

Abstract
Pain management is an important problem worldwide. The current frontline approach for pain management is the use of opioid analgesics. The primary analgesic target of opioids is the μ-opioid receptor (MOR). Deletion of phospholipase Cβ3 (PLCβ3) or selective inhibition of Gβγ regulation of PLCβ3 enhances the potency of the antinociceptive effects of morphine suggesting a novel strategy for achieving opioid-sparing effects. Here we investigated a potential mechanism for regulation of PLC signaling downstream of MOR in human embryonic kidney 293 cells and found that MOR alone could not stimulate PLC but rather required a coincident signal from a Gq-coupled receptor. Knockout of PLCβ3 or pharmacological inhibition of its upstream regulators, Gβγ or Gq, ex vivo in periaqueductal gray slices increased the potency of the selective MOR agonist [D-Ala2, N-Me-Phe4, Gly5-ol]-enkephalin acetate salt in inhibiting presynaptic GABA release. Finally, inhibition of Gq- G protein-coupled receptor coupling in mice enhanced the antinociceptive effects of morphine. These data support a model where Gq and Gβγ-dependent signaling cooperatively regulate PLC activation to decrease MOR-dependent antinociceptive potency. Ultimately, this could lead to identification of new non-MOR targets that would allow for lower-dose utilization of opioid analgesics.
SIGNIFICANCE STATEMENT
Previous work demonstrated that deletion of phospholipase Cβ3 (PLCβ3) in mice potentiates μ-opioid receptor (MOR)-dependent antinociception. How PLCβ3 is regulated downstream of MOR had not been clearly defined. We show that PLC-dependent diacylglycerol generation is cooperatively regulated by MOR-Gβγ and Gq-coupled receptor signaling through PLCβ3 and that blockade of either Gq-signaling or Gβγ signaling enhances the potency of opioids in ex vivo brain slices and in vivo. These results reveal potential novel strategies for improving opioid analgesic potency and safety.
Introduction
Pain management is an important problem worldwide. The current frontline approach for clinical pain-management is the use of opioid analgesics. While these compounds are highly effective, they come with substantial drawbacks. Prolonged use results in the development of tolerance and physical dependence, which severely limits their use in treatment of chronic pain. Hence, μ-opioid receptor (MOR) agonists produce reinforcing effects and thus have abuse liability. Severe respiratory depression as a result of opioid overdose is the major cause of opioid-related deaths.
The primary analgesic target of opioids is MOR. MORs are G protein-coupled receptors (GPCRs) that are expressed in both pre- and postsynaptic locations throughout the nervous system and can activate many different signaling pathways. As GPCRs, MORs activate G proteins and are desensitized and/or internalized through recruitment of β-arrestins (Bohn et al., 2000; Shenoy and Lefkowitz, 2011; Kliewer et al., 2020). One approach to improving opioid analgesics has been to find strategies that improve potency and efficacy of opioid agonists while limiting MOR desensitization and internalization (Schmid et al., 2017; Kandasamy et al., 2021). Work by our laboratory has identified phospholipase C signaling as a process that limits the antinociceptive effects of MOR agonists (Xie et al., 1999) and that pharmacological attenuation or blockade of activation of this pathway enhances the potency of opioid analgesics in mice (Bonacci et al., 2006; Mathews et al., 2008; Smrcka et al., 2008; Bianchi et al., 2009; Hoot et al., 2013; Campbell and Smrcka, 2018).
Phospholipase C is the upstream enzyme responsible for protein kinase C (PKC) activation, and this pathway can be activated by GPCRs. PKC has been implicated in adaptations involved in morphine tolerance through alterations in MOR signaling (Gabra et al., 2008; Bailey, Llorente et al., 2009; Bailey, Oldfield et al., 2009; Ingram and Traynor, 2009; Arttamangkul et al., 2015), but the mechanisms for upstream regulation of PKC activation in the opioid system have not been examined. Our laboratory has been interested in understanding the mechanisms for activation of phospholipase Cβ by opioid receptors. Since PLCβ3 is activated by Gβγ subunits released from Gi-coupled receptors (Smrcka and Sternweis, 1993; Li et al., 2000; Smrcka, 2008) we hypothesized that PLCβ3 would be activated downstream of MOR via a Gβγ-dependent signal transduction pathway. Indeed, inhibition of Gβγ signaling with M119 or gallein enhanced the antinociceptive effects of morphine in wild-type (WT) mice but not in PLCβ3−/− mice (Bonacci et al., 2006).
Here we further explored potential mechanisms for MOR-dependent PLC activation and the relevance of these mechanisms to presynaptic opioid-dependent inhibition of neurotransmitter (GABA) release and to antinociception in mice. In vitro, MOR activation alone did not stimulate PLC signaling but, rather, required coincident activation of a Gq-coupled receptor, consistent with the previously described property of PLCβ3 as a coincidence detector for Gαq and Gi signaling (Philip et al., 2010). In periaqueductal gray (PAG) brain slices, inhibition of either Gβγ or Gαq signaling through PLCβ3 enhanced opioid-dependent inhibition of neurotransmitter (GABA) release. Finally, blockade of either Gαq or Gβγ in mice enhanced morphine-dependent antinociception in mice. These data show that MOR signaling is inhibited in presynaptic terminals through a PLCβ3-dependent mechanism that utilizes coincident inputs from Gq-coupled receptors and MOR to modulate antinociception.
Materials and Methods
Reagents
The reagents used in this study were gallein (Tocris, Minneapolis, MN), myrGq-CT inhibitor and scrambled peptide (GenScript USA Inc., Piscataway, NJ), YM-254890 (MedChemExpress MCE, Monmouth Junction, NJ), [D-Ala2, N-Me-Phe4, Gly5-ol]-enkephalin acetate salt (DAMGO; Sigma-Aldrich, St. Louis, MO), morphine (Henry Schein, Melville, NY), carbachol (Montana Molecular, Bozeman, MT), ATP (Sigma-Aldrich), Ser-Phe-Leu-Leu-Arg-Asn-amide trifluoroacetate salt (Sigma-Aldrich), and pertussis toxin (PTX; Sigma-Aldrich).
Animals
All animal procedures were conducted at the University of Michigan according to National Institutes of Health’s Guide for the Care and Use of Laboratory Animals and with approval of Institutional Animal Care and Use Committee at the University of Michigan. WT C57BL/6 mice purchased from Envigo (Indianapolis, IN) and from an in-house breeding colony were used for these studies. PLCβ3−/− mice were purchased from Jackson Laboratories (Bar Harbor, ME) and were bred on a C57BL/6 background. Mice were housed with a maximum of 5 animals per cage in clear polypropylene cages with corn cob bedding and Nestlets as enrichment. Animals were housed in specific pathogen-free rooms maintained between 68°F and 72°F and between 30% and 70% humidity and a 12-hour light/dark cycle (lights on at 7 am and lights off at 7 pm) light/dark cycle with free access to food (Laboratory Diets, St. Louis, MO; 5L0D) and water. Experiments were conducted in the housing room during the light cycle. All mice were used between 8 and 15 weeks of age and weighed 19 to 26 g. A combination of male and female mice was used in gallein experiments, but male mice were used for myrGq inhibitor experiments. Mice were tested only once with a single dose of drug, and all analyses are between-subject.
Electrophysiology studies were done at Oregon Health & Science University. These studies used male and female WT C57BL/6 mice and PLCβ3−/− mice and WT littermates. Mice were group housed with unlimited access to food and water. Lights were maintained on a 12-hour light/dark cycle (lights on at 7:00 am). Mice were sacrificed, and cellular recordings were conducted during the light phase of this cycle. The Institutional Animal Care and Use Committee at Oregon Health & Science University approved all experimental procedures. Experiments were conducted in accordance with the US National Research Council (2011) Guide for the Care and Use of Laboratory Animals.
