

Abstract
A dopamine D2 receptor mutation was recently identified in a family with a novel hyperkinetic movement disorder. That allelic variant D2-I212F is a constitutively active and G protein–biased receptor. We now describe mice engineered using CRISPR-Cas9–mediated gene editing technology to carry the D2-I212F variant. Drd2I212F mice exhibited gait abnormalities resembling those in other mouse models of chorea and/or dystonia and had striatal D2 receptor expression that was decreased approximately 30% per Drd2I212F allele. Electrically evoked inhibitory postsynaptic conductances in midbrain dopamine neurons and striatum from Drd2I212F mice, caused by G protein activation of potassium channels, exhibited slow kinetics (e.g., approximately four- to sixfold slower decay) compared with Drd2+/+ mice. Current decay initiated by photolytic release of the D2 antagonist sulpiride from CyHQ-sulpiride was also ∼fourfold slower in midbrain slices from Drd2I212F mice than Drd2+/+ mice. Furthermore, in contrast to Drd2+/+ mice, in which dopamine is several-fold more potent at neurons in the nucleus accumbens than in the dorsal striatum, reflecting activation of Gαo versus Gαi, dopamine had similar potencies in those two brain regions of Drd2I212F mice. Repeated cocaine treatment, which decreases dopamine potency in the nucleus accumbens of Drd2+/+ mice, had no effect on dopamine potency in Drd2I212F mice. The results demonstrate the pathogenicity of the D2-I212F mutation and the utility of this mouse model for investigating the role of pathogenic DRD2 variants in early-onset hyperkinetic movement disorders.
SIGNIFICANCE STATEMENT
The first dopamine receptor mutation to cause a movement disorder, D2-I212F, was recently identified. The mutation makes receptor activation of G protein–mediated signaling more efficient. To confirm the pathogenesis of D2-I212F, this study reports that mice carrying this mutation have gait abnormalities consistent with the clinical phenotype. The mutation also profoundly alters D2 receptor expression and function in vivo. This mouse model will be useful for further characterization of the mutant receptor and for evaluation of potential therapeutic drugs.
Introduction
Many movement disorders are treated with or caused by drugs that modulate the activationof one or more dopamine receptors (Luquin-Piudo and Sanz,2011; Cepeda et al., 2014; Vaiman et al., 2021). Nevertheless, no naturally occurring mutation of a dopamine receptor was known to cause a movement disorder until the recent identification of the DRD2 variant c.634A > T, p.Ile212Phe (Fig. 1A), which cosegregates with a phenotype of progressive chorea and cervical dystonia in a Dutch family (van der Weijden et al., 2021a). The dopamine D2 receptor modulates both Gαi/o-mediated signaling pathways, such as inhibition of adenylyl cyclase and activation of G protein–regulated inwardly rectifying potassium channels (GIRKs), and arrestin-mediated signaling pathways, including regulation of the protein kinase Akt and glycogen synthase kinase-3 (Beaulieu and Gainetdinov, 2011). The mutant D2-I212F exhibits enhanced activation of G protein–mediated signaling combined with diminished binding of arrestin in human embryonic kidney (HEK) 293 cells; thus, it is a constitutively active and signaling-biased receptor relative to the reference D2 receptor (Rodriguez-Contreras et al., 2021; van der Weijden et al., 2021a).
Fig. 1.
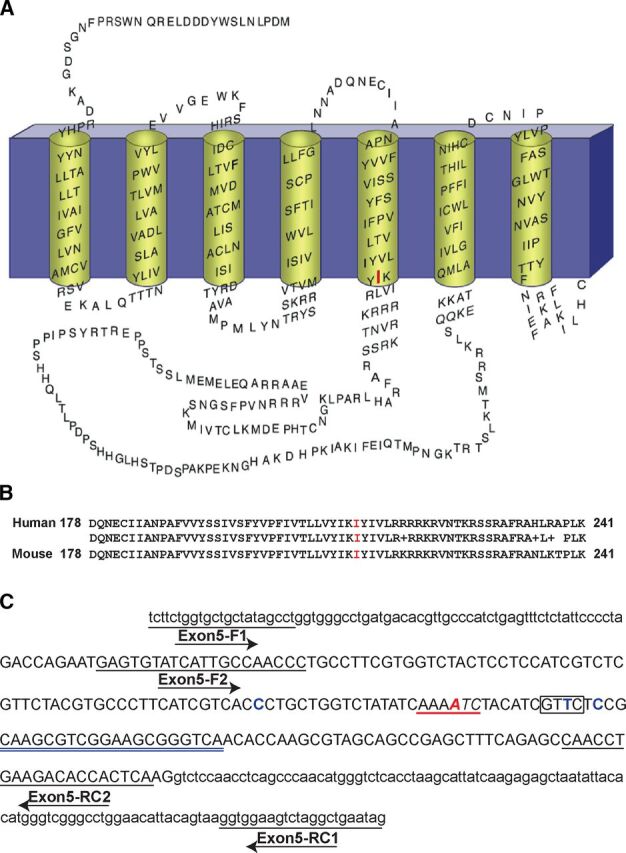
Strategy for producing Drd2I212F mice. (A) Membrane topology of the human D2L receptor, depicting the I212 position in red. (B) Alignment of amino acid sequences from exon 5 of human and mouse D2 dopamine receptor genes, corresponding to the amino acids 178–241 in both proteins. Identity between human and mouse amino acid sequences is shown. (C) Mouse Drd2 exon 5 nucleotide sequence, including intron/exon 5 junctions, showing the PCR primers (underlined) used for genotyping the mice and the position of the gRNA sequence (double underlined in blue). Exon 5 sequence is in uppercase and intron sequence in lowercase. In red is shown the position of the wild-type amino acid (A and B) and nucleotide (C) that was changed to generate the D2-I212F variant. The ssODN template contained three synonymous changes: C-A, T-A, and C-A; the WT nucleotide is shown at these positions in blue. Moreover, the underlined sequence in red (C) shows the position of the ApoI(RAATTY) site, whereas the box shows the location of the introduced RsaI (GTAC) site. Both restriction sites are present in knock-in but not in WT mice. R, purine (A/G); Y, pyrimidine (T/C).
The enhanced D2-I212F–mediated activation of G proteins is manifested in both increased basal activation of Gαi1 and GαoA and increased agonist potency for activation of Gαi1 but not GαoA (Rodriguez-Contreras et al., 2021). We speculated that this would affect striatal D2 receptor signaling. In striatal D2 receptor–expressing medium spiny neurons (D2-MSNs), the more efficient coupling of wild-type (WT) D2 receptors to Gαo in the nucleus accumbens (NAc) results in more potent dopamine activation of G protein–mediated signaling compared with that in dorsal neostriatum, which is mediated by Gαi (Marcott et al., 2018). Furthermore, repeated cocaine exposure decreases the abundance of Gαo in the NAc and eliminates high-potency dopamine activation of GIRK that is mediated by wild-type D2 receptors (Gong et al., 2021). Because the high-potency coupling of D2-I212F to Gαi1 results in similar potency for activation of Gαi1 and GαoA in HEK 293 cells, we suggested that the potency of dopamine would not differ between the nucleus accumbens and dorsal striatum (DSt) in mice expressing D2-I212F and that accumbal dopamine signaling might be unaffected by repeated cocaine treatment (Rodriguez-Contreras et al., 2021).
Gait abnormalities are common in movement disorders with choreatic and dystonic features and sometimes treated with drugs that modulate dopamine receptor activity (Koller and Trimble, 1985; Barbosa and Warner, 2018). Gait abnormalities that have been observed in mouse models of dystonia and chorea include altered stride length and frequency (Dai et al., 2009), the ratio of the time spent in propulsion to the time spent braking (Wright et al., 2015), and splaying of the hind limbs (Liu et al., 2015).
To evaluate the pathogenic role of the D2-I212F variant, we have now generated a knock-in mouse line, Drd2I212F. Both Drd2+/I212F and Drd2I212F/I212F mice exhibited gait abnormalities and decreased striatal D2 receptor expression. GIRK-mediated inhibitory postsynaptic conductances (IPSCs) in Drd2I212F mice were characterized by slow kinetics in both midbrain dopamine neurons and D2-MSNs in the basal forebrain. In addition, cocaine treatment had no effect on the potency of dopamine in the basal forebrain MSNs of Drd2I212F mice, in contrast to cocaine effects in WT mice (Marcott et al., 2018; Gong et al., 2021), which is consistent with the higher potency of D2-I212F than D2-WT for Gαi1 observed in HEK293 cells (Rodriguez-Contreras et al., 2021; van der Weijden et al., 2021a).
Materials and Methods
Animals
All studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals established by the National Institutes of Health. Protocols were approved by Institutional Animal Care and Use Committees at the VA Portland Health Care System, Oregon Health & Science University (OHSU), and the University of Colorado School of Medicine. All mice were allowed ad libitum access to food and water and were maintained on a 12-hour light/dark cycle in a climate-controlled facility.
