

Abstract
The cancer-testis antigen encoded by the MAGE-1 gene is an attractive antigen in tumor immunotherapy because it can be processed as a foreign antigen by the immune system and generate tumor-specific cellular immune response in vivo. However, increase of the potency of MAGE-1 DNA vaccines is still needed. The high degree of sequence homology and intrinsic immunogenicity of heat shock protein 70 (HSP70) have prompted the suggestion that HSP70 might have immunotherapeutic potential, as HSP70 purified from malignant and virally infected cells can transfer and deliver antigenic peptides to antigen-presenting cells to elicit peptide-specific immunity. In this research, we evaluated the enhancement of linkage of Mycobacterium tuberculosis HSP70 to MAGE-1 gene of the potency of antigen-specific immunity elicited by naked DNA vaccines. We found that vaccines containing MAGE-1-HSP70 fusion genes enhanced the frequency of MAGE-1–specific cytotoxic T cells in contract to vaccines containing the MAGE-1 gene alone. More importantly, the fusion converted a less effective DNA vaccine into one with significant potency against established MAGE-1–expressing tumors. These results indicate that linkage of HSP70 to MAGE-1 gene may greatly enhance the potency of DNA vaccines, and generate specific antitumor immunity against MAGE-1–expressing tumors.
Keywords: Cytotoxic T lymphocyte, ELISpot, Heat shock protein 70, MAGE-1, Vaccine
Introduction
Vaccination has become standard practice for the prevention of various infectious diseases. The possibility of using similar strategies for malignant tumors was suggested nearly a century ago and has been supported by a prolific body of research in animal models. A recent resurgence in cancer vaccine development has been spurred on by a better understanding of how immune responses are induced in both normal and tumor-bearing individuals. The ability to elicit antigen-specific T-cell responses appears to be of central importance in eradicating established tumors in animals and has therefore been the focus for tumor vaccines to activate cellular immune responses more efficiently and specifically in cancer patients. DNA vaccines are a novel and potentially powerful approach to preventing and treating disease. It is well demonstrated that plasmid encoding different antigens of viral, bacterial, parasitic, and tumor origin could provoke immune responses in various species [1, 2].
There is now a consensus that human tumors possess tumor-rejection antigens recognized by autologous cytotoxic T lymphocytes (CTLs), and the generation of specific CTLs against tumors represents the most competent antitumor immune responses of the tumor-bearing host. The MAGE gene family was the first reported example of these tumor-rejection antigen genes, which are expressed in a variety of malignant neoplasms, and in the testis as the only normal tissue [3]. The cancer-testis antigen encoded by MAGE-1 gene can be recognized as a foreign antigen by immune system, and generate antitumoral T-cell responses in cancer patients. In addition, the strict tumor specificity of this antigen ought to ensure the absence of damage on normal tissues following immunization, and the presence on tumors of most histological origins makes it possible to apply the same vaccine to a large number of patients. Following the identification of tumor-rejection antigens, small clinical trials of therapeutic vaccination have been carried out using these antigens, mostly in metastatic melanoma patients. Tumor regressions have been observed in only 5–20% of patients [4–6]. But most of the vaccinated patients do not display regression, a failure that could be due to either an inability of the vaccine to induce an adequate T-cell response or resistance of tumor to immune attack. However, optimal regimes to enhance immunogenicity of DNA immunization remain to be established. One approach to enhance the effectiveness of vaccination is to utilize various genes as molecular adjuvants, such as granulocyte-macrophage colony-stimulating factor (GM-CSF) [7, 8], cytokines [9], and heat shock proteins (HSPs) [10–12]. Recent studies on HSPs revealed its essential role in immunogenicity [13]. HSPs are among the major targets of the immune responses to many bacterial and parasitic pathogens [14–16]. This is perhaps best characterized in the case of mycobacterial infections, in which exposure to Mycobacterium tuberculosis or M. leprae leads to humoral and cellular immune responses to HSP70, HSP60, or other HSPs. In the past few years, studies have demonstrated that immunization with HSP complexes isolated from tumor or virus-infected cells are able to induce potent antitumor [17] or antiviral immunity [18]. Immunogenic HSP-peptide complexes can also be reconstituted in vitro by mixing the peptides with HSPs [19], and HSP-based protein vaccines can also be developed by fusing the antigen to HSPs [20]. Recent researches have demonstrated that linkage of HPV-16 E7 gene to M. tuberculosis HSP70 leads to the enhancement of DNA vaccine potency [10, 11]. Those investigations have made HSP70 attractive for use as an adjuvant in immunotherapy.
In this study, we developed a new DNA vaccine by linking M. tuberculosis HSP70 gene to the gene of cancer-testis antigen MAGE-1 to investigate whether the fusion gene can enhance the potency of DNA vaccines. We compared DNA vaccines containing full-length MAGE-1 gene with DNA containing MAGE-1 fused to M. tuberculosis HSP70 gene for their immune response to MAGE-1–expressing murine tumors and their ability to protect animal against the development of these tumors. We showed that linkage of HSP70 to MAGE-1 dramatically increases expansion and activation of MAGE-1–specific immune responses, which enhance the protective and therapeutic immunity against the MAGE-1–expressing tumor models.