Maintenance of HEK293 cell culture and Stable MOR-FLAG HEK293 cell culture
Human embryonic kidney 293 (HEK293) cells were grown in Dulbecco’s modified Eagle’s medium (Corning, Corning, NY) with 4.5 g/L glucose, L-glutamine and sodium pyruvate with 100U penicillin/streptomycin and research grade 10% FBS (ThermoFisher Scientific, Pittsburgh, PA). Cells were maintained in a 5% CO2 humid atmosphere at 37°C. HEK293 cells stably expressing MOR-FLAG tagged receptor were obtained from the Puthenveedu laboratory at the University of Michigan and were maintained with addition of 50 mg/mL geneticin (G418 sulfate; ThermoFisher Scientific, Rochester, NY).
Transduction of diacylglycerol fluorescent biosensor and M1 muscarinic acetylcholine receptor in HEK293 cells
HEK293 cells were used for these studies. Green fluorescent up diacylglycerol (DAG) assay kit (#U0300G) and CAAX-Green downward DAG kit (#D0331G) were purchased through Montana Molecular (Bozeman, MT). HEK293 cells were incubated (8–24 hours) with a viral transduction reaction including DAG Sensor BacMam, sodium butyrate, and M1 muscarinic acetylcholine BacMam (receptor control was not added for endogenous Gq-coupled receptor experiments). A 96-well black plate with transparent bottom (Corning) was used (50 µL of 500 000 cells/mL per well) with BacMam transduction reagent (100 µL per well).
Activation of DAG sensor and collection of data
Assays were conducted with a Hamamatsu μCell FDSS plate reader. Agonists were loaded into a 96-well plastic conical-bottom source plate (ThermoFisher Scientific, Rochester NY) prior to transfer by the instrument. Before placing the cells in the plate reader, transduction media was exchanged with 120 µL of warmed Gibco Dulbecco’s PBS with Ca2+ and Mg2+ (Life Technologies Co, Grand Island, NY). Cells at 80% confluency were kept in the dark inside the plate reader and incubated in Gibco Dulbecco’s PBS for 10 minutes before the agonist was added. Baseline fluorescence at 540 nM was measured each second for 30 seconds followed by 15 µL of agonist added simultaneously to each well of the plate. Fluorescence intensity measurements were acquired every second for 230 seconds. Data are normalized to baseline fluorescence (ΔF/Fo = 1) in each well, and the change in fluorescence in each well relative to baseline is monitored over time. Each condition was tested in 3 or 4 wells in at least 3 different sets of experiments.
Transient transfection of MOR in HEK293 cells
HEK293 cells were transiently transfected with flag-MOR cDNA using Lipofectamine 2000 in a 10-cm plate at 70% confluency 1 day before BacMam transduction. After 24 hours, cells were transferred to a 96-well plate at 80% confluency and incubated for 24 hours before the assay.
Immunocytochemistry
Transfected cells and stable MOR-FLAG cells were plated in a 20-mm glass bottom cell culture dish (Wuxi NEST Biotechnology, China). Cells were allowed to adhere and then fixed with 4% paraformaldehyde for 15 minutes and then incubated with 10% normal goat serum in PBS containing 0.1% Triton ×100 (PBS-T) for 1 hour at room temperature. Anti-FLAG primary antibody DYKDDDK tag polyclonal antibody (Invitrogen, Rockford, IL) was incubated at a dilution of 1:1000 in 2% goat serum in PBS-T overnight at 4°C. After 3 washes with PBS-T, cells were incubated with secondary antibody goat anti-rabbit Alexa Fluor 568 (Life Technologies, Carlsbad, CA) at a dilution of 1:1000 in PBS-T for 1.5 hour at room temperature. After 3 washes with PBS-T, cells were imaged using confocal microscopy at 63×.
Electrophysiological recordings
Mice (postnatal day > 25) were anesthetized with isoflurane, brains were removed, and brain slices containing the ventrolateral periaqueductal gray (vlPAG) were cut with a vibratome (180–220 µm thick) in sucrose cutting buffer containing 75 mM NaCl, 2.5 mM KCl, 0.1 mM CaCl2, 6 mM MgSO4, 1.2 mM NaH2PO4, 25 mM NaHCO3, 2.5 mM dextrose, and 50 mM sucrose and placed in a holding chamber with artificial cerebral spinal fluid containing126 mM NaCl, 21.4 mM NaHCO3, 11.1 mM dextrose, 2.5 mM KCl, 2.4 mM CaCl2, 1.2 mM MgCl2, and 1.2 mM NaH2PO4, pH 7.35, and equilibrated with 95% O2/5% CO2 until moved into a recording chamber. In experiments using gallein and myristoylated peptide from the C terminus of Gαq (myrGαq-CT) inhibitors, slices were incubated for at least 30 minutes in artificial cerebral spinal fluid plus inhibitor before recording. Recordings were made with electrodes pulled to 2 to 4 MOhm resistance with an internal solution consisting of 140 mM CsCl, 10 mM HEPES, 10 mM KCl, 1 mM MgCl2, 1 mM EGTA, 0.3 mM CaCl2, 4 mM MgATP, and 3 mM NaGTP, pH 7.4. Junction potentials of 5 mV were corrected at the beginning of the experiments. Access resistance was monitored throughout the experiments. Neurons were voltage-clamped at −70 mV. Miniature inhibitory postsynaptic currents (mIPSCs) were collected in the presence of tetrodotoxin (500 nM). Evoked inhibitory postsynaptic currents (eIPSCs) were stimulated with bipolar stimulating electrodes placed approximately 50 to 100 µm away from the recording site. A paired-pulse stimulation paradigm was used [2 pulses (2 milliseconds) at 50–100 millisecond intervals] and paired-pulse ratios (pulse 2/pulse 1) were determined. Data were collected with Axopatch 200B microelectrode amplifier (Molecular Devices) at 5 kHz and low-pass filtered at 2 kHz. Currents were digitized with InstruTECH ITC-18 (HEKA), collected via AxoGraph data acquisition software, and analyzed using AxoGraph (Axograph Scientific).
Intracerebroventricular pretreatment injection
Intracerebroventricular injection was done after baseline withdrawal latencies were taken and before morphine injection. Hamilton syringes of 10 µL with a 26 G (catalog no. 7804-03; point 4, 12° bevel) needle were used, with a custom-made stopper that allowed 4 mm of the needle to enter the skull. Mice were anesthetized using isoflurane until they were no longer responsive to noxious stimuli and breathing slowed down to 1 inhale per second. Injection is free-handed utilizing the ears and eyes for orientation to target the lateral ventricles of the brain. The needle was inserted through the skull using published methods (Laursen and Belknap, 1986). Immediately after the experimental procedure, mice are euthanized to confirm injection site. When the needle enters the skull, 3 µL of solution is injected into the ventricles, and then after 30 seconds, the needle is carefully removed. Mice were placed back in their home cage to recover from isoflurane anesthesia. Following recovery from anesthesia, mice were able to move and groom in a normal manner. Gallein was administered 30 minutes prior to morphine; MyrGαq-CT peptide was administered 60 minutes prior to morphine.
Warm water tail withdrawal
Withdrawal latencies were determined by briefly placing a mouse into a cylindrical plastic restrainer and immersing 2 to 3 cm of the tail tip into a water bath maintained at 55°C. The latency to tail withdrawal or rapidly flicking the tail back and forth was recorded with a maximum cutoff time of 15 seconds to prevent tissue damage; baseline latencies, 2 to 3 seconds for 55°C, were consistent for each assay. Mice were briefly habituated to handling and restrainer, injected with saline (intraperitoneally), and 30 minutes later, withdrawal latencies were recorded (baseline withdrawal latency). Thirty minutes after intracerebroventricular injections, withdrawal latencies were recorded, and then mice were injected with 3.2 mg/kg morphine (intraperitoneally). Withdrawal latencies were recorded 30, 60, 90, and 120 minutes post-morphine injection.
