

Abstract
DNA topoisomerase IIα (170 kDa, TOP2α/170) is essential in proliferating cells by resolving DNA topological entanglements during chromosome condensation, replication, and segregation. We previously characterized a C-terminally truncated isoform (TOP2α/90), detectable in human leukemia K562 cells but more abundantly expressed in a clonal subline, K/VP.5, with acquired resistance to the anticancer agent etoposide. TOP2α/90 (786 aa) is the translation product of a TOP2α mRNA that retains a processed intron 19. TOP2α/90 lacks the active-site tyrosine-805 required to generate double-strand DNA breaks as well as nuclear localization signals present in the TOP2α/170 isoform (1531 aa). Here, we found that TOP2α/90, like TOP2α/170, was detectable in the nucleus and cytoplasm of K562 and K/VP.5 cells. Coimmunoprecipitation of endogenous TOP2α/90 and TOP2α/170 demonstrated heterodimerization of these isoforms. Forced expression of TOP2α/90 in K562 cells suppressed, whereas siRNA-mediated knockdown of TOP2α/90 in K/VP.5 cells enhanced, etoposide-mediated DNA strand breaks compared with similarly treated cells transfected with empty vector or control siRNAs, respectively. In addition, forced expression of TOP2α/90 in K562 cells inhibited etoposide cytotoxicity assessed by clonogenic assays. qPCR and immunoassays demonstrated TOP2α/90 mRNA and protein expression in normal human tissues/cells and in leukemia cells from patients. Together, results strongly suggest that TOP2α/90 expression decreases drug-induced TOP2α-DNA covalent complexes and is a determinant of chemoresistance through a dominant-negative effect related to heterodimerization with TOP2α/170. Alternative processing of TOP2α pre-mRNA, and subsequent synthesis of TOP2α/90, may be an important mechanism regulating the formation and/or stability of cytotoxic TOP2α/170-DNA covalent complexes in response to TOP2α-targeting agents.
Introduction
The well-characterized human DNA topoisomerase IIα (170 kDa, TOP2α/170) enzyme is essential in proliferating cells, because this enzyme processively resolves DNA topological entanglements that occur during chromosome condensation, replication, and segregation (DeWeese and Osheroff, 2009; Vos et al., 2011; Chen et al., 2013; Pommier et al., 2016). The TOP2α/170 enzyme is comprised of homodimers with enzymatic activity regulated by the successive opening and closing of three distinct subunit dimerization interfaces or gates, the entry N-/ATP-gate, DNA-gate, and exit C-gate (Dong and Berger, 2007; Laponogov et al., 2010; Wendorff et al., 2012).
The entry N-/ATP-gate is formed by the two N-terminal ATPase domains (i.e., first 400 amino acids [aa] of each TOP2α/170 subunit). When this gate is open, one DNA duplex (i.e., designated the G-segment) is loaded into the enzyme cavity (Dong and Berger, 2007; Laponogov et al., 2010; Wendorff et al., 2012). The TOP2α/170 enzyme subsequently generates a transient double-strand DNA break (i.e., TOP2α/170-DNA covalent cleavage complex) in this segment through a transesterification reaction mediated by the active site Tyr805 residue harbored in each TOP2α/170 subunit. Upon the binding of ATP, the N-/ATP-gate closes, and a second intact DNA duplex, designated the T-segment, is captured, transported through the DNA double-strand break (i.e., the DNA gate), and exits from the open C-gate (Dong and Berger, 2007; Laponogov et al., 2010; Wendorff et al., 2012). After T-segment strand passage, ATP hydrolysis occurs, the transient double-strand break in the G-segment is resealed by reversal of the transesterification event, which restores the integrity of the DNA and regenerates the free Tyr805 residue present in each TOP2α/170 subunit. Finally, the N-/ATP-gate is reopened, and the enzyme is reset for another round of catalytic activity (Dong and Berger, 2007; Laponogov et al., 2010; Wendorff et al., 2012).
Given that TOP2α/170 enzymatic activity is necessary for cell survival and that it is highly expressed in proliferating cells (Vos et al., 2011; Chen et al., 2013; Pommier et al., 2016), this enzyme has been widely exploited as an anticancer therapeutic target (Nitiss, 2009; Pommier et al., 2010). Type IIA topoisomerase interfacial inhibitors (e.g., etoposide, mitoxantrone, amsacrine, doxorubicin, and analogs) interact within the four base region of the scissile break sites generated by TOP2α/170 on the top and bottom strands of DNA and impede the reversal of the TOP2α/170-DNA covalent cleavage complex and the subsequent resealing of the double strand break (Pommier and Marchand, 2011). Therefore, inhibitors like etoposide, exert their cytotoxic therapeutic effects through the accumulation of double-strand DNA breaks, which ultimately trigger programmed cell death (Pommier and Marchand, 2011).
Recently, we demonstrated that both parental human leukemia K562 cells and a cloned K562 cell line (K/VP.5 cells) with acquired resistance to etoposide (Ritke and Yalowich, 1993; Ritke et al., 1994b) express two TOP2α isoforms, TOP2α/170 and a novel 90 kDa protein (i.e., TOP2α/90) (Kanagasabai et al., 2017). TOP2α/170 is decreased in K/VP.5 cells to approximately one-fifth the level found in parental K562 cells (Kanagasabai et al., 2017). In contrast, K/VP.5 cells express approximately threefold greater levels of TOP2α/90 compared with parental K562 cells (Kanagasabai et al., 2017).
TOP2α/90 (786 aa) is the translation product of a TOP2α mRNA that retains a processed intron 19 (302 nt), harbors an in-frame stop codon, and contains two consensus poly(A) sites (Fig. 1). As a result of intron 19 retention, TOP2α/90 is missing the C-terminal 770 aa present in TOP2α/170 (1531 aa), which are replaced by 25 unique aa encoded by translation of the exon 19/intron 19 “read-through.” Therefore, TOP2α/90 lacks active site Tyr805, canonical dimerization domains (DD) (aa 1053–1069 and 1121–1143), and the nuclear localization signals (NLS) (aa 1259–1296 and 1454–1497) found in the TOP2α/170 isoform (Kanagasabai et al., 2017) (Fig. 1).
Fig. 1.

Schematic representation of the TOP2α/170 (A) and TOP2α/90 (B) mRNA and protein. TOP2α/90 mRNA retains a processed intron 19 and encodes a TOP2α isoform of 786 aa. The well-characterized TOP2α/170 mRNA encodes a TOP2α isoform of 1531 aa. Depicted are 1) the ATP-gate that harbors the ATPase domain; 2) the DNA-gate that is comprised of the winged-helix domain (WHD) and harbors the active site tyrosine; 3) the C-gate that is comprised of the coiled-coil region (coiled domain) and harbors the characterized dimerization sequences, DD1053–1069 and DD1121–1143 (Frère et al., 1995; Berger et al., 1996; Frère-Gallois et al., 1997; Kroll, 1997; Bjergbaek et al., 1999); and 4) the C-terminal domain (CTD) that contains the defined nuclear localization signals NLS1259–1296 and NLS1454–1497 (Mirski et al., 1997, 1999). The epitope positions and the nomenclature for the antibodies used in our studies are denoted.
Based on the increased expression of TOP2α/90 in acquired resistant K/VP.5 cells and the structural differences between the TOP2α/90 and TOP2α/170 isoforms, the potential biologic functions and pharmacological consequences related to expression of the truncated TOP2α/90 isoform were investigated. Notably, results from immunoblot, immunofluorescence, and immunoprecipitation experiments revealed that, despite the absence of NLS and DD, TOP2α/90 was detected in the nucleus and formed heterodimers with TOP2α/170. Etoposide-induced DNA damage and cytotoxicity in K562 cells were decreased after forced expression of TOP2α/90. Conversely, etoposide-induced DNA strand breaks were increased in K/VP.5 cells subsequent to siRNA knockdown of TOP2α/90. Taken together these results are consistent with dominant negative effects of the TOP2α/90 isoform as a determinant of acquired drug resistance through heterodimerization with TOP2α/170, thereby decreasing drug-induced TOP2α/170-DNA covalent cleavage complexes.
Materials and Methods
Materials.
Human K562 leukemia cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Corning, Manassas, VA) supplemented with 10% fetal bovine serum (FBS). Etoposide-resistant K/VP.5 cells were selected and cloned subsequent to intermittent and eventually continuous exposure of K562 cells to 0.5 µM etoposide as previously described (Ritke and Yalowich, 1993). K/VP.5 cells were maintained in DMEM/10% FBS with etoposide (0.5 µM) added every other week. All experiments described below were performed utilizing K562 and K/VP.5 cells growing in log phase. Human stomach and thymus tissue lysates were purchased from Protein Biotechnologies Inc. (Ramona, CA). Human cord blood mononuclear cells were purchased from ALLCELLS (Alameda, CA). Deidentified acute myeloid leukemia (AML) patient samples were obtained from The Ohio State University Comprehensive Cancer Center Leukemia Tissue Bank Shared Resource.