Design and Generation of Drd2+/I212F Mice
Knock-in Drd2+/I212F mice were produced via the electroporation of one-cell-stage C57BL6/NJ mouse embryos using a NEPA 21 electroporator (NEPA GENE Co. Ltd., Chiba, Japan) as described (Teixeira et al., 2018). Ribonucleoprotein complexes of SpCas9 protein (NEB; Ipswitch, MA), single-stranded oligodeoxynucleotide (ssODN) template, and guide RNA (gRNA) were prepared with final concentration of 200 μg/μl, 100 μg/μl, and 200 μg/μl, respectively. The ssODN template TCCATCGTCTCGTTCTACGTGCCCTTCATCGTCACaCTGCTGGTCTATATCAAATTCTACATCGTACTaCGCAAGCGTCGGAAGCGGGTCAACACCAAGCGTAGCAGCCGAGCT incorporates the desired T change (bold, italic) in exon 5 of the Drd2 gene to introduce the I212F mutation, a synonymous A mutation (italic in the underlined sequence) to introduce an RsaI site for genotyping purpose, and two additional synonymous mutations (lowercase) to remove Cas9 protospacer adjacent motif sequences. This DNA template was synthesized by Integrated DNA Technologies (Coralville, IA). The gRNA (UGACCCGCUUCCGACGCUUG) was synthesized as a chemically modified CRISPR gRNA by Synthego Corporation (Redwood City, CA). After electroporation, embryos were transplanted into pseudopregnant recipient CD-1 female mice, and founders from these litters were identified as described below.
Putative F0 founders were genotyped by polymerase chain reaction (PCR)/digestion and Sanger sequencing (see details below). Selected F0 founders were also analyzed by Sanger sequencing for potential modification of CRISPOR-predicted off-target (OT) sites (see below). Founders were crossed with C57BL/6JN mice, and F1 pups were genotyped by PCR/digestion and DNA sequencing.
Analysis of Target Site and Offspring Genotyping
Mice potentially containing the knock-in mutation were initially screened for DNA sequence changes in Drd2 exon 5 by PCR and RsaI digestion analysis as described below. The I212F mutation introduces an ApoI restriction site that was also used for genotyping with ApoI-HF. Sequence-specific forward and reverse primers (Fig. 1C; Supplemental Table 1) for amplifying exon 5 and the introns/exon 5 junctions were designed against the reference Mus musculus genome, strain C57BL/6NJ (GeneBank assembly accession: GCA_001632555.1). Extraction of genomic DNA was performed using the HotSHOT method (Truett et al., 2000). Briefly, tail snip samples were heated in 100 ul of alkaline buffer (25 mM NaOH, 0.2 mM EDTA) at 95°C for 25 minutes and neutralized with 100 ul of 40 mM Tris-HCl, pH 5. Two μl of supernatant was used per 20-ul PCR reaction. PCR was performed using the HotStarTaq Master Mix Kit (Qiagen; Germantown Road, MD) under the following conditions: 95°C for 3 minutes, 35 cycles at 95°C for 15 seconds, 58°C for 15 seconds, 72°C for 30 seconds, and a final extension step at 72°C for 5 minutes. PCR products were digested with RsaI or ApoI-HF (NEB Inc; Ipswich, MA) and were separated on 1.5% agarose gels. Mice harboring the on-target Drd2-exon 5 knock-in allele were identified based on fragment size after digestion of the PCR product. For example, PCR amplification using Exon5-F1 and Exon5-RC1 primers (Supplemental Table 1) generated a 376–base pair (bp) product (Supplemental Fig. 1A). After restriction digestion, the mutated allele generated two fragments of ∼0.19 kilobase, differing from the undigested WT fragment of ∼0.38 kilobase (Supplemental Fig. 1B). F1 offspring from selected founders crossed with C57BL/6J (WT) mice were genotyped following the same procedure.
Mutation Detection by Sanger Sequencing
To confirm positive F0 heterozygous-genotyped mice and their F1 offspring, the 181-bp and 376-bp amplicon products (Supplemental Fig. 1A) were purified using the Monarch PCR & DNA Cleanup Kit (New England Biolabs, Ipswich, MA) and sent for Sanger DNA sequencing at the OHSU Vollum Sequence Core (Portland, OR). To check the integrity of both WT and mutated DNA allelic strands, the 376-bp amplicon containing Drd2-exon5 and introns/exon 5 junctions from several F0 and F1 mice were cloned into TOPO-TA Cloning vector (Invitrogen by ThermoFisher Scientific; Waltham, MA) according to manufacturer’s instructions. Plasmids were purified using the Wizard Plus Miniprep DNA Purification System (Promega, Madison, WI) and screened for WT- and I212F-mutated allele strands by dual digestion analysis using EcoRI digestion (New England Biolabs, Ipswich, MA) to release the insert and RsaI or ApoI-HF digestion for selecting WT versus mutated strand clones. Positive clones for each allelic strand per mouse were sent for DNA sequencing using the universal T7-Forward oligo.
Molecular Characterization of Drd2 Exon 5 in Drd2I212F Mice
In founder 429 (F0-429), digestion of the PCR amplicon with ApoI-HF supported the heterozygosity of the I212F mutation. Direct DNA sequencing of the amplicon confirmed the presence of the I212F mutation and showed no mixed sequence reads in the F0-429 mouse (Fig. 2A, top). Cloning and sequencing of both alleles confirmed the heterozygosity of the I212F mutation in this founder. However, the F0-429 mouse was homozygous for the RsaI site (Fig. 2a, bottom), in agreement with the results obtained by restriction enzyme analysis and direct DNA sequencing of the PCR amplicon (Fig. 2A, top). No other modifications were observed in this founder. The positive F1 offspring of this founder (five females and two males) were heterozygous for the I212F mutation and RsaI site by both restriction enzyme analysis and DNA sequencing (example in Fig. 2B). This line is referred to as lineage A.
Fig. 2.

Production of Drd2I212F mice. (A) Chromatograms showing the Sanger sequencing confirmation of the c.634A>T; p.Ile212Phe mutation in Founder F0-429 (lineage A). Top, 181-bp product obtained by PCR amplification of gDNA sample using the Exon5-F2 and Exon5-RC2 primer pair was purified and sent for sequencing. Bottom, 376-bp product amplified by PCR using the Exon5-F1 and Exon5-RC1 primer pair was cloned into TOPO-TA vector (Invitrogen), and plasmids were sent for sequencing. For the mutated strand, the reverse-complement sequence is shown. The black arrow indicates the location of c634A in the wild-type strand. However, F0-429 was homozygous for both nucleotide changes introduced downstream of the c.634A>T position. (B) Representative chromatogram showing Sanger sequencing of the 181-bp amplicon from a F1 mouse of lineage A. Founder F0-429 (male) was crossed with a C57BL/6NJ female (WT) mouse. F1 offspring (nine females and five males) were genotyped by PCR and restriction analysis. Sanger sequencing of 181-bp amplicons from positive F1 mice (five females and two males) confirmed that they were heterozygous for all designed nucleotide changes. Wide, red arrows in both panels point to the A-T change to generate the Drd2I212F variant; blue arrows show synonymous nucleotide changes introduced downstream of the c.634A>T position. The nucleotide change in uppercase introduced to generate an RsaI site is indicated by a dashed-line arrow, and its site (GTAC) is shown in a box, whereas the nucleotide change in lowercase was introduced to remove a gRNA protospacer adjacent motif sequence. The molecular characterization of founder F0-421 (lineage B) and its F1 offspring is shown in Supplemental Fig. 2.
Founder F0-421 carried the expected I212F-mutation as well as the introduced RsaI site; however, mixed sequence reads were observed by direct DNA sequencing of the PCR product (Supplemental Fig. 2A, top). Analysis of the two alleles by TOPO-TA cloning and DNA sequencing revealed that one allele was WT for the targeted mutation site but had a nine-nucleotide deletion downstream of that I212 site, whereas only the expected nucleotide changes, with no deletions/insertions, were present in the I212F-mutated allele (Supplemental Fig. 2A, bottom). We cloned and analyzed both Drd2-exon 5 alleles from its positive F1 offspring (two females and 5 males). As expected, WT strands had no genetic modifications (Supplemental Fig. 2B), whereas the mutated strands have only the nucleotide changes described in Fig. 1C. Both Drd2I212F lineages A and B (from founders F0-429 and F0-421, respectively) were maintained as heterozygous breeding colonies at the VA Portland Health Care System. In addition, mice of lineage A were maintained at the University of Colorado Anschutz Medical Campus.