Data analysis
For HEK cell experiments, mean ± S.E.M. were calculated for each data set. For animal experiments, mean and S.D. were calculated for each data set. Where indicated, unpaired Student’s 2-tailed t tests, 1-way ANOVAs with Tukey’s or Dunnett’s multiple comparisons tests, or 2-way ANOVAs with Sidak’s multiple comparisons tests were conducted for all analyses involving the comparison of group means as indicated in the figure legends. Concentration dependent curves were fitted using nonlinear regression. Area under the curve calculations are after baseline subtraction (vehicle). %Max response was calculated as a percentage of the maximal response to phorbol 12,13-dibutyrate (PDBu) of separately analyzed wells in the same plate after vehicle subtraction. All analyses were performed using Prism 9 (GraphPad, San Diego, CA). Statistical significance was accepted at P < 0.05.
Each electrophysiological recording from a single neuron is treated as an individual observation because the vlPAG contains heterogenous cell populations; however, all data sets contain recordings from at least 3 separate animals. Drug effects were reversed by specific antagonists, and peak drug effects were measured as an increase in current from the average of baseline and washout or the presence of antagonists. Differences between groups were assessed using Student’s t test or ANOVA when appropriate (significance is denoted as *P < 0.05). All data are expressed as mean ± S.E.M. except Fig. 6, where data are mean and S.D. Data were analyzed with Prism 9 (GraphPad Software).
Results
MOR does not activate PLCβ in HEK293 cells
To measure activation of PLC in cells, HEK 293 cells were transduced with a PKC-based fluorescent reporter that detects PLC-dependent DAG production as an increase in overall fluorescence intensity (Green UP DAG assay, Montana Molecular) (Tewson et al., 2016). The sensor was expressed efficiently in the cytoplasm of the cells (Fig. 1A). Addition of the phorbol ester Pbdu as a positive control that binds directly to C1 domain of the sensor produced a strong sustained increase in fluorescence (Fig. 1A). To measure GPCR-dependent PLC activation, we transduced cells with the Gq-coupled M1 muscarinic acetylcholine receptor and stimulated cells with the muscarinic agonist carbachol. After establishing baseline fluorescence, addition of carbachol produced a strong time-dependent increase in fluorescence (Fig. 1B). To measure MOR-dependent activation of PLC activity, cells transduced with the reporter were transfected with N-terminally flag tagged MOR. MOR was detected at the plasma membrane (PM) by immunocytochemistry in the majority of the cells (Fig. 1C). Surprisingly, addition of saturating concentrations of DAMGO or morphine produced no detectable PLC activation (Fig. 1D).
Fig. 1.
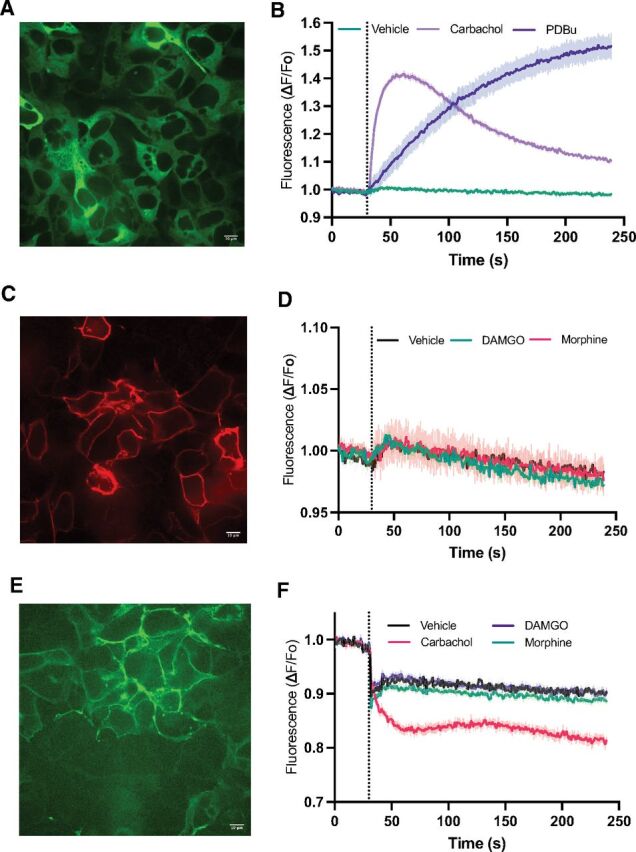
MOR activation alone does not stimulate detectable DAG production. (A) Field of HEK293 cells showing transduction of the DAG reporter. HEK293 cells were transduced with a fluorescent DAG reporter (DAG-up, Montana Molecular) and M1 muscarinic acetylcholine receptor (M1R). (B) At the dotted line, Gibco Dulbecco’s PBS vehicle (Veh), PDBu or 50 μM carbachol (Carb) were added, and the change in fluorescence intensity across the entire well of a 96-well plate relative to baseline Fo was monitored. (C) Cells were transduced with the DAG reporter, transfected with flag-MOR. Cells were fixed and stained with an antiflag antibody. (D) Cells transduced with the DAG reporter and transfected with flag-MOR were treated with Gibco Dulbecco’s PBS (veh), 1 μM DAMGO or 10 μM morphine (Morph) as in (B). (E) Cells were transduced with the CAAX-DAG reporter (DAG-down, Montana Molecular) and M1R and transfected with MOR. Shown is a field of live cells showing PM localization of the reporter. (F) Cells expressing CAAX-DAG reporter, M1R, and MOR were treated with the indicated agonists and fluorescence intensity was measured. For (B), (D). and (F), each trace is the mean ± S.E.M. of 3 or 4 separately transduced wells in a 96-well plate, representative of at least 3 separate experiments. All baseline traces were normalized to 1. The initial downward deflections at the dotted line in traces in (F) are artifacts associated with compound/vehicle addition.
We considered the possibility that the DAG sensor needs to be targeted to the PM to detect local PLC activation and DAG production (Halls et al., 2016). To target the DAG sensor to the PM, a PM targeting CAAX sequence was fused to the C-terminus of a DAG sensor that responds with decreasing fluorescence intensity upon DAG binding (CAAX-Green Down DAG assay, Montana Molecular). Localization to the PM was confirmed by monitoring GFP fluorescence of the sensor transduced into HEK293 cells (Fig. 1E). Activation of transfected MOR with either DAMGO or morphine produced no detectable change in reporter fluorescence, while transduced M1 muscarinic receptors produced robust DAG accumulation (Fig. 1F).
Synergistic stimulation of PLC activity by muscarinic Gq-coupled receptors and MOR in HEK293 cells
Synergistic activation of PLCβ3 has been implicated in cross-talk between Gi- and Gq-coupled receptor-dependent activation of PI hydrolysis in cells (Rebres et al., 2011; Pfeil et al., 2020). To test whether low-level stimulation of Gαq signaling could reveal MOR-dependent stimulation of DAG production downstream of PLC activity, we coexpressed the M1-muscarinic receptor with MOR in HEK293 cells and stimulated with either a subsaturating concentration (100 nM) of carbachol alone or carbachol with saturating concentrations of either DAMGO (100 nM) or morphine (1 μM). As before, treatment of cells with either DAMGO or morphine did not activate the DAG reporter while 100 nM carbachol led to a small increase in DAG production. When cells were costimulated with DAMGO and carbachol or morphine and carbachol together, DAG production was strongly increased relative to the signal with carbachol alone (Fig. 2A). Traces were quantified, and data are plotted in Fig. 2B. Since DAMGO and morphine gave no response on their own, anything greater than the carbachol alone response is greater than additive and thus synergistic. To confirm that DAMGO and morphine components of the synergistic responses were Gi-dependent, cells were pretreated with PTX, followed by addition of agonists. Treatment with PTX eliminated the DAMGO or morphine-dependent components of the response without affecting the response to carbachol (Fig. 2C–2E).
Fig. 2.