Cytoplasmic and Nuclear Extracts Isolation.
Cytoplasmic and nuclear extracts were isolated utilizing NE-PER Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher Scientific, Waltham, MA) according to manufacturer’s protocol. Protein determinations using a Pierce BCA Protein Assay kit (Thermo Scientific, Rockford, IL) indicated that 15% and 18% of total cellular protein were in the nuclear fractions in K562 and K/VP.5 cells, respectively.
Immunoassays.
Cellular, cytoplasmic, nuclear, or tissue extracts were subjected to Western blot analysis as previously described (Kanagasabai et al., 2017). Unless otherwise noted, 12.5 µg of protein was loaded into each well. Membranes were incubated overnight at 4°C with one of the following primary antibodies: a rabbit polyclonal antibody raised against the human TOP2α/90/170 N-terminal sequence (aa 14–27) (cat. no. ab74715; Abcam, Cambridge, MA; used at 1:1000 dilution) (Kanagasabai et al., 2017), the anti-rabbit TOP2α/170 C-terminal specific antibody (generated against a recombinant 70-kDa fragment corresponding to only the TOP2α/170 C-terminal sequence; used at 1:5000 dilution) (Ritke et al., 1994a), the anti-rabbit TOP2α/90 specific antibody (raised against a synthetic peptide corresponding to the C-terminal sequence of TOP2α/90 encoded by the exon 19/intron 19 “read-through”; used at 1:1000 dilution or 1:250) (Kanagasabai et al., 2017), a mouse monoclonal β-actin antibody (cat. no. A1978; Sigma-Aldrich, St. Louis, MO, used at 1:5000 dilution), a mouse monoclonal lamin B antibody (cat. no. sc-365962; Santa Cruz Biotechnology, Santa Cruz, CA, used at 1:1000), or a glyceraldehyde 3-phosphate dehydrogenase mouse monoclonal antibody (1:2000 dilution, cat. no. sc-47724; Santa Cruz Biotechnology). The membranes were subsequently incubated at room temperature for ∼1.5 hours with a donkey anti-rabbit or anti-mouse secondary antibody (1:8000 dilution) (Jackson Immuno Research, West Grove, PA). Finally, TOP2α isoforms were detected using the Immun-Star or Clarity Max chemiluminescence kits (Bio-Rad Laboratories, Hercules, CA). All immunoassay images were acquired with the ChemiDoc XRS+ imaging system and analyzed with ImageLab software (Bio-Rad Laboratories).
Immunofluorescence Assays.
Log phase K562 and K/VP.5 cells (2 × 106) were washed once with PBS (pH 7.4) followed by loading aliquots (200 µl; 20,000 cells) onto glass slides for centrifugation/immobilization (Cytospin 4; ThermoFisher) and fixation (4% paraformaldehyde). Cells were then permeabilized in 0.1% Triton X-100 in ice-cold PBS for 30 minutes. Incubation in 4% bovine serum albumin (1 hour, room temperature) was followed by the addition of rabbit polyclonal TOP2α/170 C-terminal specific antibody (Ritke et al., 1994b) used at 1:1000 dilution, or a TOP2α/90 specific custom antibody (Kanagasabai et al., 2017) (diluted 1:500 or 1:1000) for an additional 1 hour at room temperature. Slides were gently washed (dipped) in five successive Coplin jars containing PBS, after which cells were incubated (45 minutes, room temperature) with appropriate fluorophore conjugated secondary antibodies (Alexa-Fluor 594, red staining; Invitrogen, Waltham, MA). After washing, cells were costained with DAPI for visualization of DNA/nuclei immediately before immunofluorescence microscopy analyses (Nikon Eclipse Ti). Total and nuclear localized TOP2α were quantified using ImageJ.
Endogenous TOP2α/170 and TOP2α/90 Coimmunoprecipitation Assays.
K562 and K/VP.5 cells (2 × 106) were washed once with PBS and lysed in 500 µl of immunoprecipitation lysis buffer containing 20 mM HEPES, 140 mM NaCl, 2 mM EDTA, 10% glycerol, 1% NP-40 with 0.1% phosphatase inhibitors, 10 mM Na3VO4, and a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN) for 30 minutes on ice. The TOP2α/170 C-terminal specific antibody or the TOP2α/90 specific custom antibody were added to the cell lysates and incubated for 2 hours at 4°C. Protein A agarose (20 μl/sample) (ThermoFisher) was added to the cell lysates and incubated overnight at 4°C. After washing, the precipitated immune complexes were released in 100 μl of SDS-PAGE sample buffer (6×) by boiling for 5 minutes. The resulting supernatant was then subjected to immunoassay analysis as described above. Identical control immunoprecipitation experiments were performed using preimmune serum.
Transfection Experiments.
Briefly, for TOP2α/90 gain-of-function experiments, K562 cells (2.25 × 106 cells in 100 µl per condition) were transfected with empty vector (pcDNA3.1, negative control) or pcDNA/TOP2α/90 (7.5 µg plasmid) by electroporation technology (Nucleofector Kit V; Lonza, Basel, Switzerland) according to manufacturer’s instructions and as reported previously (Kanagasabai et al., 2017). Forty-eight hours later, transfected K562 cells were used for comet and colony forming assays (see below).
Alternatively, for TOP2α/90 loss-of-function experiments, K/VP.5 cells (2.25 × 106 cells in 100 µl per condition) were transfected with Silencer Select Negative Control siRNA #1 (#4390843; ThermoFisher) or TOP2α/90 specific Silencer Select Custom Designed siRNAs (siRNA #1, sense 5′-GUAUAAGCUCAUAUAUUCAtt-3′, antisense 5′-UGAAUAUAUGAGCUUAUACtt-3′; siRNA #2 sense 5′-GGAUAUUCAUAAGUCUGCAtt-3′, antisense 5′-UGCAGACUUAUGAAUAUCCtt-3′; ThermoFisher) at 10 and 50 nM final concentrations utilizing electroporation technology (Nucleofector Kit V; Lonza) according to the manufacturer’s instructions. Twenty-four hours after transfection, cellular extracts were prepared and immunoblotting experiments were performed as outlined above. siRNA transfected K/VP.5 cells were also used for comet assays (see below).
Comet Assays.
The neutral (pH. 9, detects double-stranded breaks) and alkaline (pH 13, detects primarily single-strand breaks) single-cell gel electrophoresis (Comet) assays were performed according to the manufacturer’s protocol (CometAssay Kit, cat. no. 4250-050-K; Trevigen, Gaithersburg, MD) and as previously described by our laboratory (Vlasova et al., 2011; Kanagasabai et al., 2017). Briefly, K562 and K/VP.5 cells (2.25 × 106 cells in 100 µl per condition) were transfected as described above. Forty-eight hours after transfection with pcDNA/TOP2α/90 (for forced expression of TOP2α/90) or 24 hours after transfection with TOP2α/90 siRNAs (for TOP2α/90 knockdown), cells were washed and resuspended in buffer (25 mM HEPES, 10 mM glucose, 1 mM MgCl2, 5 mM KCl, 130 mM NaCl, 5 mM NaH2PO4, pH 7.4). The transfected K562 and K/VP.5 cells were subsequently incubated with 1, 2, or 25 μM etoposide or DMSO (solvent control) for 1 hour at 37°C. The transfected and treated cells were washed with ice-cold buffer and resuspended to 0.28 × 106 cells/ml. Following neutral or alkaline electrophoresis and subsequent staining with a fluorescent DNA intercalating dye, SYBR Gold, the migrating fragments (comet tail) from the nucleoid (comet head) were visualized and the images captured by fluorescence microscopy. The Olive tail moment, which is defined as the product of the comet tail length and the fraction of total DNA in the comet tail (Olive, 2002), was quantified by the ImageJ processing program with the open-source software tool OpenComet (Gyori et al., 2014; www.cometbio.org). Olive tail moments from greater than 100 cells per sample condition were determined.
Colony-Forming Assays.
Colony-forming assays were performed by a modification of the procedure previously described by our laboratory (Yalowich et al., 1985). Briefly, K562 cells (2.25 × 106 cells in 100 µl per condition) were transfected with empty vector or pcDNA/TOP2α/90 as described above. Forty-eight hours later, transfected cells were adjusted to a concentration of 1 × 106 cell/ml and incubated with etoposide (10 and 25 µM) or DMSO (solvent control, 0.1% v/v) for 1 hour at 37°C. Drug/solvent were then removed by washing cells twice in complete DMEM media. Cells were resuspended in fresh media at 2 × 105 cell/ml and incubated overnight at 37°C. Cells were then plated in 35 mm dishes (5000 cell/dish) in duplicate in 0.35% agar containing DMEM/10% FBS and incubated for 10–12 days in a humidified atmosphere of 5% CO2 at 37°C. Colonies were then counted on an inverted microscope. Cytotoxicity was determined by comparing drug-treated colony formation with colony counts from cells incubated with DMSO solvent alone. For the six experiments performed, cloning efficiency for DMSO controls was 7.7% ± 1.9%.