Analysis of Off-Target Sites
The Web-based tool CRISPOR (www.crispor.tefor.net; Concordet and Haeussler, 2018) was used to predict potential OT sites for the gRNA UGACCCGCUUCCGACGCUUG. No off-target locus with fewer than four nucleotide mismatches to the gRNA was found in the C57BL/6J reference genome. Five sites with four nucleotide mismatches were identified (Supplemental Table 2). However, only OT1 [cutting frequency determination (CFD) score 0.45918] and OT2 (CFD score 0.13333) sites had a CFD score higher than 0.1 (Doench et al., 2016). For OT3-5 sites, the CFD scores were 0.015278, 0.01500, and 0.00824, respectively (Supplemental Table 2). Despite the low CFD score, we characterized OT4 because it is on the same chromosome as Drd2, and therefore, an unintended mutation would not be quickly eliminated in subsequent generations. We also characterized OT3 because its CFD score was slightly higher than OT4.
To identify any OT mutations in the founders for the two lineages, PCR reactions were carried out using primer pairs designed to flank the potential OT1 to OT4 sites (Supplemental Table 1). Three to four independently generated PCR products for each OT1-4/founder were purified using the Monarch PCR & DNA Cleanup Kit (NEB Inc.) and sent for Sanger sequencing at the OHSU Vollum Sequence Core. We found no modifications in the studied off-target sequences when compared with the reference M. musculus genome, strain C57BL/6NJ.
Gait Analysis
We used the DigiGait system (Mouse Specifics, Inc., Framingham, MA) to quantify gait abnormalities in Drd2I212F mice. DigiGait uses a variable-speed motorized transparent treadmill belt and a ventral video camera capturing 150 frames/s to calculate over 35 indices of gait for each limb. For these experiments, the rear of the device was raised so that the treadmill was 9 degrees below horizontal. Each mouse was placed in a plexiglas compartment that sits on top of the transparent treadmill and is illuminated from above and below. The mouse was allowed to explore the compartment for several minutes, then the camera was turned on and the treadmill was started at 24 cm/s. After 4 to 5 seconds, the treadmill was turned off, the video clip was saved, and the mouse was returned to its home cage. Videos were processed by an individual blind to mouse genotype.
Thirty-eight animals at 10–12 months of age were tested on the DigiGait System, in which the mouse is recorded via a ventrally mounted camera while running on a transparent treadmill. Relatively old mice were used because, in humans, the clinical symptoms worsen with age. The genotype distribution was 13 Drd2+/+ (eight male and five female), 16 Drd2+/I212F mice (nine male and seven female), and 9 Drd2I212F/I212F mice (four male and five female). Results from left and right limbs were pooled (except for stance width), whereas fore- and hind limbs were analyzed separately.
D2 Receptor Radioligand Binding
To measure membrane expression of striatal D2 receptors, pooled striata from each mouse were homogenized for 10 seconds in 4 ml of Tris-buffered saline (TBS: 50 mM Tris, 120 mM NaCl, pH 7.4) using a Polytron homogenizer (Brinkmann Instruments, Westbury, NY), then centrifuged at 17,000g at 4°C for 20 minutes. The resulting pellet was resuspended in 4 ml TBS + 2 mM Na-EDTA, incubated for 30 minutes at 25°C, and centrifuged again at 17,000g for 20 minutes. The final pellet was resuspended in 3 ml TBS. Protein determination was performed using the BCA Protein Assay Kit (Thermo Scientific Inc; Waltham, MA). Saturation analyses were carried out by preparing ∼0.4 nM [3H]spiperone in 40 nM ketanserin (final concentrations in assay; ketanserin added to inhibit binding to 5-HT2 receptors), then performing 2× dilutions in assay buffer (TBS containing 0.002% bovine serum albumin). Assays containing tissue samples, various concentrations of [3H]spiperone/ketanserin, and (+)-butaclamol (2 µM; to define nonspecific binding) or assay buffer were incubated at 37°C for 1 hour in a final volume of 1 ml before addition of ice-cold buffer and vacuum filtration. Thirty mice were used for radioligand binding experiments (23 males and seven females, 17 of the lineage A and 13 of the lineage B). The density of binding sites (Bmax) and affinity (Kd) of the receptors for [3H]spiperone were determined from saturation curves analyzed by nonlinear regression using GraphPad Prism 9 (San Diego, California). One heterozygous mouse was excluded from Fig. 3 (for both Kd and Bmax) because its Bmax value was more than two standard deviations from the mean. Comparisons of the heterozygous mutant group with that mouse to wild-type and homozygous mutant groups for Bmax were both statistically significant at P < 0.05.
Fig. 3.
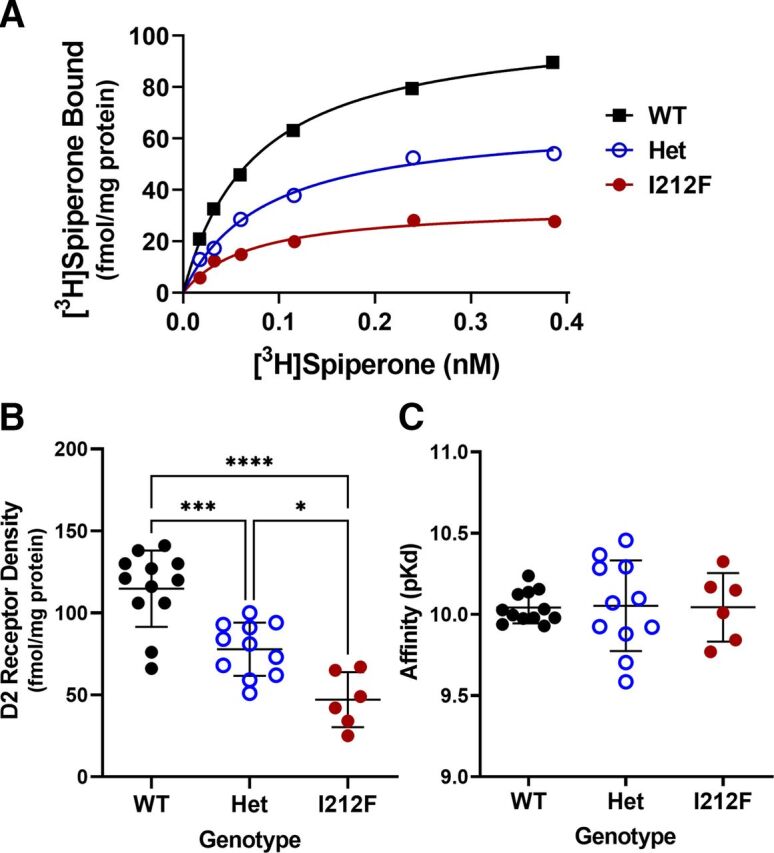
Characterization of striatal D2 receptors. Results are shown for [3H]spiperone binding to striatal membranes from 11–13 month old Drd2+/+ (WT), Drd2+/I212F (Het), and Drd2I212F/I212F (I212F) mice. Saturation binding analysis was carried out using the D2 antagonist radioligand [3H]spiperone, and the Bmax and affinity of D2 receptors for the radioligand (Kd) was obtained by nonlinear regression. (A) Representative binding curves from one mouse of each genotype. (B) Mean Bmax values ± S.D. (C) Mean Kd values ± S.D. *P < 0.05; *** P < 0.001; **** P < 0.0001 by Tukey’s post hoc test.
Stereotaxic Surgery
To express GIRK2 in MSNs, Drd2I212F/I212F (nine males and seven females) and control littermate mice (three males and four females, 3–5 weeks old) were anesthetized with inhaled isoflurane (2%) and positioned in a stereotaxic apparatus. Mice were bilaterally injected with 400 nl AAV9.hSyn.tdTomato.T2A.GIRK2 (University of Pennsylvania Viral Core, V3992) into the dorsal (coordinates in millimeter from bregma: +1.2 anterior, ±1.85 lateral, −3.3 ventral) and ventral (+1.5 anterior, ±1.15 lateral, −4.3 ventral) part of the striatum using a Nanoject III at 100 nl/min. The pipette was kept at the site for 5 minutes and then slowly withdrawn. Mice were allowed to recover for 3 to 4 weeks following surgery.
Midbrain Slice Preparation
Mice were anesthetized with isoflurane and euthanized by rapid decapitation. Brains were removed and placed in warm (30°C) modified Krebs buffer containing NaCl (126 mM), KCl (2.5 mM), MgCl2 (1.2 mM), CaCl2 (2.4 mM), NaH2PO4 (1.4 mM), NaHCO3 (25 mM), and D-glucose (11 mM) with MK-801 (3 μM). Horizontal slices containing the substantia nigra were cut at 222 μM in Krebs buffer bubbled with 95%/5% O2/CO2 using a vibrating microtome (Leica). Slices were allowed to recover at 30°C in vials with 95%/5% O2/CO2 Krebs with MK801 (10 μM) for at least 30 minutes prior to recording. Slices were hemisected and mounted in the recording chamber of an upright microscope (Olympus). The temperature was maintained at 34 − 36°C, and modified Krebs buffer was perfused over the slices at 1 to 2 ml/min. Twenty-four mice were used in these experiments, 17 of lineage A and seven of lineage B (11 males, 13 females; 46–90 days old).
