteoarthritis progression and relieves pain in rats via upregulation of Nrf2/PRDX1. Acta Pharmacol Sin. 2022;43:417‐428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wan Y‐W, Al‐Ouran R, Mangleburg CG, et al. Meta‐analysis of the Alzheimer's disease human brain transcriptome and functional dissection in mouse models. Cell Rep. 2020;32:107908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mukherjee S, Heath L, Preuss C, et al. Molecular estimation of neurodegeneration pseudotime in older brains. Nat Commun. 2020;11:5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Milind N, Preuss C, Haber A, et al. Transcriptomic stratification of late‐onset Alzheimer's cases reveals novel genetic modifiers of disease pathology. PLoS Genet. 2020;16:e1008775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johnson ECB, Carter EK, Dammer EB, et al. Large‐scale deep multi‐layer analysis of Alzheimer's disease brain reveals strong proteomic disease‐related changes not observed at the RNA level. Nat Neurosci. 2022;25:213‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wingo AP, Liu Y, Gerasimov ES, et al. Integrating human brain proteomes with genome‐wide association data implicates new proteins in Alzheimer's disease pathogenesis. Nat Genet. 2021;53:143‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Friedrich JO, Adhikari NK, Beyene J, The ratio of means method as an alternative to mean differences for analyzing continuous outcome variables in meta‐analysis: a simulation study. BMC Med Res Methodol. 2008;8:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu G, Wang Li‐G, Han Y, He Q‐Y. He, clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16:284‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Neuner SM, Tcw J, Goate AM. Genetic architecture of Alzheimer's disease. Neurobiol Dis. 2020;143:104976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ghoussaini M, Mountjoy E, Carmona M, et al. Open Targets Genetics: systematic identification of trait‐associated genes using large‐scale genetics and functional genomics. Nucleic Acids Res. 2021;49:D1311‐d1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ladia BHK. synapser: R language bindings for Synapse API. 2021. R package version 0.10.101. https://github.com/Sage-Bionetworks/synapser/
- 18. Ochoa D, Karim M, Ghoussaini M, Hulcoop DG, Mcdonagh EM, Dunham I. Human genetics evidence supports two‐thirds of the 2021 FDA‐approved drugs. Nat Rev Drug Discov. 2022;21(8):551. [DOI] [PubMed] [Google Scholar]
- 19. Sollis E, Mosaku A, Abid A, et al. The NHGRI‐EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 2023;51:D977‐D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sieberts SK, Perumal TM, Carrasquillo MM, et al. Large eQTL meta‐analysis reveals differing patterns between cerebral cortical and cerebellar brain regions. Sci Data. 2020;7:340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robins C, Liu Y, Fan W, et al. Genetic control of the human brain proteome. Am J Hum Genet. 2021;108:400‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ochoa D, Hercules A, Carmona M, et al. Open Targets Platform: supporting systematic drug‐target identification and prioritisation. Nucleic Acids Res. 2021;49:D1302‐d1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Axtman AD, Brennan PE, Frappier‐Brinton T, et al. Open drug discovery in Alzheimer's disease. Alzheimers Dement (N Y). 2023;9:e12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hodes RJ, Buckholtz N. Accelerating medicines partnership: Alzheimer's Disease (AMP‐AD) knowledge portal aids alzheimer's drug discovery through open data sharing. Expert Opin Ther Targets. 2016;20:389‐391. [DOI] [PubMed] [Google Scholar]
- 25. Liu A, Manuel AM, Dai Y, et al. Identifying candidate genes and drug targets for Alzheimer's disease by an integrative network approach using genetic and brain region‐specific proteomic data. Hum Mol Genet. 2022;31(19):3341‐3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baloni P, Arnold M, Buitrago L, et al. Multi‐Omic analyses characterize the ceramide/sphingomyelin pathway as a therapeutic target in Alzheimer's disease. Commun Biol. 2022;5:1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou Y, Fang J, Bekris LM, et al. AlzGPS: a genome‐wide positioning systems platform to catalyze multi‐omics for Alzheimer's drug discovery. Alzheimers Res Ther. 2021;13:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wörheide MK, Krumsiek J, Nataf S, et al. An integrated molecular atlas of Alzheimer's disease. medRxiv. 2022. [Google Scholar]
- 29. Wörheide M, Krumsiek J, Nho K, et al. A network‐based, multi‐omics atlas for target identification and prioritization in Alzheimer's disease. Alzheimer's & Dementia. 2020;16:e045594. [Google Scholar]