Human TOP2α/170 and TOP2α/90 Real-Time Polymerase Chain Reaction Assays.
All total RNA samples were treated with DNAse I (RNase-Free DNase Set; Qiagen (Germantown, MD); or TURBO DNA-free Kit; ThermoFisher Scientific). Quantitative real-time PCR experiments (total reaction volume 20 µl) were performed using TaqMan Gene Expression hydrolysis probes (ThermoFisher Scientific) as previously described (Kanagasabai et al., 2017). TOP2α/90 expression levels were measured using a custom hydrolysis probe that spans the exon 19/intron 19 boundary (5′-TCATGGTGAGGTAAACACACAATCC-3′). TOP2α/170 expression levels were measured using a hydrolysis probe spanning the exon 34/35 boundary (TaqMan assay Hs01032151_g1), which is only present in TOP2α/170 cDNAs. The relative gene expression levels of TOP2α/90 or TOP2α/170 in each cell line or human tissue were normalized to TATA-box binding protein (TBP, TaqMan assay Hs99999910_m1) expression using the 2−ΔΔCt method (Schmittgen and Livak, 2008).
Statistical Analysis.
The results are presented as the mean values ± S.E.M. values for n ≥ 3 separate experiments. Statistical analysis was performed by using a two-tailed paired Student’s t-with statistical significance of differences set at P < 0.05 or P < 0.025 with a Bonferroni correction. SAS 9.4 (The SAS Institute, Cary, NC) was used in all statistical analyses.
Results
Nuclear Localization of the Truncated TOP2α/90 Isoform.
Our laboratory recently established that TOP2α/90 is the truncated translation product of an alternatively processed TOP2α mRNA (Fig. 1B) (Kanagasabai et al., 2017). Although TOP2α/90 does not harbor the well-characterized NLS1259–1296 and NLS1454–1497 present in TOP2α/170 (Fig. 1A) (Mirski et al., 1997, 1999), this truncated TOP2α does contain two additional putative bipartite NLS sequences, NLS606–676 and NLS727–743 (Mirski et al., 1997). Fusion-truncated forms of human TOP2α, which contained these NLS sequences, were excluded from the nucleus, suggesting that these NLS motifs were nonfunctional (Mirski et al., 1997; Ernst et al., 2000). TOP2α/90 was analyzed for additional NLS sequences due to the inclusion of the unique 25-aa C-terminal sequence encoded by a processed intron 19 (i.e., aa 762–786: VNTQSMFPESIISEIPAESFSKQIW) (Fig. 1B) (Kanagasabai et al., 2017). Algorithm analyses (Brameier et al., 2007; Kosugi et al., 2009; Nguyen Ba et al., 2009) revealed that the unique TOP2α/90 sequence does not harbor a classic NLS sequence (data not shown). Together, these results cast doubt that this truncated protein gains entry to or functions in the nucleus.
To investigate this question, total cellular, cytoplasmic, and nuclear specific lysates were isolated from proliferating K562 and K/VP.5 cells to evaluate the distribution of TOP2α/90 and TOP2α/170 by immunoassays. As previously demonstrated (Kanagasabai et al., 2017), immunoblotting of total cell lysates using an antibody raised against the N-terminal portion of TOP2α (TOP2α/90/170 Ab) (Fig. 1) demonstrated that, compared with parental K562 cells, the expression level of TOP2a/170 was attenuated and that of TOP2a/90 was increased in K/VP.5 cells (Fig. 2A). Surprisingly, immunoassays using cytoplasmic and nuclear extracts revealed that TOP2α/90 could be detected in both cellular compartments (Fig. 2B). Additionally, these experiments demonstrated that TOP2α/90 levels were greater in K/VP.5 compared with parental K562 nuclei (Fig. 2B). In contrast, TOP2α/170 was more abundant in K562 compared with K/VP.5 nuclei (Fig. 2B). Immunoassays using β-actin and lamin B antibodies demonstrated that the cytoplasmic and nuclear extracts were not cross-contaminated and that the lanes were loaded equally (Fig. 2B).
Fig. 2.

TOP2α/90 and TOP2α/170 can be detected in both nuclear and cytoplasmic extracts. (A) Representative immunoassay using K562 and K/VP.5 cellular lysates. Immunoblots were probed with the TOP2α/90/170 Ab (see Fig. 1 for epitope position) or β-actin Ab. (B) Representative immunoassay using K562 and K/VP.5 cytoplasmic (19-second exposure) and nuclear lysates (9-second exposure). Immunoblots were probed with the TOP2α/90/170 Ab or β-actin Ab. (C) Representative immunofluorescence experiment using fixed K562 and K/VP.5 cells probed with the TOP2α/90 Ab (see Fig. 1B for epitope position). (D) Representative immunofluorescence experiment using fixed K562 and K/VP.5 cells probed with the TOP2α/170 Ab (see Fig. 1A for epitope position).
To validate the nuclear and cytoplasmic localization of truncated and full-length TOP2α, immunofluorescence experiments were performed using a TOP2α/90 specific antibody generated against the unique 25 aa (TOP2α/90 Ab) (Fig. 1B) (Kanagasabai et al., 2017) and a C-terminal antibody specific for TOP2α/170 (TOP2α/170 Ab) (Fig. 1A) (Ritke et al., 1994a; Kanagasabai et al., 2017). Results confirmed that TOP2α/90 (Fig. 2C) and TOP2α/170 (Fig. 2D) are located in both the nucleus and cytoplasm of K562 and K/VP.5 cells. Total cellular and nuclear TOP2α/90 and TOP2α/170 levels were assessed in 25 cells from both K562 and K/VP.5 lines, in each of three separate experiments (data not shown). For total cellular content, truncated TOP2α/90 levels were 2.4 ± 0.4-fold greater in resistant K/VP.5 compared with K562 cells (P < 0.05) (Fig. 2C). Conversely, full-length TOP2α/170 levels were 3.3 ± 0.2-fold greater in K562 compared with resistant K/VP.5 cells (P < 0.001) (Fig. 2D). In both K562 and K/VP.5 cells, the percent of total cellular TOP2α found in the nucleus was ∼65% for TOP2α/90 and ∼60% for TOP2α/170. Taken together, these studies support our previous observations that K/VP.5 cells express higher levels of TOP2α/90 compared with K562 cells (Fig. 2A) (Kanagasabai et al., 2017) and suggest that increased nuclear content of truncated TOP2α/90 in K/VP.5 cells may play an important role in acquired resistance to etoposide and other TOP2α-targeting drugs.
TOP2α/90 and TOP2α/170 Isoforms Heterodimerize.
Although TOP2α/90 lacks the functional C-terminal NLS (Mirski et al., 1997, 1999) and DD sequences (Frère et al., 1995; Berger et al., 1996; Frère-Gallois et al., 1997; Kroll, 1997; Bjergbaek et al., 1999) harbored in TOP2α/170 (Fig. 1B), the presence of this truncated isoform in the nucleus (Fig. 2, B and C) is consistent with the concept that TOP2α/90 binds with TOP2α/170 and gains entry to the nucleus as a heterodimer. To test this theory, coimmunoprecipitations of endogenous TOP2α isoforms were performed using our three well-characterized antibodies, TOP2α/90/170 Ab (cross-reacts with both TOP2α/90 and TOP2α/170) (Fig. 1; Fig. 2, A and B; Fig. 3A; Fig. 4A; Fig. 5A; and Fig. 7) (Kanagasabai et al., 2017), TOP2α/90 Ab (reacts with only TOP2α/90) (Fig. 1B, Fig. 2C, and Fig. 3B) (Kanagasabai et al., 2017), and TOP2α/170 Ab (reacts with only TOP2α/170) (Fig. 1A, Fig. 2D, and Fig. 3C) (Kanagasabai et al., 2017). Whole cell lysates from nontransfected proliferating K562 and K/VP.5 cells were immunoprecipitated with the TOP2α/170 specific antibody and subsequently immunoblotted with the TOP2α/90/170 Ab. Immunoreactive TOP2α/90 and TOP2α/170 were detected in both lysates (Fig. 3D), suggesting that TOP2α/170 homodimers and TOP2α/90:TOP2α/170 heterodimers are formed in both cell lines. Similarly, when lysates were immunoprecipitated with the TOP2α/170 Ab and immunoblotted with the TOP2α/90 Ab (Fig. 3E), only immunoreactive TOP2α/90 was detected, again indicating that TOP2α/90 and TOP2α/170 heterodimerize. Finally, a reciprocal coimmunoprecipitation experiment was performed where lysates were immunoprecipitated with the TOP2α/90 Ab and subsequently immunoblotted with the TOP2α/170 Ab. Immunoreactive TOP2α/170 was detected (Fig. 3F), demonstrating once more that TOP2α/90:TOP2α/170 heterodimers are present in both K562 and K/VP.5 cells. Control immunoprecipitation experiments were performed using preimmune serum followed by immunoblotting with the TOP2α/90/170 Ab. Neither TOP2α/90 nor TOP2α/170 was detected (data not shown). Taken together, these results indicate that TOP2α/90:TOP2α/170 heterodimers form endogenously (within nontransfected cells).