Forebrain Slice Preparation
Mice were anesthetized with isoflurane and transcardially perfused with 10 ml of ice-cold carbogenated (95% v/v O2, 5% v/v CO2) sucrose cutting solution containing (in mM) 75 NaCl, 2.5 KCl, 6 MgCl2, 1.2 NaH2PO4, 25 NaHCO3, 0.1 CaCl2, 11.1 D-glucose, and 1 kynurenic acid. The brain was subsequently removed and coronally sectioned (240 μM) on a vibratome. Slices were transferred to an oxygenated 34°C chamber filled with artificial cerebrospinal fluid (aCSF) solution, consisting of (in mM) 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.5 CaCl2, 1.2 NaH2PO4, 21.4 NaHCO3, 11.1 D-glucose, and 10 μM MK-801 to prevent excitotoxicity, for at least 1 hour. Afterward, slices were placed in a recording chamber, which was constantly perfused with aCSF solutions at a flow rate of 2 ml/min. Solutions also contained SCH23390 (1 μM), scopolamine (200 nM), picrotoxin (100 μM), CGP55845 (300 nM), DNQX (10 μM), and dihydro-β-erythrosine hydrobromide (DhβE; 1 μM). Tissue was visually identified using a BXWI51 microscope (Olympus) with custom-built infrared gradient contrast optics. Fluorescence was visualized with light-emitting diodes (Thorlabs).
Electrophysiology—Midbrain
Recordings were obtained using glass electrodes with a starting resistance of 1.3–1.9 MΩ when filled with an internal solution containing potassium methanesulfonate (75 mM), NaCl (20 mM), MgCl2 (1.5 mM), HEPES potassium salt (5 mM), ATP (2 mM), GTP (0.2 mM), phosphocreatine (10 mM), and BAPTA-tetrapotassium salt (pH 7.35−7.45, 10 mM) at 275 − 288 mOsm. Cells were voltage-clamped at −60 mV using an Axopatch 200A integrating patch clamp (Molecular Devices, San Jose, CA). Recordings were made using Axograph 10 and Chart 5.5 (AD Instruments, Sydney Australia). D2 receptor–expressing dopamine neurons in the substantia nigra were identified by location, size, and firing properties. These studies were conducted by experimenters blind to mouse genotype. D2-IPSCs were elicited using a bipolar electrode and a constant current stimulus isolator (Warner Instruments, Hamden CT). 1-((8-Cyano-7-hydroxyquinolin-2-yl)methyl)-1-ethyl-2-((2-methoxy-5-sulfamoylbenzamido)methyl)pyrrolidin-1-ium 2,2,2-trifluoroacetate (CyHQ-sulpiride) (Asad et al., 2020) was kept as a stock solution in DMSO (10 mM) and recirculated at 15 μM. A ThorLabs M365LP1-C1 LED (Newton, NJ) was used to photolyze CyHQ-sulpiride by means of a 1-second flash (365 nm) at 6.5 mW.
Electrophysiology—Forebrain
MSNs were voltage clamped in whole-cell configuration at −60 mV using Axopatch 200B amplifiers (Molecular Devices, San Jose, CA), and signals were acquired with Axograph X at 5 kHz and filtered to 2 kHz or acquired with LabChart (ADInstruments; Colorado Springs, CO) at 1 kHz. Recording glass pipettes with a tip of resistance of 1.5-2 MΩ (World Precision Instruments; Sarasota, FL) were filled with potassium-based intracellular solution (in mM): 115 K-methylsulfate, 20 NaCl, 1.5 MgCl2, 10 HEPES(K), 10 BAPTA-tetrapotassium, 1 mg/ml ATP, 0.1 mg/ml GTP, and 1.5 mg/ml sodium phosphocreatine (pH 7.4, 275 mOsm). Dopamine release was triggered by electrical stimulation (1 ms) using a monopolar glass stimulating electrode filled with aCSF. For concentration-response curve experiments, cocaine (10 μM) was included in the recording solution to block dopamine reuptake. Dopamine was bath applied via perfusion. D2-IPSCs were evoked once per minute.
Statistical Analysis
Gait data were analyzed first by two-way ANOVA (genotype and sex). There were no main effects of sex. A significant interaction between genotype and sex was observed for forelimb propel time [F(2,70) = 3.24, P = 0.0449]. The other measures had no significant interaction between genotype and sex, so the effect of genotype was assessed by two-way ANOVA followed by Dunnett’s multiple comparisons test.
Statistical analyses of forebrain D2-IPSC data were performed in Prism 8 (GraphPad). Statistical significance was determined using the Mann-Whitney U test, Sidak’s multiple comparisons test, or one-way ANOVA with Tukey’s post hoc analysis.
Results
Generation of Drd2I212F Mice
To mimic the human D2-I212F variant, we introduced the c.634A>T mutation in mice (Fig. 1, A and B) using the CRISPR-Cas9–mediated gene-editing system (Jinek et al., 2012). The strategy for producing the Drd2I212F knock-in mouse line, the genetic characterization of the mice, and confirmation of the probable absence of off-target effects are described in Materials and Methods. We amplified and sequenced the exon 5 target locus (Fig. 1C) from several heterozygous Drd2+/I212F F0 mice and selected two founders (F0-429, lineage A, and F0-421, lineage B; see Fig. 2 and Supplemental Fig. S2, respectively). Both founders, heterozygous for the mutation c.634A>T, were crossed with inbred control mice, and F1 pups were also genotyped by PCR/digestion and DNA sequencing.
Drd2I212F/I212F and Drd2+/I212F mice (from both lineages) in their home cages were not readily distinguishable from Drd2+/+ mice, showing no obvious defects in size or morphology. Of 179 mice born at the VA Portland Health Care System of heterozygote crosses prior 5/16/2022, the genotype distribution (Drd2+/+:Drd2+/I212F:Drd2I212F/I212F) was 32:51:30 for mice of lineage A and 16:35:15 for mice of lineage B, close to the expected 1:2:1 distribution.
Gait Abnormality in Drd2I212F Mice
In humans, the novel DRD2I212F variant cosegregates with a unique hyperkinetic movement disorder characterized by adolescent onset and progressive chorea and cervical dystonia. We studied gait because it is a quantitative measure for which some parameters are affected in other mouse models of chorea or dystonia (Dai et al., 2009; Liu et al., 2015; Wright et al., 2015). Thirty-eight animals at 10–12 months of age were tested on the DigiGait System, in which the mouse is recorded via a ventrally mounted camera while running on a transparent treadmill. There was a significant effect of genotype (ANOVA) for five of the seven measures for forelimbs and three for hind limbs, with increased stride length and decreased stride frequency being the most robust genotype-dependent effects observed (Table 1). For Drd2+/I212F mice, post hoc analysis identified significant increases in forelimb swing time, hind limb propel time, and forelimb and hind limb stride length, as well as decreased forelimb and hind limb stride frequency. Homozygous mutant Drd2I212F/I212F exhibited the same changes and also significantly increased forelimb propel time and propel/brake ratio (Table 1).
TABLE 1.
Gait analysis in Drd2I212F mice.
Results are shown for selected measures of gait obtained using a DigiGait treadmill. Time measurements (swing, time spent swinging the limb forward; propel, time spent in propulsion with the paw pushing backward; brake) are in milliseconds, length measurements (stride length; stance width) are in centimeters, and stride frequency is strides per second. A spreadsheet with the data for 42 measures of gait, including the seven shown here, is available in the Supplemental material (raw digigait data.xlsx).