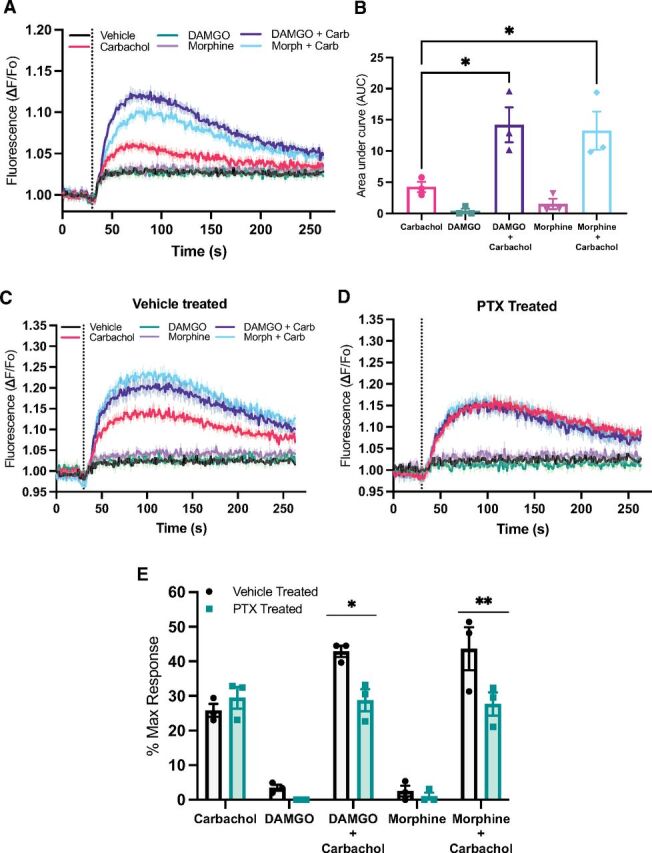
Coactivation of MOR and Gq-coupled muscarinic receptors reveals synergistic PLC activation. (A) HEK293 cells stably expressing flag-MOR were transduced with the DAG reporter and M1 receptor as in Fig. 1A and 1B. Cells were treated (compounds added at the dashed vertical line) with Gibco Dulbecco’s PBS, 100 nM carbachol, 500 nM DAMGO, 1 μM morphine, carbachol + DAMGO, or carbachol + morphine at the same concentrations. (B) To quantify these responses the area under the curve with vehicle subtracted for each curve was calculated; 1-way ANOVA F(4,10) = 11.5, P = 0.0009. (C and D) Stable MOR, HEK293 cells transduced, transfected, and treated as in (A) were treated for 16 hours without (C) and with (D) 100 ng/mL PTX for 16 hours. (E) Peak DAG production at each concentration of agonist relative to maximum PDBu-dependent DAG production was calculated from 3 independent experiments, each performed with 4 replicates. All data are mean ± S.E.M. P values were calculated with an ordinary 1-way ANOVA with Tukey’s post hoc test and 2-way ANOVA with Sidak’s post hoc test. *P < 0.05 and **P < 0.005.
Cooperation of MOR with Gq-coupled receptors in PLC activation is generalizable
To explore synergy with endogenous Gαq-coupled receptors, we used HEK293 cells with stable expression of MOR but without transfected M1-muscarinic receptor. HEK293 cells have been reported to endogenously express the M3 muscarinic receptor (Atwood et al., 2011). Stimulation with saturating concentrations of carbachol (50 μM) gave a barely detectable signal likely due to low-level endogenous expression of the M3 receptor (Fig. 3A and 3B). However, costimulation with carbachol and DAMGO resulted in strong PLC activation (Fig. 3A and 3B). We performed concentration response analysis for both DAMGO and morphine in the presence of a fixed 50 μM concentration of carbachol (a representative experiment for DAMGO is shown in Fig. 3C). EC50 for DAMGO and morphine were calculated from multiple experiments (Fig. 3D). HEK293 cells have also been reported to endogenously express other Gq-coupled receptors including P2Y11 and P2Y12 purinergic receptors and the protease-activated receptor F2R (PAR-1) (Atwood et al., 2011). Stimulation of HEK293 cells with either the purinergic agonist ATP (100 μM) or the PAR-1 agonist PAR-1 activating peptide (3 μM) resulted in very low levels of detectable DAG production that was strongly enhanced in the presence of DAMGO (Fig. 3E–3H). Both PAR and purinergic receptors can also couple to other G proteins; thus, it remains possible that other G proteins, including Gi and G12/13, could be involved in this process. Nevertheless, the most straightforward interpretation of the data, consistent with prior literature, is a model where low-level Gq activation, regardless of the nature of the activating receptor, synergizes with MOR to reveal MOR-dependent PLC activation in HEK293 cells.
Fig. 3.
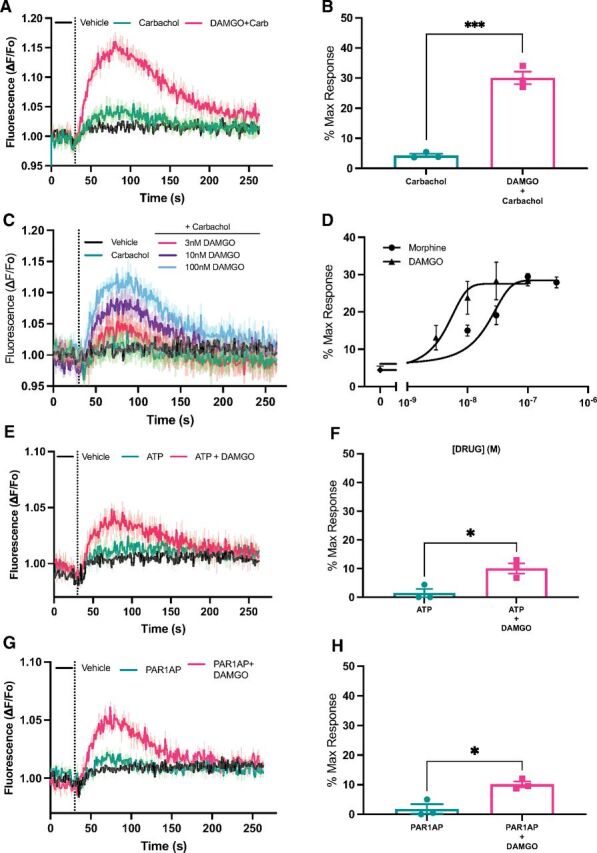
Gq synergy with MOR for PLC activation is independent of the nature of the stimulating Gq-coupled GPCR. (A) HEK cells stably expressing MOR and transduced with the DAG reporter without transduction of the M1 muscarinic receptor were treated with vehicle, 50 μM carbachol, or 50 μM carbachol + 100 nM DAMGO. Data are mean ± S.E.M. from 1 representative experiment. (B) Peak DAG production relative to maximum PDBu-dependent DAG production was calculated from 3 independent experiments each performed in 4 independent wells as in (A); unpaired t test t(4) = 12.03, P = 0.0003. (C) HEK293 cells stably expressing MOR were as in (A) treated with vehicle, 50 μM carbachol, or carbachol + varying concentrations of DAMGO. Representative traces from 1 experiment with 4 replicates each condition. (D) Peak DAG production at each concentration of agonist relative to maximum PDBu-dependent DAG production was calculated from 3 independent experiments each performed in 4 independent wells for each concentration. Nonlinear regression curve fitting: morphine EC50= 17 nM (95% CI, 7–27 nM) and DAMGO EC50= 3.8 nM (95% CI, 1.6–7.7 nM). (E) Experiments were performed as in (A) except 100 μM ATP instead of carbachol was used as the agonist for activation of Gq. Data are mean ± S.E.M. from 1 representative experiment. (F) Peak DAG production relative to maximum PDBu-dependent DAG production was calculated from 3 independent experiments each performed in 4 independent wells as in B; unpaired t test t(4) = 3.7, P = 0.02. (G) Experiments were performed as in (A) except 3 μM PAR-1 activating peptide (SSFLRN) was used as the agonist for activation of Gq. Data are mean ± S.E.M. from 1 representative experiment. (H) Peak DAG production relative to maximum PDBu-dependent DAG production was calculated from 3 independent experiments each performed in 4 independent wells as in B; unpaired t test t(4) = 4.45, P = 0.01. *P < 0.05, ***P < 0.0005.