Fig. 3.

TOP2α/90 heterodimerizes with TOP2α/170 in both K562 and K/VP.5 cells. (A–C) Representative immunoassay using K562 and K/VP.5 whole cell extracts. Immunoblots were probed with the TOP2α/90/170 Ab, TOP2α/90 Ab, or TOP2α/170 Ab. (D–F) Immunoprecipitation (IP) experiments were performed using K562 and K/VP.5 whole cell extracts. The precipitated immune complexes were released in SDS-PAGE sample buffer, subjected to SDS-PAGE, and immunoblotted (IB), using the indicated antibodies. Input immunoblots are also shown for each experiment and β-actin Ab loading controls.
Fig. 4.
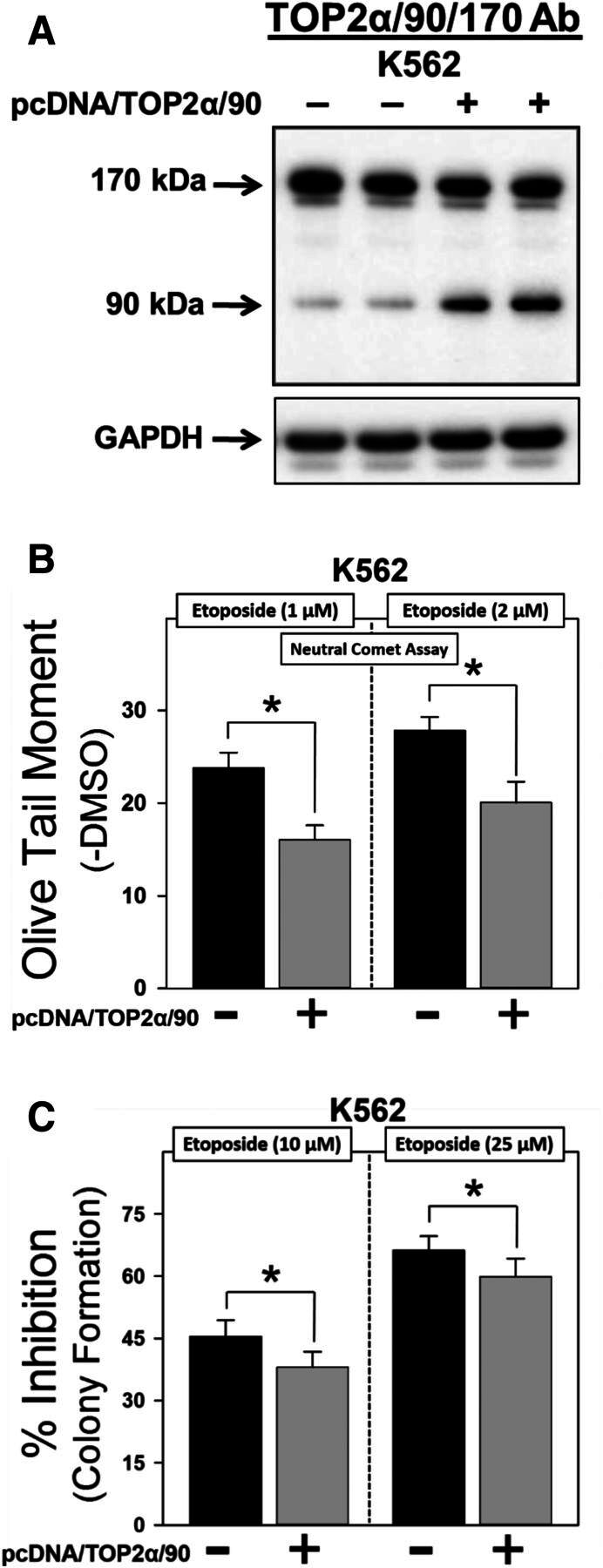
Forced expression of TOP2α/90 in K562 cells reduces etoposide sensitivity. (A) Representative immunoassay using cellular lysates isolated from empty vector or pcDNA/TOP2a/90-transfected K562 cells. (B) Etoposide (1 and 2 μM)-induced DNA damage in empty vector and pcDNA/TOP2α/90-transfected K562 cells was determined by neutral comet assays (assessing DNA double-strand breaks) after a 1-hour incubation and subtraction of DMSO vehicle controls, as described in Materials and Methods. Results shown are the mean ± S.E.M. for three experiments run on separate days. For all experimental conditions in each experiment, greater than 100 cells were evaluated by OpenComet software. *P < 0.025, comparing pcDNA/TOP2α/90-transfected to empty vector-transfected K562 cells. (C) Etoposide (10 and 25 µM)-induced cytotoxicity in empty vector- and pcDNA/TOP2α-transfected K562 cells was assessed by clonogenic assays. Results shown are the mean ± S.E.M. for six experiments run on separate days. *P < 0.025, comparing pcDNA/TOP2α/90-transfected to empty vector-transfected K562 cells.
Fig. 5.
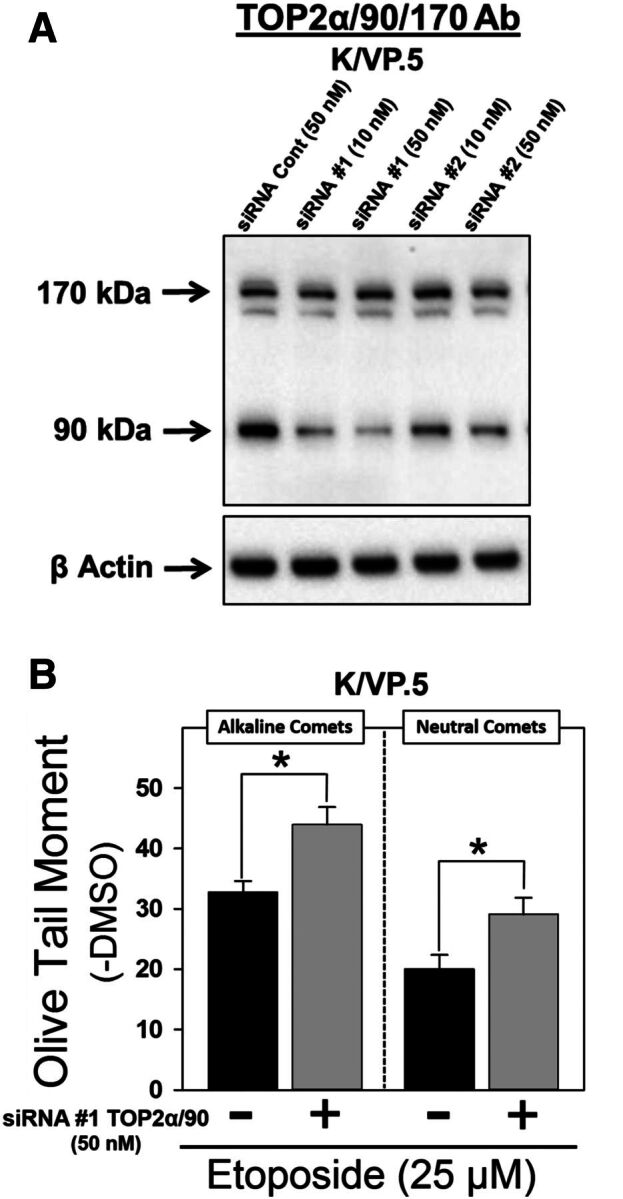
TOP2α/90 siRNA knockdown in K/VP.5 cells enhances etoposide-induced DNA damage. (A) K/VP.5 cells were transfected with negative control or TOP2α/90-specific Silencer Select Custom Designed siRNAs at the concentrations indicated. Twenty-four hours after transfection, cellular extracts were prepared and immunoblotting experiments were performed using the TOP2α/90/170 Ab. (B) K/VP.5 cells were transfected as above in (A). Twenty-four hours later cells were incubated for 1 hour with etoposide (25 µM) or DMSO (control) and alkaline or neutral comet assays were performed. Results shown are the mean ± S.E.M. for five to six experiments run on separate days. For all experimental conditions in each experiment, greater than 100 cells were evaluated by OpenComet software, *P < 0.025, comparing TOP2α/90 siRNA-transfected to negative control siRNA transfected K/VP.5 cells.
TOP2α/90 Exhibits Dominant-Negative Properties.