| Swing Time | Propel Time | Brake Time | Propel/Brake | Stride Freq. | Stride Length | Stance Width | |
|---|---|---|---|---|---|---|---|
| Forelimbs | |||||||
| WTa | 94 ± 4 | 93 ± 5 | 65 ± 4 | 1.63 ± 0.16 | 4.07 ± 0.09 | 6.07 ± 0.13 | 1.5 ± 0.07 |
| Het | 109 ± 2*** | 100 ± 3c | 62 ± 3 | 1.71 ± 0.09 | 3.78 ± 0.07** | 6.49 ± 0.15b | 1.6 ± 0.04 |
| I212F | 109 ± 3** | 114 ± 4** | 56 ± 3 | 2.19 ± 0.21b | 3.63 ± 0.06*** | 6.72 ± 0.10*** | 1.5 ± 0.06 |
| ANOVA | F (2,73) = 9.9 | F (2,70) = 8.8 | F (2,73) = 1.7 | F (2,73) = 3.5 | F (2,73) = 7.8 | F (2,73) = 7.5 | F (2,35) = 0.39 |
| P = 0.0002 | P = 0.0004 | P = 0.19 | P = 0.035 | P = 0.0009 | P = 0.001 | P = 0.68 | |
| Hind Limbs | |||||||
| WT | 98 ± 3 | 110 ± 5 | 36 ± 2 | 3.10 ± 0.14 | 4.20 ± 0.09 | 5.85 ± 0.11 | 3.0 ± 0.07 |
| Het | 107 ± 3 | 126 ± 3** | 40 ± 2 | 3.49 ± 0.23 | 3.76 ± 0.06*** | 6.54 ± 0.09*** | 2.8 ± 0.04 |
| I212F | 101 ± 3 | 136 ± 4*** | 41 ± 2 | 3.62 ± 0.32 | 3.71 ± 0.09*** | 6.67 ± 0.13*** | 2.9 ± 0.05 |
| ANOVA | F (2,73) = 2.53 | F (2,73) = 10.9 | F (2,73) = 1.19 | F (2,73) = 1.26 | F (2,73) = 12.2 | F (2,73) = 15.7 | F (2,35) = 1.97 |
| P = 0.087 | P < 0.0001 | P = 0.31 | P = 0.29 | P < 0.0001 | P < 0.0001 | P = 0.154 | |
aWT, Drd2+/+; Het, Drd2+/I212F; I212F, Drd2I212F/I212F.
bP < 0.05; **P < 0.01; ***P < 0.001 compared with Drd2+/+ mice.
cP < 0.05 compared with Drd2I212F/I212F.
Thirty-two of these mice were assessed a few weeks later for basal and cocaine-stimulated locomotor activity (Supplemental Fig. 3). The mice traveled longer distances within 30 minutes after cocaine (10 mg/kg i.p.) than after saline treatment, but there was no significant effect of genotype.
Striatal D2 Receptor Density
D2-I212F is expressed at 35%–40% of the density of D2-WT after transfection of a given amount of DNA in HEK293 cells (Rodriguez-Contreras et al., 2021; van der Weijden et al., 2021a). We carried out radioligand binding studies with striatal tissue from a subset of the mice used for gait analysis to determine if D2 receptor density was also reduced in Drd2I212F mice. Genotype significantly affected the density of neostriatal D2 receptors [F(2,26) = 25.71; P < 0.0001]. The density of receptors was decreased by 32% in Drd2+/I212F mice and by 59% in Drd2I212F/I212F mice (Fig. 3, A and B). There was no effect of genotype on the affinity of the receptors for [3H]spiperone [F(2,26) = 0.008; P = 0.99; Fig. 3C].
Midbrain D2 Autoreceptors
We used Drd2I212F mice to confirm some of the results obtained after adeno-associated virus–mediated expression of the variant in midbrain dopamine neurons (Rodriguez-Contreras et al., 2021; van der Weijden et al., 2021a). Midbrain slices prepared from mice of three genotypes [Drd2+/+ (WT), Drd2+/I212F (HET), and Drd2I212F/I212F (I212F)] were electrically stimulated (five stimuli at 40 Hz) to elicit D2 receptor–GIRK IPSCs in response to somatodendritic dopamine release. Electrically evoked IPSCs from Drd2I212F mice were slow compared with those from Drd2+/+ mice (Fig. 4A). The amplitudes of evoked IPSCs were not significantly different among genotypes (Fig. 4B: Kruskal-Wallis test, H = 2.93, P < 0.05). However, the duration of the IPSCs was increased in both Drd2I212F/I212F and Drd2+/I212F mice relative to controls [Fig. 4C; one-way ANOVA, F(2,29) = 192.7, P < 0.0001]. We assessed the role of signal termination on this widening of the IPSC by calculating the τ of decay. Drd2+/+ and Drd2I212F/I212F mice were fit by a single exponential, with the latter showing a considerable slowing of the decay back to baseline (Fig. 4D; Kruskal-Wallis test, H = 35.22, P < 0.0001). Interestingly, the evoked IPSCs in dopamine neurons from the Drd2+/I212F mice were fit by analyzing in terms of two decay constants. Each component of this double exponential was similar to the τ measured in the corresponding homozygous genotype, reflecting the presence of distinct components arising from the two variants.
Fig. 4.

Prolonged signaling kinetics in midbrain dopamine neurons from Drd2I212F mice. (A) Representative recordings of D2-IPSCs elicited by five electrical pulses delivered at 40 Hz in midbrain slices from Drd2+/+ (WT, black), Drd2+/I212F (HET, cyan), and Drd2I212F/I212F (I212F, red) mice. (B) Average amplitude of D2-IPSCs elicited with five pulses in each genotype. (WT, 93 ± 20 pA; HET, 63 ± 8 pA; and I212F, 53 ± 8 pA; Kruskal-Wallis test, H = 2.93, P > 0.05). (C) Average width of the D2-IPSC measured at 50% of the peak (half width) (WT, 0.40 ± 0.01 seconds; HET, 0.63 ± 0.03 seconds; and I212F, 1.33 ± 0.05 seconds). (D) τ values obtained with either a single (WT, I212F) or double (HET) exponential fit. Drd2+/I212F mice displayed a two-component decay, with a fast component (HET-F) resembling Drd2+/+ and a slow component (HET-S) resembling τ values for Drd2I212F/I212F (WT, 0.3 ± 0.2 seconds; Het-F, 0.43 ± 0.03 seconds; Het-S, 2.1 ± 0.4 seconds; and I212F, 1.3 ± 0.1 seconds). (E) Normalized (scaled-to-peak) D2 currents terminated by photolytic release of sulpiride in Drd2+/+ (WT, black), Drd2+/I212F (HET, cyan), and Drd2I212F/I212F (I212F, red) animals. (F) Amplitude of the current induced by 10 µM dopamine across genotypes. Homozygous mutants displayed a significant decrease compared with both WT and heterozygous animals (WT, 287 ± 27 pA; HET, 264 ± 38 pA; I212F, 130 ± 27 pA. (G) Quantification of the decay τ following photolytic release of sulpiride. Homozygous mutants exhibit the slowest kinetics, with heterozygous animals showing an intermediate slowing effect compared with WT animals (WT, 310 ± 20 milliseconds; HET, 600 ± 42 milliseconds; I212F, 1132 ± 191 milliseconds). All values plotted are mean ± S.E.M. (B–D) N = 10 cells/3 mice (WT), 13 cells/4 mice (HET), and 10 cells/3 mice (I212F). (F and G) N = 18 cells/6 mice (WT), 11 cells/4 mice (HET), and 7 cells/4 mice (I212F). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 using Dunn’s after Kruskal-Wallis or Tukey's after ANOVA. ns, not significant.
We also examined the termination of signaling kinetics using exogenously applied dopamine (10 µM) and photolytic release of the competitive inverse agonist sulpiride from CyHQ-sulpiride (Asad et al., 2020) since the amplitude and kinetics of D2-IPSCs may be influenced by changes in evoked dopamine release. Cells from all genotypes exhibited an outward current induced by 10 µM dopamine (Fig. 4E) that returned to baseline following photolytic release of sulpiride (Fig. 4B). The maximum current induced by 10 µM dopamine in cells from Drd2I212F/I212F animals was reduced to ∼50% compared with cells from Drd2+/+ or Drd2+/I212F mice [Fig. 4F, one-way ANOVA; F(2,33) = 4.994, P < 0.05]. Photolytic release of sulpiride produced a fast decay of signaling in cells from all three genotypes. However, as for the electrically evoked IPSCs, the rate of decay was gene dosage–dependent [Fig. 4G, one-way ANOVA: F(2,33) = 30.73, P < 0.0001].
Postsynaptic D2 Receptors in Basal Forebrain
D2-I212F receptors expressed in HEK293 cells or dopamine neurons in the mouse midbrain differ in sensitivity to agonist and kinetics of GIRK regulation compared with D2-WT receptors (Fig. 4; also Rodriguez-Contreras et al., 2021; van der Weijden et al., 2021a). Here, we examine whether the kinetics and sensitivity of D2-I212F receptors also differ from D2-WT in D2-MSNs. An adeno-associated virus encoding a GIRK2 channel and a tdTomato fluorophore was injected into both the DSt and the NAc shell of Drd2I212F/I212F mice or their wild-type littermates (Fig. 5A), as described previously (Marcott et al., 2018; Gong et al., 2021). Three weeks later, coronal brain slices containing the DSt and NAc were cut for electrophysiological recordings. Recordings were made in the presence of NMDA, GABAA, GABAB, muscarinic, and D1 receptor antagonists to isolate D2 receptor–mediated GIRK2 currents. In a whole-cell voltage clamp, a single electrical stimulus evoked D2-IPSCs in D2-MSNs (Fig. 5B). We observed that the amplitude of D2-IPSCs was smaller in both the DSt and NAc of Drd2I212F/I212F mice compared with littermate controls (Fig. 5C). Similar to somatodendritic IPSCs recorded from dopamine neurons, electrically evoked D2-IPSCs in both DSt and NAc D2-MSNs from Drd2I212F mice were slower to activate (Fig. 5D) and were slower to decay than littermate controls (Fig. 5E). The percent changes between D2-IPSCs recorded in Drd2I212F versus D2-WT mice were similar in the DSt and NAc (10%–90% rise: DSt, 69% increase versus D2-WT; NAc, 61% increase versus D2-WT; τ decay: DSt, 650% increase versus D2-WT; NAc, 573% increase versus D2-WT), indicating that our previous observation that D2 receptor kinetics are slower in the NAc than in the DSt (Marcott et al., 2018) also holds for mice expressing D2-I212F. The results suggest that the D2-I212F mutation causes slower D2 receptor signaling kinetics in both the DSt and the NAc.