DAMGO-mediated inhibition of GABA release in the PAG is greater in PLCβ3 KO mice or with blockade of Gαq or Gβγ signaling
To examine the role of synergistic PLCβ activation via Gq and MOR-dependent Gi/Gβγ signaling in a physiologic setting, we blocked each of these components individually in PAG brain slices. Inhibition of GABA release by presynaptic MORs in the PAG produces antinociception in the descending pain pathway (Vaughan et al., 1997; Lau and Vaughan, 2014; Morgan et al., 2020). Since the only PLCβ isoform that is synergistically regulated by Gαq and Gi/Gβγ is PLCβ3 and since MOR-mediated antinociception is enhanced in PLCβ3−/− mice, we first tested whether MOR-dependent inhibition of GABA release was potentiated in vIPAG slices from these mice. eIPSCs were isolated in the presence of NBQX, an inhibitor of AMPA glutamate receptor-mediated synaptic currents (Fig. 4A). The concentration-response curve for DAMGO-mediated inhibition of the GABAergic eIPSCs was shifted to the left in recordings from PLCβ3−/− slices compared with recordings from PLCβ3+/+ slices (Fig. 4B). Thus, lower concentrations of DAMGO were sufficient to inhibit GABAergic eIPSCs when PLCβ3 was deleted.
Fig. 4.

DAMGO inhibition of GABAergic eIPSCs is potentiated in slices from PLCβ3−/− mice and with Gβγ inhibition. (A) Representative eIPSCs from a recording from a slice from a WT mouse showing the effect of DAMGO (1 µM) and reversal with naloxone. (B) Concentration-response curves for DAMGO-mediated inhibition in slices from WT compared with PLCβ3−/−mice. The EC50 for DAMGO is shifted to the left in slices from PLCβ3−/−mice (238 nM; 95% CI, 140–376 nM) compared with WT mice (1.3 µM; 95% CI, 732 nM–2.8 µM; F(1,79) = 22.8, P < 0.0001. Recordings were from 4 to 6 cells from at least 3 mice per data point. (C) Gallein shifted the DAMGO concentration-response curve in slices from PLCβ3+/+ mice (control, 0.8 μM (95% CI 0.52–1.54 μM), gallein, 0.12 μM (95% CI, 0.08–0.18 μM)]. (D) Gallein does not shift the DAMGO concentration-response curve in slices from PLCβ3−/− mice. EC50 = 162 nM (95% CI 81–367 nM). PLCβ3+/+ and PLCβ3−/− curves are the same as in (B). Recordings were from 4 to 6 cells from at least 3 mice per data point. Data are mean ± S.E.M.
To test whether inhibition of Gβγ signaling could potentiate MOR-dependent inhibition of GABA release, we incubated slices from WT mice with gallein (Fig. 4C). Gallein is an inhibitor of Gβγ that selectively blocks activation of a subset of effectors including PLCβ3 (Bonacci et al., 2006). We have previously demonstrated that gallein enhances the antinociceptive potency of morphine in mice (Bonacci et al., 2006; Lehmann et al., 2008; Mathews et al., 2008; Bianchi et al., 2009; Campbell and Smrcka, 2018) supporting the idea that gallein inhibits PLCβ3 activation by Gβγ without inhibiting interaction of Gβγ with other targets relevant to MOR actions including Ca2+ and K+ channels . Gallein (10 µM) potentiated inhibition at various concentrations of DAMGO, leading to a left shift in the DAMGO concentration-response curve with a minor effect on efficacy (Fig. 4C). Gallein also potentiated the ability of DAMGO (50 nM) to inhibit the frequency of mIPSCs (43% ± 4%, n = 6) compared with control (6% ± 4%, n = 5), without changing mIPSC amplitude (2% ± 5%) indicating a presynaptic effect of gallein on MOR signaling. Gallein had no effect in slices from PLCβ3−/− mice (Fig. 4D), indicating that gallein enhances MOR-dependent inhibition of GABA release through blockade of Gβγ-dependent regulation of PLCβ3. This provides evidence for a synaptic mechanism underlying gallein’s ability to enhance the nociceptive potency of morphine in mice.
To examine the role of Gαq signaling in the PAG we used a myrGαq-CT that competes for Gα subunit interactions with endogenous GPCRs to prevent G protein activation (Gilchrist et al., 2002). Slices pretreated with myrGαq-CT revealed DAMGO-dependent inhibition of eIPSCs at 50 nM DAMGO to an extent similar to treatment with gallein (Fig. 5A). A similar potentiation was produced after incubating slices in a small molecule inhibitor of Gαq YM-254890 (500 nM) (Schmitz et al., 2014). The paired-pulse ratios for eIPSCs in the presence of DAMGO compared with baseline were changed in both inhibitors (myrGαq-CT: t(5) = 5.4, P = 0.003; YM-254890: t(5) = 3.1, P = 0.03), indicating that the DAMGO-mediated inhibition is via presynaptic MORs. Neither gallein nor myrGαq-CT had any effect on DAMGO dependent inhibition of eIPSCs in slices from PLCβ3−/− mice (Fig. 5B) or in the absence of DAMGO stimulation (Fig. 5C). These results indicate that blocking either Gβγ or Gαq is sufficient to enhance MOR inhibition of GABA release at low concentrations of a MOR agonist and that these G proteins dampen MOR signaling in vlPAG terminals via PLCβ3.
Fig. 5.

Gq inhibition potentiates DAMGO inhibition of GABAergic eIPSCs in PAG slices. (A) Bar graph comparing effects of Gβγ and Gq inhibitors on inhibition of eIPSCs produced by a single dose of DAMGO (50 nM). All samples were preincubated with either vehicle control (DMSO) or the indicated inhibitors at 10 μM for 30 minutes, followed by addition of DAMGO. One-way ANOVA, F(3, 21) = 14.4, P < 0.0001; Dunnett’s multiple comparisons, **P < 0.01, ****P < 0.0001. (B) Gallein (10 μM) and mGq-CT (10 μM) had no effect on DAMGO-dependent inhibition of eIPSCs in PAG slices isolated from PLCβ3−/− mice. Experiments were performed as in (A) except PAG slices from PLCβ3−/− mice were used. Symbols denote number of recordings and numbers in bars denote number of animals. (C) Gallein (10 μM) and mGq-CT (10 μM) had no effect on eIPSCs in the absence of MOR activation. eIPSCs were measured before and after addition of the indicated inhibitors without addition of DAMGO. Data are mean ± S.E.M. of all recordings. Numbers in bar graphs indicate the number of animals tested.
Since either Gαq or Gβγ inhibition alone is sufficient to enhance MOR potency, these data together indicate that signaling via both subunits simultaneously is required to maintain inhibition of MOR-dependent regulation of neurotransmitter release via PLCβ3.
Gq signaling and antinociception in mice
Since simultaneous activation of PLCβ3 by Gαq and Gβγ is required for inhibition of MOR-dependent regulation of GABA release in the PAG we examined whether either inhibition of Gαq or Gβγ is sufficient to enhance MOR-dependent antinociception. As previously discussed, we have previously demonstrated that Gβγ inhibition with gallein enhances morphine-dependent antinociception (Bonacci et al., 2006; Mathews et al., 2008). To test whether Gq inhibition in vivo in the PAG would enhance MOR-dependent antinociception, we injected mice intracerebroventricularly with either myrGαq-CT or control myrGαq-scrambled peptide or with gallein as a reference and measured morphine effects (3.2 mg/kg) in the warm-water tail withdrawal assay. At this dose, morphine alone had very little, if any, effect on tail withdrawal latencies as compared with baseline and post-intracerebroventricular withdrawal latencies. As previously described (Mathews et al., 2008), gallein (100 nmoles) had no effect on antinociception in the absence of morphine (post-intracerebroventricularly on graph) but strongly increased the effects of morphine, in terms of magnitude and duration of antinociception (Fig. 6A). Similarly, mice injected with myrGαq-CT did not have altered withdrawal latencies compared with baseline latencies and compared with myrGαq-scrambled control peptide alone but showed enhanced morphine-induced antinociception compared with DMSO (vehicle) or myrGαq-scrambled peptide injected mice (Fig. 6B).