Previously our laboratory demonstrated that transfection of a TOP2α/90 expression plasmid into K562 cells, which express low levels of endogenous TOP2α/90, resulted in a dramatic increase in the levels of this truncated isoform (Kanagasabai et al., 2017). Importantly, as TOP2α/90 expression levels increased, a corresponding decrease in etoposide-induced TOP2α-DNA covalent complexes and DNA single-strand breaks [as assessed by alkaline (pH 13) single cell gel electrophoresis (comet) assays] was observed (Kanagasabai et al., 2017). Given that TOP2α/90 lacks the active site Tyr805 required for the formation of TOP2α-DNA covalent complexes (Fig. 1) (Vos et al., 2011; Chen et al., 2013; Pommier et al., 2016), and the proclivity for etoposide to stabilize these complexes (Pommier and Marchand, 2011; Ketron and Osheroff, 2014), we posit that the dominant-negative effects of TOP2α/90, observed previously in the presence of etoposide (Kanagasabai et al., 2017), occur through the generation of TOP2α/90:TOP2α/170 heterodimers (Fig. 3, D–F).
To test this hypothesis, we performed both TOP2α/90 “gain-of-inhibitory-function” and “loss-of-inhibitory-function” experiments by transfecting K562 cells with pcDNA/TOP2α/90 and transfecting K/VP.5 cells with TOP2α/90-specific siRNAs, respectively (Figs. 4 and 5).
Initial studies were performed by transfecting K562 cells with empty vector or the pcDNA/TOP2α/90 expression construct (Fig. 4A) followed 48 hours later by a 1-hour etoposide or DMSO vehicle treatment and immediate evaluation of DNA damage. By using neutral (pH 9) single cell gel electrophoresis (comet) assays, etoposide-induced DNA double-strand breaks were attenuated in TOP2α/90-transfected compared with empty vector transfected K562 cells (Fig. 4B). Averaging results from three experiments run on separate days, there was a reduction in DNA damage to 67.3% ± 3.2% and 71.6% ± 4.6% of the pcDNA control, in cells treated with 1 μM etoposide (P = 0.012) and 2 μM etoposide (P = 0.011), respectively.
As a complement to these DNA damage studies, clonogenic assays were performed in K562 cells transfected with empty vector or the pcDNA/TOP2α/90 expression construct followed 48 hours later by a 1-hour incubation with etoposide (10 and 25 µM) or DMSO vehicle. After 1 hour, cells were washed free of drug, resuspended in fresh media overnight, then plated in soft agar and grown for 10–12 days. Etoposide exhibited a concentration-dependent cytotoxic effect (10 vs. 25 μM) in empty vector transfected K562 cells (Fig. 4C). Notably, TOP2α/90-transfected K562 cells showed a reduced sensitivity to etoposide compared with the empty vector transfected cells (Fig. 4C). Averaging results from six experiments run on separate days, there was a reduction in the percent inhibition of colony formation to 83.7% ± 3.1% and 89.9% ± 2.8% of the pcDNA control in cells treated with 10 μM etoposide (P = 0.006) and 25 μM etoposide (P = 0.012), respectively (Fig. 4C). These “gain-of-inhibitory-function” results are consistent with a dominant-negative activity of TOP2α/90 and further suggest a role for this isoform in acquired resistance.
Next, the reciprocal TOP2α/90 “loss-of-inhibitory-function” experiments were initiated in K/VP.5 cells (which express high levels of the truncated TOP2α isoform) where two TOP2α/90 specific siRNAs (siRNA/90 #1 and #2) targeting retained intron 19 sequences, were used to determine whether knockdown of TOP2α/90 would enhance etoposide-induced DNA damage. TOP2α/90 protein levels were reduced in a concentration-dependent manner by siRNA/90 #1 and siRNA/90 #2 (10 and 50 nM) in K/VP.5 whole cell lysates isolated 24 hours after transfection of K/VP.5 cells (Fig. 5A). Averaging results from six experiments, there was a decrease in TOP2α/90 protein expression of 40.0% ± 11.3% (10 nM siRNA/90 #1, P < 0.01) and 52.4% ± 7.3% (50 nM siRNA/90 #1, P < 0.002) compared with the negative control siRNA transfected K/VP.5 cells. Although siRNA/90 #2 (50 nM) reduced TOP2α/90 protein expression levels (i.e., 34.4% ± 6.8%, P < 0.002), a significant decrease of the truncated isoform was not observed in K/VP.5 cells transfected with 10 nM of siRNA/90 #2. No further knockdown of TOP2α/90 protein expression was observed in cells transfected with the combination of siRNA/90 #1 and siRNA/90 #2 at 50 nM or in cells harvested 48 hours after individual siRNA transfection (data not shown). Notably, TOP2α/170 protein levels were unchanged in cells transfected with either siRNA/90 #1 or siRNA/90 #2 (Fig. 5A), indicating the specificity for TOP2α/90 knockdown with these siRNAs.
For loss-of-inhibitory-function TOP2α/90 studies, K/VP.5 cells transfected with 50 nM negative control siRNA or siRNA #1 were incubated with DMSO solvent control or etoposide (25 µM) for 1 hour followed by analysis of DNA damage by alkaline single-cell gel electrophoresis (comet) assays for primarily single-strand breaks and by neutral comet assays for DNA double-strand breaks. Both alkaline and neutral comet assays revealed that etoposide-induced DNA damage was augmented in TOP2α/90 siRNA transfected K/VP.5 cells compared with negative control siRNA transfected cells (Fig. 5B). Since TOP2α/170 levels in K/VP.5 cells are much lower compared with K562 cells (Fig. 2A), greater concentrations of etoposide (25 µM) were required to observe DNA damage (Fig. 5B) compared with those used in K562 cells (1 and 2 µM) (Fig. 4C). Averaging results from five separate experiments performed on separate days, DNA damage was increased 36.1% ± 10.9% (P = 0.004) for alkaline comet assays. Averaging results from six separate experiments performed on separate days, DNA damage was increased 50.2% ± 15.3% (P = 0.020) for neutral comet assays. Increased etoposide activity in K/VP.5 cells upon TOP2α/90 knockdown demonstrates loss-of-inhibitory-function consistent with our hypothesis that this truncated TOP2α isoform acts in a dominant-negative fashion.
Overall, the studies described above strongly suggest that TOP2α/90:TOP2α/170 heterodimers produce dominant negative effects by reducing the number of TOP2α/170-DNA covalent cleavage complexes that can be “trapped” by etoposide treatment, which decreases drug-induced DNA damage and cytotoxicity associated with drug resistance.
TOP2α/90 mRNAs/Proteins are Expressed in Normal Human Tissues and in AML Patient Blasts.
Given that TOP2α/90 mRNA was present in parental K562 cells and overexpressed in etoposide resistant K/VP.5 cells (Kanagasabai et al., 2017), we investigated whether this alternatively processed TOP2α transcript was expressed in normal human tissues/cells. Blast cells from an AML patient at diagnosis and after treatment relapse were also examined. Real-time PCR experiments performed using TaqMan hydrolysis probes specific for human TOP2α/90 (i.e., exon 19/intron 19 boundary) or TOP2α/170 mRNA (i.e., exon 34/35 boundary) demonstrated that TOP2α/90 and TOP2α/170 mRNAs were detectable in all tissues/cells investigated (Fig. 6). Relative TOP2α/90 and TOP2α/170 mRNA levels in K562 and K/VP.5 cells are included for comparison (Fig. 6, A and B). Notably, normal bone marrow, stomach, thymus, testis, colon, and blasts from AML patients expressed comparable or higher levels of TOP2α/90 mRNA than K/VP.5 cells (Fig. 6A). Finally, Fig. 6C lists the nonnormalized Ct values for TOP2α/90 and TOP2α/170 in the tissues investigated to gain an appreciation of the expression levels of these mRNAs.
Fig. 6.

TOP2α/90 mRNAs are expressed in normal human tissues. A “Human Total RNA Master Tissue Panel” was purchased from Clontech. Additionally, total RNA was isolated from K562, K/VP.5 cells, blast cells from an AML patient at diagnosis and after relapse. All RNAs were treated with DNAse I, reverse transcribed, and subjected to qPCR using TaqMan Gene Expression hydrolysis probes specific for TOP2α/90 (A) or TOP2α/170 (B). Real-time PCR data were normalized to TATA Box expression using the 2−ΔCt method. (C) List of nonnormalized CT values for TOP2α/90 and TOP2α/170 mRNA expression of the tissues/cells investigated.
To verify that the TOP2α/90 mRNA detected in tissues/cells was translated, immunoblotting experiments were performed using protein lysates isolated from select human tissues and cells. Significantly, immunoreactive TOP2α/90 were detectable in the human thymus, cord blood mononuclear cells (MNC), and in blasts isolated from the same patient at diagnosis and at relapse (Fig. 7). It is also important to note that although TOP2α/90 is barely detectable in the human stomach lysate (Fig. 7), this sample may have experienced degradation given that the β-actin signal is also weak, although the same amount of thymus protein lysate (15 μg) was subjected to immunoblot analysis. Additionally, 30 µg of MNC lysate and 20 µg of AML blast lysates were loaded for immunoblot analysis, which may account for differing β-actin signals (Fig. 7). Regardless of the potential loading and/or degradation differences between these human samples, it is clear that the TOP2α/90 isoform is expressed in a number of human tissue and cell types, suggesting that, aside from a role in drug resistance, TOP2α/90 (and formation of TOP2α/90:TOP2α/170 heterodimers) could potentially play a role in certain normal tissues to protect against xenobiotics or phytochemicals targeting TOP2α.