Fig. 5.

Characterization of D2-IPSCs in striatal medium spiny neurons. (A) Injection schematic of AAV9.hSyn.tdTomato.GIRK2 into the NAc (medial shell) and DSt (dorsomedial) of the Drd2+/+ (WT) or Drd2I212F/I212F (I212F) mice. (B) Representative traces of electrical stimulation-evoked D2-IPSCs from the DSt and NAc of WT and I212F mice. (C) Quantification of the D2-IPSC amplitudes (DSt: D2-WT, 377 ± 67 pA; D2-I212F, 206 ± 21 pA; NAc: D2-WT, 358 ± 64 pA; D2-I212F, 156 ± 36 pA. (D) Quantification of 10%–90% rise time of D2-IPSCs [DSt: D2-WT, 120 ± 6 milliseconds; D2-I212F, 203 ± 5 milliseconds; NAc: D2-WT, 169 ± 4 milliseconds; D2-I212F, 272 ± 25 milliseconds; one-way ANOVA: F(3,37) = 87.21, P < 0.001]. (E) Quantification of τ of decay of D2-IPSCs [DSt: D2-WT, 261 ± 14 milliseconds; D2-I212F, 1702 ± 95 milliseconds; NAc: D2-WT, 453 ± 34 milliseconds; D2-I212F, 2595 ± 203 milliseconds; one-way ANOVA: F(3,37) = 23.78, P < 0.001]. (C–E) Summary data are mean ± S.E.M. from nine cells (DSt) and 10 cells (NAc)/6 mice (D2-WT) or 12 cells (DSt) and 10 cells (NAc)/7 mice (D2-I212F). *P < 0.05; **P < 0.01; ***P < 0.001 by Mann-Whitney U test (C) or Sidak’s multiple comparisons test (D and E).
Given that D2-IPSCs evoked by the release of dopamine differed in Drd2I212F/I212F mice, we next examined if the sensitivity of D2 receptor signaling also differed. Concentration-response relationships for dopamine in the DSt and NAc were constructed by measuring D2 receptor–mediated GIRK2 currents evoked by bath application of dopamine in the presence of cocaine (10 μM) to block dopamine reuptake (Fig. 6A). Similar to our previous findings (Marcott et al., 2018; Gong et al., 2021), the concentration of dopamine needed to achieve 50% of the maximal effect (EC50) in D2-WT mice was significantly lower in the NAc than the DSt (Fig. 6, B and C), confirming that D2 receptors in the NAc have a higher sensitivity for dopamine than in the DSt. The D2-I212F mutation led to a selective increase in the sensitivity of D2 receptors in the DSt, as reflected in a decrease in the EC50 value, but had no effect on the sensitivity of D2R signaling in the NAc (Fig. 6C). The mutation also decreased the amplitude of the outward current evoked by a saturating concentration of dopamine (300 μM) (Emax) in both regions, an effect that was greater in the DSt than the NAc (Fig. 6D). As the difference in D2 receptor sensitivity between the DSt and NAc in wild-type mice results from differential coupling to Gαo over Gαi in the NAc (Marcott et al., 2018; Gong et al., 2021), these results are consistent with our prior work demonstrating that agonist potency at D2-I212F is similar for activation of Gαo and Gαi in HEK293 cells because of selectively increased potency at Gαi (Rodriguez-Contreras et al., 2021).
Fig. 6.

Dopamine potency in striatal D2-MSNs. (A) Representative traces after bath application of dopamine (DA, 3 μM) depict dopamine-induced D2R-mediated outward currents from D2-MSNs in the DSt and NAc from Drd2+/+ (WT) or Drd2I212F/I212F (I212F) mice. Recordings were performed in the presence of cocaine (10 μM) to block dopamine uptake. D2-IPSCs were evoked once per minute and have been blanked for clarity. (B) Dopamine concentration-response relationships for D2 receptor–mediated outward GIRK2 current. (C) EC50 values calculated from dopamine concentration-response curves [shown in (B)] [DSt: D2-WT, 12.1 ± 1.7 μM; D2-I212F, 2 ± 0.7 μM; NAc: D2-WT, 3.6 ± 0.7 μM; D2-I212F, 2.4 ± 0.9 μM; Tukey’s test following one-way ANOVA: F(3,23) = 18.87, P < 0.001]. Summary data are mean ± S.E.M. from seven repetitions for D2-WT (DSt and NAc) and D2-I212F (DSt) and six repetitions for D2-I212F (NAc). (D) Maximum outward currents evoked by 300 μM dopamine [from (B)] (DSt: D2-WT, 624 ± 89 pA; D2-I212F, 201 ± 27 pA; NAc: D2-WT, 606 ± 78 pA; D2-I212F, 336 ± 25 pA). (C and D) Summary data are mean ± S.E.M. of six to seven cells from each brain region in six to seven mice of each genotype, with statistical comparisons by Mann-Whitney U test. (E) Schematic of the timeline of AAV.GIRK2 injection and 7-day cocaine administration. (F) Dopamine concentration-response relationships for D2 receptor–mediated outward GIRK2 current from D2-MSNs in the DSt and NAc from Drd2I212F/I212F (I212F) mice after 7 days of cocaine exposure or from control untreated animals [from (B)]. (G) EC50 values from (F). (H) Maximum outward currents evoked by 300 μM dopamine from (F). Summary data are mean ± S.E.M. n.s., P > 0.05, **P < 0.01, *** P < 0.001.
We reported previously that chronic cocaine exposure selectively decreases D2 receptor sensitivity in the NAc via the reduction of Gαo levels to gate cocaine-conditioned behaviors, whereas D2 receptor sensitivity for the Gαi-mediated response in the DSt is unaffected by prior cocaine exposure (Gong et al., 2021). Because agonist potency at D2-I212F is similar for Gαo and Gαi, we predicted that prior cocaine exposure would have no effect on the potency of dopamine in DSt or NAc of Drd2I212F/I212F mice. To test this, Drd2I212F/I212F mice were treated with cocaine for 7 days (20 mg/kg i.p.) (Fig. 6E). As predicted, this cocaine exposure had no effect on D2 receptor sensitivity to dopamine in either the DSt or NAc of Drd2I212F/I212F mice (Fig. 6, F and G). Interestingly, chronic exposure to cocaine selectively reduced the outward current evoked by a saturating concentration of dopamine (300 μM) in the NAc of Drd2I212F/I212F mice, without having an effect in the DSt (Fig. 6H). In wild-type mice, prior cocaine treatment did not change the maximum response in either DSt or NAc (Gong et al., 2021).
Discussion
Human carriers of the dopamine D2 receptor mutation c.634A>T, p.Ile212Phe have a hyperkinetic movement disorder characterized by symptoms of chorea and cervical dystonia that appear in adolescence and worsen with increasing age (van der Weijden et al., 2021a, 2021b). This novel mutation in exon 5 of DRD2 changes residue 212 at the cytoplasmic face of helix 5 of the D2 receptor from Ile to Phe (Fig. 1A).
The clinical phenotype associated with heterozygosity for D2-I212F has features in common with other genetic disorders characterized by childhood-onset chorea, frequently accompanied by dystonia and/or myclonus. One such disorder, called either familial dyskinesia with facial myokymia (Fernandez et al., 2001; Chen et al., 2012) or ADCY5-related dyskinesia (Carecchio et al., 2017), is caused by activating mutations of adenylyl cyclase 5 (Chen et al., 2014; Doyle et al., 2019), a striatum-enriched form of adenylyl cyclase that mediates D1 dopamine receptor stimulation and D2 receptor inhibition of adenylyl cyclase in striatal MSNs (Lee et al., 2002). A second pathogenic DRD2 mutation, M374R, which changes a residue at the cytoplasmic face of transmembrane domain 6, causes an early childhood-onset hyperkinetic movement disorder that is qualitatively similar to that caused by DRD2I212F but much more severe (Mencacci et al., 2021; van der Weijden et al., 2021b). One of the carriers of this second mutation was part of a subject population that had previously been tested for ADCY5 mutations, highlighting the similar clinical phenotypes (Mencacci et al., 2021). Similarly, activating mutations of GαoA cause early-onset dyskinesia including choreatic features (Feng et al., 2018). Because Gαo mediates many effects of D2 receptor signaling (Jiang et al., 2001; Marcott et al., 2018), it might be expected that activating mutations of the two proteins would cause overlapping phenotypes.