Fig. 6.
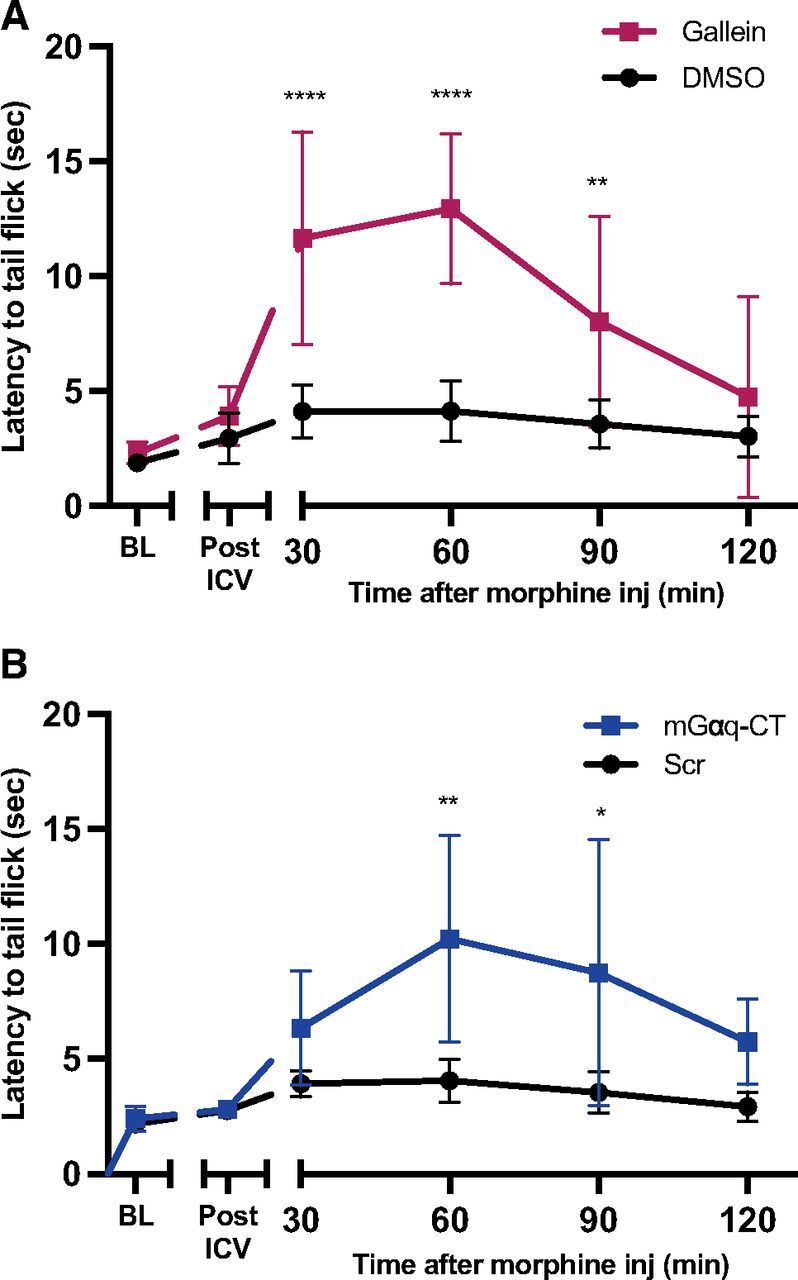
Gβγ and Gq inhibition enhance morphine-induced antinociception in mice. (A) Mice were injected intracerebroventricularly with gallein (100 nmoles) or DMSO (8 mice per condition) and allowed to recover for 30 minutes, and post-intracerebroventricular tail flick latency was measured. Then 3.2 mg/kg of morphine was injected at time 0, and tail flick latency was measured at the indicated times; mixed effects 2-way ANOVA with Sidak’s multiple comparisons, significant interaction of time X pretreatment effect F(5,69) = 9.2, P ≤ 0.0001 (B) Same as (A) except mGαq-CT (5 male mice) or myrGαq-scrambled (4 male mice) were injected intracerebroventricularly at 30 nmoles each; mixed effects 2-way ANOVA with Sidak’s multiple comparisons, significant interaction of time X pretreatment effect F(5,35) = 2.78, P = 0.03. Data were analyzed with a mixed effects ANOVA followed by Sidak’s multiple comparisons test. *P < 0.05, **P < 0.005, ****P < 0.0001 at each time point comparing treatment to control (gallein vs. DMSO or mGαq-CT vs. Scr peptide). Data are mean ± S.D.
These data, together with prior data demonstrating that MOR-dependent antinociception is enhanced in PLCβ3−/− mice, support the idea that Gαq signaling in cooperation with Gβγ signaling via PLCβ3 in the central nervous system opposes MOR-dependent antinociception (Fig. 7). This model explains how blockade of any of these components enhances morphine-dependent antinociception in vivo.
Fig. 7.

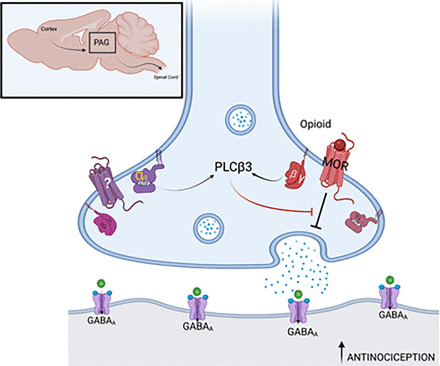
Model for mechanism of MOR-Gq coincidence detection in feedback inhibition of MOR-dependent antinociception in presynaptic PAG input neurons in the descending pain pathway. Pictured is a GABAergic synapse between a PAG input and output neuron. The boxed inset shows the anatomic location of the PAG in the rodent brain with inputs from the cortex and outputs to the spinal cord. MOR activation in the presynaptic neuron inhibits GABA release resulting in activation of output neurons that ultimately suppress afferent pain transmission in the spinal cord. PLCβ3 activation suppresses MOR actions in presynaptic neuron, and activation of PLCβ3 requires inputs from both Gi/βγ from MOR and Gq from an unknown Gq-coupled receptor. Since PLCβ3 activation requires simultaneous Gαq and Gβγ binding, blockade of either Gαq or Gβγ is sufficient to relieve the PLC-dependent inhibition of MOR signaling, leading to enhanced MOR potency and increased antinociception. Figure created with Biorender.com.
Discussion
Previous work identified negative regulatory effects of PLCβ3 on opioid antinociception (Xie et al., 1999). Our prior studies showed that inhibitors of Gβγ (M119 and gallein) enhance opioid-mediated antinociception (Bonacci et al., 2006; Mathews et al., 2008; Hoot et al., 2013) and that the effects of M119 were occluded in mice with PLCβ3 deletion supporting the idea that gallein and M119 block Gβγ-PLCβ3 interactions (Bonacci et al., 2006). Based on this information, we proposed that Gβγ released from Gi-coupled MORs activates PLCβ3, which opposes MOR-stimulated analgesia. Importantly, our results presented here show that opioids do not appreciably activate PLC and subsequent DAG production on their own unless there is coincident signaling from Gq-coupled receptors. Cross-talk between Gq-coupled receptors and MOR has previously been described for regulation of Ca2+ signaling in MOR expressing cell lines, but the mechanism for this cross-talk and its role in MOR biology have not been clearly defined (Connor and Henderson, 1996; Yeo et al., 2001; Samways et al., 2003; Werry et al., 2003; Samways and Henderson, 2006). Here we provide evidence for coincident regulation of PLC activation by Gq and Gi/Gβγ signaling MOR in HEK293 cells and show that this synergistic pathway operates in PAG synapses, a critical brain region involved in MOR-dependent antinociception (Fig. 7). Finally we show that Gβγ and Gq signaling both oppose MOR-dependent antinociception in mice. These data, together with previous data from PLCβ3−/− mice, implicate PLCβ3 as a source of Gq-MOR cross-talk in the central nervous system for MOR-dependent antinociception.