Fig. 7.

The TOP2α/90 isoform is expressed in human tissues and bone marrow cells. Representative TOP2α immunoblotting experiments using the indicated tissue or cellular lysates. The immunoblots were probed with TOP2α/90/170 Ab or β-actin Ab. Immunoblots were subsequently incubated with a secondary antibody, and cross-reactive proteins were detected using chemiluminescence (Kanagasabai et al., 2017). The following amount of protein was loaded into each well: K562 cellular lysate, 7 µg; K/VP.5 cellular lysate, 7 µg; human cord blood mononuclear cell (MNC) lysate, 30 µg; AML patient blast cell lysates (pretreatment and after relapse), 20 µg; normal human stomach tissue lysate, 15 µg; normal human thymus tissue lysate, 15 µg.
Discussion
TOP2α interfacial inhibitors/poisons (e.g., etoposide, mitoxantrone, amsacrine, doxorubicin, and analogs) are a diverse group used to treat a variety of human malignancies (Deweese and Osheroff, 2009; Nitiss, 2009; Pommier et al., 2010; Pommier and Marchand, 2011; Ketron and Osheroff, 2014). Intrinsic and acquired chemoresistance to these drugs continues to be a major complication in cancer treatment. Although many resistance mechanisms have been defined (Vassetzky et al., 1995; Pilati et al., 2012; Ganapathi and Ganapathi, 2013), acquired resistance to TOP2α inhibitors is frequently associated with decreased TOP2α/170 nuclear levels (Mirski et al., 1993, 2000; Harker et al., 1995; Mirski and Cole, 1995; Wessel et al., 1997; Yu et al., 1997; Burgess et al., 2008). In addition, our laboratory previously demonstrated reduction in TOP2α/170 levels in etoposide-resistant K/VP.5 compared with parental K562 cells (Ritke et al., 1994a; Kanagasabai et al., 2017) due in part to changes in TOP2α/170 mRNA stability (Ritke and Yalowich, 1993).
We recently identified a unique truncated TOP2α isoform, designated TOP2α/90, which is overexpressed in the etoposide-resistant K/VP.5 cell line (Fig. 2; Fig. 3, A and B; and Fig. 7) (Kanagasabai et al., 2017). TOP2α/90 is comprised of 786 aa and is the translation product of a TOP2α mRNA, which retains a processed segment of intron 19 (Fig. 1) (Kanagasabai et al., 2017). Hence, the decrease in TOP2α/170 protein levels (and resultant resistance) in K/VP.5 cells may be due to intron 19 retention as well as posttranscriptional alterations. Our present work focused on the role of TOP2α/90, produced as a result of intron 19 retention, in resistance to etoposide.
TOP2α/90 does not harbor the well-characterized TOP2α/170 NLS sequences (Mirski et al., 1997, 1999) (Fig. 1). We hypothesized, therefore, that TOP2α/90 would be predominantly located in the cytoplasm like other published drug-resistant human cancer cell lines containing TOP2α isoforms with NLS deletions (Harker et al., 1995; Mirski and Cole, 1995; Wessel et al., 1997; Yu et al., 1997; Mirski et al., 2000). Remarkably, however, immunoassays using fractionated cytoplasmic and nuclear extracts (Fig. 2B) and immunofluorescence experiments (Fig. 2C) clearly demonstrated that TOP2α/90 was detected in both the nucleus and cytoplasm of K562 and K/VP.5 cells with enrichment in the nuclear compartment.
TOP2α/170 localizes and functions in the nucleus as a homodimer (DeWeese and Osheroff, 2009). Given that TOP2α/90 was detected in the nucleus (Fig. 2) and exhibits dominant-negative properties (Figs. 4 and 5) (Kanagasabai et al., 2017), we tested the hypothesis that the dominant-negative effects of the truncated isoform result from the presence of TOP2α/90:TOP2α/170 heterodimers. In cellular extracts, coimmunoprecipitation experiments using multiple combinations of TOP2α antibodies clearly demonstrated that endogenous TOP2α/90 and TOP2α/170 proteins form heterodimers in K562 and in K/VP.5 cells (Fig. 3, D–F). Although it is not clear why using TOP2α/170 antibody for immunoprecipitation resulted in “pull-down” of TOP2α/90 in a ratio greater than 1:1 for K/VP.5 cell extracts (Fig. 3D), this result was reproduced in multiple experiments (n = 5, data not shown). Future studies will address this observation more closely. One speculation is that the stoichiometry of TOP2α/90 binding to TOP2α/170 is greater than 1:1 based on novel conformations of the truncated isoform influencing protein-protein interactions. Regardless, overall results from Fig. 3, D–F, indicate that endogenous TOP2α/90 and TOP2α/170 proteins can associate/dimerize.
Currently, it is not known how and where TOP2α/90:TOP2α/170 dimers form. Nevertheless, several studies have shown that at least two regions (DD1053–1069 and DD1121–1143) in human TOP2α/170 are essential for TOP2α/170:TOP2α/170 homodimerization (Frère et al., 1995; Berger et al., 1996; Frère-Gallois et al., 1997; Kroll, 1997; Bjergbaek et al., 1999). Because the 770 aa C terminus of TOP2α/170 is absent in TOP2α/90, this truncated isoform does not harbor these well-characterized dimerization domains (Fig. 1). Importantly, however, several studies demonstrated that human N-terminal TOP2α fragments, which encompass just the ATPase domain (i.e., aa 1–435), dimerize in vitro under the appropriate conditions (Gardiner et al., 1998; Bjergbaek et al., 1999; Campbell and Maxwell, 2002; Hu et al., 2002). For example, Gardiner et al. (1998) demonstrated by protein crosslinking experiments that N-terminal TOP2α fragments formed dimers in the absence or presence of nucleotides. Hu et al. (2002) showed by both protein crosslinking and sedimentation equilibrium analysis that N-terminal TOP2α fragments exist as monomers in the absence of cofactors but can readily dimerize in the presence of ATP, ADP, or nonhydrolyzable ATP analogs. Campbell and Maxwell (2002) demonstrated that N-terminal TOP2α fragments dimerize in the presence of ATP and in the dimeric form can interact with DNA. Finally, Bjergbaek et al. (1999) established that, if only the TOP2α/170 C-terminal primary dimerization regions (i.e., DD1053–1069 and DD1121–1143) were deleted, dimerization could still occur in the presence of DNA and an ATP analog. Taken together, these studies are consistent with the concept that TOP2α/90 and TOP2α/170 can form heterodimers even in the absence of canonical dimerization domains. Moreover, these previous N-terminal TOP2α fragment dimerization studies also suggest that TOP2α/90:TOP2α/90 homodimers may form within cells. Also of note, the 180-kDa type II topoisomerase isoform (TOP2 β/180) (Chung et al., 1989), thought to be important for transcription of developmental and hormonally regulated genes and in DNA repair (Yang et al., 2000; Ju et al., 2006; Lyu et al., 2006; McNamara et al., 2008; Cowell et al., 2012; Tiwari et al., 2012), is capable of forming heterodimers with TOP2/α/170 (Biersack et al., 1996; Gromova et al., 1998). Therefore, studies are underway to evaluate putative TOP2α/90:TOP2α/90 homodimerization and TOP2α/90 heterodimerization with TOP2β/180 and to assess potential biologic/pharmacological consequences of these protein-protein interactions.
It is possible that TOP2α/90 gains entry into the nucleus by a “piggy-back” mechanism (Christie et al., 2016) with TOP2α/170, since the full-length isoform harbors functional NLS sequences. In contrast, if dominant-negative TOP2α/90:TOP2α/170 heterodimers form in the nucleus, then this would suggest that the truncated isoform is imported by an uncharacterized NLS sequence. Also, since C-terminal truncated TOP2α isoforms, which lack NLS, are mostly excluded from the nucleus (Vassetzky et al., 1996, Soltermann et al., 1999; Ernst et al., 2000), yet can bind to and inhibit etoposide activity (Leroy et al., 2001; Vilain et al., 2003), TOP2α/90 may sequester etoposide prior to nuclear entry. Additional studies are ongoing to investigate these potential mechanisms.