Using expression in HEK 293 cells, we previously determined that basal G protein–mediated signaling and agonist potency are enhanced for D2-I212F, with the magnitude of effects depending on the Gα subtype, whereas agonist-induced binding of arrestin to D2-I212F is decreased. Molecular dynamics simulations (Rodriguez-Contreras et al., 2021) suggest that these functional changes may be the result of weakening an intramolecular salt bridge that maintains some G protein–coupled receptors in an inactive conformation (Ballesteros et al., 2001). We confirmed the effects of the mutation on G protein–mediated signaling in midbrain dopamine neurons after viral expression of D2-I212F, and also determined that a consistent difference between the wild-type D2 receptor and D2-I212F is that the rate at which Gβγ-mediated activation of GIRKs is terminated is many-fold slower for the pathogenic variant (Rodriguez-Contreras et al., 2021; van der Weijden et al., 2021a).
We have now produced a mouse constitutively expressing D2-I212F to confirm the pathogenicity of this variant and as a tool to further evaluate the functional consequences of the mutation. We found that several gait measures were altered in Drd2I212F mice tested at 10–12 months of age. Drd2212F/I212F mice took longer to swing forelimbs forward, took longer strides with hind and forelimbs, and had decreased stride frequency for both hind and forelimbs. The gait analysis also estimates the proportion of the time that paws are in contact with the treadmill that is spent propelling the mouse forward versus braking. Homozygous mutant mice spent more time in hind limb and forelimb propulsion and had an increased ratio of forelimb time spent in propulsion to time spent braking. These gait alterations were present in the absence of changes in basal locomotor activity. Importantly, considering that all humans known to have this variant are heterozygous at the DRD2 locus, Drd2+/I212F mice exhibited most of the same gait abnormalities, supporting the pathogenic role of the D2-I212F variant in humans.
In HEK 293 cells, D2-I212F is expressed at a lower density than D2-WT after transfection of identical amounts of DNA (van der Weijden et al., 2021a). Here, we show that the density of neostriatal D2 receptors was decreased in Drd2I212F mice at approximately 1 year of age, with the magnitude of the decrease dependent on the number of Drd2I212F alleles. It is noteworthy that the magnitude of the decrease was ∼30% per mutant allele, which is very close to the magnitude of the decrease after transient transfection of HEK 293 cells (Rodriguez-Contreras et al., 2021; van der Weijden et al., 2021a). This may be a consequence of receptor constitutive activity, as constitutively active GPCRs have often been shown to be less stable and therefore degraded more rapidly (Gether et al., 1997; Rasmussen et al., 1999; Alewijnse et al., 2000), but it is also possible that trafficking of the receptor to the plasma membrane is delayed by the mutation. It was previously reported that striatal binding potentials for the D2/D3 receptor positron emission tomography ligand [11C]raclopride in three subjects heterozygous for DRD2I212F were within the normal range (van der Weijden et al., 2021a). The modest decrease that the results from Drd2+/I212F mice suggest should be found in heterozygous subjects may be difficult to detect in a small number of human subjects in the context of aging-related declines in D2 receptor density.
We confirmed the slow kinetics of D2-I212F–mediated GIRK conductances seen in viral overexpression experiments using midbrain slices from homozygous Drd2I212F mice. Importantly, the two components mediated by D2-WT and D2-I212F in the heterozygous mice combined to produce GIRK currents that were substantially prolonged compared with those in wild-type mice, whether assessed by the decay of evoked IPSCs or by photolytic release of sulpiride.
Slow kinetics are not limited to D2-I212F receptors expressed in dopamine neurons, as we observed similar results in striatal D2 receptor–expressing MSNs. The finding that sulpiride-induced termination of the GIRK response to bath-applied dopamine (Fig. 4) or in the absence of dopamine (Rodriguez-Contreras et al., 2021) is slower for D2-I212F–mediated currents indicates that the slow kinetics probably reflect postsynaptic mechanisms. Prolonged GIRK responses indicate that activation of the G protein heterotrimer by D2-I212F results in availability of free Gβγ that is prolonged relative to activation by D2-WT, but the mechanistic basis for prolonged elevation of Gβγ is yet to be determined. Although one hypothesis could be that D2-I212F desensitizes more slowly because of decreased arrestin binding, we found no evidence of reduced desensitization in response to bath-applied quinpirole (van der Weijden et al., 2021a). A second hypothesis is that the higher affinity of D2-I212F receptors for dopamine slows competitive inhibition by sulpiride, although our previous finding that decay is slower for D2-I212F even in reserpine-treated slices argues against an exclusive role for slow unbinding of dopamine (Rodriguez-Contreras et al., 2021). Experiments using sulpiride uncaging with agonists of varying affinities will be required to definitively distinguish slow unbinding from downstream signaling changes (Condon et al., 2021).
Prior work has shown that differential coupling to G protein subtypes leads to higher potency D2 receptor signaling in D2-MSNs of the NAc than the DSt and that the high potency signaling is lost after repeated daily treatment with cocaine (Marcott et al., 2018; Gong et al., 2021). The high- and low-potency signaling in untreated mice reflects D2 receptor coupling to Gαo and Gαi, respectively, and the loss of accumbal high-potency signaling results from a cocaine-induced reduction in the abundance of Gαo. In the current study, we demonstrated that the potency of dopamine is similar in NAc and DSt and unaffected by prior cocaine treatment in homozygous Drd2I212F mice. In these mice, however, cocaine treatment selectively decreased the maximal response to dopamine in NAc, a response that was not observed in wild-type mice (Gong et al., 2021). It may be that the combined effect of the cocaine-induced decrease in Gαo and the lower expression of D2-I212F is to decrease the maximal response instead of decreasing potency as observed for D2-WT (Gong et al., 2021). This mutation did not affect the acute locomotor response to cocaine, but it will be important to assess the consequences of these differing electrophysiological sequelae of repeated cocaine treatment for behavior such as cocaine-conditioned place preference. Also, if future studies demonstrate that the disease progression in mice is similar to that in humans, in whom symptoms first appear in adolescence and worsen with increasing age, it may be fruitful to assess the behavioral effects of repeated cocaine treatment at multiple ages.
We have speculated that the constitutive activity and G protein bias of D2-I212F both contribute to the clinical phenotype and that an effective treatment would target these characteristics (Rodriguez-Contreras et al., 2021; van der Weijden et al., 2021a). Thus, orthosteric or allosteric agonists with arrestin bias (i.e., G protein–biased antagonists) would be promising compounds to test. We anticipate that this novel mouse model will be useful for such drug-testing studies as well as for investigations into the mechanisms of early-onset hyperkinetic movement disorders.
Acknowledgments
We appreciate the advice of Dr. Dineke Verbeek (University of Groningen) regarding gRNA and DNA template design and genetic analysis of putative founders, and we thank David Buck and Paul Bui for genotyping and maintenance of the two Drd2I212F lineages at the VA Portland Health Care System. We also appreciate the assistance of Dr. Cheryl Reed in the planning of the cocaine locomotor study and Ms. Sara Aldrich for cocaine locomotor activity study data collection.
Abbreviations
- aCSF
artificial cerebrospinal fluid
- bp
base pair
- CFD
cutting frequency determination
- CyHQ-sulpiride
1-((8-cyano-7-hydroxyquinolin-2-yl)methyl)-1-ethyl-2-((2-methoxy-5-sulfamoylbenzamido)methyl)pyrrolidin-1-ium 2, 2, 2-trifluoroacetate
- D2-MSNs
D2 receptor-expressing MSNs
- D2-IPSC
D2 receptor-mediated IPSC
- Emax
maximum response
- gRNA
guide RNA
- GIRK
G protein–regulated inwardly rectifying potassium channel
- HEK
human embryonic kidney
- IPSC
inhibitory postsynaptic conductance
- MSN
medium spiny neuron
- NAc
nucleus accumbens
- OHSU
Oregon Health & Science University
- OT
off target
- PCR
polymerase chain reaction
- ssODN
single-stranded oligodeoxynucleotide
- TBS
Tris-buffered saline
- WT
wild type
Authorship Contributions
Participated in research design: Rodriguez-Contreras, Gong, Lebowitz, Fedorov, Phillips, Ford, Williams, Neve.
Conducted experiments: Rodriguez-Contreras, Gong, Lebowitz, Williams, Neve.
Contributed new reagents or analytic tools: Fedorov, Asad, Dore.
Performed data analysis: Rodriguez-Contreras, Gong, Lebowitz, Phillips, Ford, Williams, Neve.