- 30. King EA, Davis JW, Degner JF. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 2019;15:e1008489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Crane PK, Trittschuh E, Mukherjee S, et al. Incidence of cognitively defined late‐onset Alzheimer's dementia subgroups from a prospective cohort study. Alzheimers Dement. 2017;13:1307‐1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Neff RA, Wang M, Vatansever S, et al. Molecular subtyping of Alzheimer's disease using RNA sequencing data reveals novel mechanisms and targets. Sci Adv. 2021;7:eabb5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dejanovic B, Huntley MA, De Mazière A, et al. Changes in the synaptic proteome in tauopathy and rescue of tau‐induced synapse loss by C1q antibodies. Neuron. 2018;100:1322‐1336.e1327. [DOI] [PubMed] [Google Scholar]
- 34. Shao CY, Mirra SS, Sait HBR, Sacktor TC, Sigurdsson EM. Postsynaptic degeneration as revealed by PSD‐95 reduction occurs after advanced Abeta and tau pathology in transgenic mouse models of Alzheimer's disease. Acta Neuropathol. 2011;122:285‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yuesong Gong, Lippa CF. Review: disruption of the postsynaptic density in Alzheimer's disease and other neurodegenerative dementias. Am J Alzheimers Dis Other Demen. 2010;25:547‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ashleigh T, Swerdlow RH, Beal MF. The role of mitochondrial dysfunction in Alzheimer's disease pathogenesis. Alzheimers Dement. 2023; 19(1): 333‐342. [DOI] [PubMed] [Google Scholar]
- 37. Torres AK, Jara C, Park‐Kang HS, et al. Synaptic mitochondria: an early target of amyloid‐β and tau in Alzheimer's disease. J Alzheimers Dis. 2021;84:1391‐1414. [DOI] [PubMed] [Google Scholar]
- 38. Fessel J. Does synaptic hypometabolism or synaptic dysfunction, originate cognitive loss? Analysis of the evidence. Alzheimers Dement (N Y). 2021;7:e12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Swerdlow RH. The mitochondrial hypothesis: dysfunction, bioenergetic defects, and the metabolic link to Alzheimer's disease. Int Rev Neurobiol. 2020;154:207‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Perez Ortiz JM, Swerdlow RH. Mitochondrial dysfunction in Alzheimer's disease: role in pathogenesis and novel therapeutic opportunities. Br J Pharmacol. 2019;176:3489‐3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wingo AP, Dammer EB, Breen MS, et al. Large‐scale proteomic analysis of human brain identifies proteins associated with cognitive trajectory in advanced age. Nat Commun. 2019;10:1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morton H, Kshirsagar S, Orlov E, et al. Defective mitophagy and synaptic degeneration in Alzheimer's disease: focus on aging, mitochondria and synapse. Free Radic Biol Med. 2021;172:652‐667. [DOI] [PubMed] [Google Scholar]
- 43. Pons V, Rivest S. Targeting systemic innate immune cells as a therapeutic avenue for Alzheimer disease. Pharmacol Rev. 2022;74:1‐17. [DOI] [PubMed] [Google Scholar]
- 44. Mcmanus RM. The role of immunity in Alzheimer's disease. Adv Biol (Weinh). 2022;6:e2101166. [DOI] [PubMed] [Google Scholar]
- 45. De Sousa RAL. Reactive gliosis in Alzheimer's disease: a crucial role for cognitive impairment and memory loss. Metab Brain Dis. 2022;37:851‐857. [DOI] [PubMed] [Google Scholar]
- 46. Johnson ECB, Dammer EB, Duong DM, et al. Large‐scale proteomic analysis of Alzheimer's disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med. 2020;26:769‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Abubakar MB, Sanusi KO, Ugusman A, et al. Alzheimer's disease: an update and insights into pathophysiology. Front Aging Neurosci. 2022;14:742408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sriwichaiin S, Chattipakorn N, Chattipakorn SC. Metabolomic alterations in the blood and brain in association with Alzheimer's disease: evidence from in vivo to clinical studies. J Alzheimers Dis. 2021;84:23‐50. [DOI] [PubMed] [Google Scholar]
- 49. Arnold M, Nho K, Kueider‐Paisley A, et al. Sex and APOE ε4 genotype modify the Alzheimer's disease serum metabolome. Nat Commun. 2020;11:1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fernández D, Geisse A, Bernales JI, Lira A, Osorio F. The unfolded protein response in immune cells as an emerging regulator of neuroinflammation. Front Aging Neurosci. 2021;13:682633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bourdenx M, Gavathiotis E, Cuervo AM. Chaperone‐mediated autophagy: a gatekeeper of neuronal proteostasis. Autophagy. 2021;17:2040‐2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Koopman MB, Rüdiger SGD. Alzheimer cells on their way to derailment show selective changes in protein quality control network. Front Mol Biosci. 2020;7:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information