Given that TOP2α/90 lacks the active site Tyr805 required to form TOP2α–DNA covalent complexes (Fig. 1) (Kanagasabai et al., 2017) and can heterodimerize with TOP2α/170 (Fig. 3), we hypothesized that etoposide-induced DNA damage in K562 or K/VP.5 cells could be influenced by experimentally increasing or decreasing the cellular levels of TOP2α/90. In support of this hypothesis, etoposide-induced DNA damage after 1 hour in K562 cells (express low levels of TOP2α/90) was reduced when TOP2α/90 levels were increased by transfection (gain-of-inhibitory-function) (Fig. 4B). Additionally, clonogenic assays demonstrated that etoposide-mediated cytotoxicity (assessed by colony forming assays) was decreased after forced expression of TOP2α/90 (Fig. 4C). Lower etoposide concentrations (1 and 2 µM) were used for DNA damage studies in K562 cells (Fig. 4B) compared with those in the clonogenic assays (10, 25 µM, Fig. 4C), because DNA damage was too severe to measure accurately at the higher concentrations.
Conversely, etoposide (25 µM)-induced DNA damage in K/VP.5 cells (expressing high levels of TOP2α/90) was increased when TOP2α/90 levels were decreased by siRNA knockdown (loss-of-inhibitory-function) (Fig. 5B). Taken together these studies indicate that TOP2α/90:TOP2α/170 heterodimers produce dominant negative effects by reducing the number of TOP2α/170-DNA covalent cleavage complexes that can be “trapped” by etoposide treatment, which ultimately leads to fewer double-strand DNA breaks and decreased etoposide cytotoxicity. Thus we conclude that enhanced expression of TOP2α/90 in K/VP.5 cells is a determinant of chemoresistance through a dominant negative effect related to heterodimerization with TOP2α/170.
In AML samples from the same patient (pretreatment and relapse), the ratio of TOP2α/90 to TOP2α/170 protein appears greater after treatment relapse (Fig. 7). In addition, the ratio of TOP2α/90 to TOP2α/170 mRNA was 0.9 and 0.4 for the matched relapse and pretreatment AML patient samples, respectively (comparing Fig. 6, A and B), suggesting a role for TOP2α/90 in resistance/relapse. Studies are underway to evaluate additional matched AML patient samples at diagnosis and relapse (subsequent to TOP2α inhibitor treatment) for TOP2α/90 to TOP2α/170 mRNA and protein ratios as potential biomarkers for acquired drug resistance.
Finally, qPCR data demonstrated that TOP2α/90 mRNA was detectable in the human tissues/cells investigated, with the highest expression levels found in rapidly dividing tissues/cells (Fig. 6). Immunoreactive TOP2α/90 was detectable in human thymus lysates and cord blood mononuclear cells (MNC) (Fig. 7). These results suggest that, in certain normal tissues, the expression of TOP2α/90 (and formation of TOP2α/90:TOP2α/170 heterodimers) may “fine-tune” levels of cleavage complexes to protect against environmental toxicants and/or other natural phenolic nutrients known to target TOP2α (Ketron and Osheroff, 2014). TOP2α/90:TOP2α/170 heterodimerization may also result in noncanonical topoisomerase functions for this unique complex, especially because the last 25 aa of TOP2α/90 (containing five serines and one threonine; Kanagasabai et al., 2017) may provide binding/modification sites for diverse regulatory proteins. Hence, future studies will focus on the functional role of TOP2α/90:TOP2α/170 dimers in physiological and pathophysiological conditions, including their role in acquired drug resistance.
Acknowledgments
The authors thank Dr. Junan Li, The Ohio State University College of Pharmacy, for excellent biostatistical support.
Abbreviations
- aa
amino acids
- Ab
antibody
- AML
acute myeloid leukemia
- DD
dimerization domain
- DMEM
Dulbecco’s modified Eagle’s medium
- DMSO
dimethylsulfoxide
- FBS
fetal bovine serum
- K562
human leukemia cell line
- kb
kilobase
- K/VP.5
etoposide (VP-16)-resistant human K562 leukemia cell line
- MNC
mononuclear cells
- NLS
nuclear localization signal
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- TOP2α
DNA topoisomerase IIα protein
- TOP2α/170
TOP2α 170 kDa
- TOP2α/90
TOP2α 90 kDa
Authorship Contributions
Participated in research design: Kanagasabai, Karmahapatra, Kania, Kientz, Hernandez, Yu, Elton, Yalowich.
Conducted experiments: Kanagasabai, Karmahapatra, Kania, Kientz, Hernandez, Yu, Hernandez, Kania, Elton, Yalowich.
Performed data analysis: Kanagasabai, Karmahapatra, Kientz, Elton, Yalowich.
Wrote or contributed to the writing of the manuscript: Kanagasabai, Karmahapatra, Elton, Yalowich.
Footnotes
This work was funded by The Ohio State University Comprehensive Cancer Center Support Grant (NCI P30 CA016058).
References
- Berger JM, Gamblin SJ, Harrison SC, Wang JC (1996) Structure and mechanism of DNA topoisomerase II. Nature 379:225–232. [DOI] [PubMed] [Google Scholar]
- Biersack H, Jensen S, Gromova I, Nielsen IS, Westergaard O, Andersen AH (1996) Active heterodimers are formed from human DNA topoisomerase II alpha and II beta isoforms. Proc Natl Acad Sci USA 93:8288–8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjergbaek L, Jensen S, Westergaard O, Andersen AH (1999) Using a biochemical approach to identify the primary dimerization regions in human DNA topoisomerase IIalpha. J Biol Chem 274:26529–26536. [DOI] [PubMed] [Google Scholar]
- Brameier M, Krings A, MacCallum RM (2007) NucPred--predicting nuclear localization of proteins. Bioinformatics 23:1159–1160. [DOI] [PubMed] [Google Scholar]
- Burgess DJ, Doles J, Zender L, Xue W, Ma B, McCombie WR, Hannon GJ, Lowe SW, Hemann MT (2008) Topoisomerase levels determine chemotherapy response in vitro and in vivo. Proc Natl Acad Sci USA 105:9053–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S, Maxwell A (2002) The ATP-operated clamp of human DNA topoisomerase IIalpha: hyperstimulation of ATPase by “piggy-back” binding. J Mol Biol 320:171–188. [DOI] [PubMed] [Google Scholar]
- Chen SH, Chan NL, Hsieh TS (2013) New mechanistic and functional insights into DNA topoisomerases. Annu Rev Biochem 82:139–170. [DOI] [PubMed] [Google Scholar]
- Christie M, Chang CW, Róna G, Smith KM, Stewart AG, Takeda AA, Fontes MR, Stewart M, Vértessy BG, Forwood JK, et al. (2016) Structural biology and regulation of protein import into the nucleus. J Mol Biol 428 (10, Pt A):2060–2090. [DOI] [PubMed] [Google Scholar]
- Chung TD, Drake FH, Tan KB, Per SR, Crooke ST, Mirabelli CK (1989) Characterization and immunological identification of cDNA clones encoding two human DNA topoisomerase II isozymes. Proc Natl Acad Sci USA 86:9431–9435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell IG, Sondka Z, Smith K, Lee KC, Manville CM, Sidorczuk-Lesthuruge M, Rance HA, Padget K, Jackson GH, Adachi N, et al. (2012) Model for MLL translocations in therapy-related leukemia involving topoisomerase IIβ-mediated DNA strand breaks and gene proximity. Proc Natl Acad Sci USA 109:8989–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deweese JE, Osheroff N (2009) The DNA cleavage reaction of topoisomerase II: wolf in sheep’s clothing. Nucleic Acids Res 37:738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong KC, Berger JM (2007) Structural basis for gate-DNA recognition and bending by type IIA topoisomerases. Nature 450:1201–1205. [DOI] [PubMed] [Google Scholar]
- Ernst AI, Soltermann A, Sigrist JA, Widmer L, Gasser SM, Stahel RA (2000) Ectopic expression of human topoisomerase IIalpha fragments and etoposide resistance in mammalian cells. Int J Cancer 88:99–107. [DOI] [PubMed] [Google Scholar]
- Frère V, Sourgen F, Monnot M, Troalen F, Fermandjian S (1995) A peptide fragment of human DNA topoisomerase II alpha forms a stable coiled-coil structure in solution. J Biol Chem 270:17502–17507. [DOI] [PubMed] [Google Scholar]
- Frère-Gallois V, Krebs D, Scala D, Troalen F, Fermandjian S (1997) Peptide fragments of DNA topoisomerase II with helix-forming and coiled-coil-forming properties act as inhibitors of the enzyme. Eur J Biochem 249:142–148. [DOI] [PubMed] [Google Scholar]
- Ganapathi RN, Ganapathi MK (2013) Mechanisms regulating resistance to inhibitors of topoisomerase II. Front Pharmacol 4:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner LP, Roper DI, Hammonds TR, Maxwell A (1998) The N-terminal domain of human topoisomerase IIalpha is a DNA-dependent ATPase. Biochemistry 37:16997–17004. [DOI] [PubMed] [Google Scholar]
- Gromova I, Biersack H, Jensen S, Nielsen OF, Westergaard O, Andersen AH (1998) Characterization of DNA topoisomerase II alpha/beta heterodimers in HeLa cells. Biochemistry 37:16645–16652. [DOI] [PubMed] [Google Scholar]
- Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, Clement MV (2014) OpenComet: an automated tool for comet assay image analysis. Redox Biol 2:457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harker WG, Slade DL, Parr RL, Holguin MH (1995) Selective use of an alternative stop codon and polyadenylation signal within intron sequences leads to a truncated topoisomerase II alpha messenger RNA and protein in human HL-60 leukemia cells selected for resistance to mitoxantrone. Cancer Res 55:4962–4971. [PubMed] [Google Scholar]
- Hu T, Sage H, Hsieh TS (2002) ATPase domain of eukaryotic DNA topoisomerase II. Inhibition of ATPase activity by the anti-cancer drug bisdioxopiperazine and ATP/ADP-induced dimerization. J Biol Chem 277:5944–5951. [DOI] [PubMed] [Google Scholar]
- Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG (2006) A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 312:1798–1802. [DOI] [PubMed] [Google Scholar]
- Kanagasabai R, Serdar L, Karmahapatra S, Kientz CA, Ellis J, Ritke MK, Elton TS, Yalowich JC (2017) Alternative RNA processing of topoisomerase IIα in etoposide-resistant human leukemia K562 cells: intron retention results in a novel C-terminal truncated 90-kDa isoform. J Pharmacol Exp Ther 360:152–163. [DOI] [PubMed] [Google Scholar]
- Ketron AC, Osheroff N (2014) Phytochemicals as anticancer and chemopreventive topoisomerase II poisons. Phytochem Rev 13:19–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi S, Hasebe M, Tomita M, Yanagawa H (2009) Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci USA 106:10171–10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroll DJ (1997) Homologous and heterologous protein-protein interactions of human DNA topoisomerase IIalpha. Arch Biochem Biophys 345:175–184. [DOI] [PubMed] [Google Scholar]
- Laponogov I, Pan XS, Veselkov DA, McAuley KE, Fisher LM, Sanderson MR (2010) Structural basis of gate-DNA breakage and resealing by type II topoisomerases. PLoS One 5:e11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy D, Kajava AV, Frei C, Gasser SM (2001) Analysis of etoposide binding to subdomains of human DNA topoisomerase II alpha in the absence of DNA. Biochemistry 40:1624–1634. [DOI] [PubMed] [Google Scholar]
- Lyu YL, Lin CP, Azarova AM, Cai L, Wang JC, Liu LF (2006) Role of topoisomerase IIbeta in the expression of developmentally regulated genes. Mol Cell Biol 26:7929–7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara S, Wang H, Hanna N, Miller WH Jr (2008) Topoisomerase IIbeta negatively modulates retinoic acid receptor alpha function: a novel mechanism of retinoic acid resistance. Mol Cell Biol 28:2066–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirski SE, Cole SP (1995) Cytoplasmic localization of a mutant M(r) 160,000 topoisomerase II alpha is associated with the loss of putative bipartite nuclear localization signals in a drug-resistant human lung cancer cell line. Cancer Res 55:2129–2134. [PubMed] [Google Scholar]
- Mirski SE, Evans CD, Almquist KC, Slovak ML, Cole SP (1993) Altered topoisomerase II alpha in a drug-resistant small cell lung cancer cell line selected in VP-16. Cancer Res 53:4866–4873. [PubMed] [Google Scholar]
- Mirski SE, Gerlach JH, Cole SP (1999) Sequence determinants of nuclear localization in the alpha and beta isoforms of human topoisomerase II. Exp Cell Res 251:329–339. [DOI] [PubMed] [Google Scholar]
- Mirski SE, Gerlach JH, Cummings HJ, Zirngibl R, Greer PA, Cole SP (1997) Bipartite nuclear localization signals in the C terminus of human topoisomerase II alpha. Exp Cell Res 237:452–455. [DOI] [PubMed] [Google Scholar]
- Mirski SE, Sparks KE, Yu Q, Lang AJ, Jain N, Campling BG, Cole SP (2000) A truncated cytoplasmic topoisomerase IIalpha in a drug-resistant lung cancer cell line is encoded by a TOP2A allele with a partial deletion of exon 34. Int J Cancer 85:534–539. [DOI] [PubMed] [Google Scholar]
- Nguyen Ba AN, Pogoutse A, Provart N, Moses AM (2009) NLStradamus: a simple hidden Markov model for nuclear localization signal prediction. BMC Bioinformatics 10:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss JL (2009) Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 9:338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive PL (2002) The comet assay. An overview of techniques. Methods Mol Biol 203:179–194. [DOI] [PubMed] [Google Scholar]
- Pilati P, Nitti D, Mocellin S (2012) Cancer resistance to type II topoisomerase inhibitors. Curr Med Chem 19:3900–3906. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Leo E, Zhang H, Marchand C (2010) DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol 17:421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, Marchand C (2011) Interfacial inhibitors: targeting macromolecular complexes. Nat Rev Drug Discov 11:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, Sun Y, Huang SN, Nitiss JL (2016) Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol 17:703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritke MK, Allan WP, Fattman C, Gunduz NN, Yalowich JC (1994a) Reduced phosphorylation of topoisomerase II in etoposide-resistant human leukemia K562 cells. Mol Pharmacol 46:58–66. [PubMed] [Google Scholar]
- Ritke MK, Roberts D, Allan WP, Raymond J, Bergoltz VV, Yalowich JC (1994b) Altered stability of etoposide-induced topoisomerase II-DNA complexes in resistant human leukaemia K562 cells. Br J Cancer 69:687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritke MK, Yalowich JC (1993) Altered gene expression in human leukemia K562 cells selected for resistance to etoposide. Biochem Pharmacol 46:2007–2020. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. [DOI] [PubMed] [Google Scholar]
- Soltermann A, Ernst A, Leroy D, Stahel RA, Gasser SM (1999) The cytochrome b5 tail anchors and stabilizes subdomains of human DNA topoisomerase II alpha in the cytoplasm of retrovirally infected mammalian cells. Exp Cell Res 249:308–319. [DOI] [PubMed] [Google Scholar]
- Tiwari VK, Burger L, Nikoletopoulou V, Deogracias R, Thakurela S, Wirbelauer C, Kaut J, Terranova R, Hoerner L, Mielke C, et al. (2012) Target genes of topoisomerase IIβ regulate neuronal survival and are defined by their chromatin state. Proc Natl Acad Sci USA 109:E934–E943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassetzky YS, Alghisi GC, Gasser SM (1995) DNA topoisomerase II mutations and resistance to anti-tumor drugs. BioEssays 17:767–774. [DOI] [PubMed] [Google Scholar]
- Vassetzky YS, Alghisi GC, Roberts E, Gasser SM (1996) Ectopic expression of inactive forms of yeast DNA topoisomerase II confers resistance to the anti-tumour drug, etoposide. Br J Cancer 73:1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilain N, Tsai-Pflugfelder M, Benoit A, Gasser SM, Leroy D (2003) Modulation of drug sensitivity in yeast cells by the ATP-binding domain of human DNA topoisomerase IIalpha. Nucleic Acids Res 31:5714–5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasova II, Feng WH, Goff JP, Giorgianni A, Do D, Gollin SM, Lewis DW, Kagan VE, Yalowich JC (2011) Myeloperoxidase-dependent oxidation of etoposide in human myeloid progenitor CD34+ cells. Mol Pharmacol 79:479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SM, Tretter EM, Schmidt BH, Berger JM (2011) All tangled up: how cells direct, manage and exploit topoisomerase function. Nat Rev Mol Cell Biol 12:827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendorff TJ, Schmidt BH, Heslop P, Austin CA, Berger JM (2012) The structure of DNA-bound human topoisomerase II α: conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J Mol Biol 424:109–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel I, Jensen PB, Falck J, Mirski SE, Cole SP, Sehested M (1997) Loss of amino acids 1490Lys-Ser-Lys1492 in the COOH-terminal region of topoisomerase IIalpha in human small cell lung cancer cells selected for resistance to etoposide results in an extranuclear enzyme localization. Cancer Res 57:4451–4454. [PubMed] [Google Scholar]
- Yang X, Li W, Prescott ED, Burden SJ, Wang JC (2000) DNA topoisomerase IIbeta and neural development. Science 287:131–134. [DOI] [PubMed] [Google Scholar]
- Yalowich JC, Zucali JR, Gross M, Ross WE (1985) Effects of verapamil on etoposide, vincristine, and adriamycin activity in normal human bone marrow granulocyte-macrophage progenitors and in human K562 leukemia cells in vitro. Cancer Res 45:4921–4924. [PubMed] [Google Scholar]
- Yu Q, Mirski SE, Sparks KE, Cole SP (1997) Two COOH-terminal truncated cytoplasmic forms of topoisomerase II alpha in a VP-16-selected lung cancer cell line result from partial gene deletion and alternative splicing. Biochemistry 36:5868–5877. [DOI] [PubMed] [Google Scholar]