Wrote or contributed to the writing of the manuscript: Rodriguez-Contreras, Gong, Lebowitz, Fedorov, Dore, Ford, Williams, Neve.
Footnotes
Support for this research was provided by National Institutes of Health National Institute of Neurologic Disorders and Stroke [Grant R21NS117713] (to K.A.N.); National Institute on Drug Abuse [Grant T32DA007262] (to J.L.L.), [Grant R01DA004523] (to J.T.W.), [Grant R01DA035821] (to C.P.F.), [Grant R01DA046081] (to T.J.P.), and [Grant U01DA041579] (to T.J.P.]; and National Institute of Mental Health [Grant R21MH123085] (to C.P.F.); New York University Abu Dhabi; the OHSU Shared Resources Pilot program; and by a Merit Review Award [BX003279] and a Senior Research Career Scientist Award [15F-RCS-009] from the US Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development.
No author has an actual or perceived conflict of interest with the contents of this article.
1D.R.-C. and S.G. contributed equally to this work.
A preprint of this article was deposited in bioRxiv: https://www.biorxiv.org/content/10.1101/2022.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Alewijnse AE, Timmerman H, Jacobs EH, Smit MJ, Roovers E, Cotecchia S, Leurs R (2000) The effect of mutations in the DRY motif on the constitutive activity and structural instability of the histamine H(2) receptor. Mol Pharmacol 57:890–898. [PubMed] [Google Scholar]
- Asad N, McLain DE, Condon AF, Gore S, Hampton SE, Vijay S, Williams JT, Dore TM (2020) Photoactivatable dopamine and sulpiride to explore the function of dopaminergic neurons and circuits. ACS Chem Neurosci 11:939–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SG, Shi L, Gether U, Javitch JA (2001) Activation of the β 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem 276:29171–29177. [DOI] [PubMed] [Google Scholar]
- Barbosa P, Warner TT (2018) Dystonia. Handb Clin Neurol 159:229–236. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63:182–217. [DOI] [PubMed] [Google Scholar]
- Carecchio MMencacci NEIodice APons RPanteghini CZorzi GZibordi FBonakis ADinopoulos AJankovic J, et al. (2017) ADCY5-related movement disorders: Frequency, disease course and phenotypic variability in a cohort of paediatric patients. Parkinsonism Relat Disord 41:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Murphy KP, Parent M, Levine MS (2014) The role of dopamine in Huntington’s disease. Prog Brain Res 211:235–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-ZMatsushita MMRobertson PRieder MGirirajan SAntonacci FLipe HEichler EENickerson DABird TD, et al. (2012) Autosomal dominant familial dyskinesia and facial myokymia: single exome sequencing identifies a mutation in adenylyl cyclase 5. Arch Neurol 69:630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YZFriedman JRChen DHChan GCBloss CSHisama FMTopol SECarson ARPham PHBonkowski ES, et al. (2014) Gain-of-function ADCY5 mutations in familial dyskinesia with facial myokymia. Ann Neurol 75:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concordet JP, Haeussler M (2018) CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res 46 (W1):W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condon AF, Robinson BG, Asad N, Dore TM, Tian L, Williams JT (2021) The residence of synaptically released dopamine on D2 autoreceptors. Cell Rep 36:109465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Dudek NL, Li Q, Fowler SC, Muma NA (2009) Striatal expression of a calmodulin fragment improved motor function, weight loss, and neuropathology in the R6/2 mouse model of Huntington’s disease. J Neurosci 29:11550–11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JGFusi NSullender MHegde MVaimberg EWDonovan KFSmith ITothova ZWilen COrchard R, et al. (2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle TB, Hayes MP, Chen DH, Raskind WH, Watts VJ (2019) Functional characterization of AC5 gain-of-function variants: Impact on the molecular basis of ADCY5-related dyskinesia. Biochem Pharmacol 163:169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Khalil S, Neubig RR, Sidiropoulos C (2018) A mechanistic review on GNAO1-associated movement disorder. Neurobiol Dis 116:131–141. [DOI] [PubMed] [Google Scholar]
- Fernandez M, Raskind W, Wolff J, Matsushita M, Yuen E, Graf W, Lipe H, Bird T (2001) Familial dyskinesia and facial myokymia (FDFM): a novel movement disorder. Ann Neurol 49:486–492. [PubMed] [Google Scholar]
- Gether U, Ballesteros JA, Seifert R, Sanders-Bush E, Weinstein H, Kobilka BK (1997) Structural instability of a constitutively active G protein-coupled receptor. Agonist-independent activation due to conformational flexibility. J Biol Chem 272:2587–2590. [DOI] [PubMed] [Google Scholar]
- Gong S, Fayette N, Heinsbroek JA, Ford CP (2021) Cocaine shifts dopamine D2 receptor sensitivity to gate conditioned behaviors. Neuron 109:3421–3435.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Spicher K, Boulay G, Wang Y, Birnbaumer L (2001) Most central nervous system D2 dopamine receptors are coupled to their effectors by Go. Proc Natl Acad Sci USA 98:3577–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller WC, Trimble J (1985) The gait abnormality of Huntington’s disease. Neurology 35:1450–1454. [DOI] [PubMed] [Google Scholar]
- Lee KWHong JHChoi IYChe YLee JKYang SDSong CWKang HSLee JHNoh JS, et al. (2002) Impaired D2 dopamine receptor function in mice lacking type 5 adenylyl cyclase. J Neurosci 22:7931–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YB, Tewari A, Salameh J, Arystarkhova E, Hampton TG, Brashear A, Ozelius LJ, Khodakhah K, Sweadner KJ (2015) A dystonia-like movement disorder with brain and spinal neuronal defects is caused by mutation of the mouse laminin β1 subunit, Lamb1. eLife 4:e11102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luquin-Piudo MR, Sanz P (2011) Dopamine receptors, motor responses, and dopaminergic agonists. Neurologist 17(6, Suppl 1)S2–S8. [DOI] [PubMed] [Google Scholar]
- Marcott PF, Gong S, Donthamsetti P, Grinnell SG, Nelson MN, Newman AH, Birnbaumer L, Martemyanov KA, Javitch JA, Ford CP (2018) Regional heterogeneity of D2-receptor signaling in the dorsal striatum and nucleus accumbens. Neuron 98:575–587.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mencacci NESteel DMagrinelli FHsu JKeller Sarmiento IJTroncoso Schifferli MMuñoz DStefanis LLubbe SJWood NW, et al. (2021) Childhood-onset chorea caused by a recurrent de novo DRD2 variant. Mov Disord 36:1472–1473. [DOI] [PubMed] [Google Scholar]
- Rasmussen SGF, Jensen AD, Liapakis G, Ghanouni P, Javitch JA, Gether U (1999) Mutation of a highly conserved aspartic acid in the β2 adrenergic receptor: constitutive activation, structural instability, and conformational rearrangement of transmembrane segment 6. Mol Pharmacol 56:175–184. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Contreras D, Condon AF, Buck DC, Asad N, Dore TM, Verbeek DS, Tijssen MAJ, Shinde U, Williams JT, Neve KA (2021) Signaling-biased and constitutively active dopamine D2 receptor variant. ACS Chem Neurosci 12:1873–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira M, Py BF, Bosc C, Laubreton D, Moutin MJ, Marvel J, Flamant F, Markossian S (2018) Electroporation of mice zygotes with dual guide RNA/Cas9 complexes for simple and efficient cloning-free genome editing. Sci Rep 8:474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML (2000) Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques 29:52–, 54.. [DOI] [PubMed] [Google Scholar]
- Vaiman EE, Shnayder NA, Novitsky MA, Dobrodeeva VS, Goncharova PS, Bochanova EN, Sapronova MR, Popova TE, Tappakhov AA, Nasyrova RF (2021) Candidate genes encoding dopamine receptors as predictors of the risk of antipsychotic-induced parkinsonism and tardive dyskinesia in schizophrenic patients. Biomedicines 9:879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Weijden MCMRodriguez-Contreras DDelnooz CCSRobinson BGCondon AFKielhold MLStormezand GNMa KYDufke CWilliams JT, et al. (2021a) A gain-of-function variant in dopamine D2 receptor and progressive chorea and dystonia phenotype. Mov Disord 36:729–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Weijden MCM, Rodriguez-Contreras D, Neve KA, Verbeek DS, Tijssen MAJ (2021b) Reply to: "Childhood Onset Chorea Caused by a Recurrent De Novo DRD2 Variant". Mov Disord 36:1473–1474. [DOI] [PubMed] [Google Scholar]
- Wright DJ, Renoir T, Smith ZM, Frazier AE, Francis PS, Thorburn DR, McGee SL, Hannan AJ, Gray LJ (2015) N-Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl Psychiatry 5:e492. [DOI] [PMC free article] [PubMed] [Google Scholar]