PLCβ3, but not PLCβ1 or PLCβ4, is activated by Gβγ, and the vast majority of effector regulation by Gβγ subunits occurs downstream of Gi-coupled receptors (Campbell and Smrcka, 2018; Smrcka and Fisher, 2019). PLCβ3 is unique in that it is strongly synergistically regulated by Gαq and Gβγ, and it was proposed that this could serve as a coincidence detector for cells to respond to simultaneous signals from Gi- and Gq-coupled receptors (Philip et al., 2010; Rebres et al., 2011). This was initially demonstrated in detailed in vitro biochemical reconstitution experiments and later confirmed downstream of Gq- and Gi-coupled receptors in bone marrow derived macrophages and in NIH3T3 cells. A recent study confirmed and extended these observations with a broader range of receptors (Pfeil et al., 2020).
In the HEK cell-based studies we tested several examples of Gq-coupled receptors and observed synergistic activation of PLC indicating that the negative regulation exerted on MORs originates from the biochemical properties of PLCβ3. Thus, we propose that any Gq-coupled receptor would synergize with MOR in this system. The myrGαq-CT used as an inhibitor of Gq signaling in these studies does not inhibit Gαq directly but rather competes for interactions between Gq-coupled GPCRs and Gαq preventing activation of Gαq by GPCRs. The effectiveness of this inhibitor in our experiments indicates that tonic Gq-coupled receptor activation in the PAG is limiting MOR-mediated analgesia via this mechanism. Future experiments will determine the nature of this receptor or possibly multiple receptors. A recent C. elegans screen identified GPR139 as a Gq-coupled GPCR that opposes opioid analgesia (Wang et al., 2019) and is one possible candidate.
In contrast to our results, Halls et al. (2016) reported that morphine, but not DAMGO, activated a PM targeted PKC sensor, pmCKAR, without a requirement for coincident Gq activation. We see robust responses to both DAMGO and morphine in the presence of a Gq stimulus regardless of the localization of the sensor. It is possible that the DAG sensor used in our study is less sensitive than pmCKAR. CKAR is a PKCα-based FRET reporter, which contains both Ca2+ and DAG binding sites that interact cooperatively, which may sensitize CKAR to local generation of DAG in the presence of elevated Ca2+. It is also possible that in the HEK cell line used in that study, there is a tonic Gq signal that does not translate across different HEK cell lines. Thus, while MOR may stimulate low-level PLC activation in the absence of Gq signaling, coincident Gq activation results in robust MOR-dependent PLC activation that we demonstrate to have physiologic relevance.
One strategy to avoid development of opioid tolerance for treatment of chronic pain and to reduce the potential for addiction would be to lower the doses of morphine needed to produce analgesia. We and others have previously shown that inhibition of Gβγ subunits increases the antinociceptive potency of morphine without enhancing side effects such as constipation and respiratory depression (Hoot et al., 2013) suggesting that inhibition of a subset of effectors downstream of Gβγ is a possible strategy to reduce morphine doses required for pain management (Campbell and Smrcka, 2018). Targeting Gαq signaling is a possible alternative strategy. Since Gαq signaling appears to be tonically activated by a yet to be unidentified Gq-coupled GPCR, either an antagonist or inverse agonist targeting this receptor would likely enhance the antinociceptive effects of morphine.
Highly potent MOR agonists such as fentanyl already exist and are very dangerous drugs. Since these drugs target MOR itself, their potency with respect to causing side effects severely limits their usefulness. MOR-dependent G protein activation is relatively cell context independent, while signaling downstream of G protein activation is highly cell context dependent. Gβγ signaling depends on the cell type–specific expression of Gβγ-regulated effectors and cell type–specific responses to regulation of those receptors (Lin and Smrcka, 2011; Campbell and Smrcka, 2018; Fisher et al., 2020). Thus, targeting PLCβ3 or its regulators may enhance the potency of MOR antinociceptive effects relative to side effects because the neurons responsible for antinociception may have different downstream signaling responses that are more sensitive to PLCβ3 than the neurons responsible for respiratory depression or constipation. The signaling mechanisms downstream of PLCβ3 that oppose opioid analgesia have not yet been identified. One possibility is through PKC-dependent phosphorylation of key targets such as MOR itself. Both of these issues will be the subject of further investigation.
Author Contributions
Participated in research design: Smrcka, Jutkiewicz, Ingram.
Conducted experiments: Sanchez, Ingram.
Performed data analysis: Sanchez, Smrcka, Jutkiewicz, Ingram.
Wrote or contributed to the writing of the manuscript: Sanchez, Smrcka, Jutkiewicz, Ingram.
Abbreviations
- DAG
diacylglycerol
- DAMGO
[D-Ala2, N-Me-Phe4, Gly5-ol]-enkephalin acetate salt
- eIPSCs
evoked GABAergic inhibitory postsynaptic currents
- GPCRs
G protein-coupled receptors
- HEK293
human embryonic kidney 293 cell line
- mIPSCs
miniature inhibitory postsynaptic currents
- MOR
μ-opioid receptor
- myrGαq-CT
myristoylated peptide from the C terminus of Gαq
- PAG
periaqueductal gray
- PAR-1
protease-activated receptor F2R
- PDBu
phorbol 12,13-dibutyrate
- PKC
protein kinase C
- PLC
phospholipase C
- PM
plasma membrane
- PTX
pertussis toxin
- PBS-T
PBS containing 0.1% Triton ×100
- vlPAG
ventrolateral periaqueductal gray
- WT
wild-type
Footnotes
This work was supported by National Institutes of Health Institute of Drug Abuse [Grants R01DA048625; R01DA048625-S1].
No author has an actual or perceived conflict of interest with the contents of this article.
Department of Pharmacology, University of Michigan, Ann Arbor, Michigan (G.A.S., E.M.J., A.V.S.) and Department of Neurologic Surgery, Oregon Health Sciences University, Portland, Oregon (S.I.)
Current affiliation: Department of Anesthesiology, University of Colorado, Anschutz Medical Campus, Aurora, Colorado.
References
- Arttamangkul S, Birdsong W, Williams JT (2015) Does PKC activation increase the homologous desensitization of μ opioid receptors? Br J Pharmacol 172:583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Lopez J, Wager-Miller J, Mackie K, Straiker A (2011) Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics 12:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Llorente J, Gabra BH, Smith FL, Dewey WL, Kelly E, Henderson G (2009) Role of protein kinase C and mu-opioid receptor (MOPr) desensitization in tolerance to morphine in rat locus coeruleus neurons. Eur J Neurosci 29:307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CPOldfield SLlorente JCaunt CJTeschemacher AGRoberts LMcArdle CASmith FLDewey WLKelly E, et al. (2009) Involvement of PKC α and G-protein-coupled receptor kinase 2 in agonist-selective desensitization of mu-opioid receptors in mature brain neurons. Br J Pharmacol 158:157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi E, Norcini M, Smrcka A, Ghelardini C (2009) Supraspinal Gbetagamma-dependent stimulation of PLCbeta originating from G inhibitory protein-mu opioid receptor-coupling is necessary for morphine induced acute hyperalgesia. J Neurochem 111:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG (2000) Mu-opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature 408:720–723. [DOI] [PubMed] [Google Scholar]
- Bonacci TM, Mathews JL, Yuan C, Lehmann DM, Malik S, Wu D, Font JL, Bidlack JM, Smrcka AV (2006) Differential targeting of Gbetagamma-subunit signaling with small molecules. Science 312:443–446. [DOI] [PubMed] [Google Scholar]
- Campbell AP, Smrcka AV (2018) Targeting G protein-coupled receptor signalling by blocking G proteins. Nat Rev Drug Discov 17:789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor M, Henderson G (1996) delta- and mu-opioid receptor mobilization of intracellular calcium in SH-SY5Y human neuroblastoma cells. Br J Pharmacol 117:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher IJ, Jenkins ML, Tall GG, Burke JE, Smrcka AV (2020) Activation of phospholipase C β by Gβγ and Gαq involves C-terminal rearrangement to release autoinhibition. Structure 28:810–819.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabra BH, Bailey CP, Kelly E, Smith FL, Henderson G, Dewey WL (2008) Pre-treatment with a PKC or PKA inhibitor prevents the development of morphine tolerance but not physical dependence in mice. Brain Res 1217:70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist A, Li A, Hamm HE (2002) G alpha COOH-terminal minigene vectors dissect heterotrimeric G protein signaling. Sci STKE 2002:pl1. [DOI] [PubMed] [Google Scholar]
- Halls ML, Yeatman HR, Nowell CJ, Thompson GL, Gondin AB, Civciristov S, Bunnett NW, Lambert NA, Poole DP, Canals M (2016) Plasma membrane localization of the μ-opioid receptor controls spatiotemporal signaling. Sci Signal 9:ra16. [DOI] [PubMed] [Google Scholar]
- Hoot MR, Sypek EI, Reilley KJ, Carey AN, Bidlack JM, McLaughlin JP (2013) Inhibition of Gβγ-subunit signaling potentiates morphine-induced antinociception but not respiratory depression, constipation, locomotion, and reward. Behav Pharmacol 24:144–152. [DOI] [PubMed] [Google Scholar]
- Ingram SL, Traynor JR (2009) Role of protein kinase C in functional selectivity for desensitization at the mu-opioid receptor: from pharmacological curiosity to therapeutic potential. Br J Pharmacol 158:154–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandasamy RHillhouse TMLivingston KEKochan KEMeurice CEans SOLi MHWhite ADRoques BPMcLaughlin JP, et al. (2021) Positive allosteric modulation of the mu-opioid receptor produces analgesia with reduced side effects. Proc Natl Acad Sci USA 118:e2000017118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer A, Gillis A, Hill R, Schmiedel F, Bailey C, Kelly E, Henderson G, Christie MJ, Schulz S (2020) Morphine-induced respiratory depression is independent of β-arrestin2 signalling. Br J Pharmacol 177:2923–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau BK, Vaughan CW (2014) Descending modulation of pain: the GABA disinhibition hypothesis of analgesia. Curr Opin Neurobiol 29:159–164. [DOI] [PubMed] [Google Scholar]
- Laursen SE, Belknap JK (1986) Intracerebroventricular injections in mice. Some methodological refinements. J Pharmacol Methods 16:355–357. [DOI] [PubMed] [Google Scholar]
- Lehmann DM, Seneviratne AMPB, Smrcka AV (2008) Small molecule disruption of G protein β γ subunit signaling inhibits neutrophil chemotaxis and inflammation. Mol Pharmacol 73:410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D (2000) Roles of PLC-β2 and -β3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science 287:1046–1049. [DOI] [PubMed] [Google Scholar]
- Lin Y, Smrcka AV (2011) Understanding molecular recognition by G protein βγ subunits on the path to pharmacological targeting. Mol Pharmacol 80:551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews JL, Smrcka AV, Bidlack JM (2008) A novel Gbetagamma-subunit inhibitor selectively modulates μ-opioid-dependent antinociception and attenuates acute morphine-induced antinociceptive tolerance and dependence. J Neurosci 28:12183–12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MM, Tran A, Wescom RL, Bobeck EN (2020) Differences in antinociceptive signalling mechanisms following morphine and fentanyl microinjections into the rat periaqueductal gray. Eur J Pain 24:617–624. [DOI] [PubMed] [Google Scholar]
- Pfeil EMBrands JMerten NVögtle TVescovo MRick UAlbrecht IMHeycke NKawakami KOno Y, et al. (2020) Heterotrimeric G protein subunit Gαq is a master switch for Gβγ-mediated calcium mobilization by Gi-coupled GPCRs. Mol Cell 80:940–954.e6. [DOI] [PubMed] [Google Scholar]
- Philip F, Kadamur G, González Silos R, Woodson J, Ross EM (2010) Synergistic activation of phospholipase C-β3 by Galpha(q) and Gbetagamma describes a simple two-state coincidence detector. Curr Biol 20:1327–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebres RARoach TIAFraser IDCPhilip FMoon CLin KMLiu JSantat LCheadle LRoss EM, et al. (2011) Synergistic Ca2+ responses by Gαi- and Gαq-coupled G-protein-coupled receptors require a single PLCβ isoform that is sensitive to both Gβγ and Gαq. J Biol Chem 286:942–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samways DS, Henderson G (2006) Opioid elevation of intracellular free calcium: possible mechanisms and physiological relevance. Cell Signal 18:151–161. [DOI] [PubMed] [Google Scholar]
- Samways DS, Li WH, Conway SJ, Holmes AB, Bootman MD, Henderson G (2003) Co-incident signalling between μ-opioid and M3 muscarinic receptors at the level of Ca2+ release from intracellular stores: lack of evidence for Ins(1,4,5)P3 receptor sensitization. Biochem J 375:713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CL, Kennedy NM, Ross NC, Lovell KM, Yue Z, Morgenweck J, Cameron MD, Bannister TD, Bohn LM (2017) Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 171:1165–1175.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz ALSchrage RGaffal ECharpentier THWiest JHiltensperger GMorschel JHennen SHäußler DHorn V, et al. (2014) A cell-permeable inhibitor to trap Gαq proteins in the empty pocket conformation. Chem Biol 21:890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ (2011) β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci 32:521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrcka AV (2008) G protein βγ subunits: central mediators of G protein-coupled receptor signaling. Cell Mol Life Sci 65:2191–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrcka AV, Fisher I (2019) G-protein βγ subunits as multi-functional scaffolds and transducers in G-protein-coupled receptor signaling. Cell Mol Life Sci 76:4447–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrcka AV, Lehmann DM, Dessal AL (2008) G protein betagamma subunits as targets for small molecule therapeutic development. Comb Chem High Throughput Screen 11:382–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrcka AV, Sternweis PC (1993) Regulation of purified subtypes of phosphatidylinositol-specific phospholipase C β by G protein α and β γ subunits. J Biol Chem 268:9667–9674. [PubMed] [Google Scholar]
- Tewson PH, Martinka S, Shaner NC, Hughes TE, Quinn AM (2016) New DAG and cAMP sensors optimized for live-cell assays in automated laboratories. J Biomol Screen 21:298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Connor MA, Christie MJ (1997) How opioids inhibit GABA-mediated neurotransmission. Nature 390:611–614. [DOI] [PubMed] [Google Scholar]
- Wang DStoveken HMZucca SDao MOrlandi CSong CMasuho IJohnston COpperman KJGiles AC, et al. (2019) Genetic behavioral screen identifies an orphan anti-opioid system. Science 365:1267–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werry TD, Wilkinson GF, Willars GB (2003) Mechanisms of cross-talk between G-protein-coupled receptors resulting in enhanced release of intracellular Ca2+. Biochem J 374:281–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Samoriski GM, McLaughlin JP, Romoser VA, Smrcka A, Hinkle PM, Bidlack JM, Gross RA, Jiang H, Wu D (1999) Genetic alteration of phospholipase C β3 expression modulates behavioral and cellular responses to mu opioids. Proc Natl Acad Sci USA 96:10385–10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo A, Samways DS, Fowler CE, Gunn-Moore F, Henderson G (2001) Coincident signalling between the Gi/Go-coupled δ-opioid receptor and the Gq-coupled m3 muscarinic receptor at the level of intracellular free calcium in SH-SY5Y cells. J Neurochem 76:1688–1700. [DOI] [PubMed] [Google Scholar]