Material and methods
Plasmid DNA constructs and preparation
pcDNA3.1(+) expression vector (Invitrogen, Carlsbad, CA, USA) with the cytomegalovirus (CMV) promoter were the basic plasmids used for the preparation of DNA vaccines. The MAGE-1 gene was amplified by RT-PCR from the total RNA of human hepatocellular carcinoma HepG2 (American Tissue Culture Collection, Rockville, MD, USA) cells. The following primers were used: 5′-GCGGAATTCATGTCTCTTGAGCAG-3′ and 5′-CGGCTCGAGTTAGACTCCCTCTTCCTCCT-3′. The amplified product was digested with EcoRI and XhoI, and cloned into pcDNA3.1(+) downstream of the CMV promoter. The resulting plasmid was named pcDNA3-MAGE-1. The M. tuberculosis HSP70 gene was amplified from the genome DNA of M. tuberculosis H37Rv (provided by Prof. Yuan Li, Fourth Military Medical University, Xi’an, China) by PCR using a pair of primers: 5′-GGCGGATCCATGGCTCGTCGGGTCGGGATC-3′ and 5′-GCGCTCGAGTCACTTGGCCTCCCGGCCGTC-3′. The amplified product was further cloned into the BamHI and XhoI sites of pcDNA3.1(+) vector to construct the plasmid pcDNA3-HSP70. For the generation of MAGE-1 and HSP70 fusion gene, MAGE-1 gene was amplified by PCR using primers designed to generate HindIII and BamHI restriction sites at 5′ and 3′ ends of the amplified fragments, respectively. Then the MAGE-1 gene was subcloned to pcDNA3-HSP70 in the responding restriction site’s 5′-flank to the HSP70 gene. To construct the retroviral vector pLXSN-MAGE-1, MAGE-1 DNA was amplified by PCR, and the fragment was subcloned into the EcoRI and XhoI sites of pLXSN plasmid (Clontech, Palo Alto, CA, USA). The presence of the inserted fragment was confirmed by restriction enzyme digestion and gel electrophoresis. All constructs was confirmed by DNA sequencing.
Plasmid DNA harboring MAGE-1, HSP70, or MAGE-1-HSP70 gene and the “empty” plasmid vector were introduced into E. coli DH5α competent cells. The DNA was prepared using the Maxipreps DNA Purification System (Promega, Madison, WI, USA). DNA concentration was determined by the absorbance at 260 nm (Fig. 1).
Fig. 1.

Plasmid DNA constructions. pcDNA3.1(+) expression vector with the CMV promoter were used as the basic plasmids. Plasmids pcDNA3-MAGE-1 and pcDNA3-HSP70 were constructed by subcloning the MAGE-1 and HSP70 genes into the downstream of CMV promoters, respectively. The MAGE-1 genes were inserted into the 5′ of M. tuberculosis HSP70 gene of pcDNA3-HSP70 plasmid to construct HSP70 and MAGE-1 fusion DNA tumor vaccine
Cell culture
The cultures were free of Mycoplasma and were maintained in complete medium (DMEM; Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum, 2-mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (Life Technologies). A mouse melanoma cell line B16 was used for generation of cells stably expressing human MAGE-1 antigen. To establish the B16-MAGE-1 cell line, recombinant retroviruses were produced by transfecting 2×105 PA317 cells (retroviral packaging cell line) with 10-μg plasmid DNA pLXSN-MAGE-1. Viruses were harvested 2 days later, and the filtered culture supernatant was used to infect the B16 cells in presence of polybrene (8 μg/ml; Sigma, St Louis, MO, USA). Infected cells were grown in a medium containing G418 (600 μg/ml; Sigma) until colonies appeared. The expression of MAGE-1 in B16-MAGE-1 cells was determined by Western blot using the anti-MAGE-1 antibody (MA454; NeoMarkers, Frenmont, CA, USA).
Mice and vaccinations
C57BL/6 mice (6–8 weeks old) were obtained from the Laboratory Animal Center of the Fourth Military Medical University (Xi’an, China). Mice were housed in microisolation in a dedicated, pathogen-free facility, and all animal experimentation was conducted in accordance with animal ethics guidelines.
Groups of six mice were injected intramuscularly (i.m.) with 100 μg / mouse empty pcDNA3.1 plasmids, pcDNA3-MAGE-1, pcDNA3-HSP70, pcDNA3-MAGE-1-HSP70, or 50 μg / mouse pcDNA3-MAGE-1 mixed with 50 μg / mouse pcDNA3-HSP70 in PBS, and mice intramuscularly injected with 100-μl PBS were used as control. A second vaccinatoin was performed i.m. 2 weeks later. The mice were sacrificed 2 weeks after the booster, and splenocytes within groups were pooled.
INF-γ enzyme-linked immunosorbent spot (ELISpot) assays
Mouse IFN-γ ELISpot assay was performed in PVDF-bottomed 96-well plates (Millipore, Bedford, MA, USA) by using a murine IFN-γ ELISpot kit (Diaclone, Besancone, France) according to the manufacturer’s instructions with minor modifications. Briefly, plates were coated overnight at 4°C with anti-IFN-γ capture antibody and washed three times with PBST (PBS + 0.05% Tween 20). Plates were blocked for 2 h with 2% skimmed dry milk. Splenocytes (1×106 cells/well) were then added together with the indicated number of lethally irradiated (10,000 cGy) B16 or B16-MAGE-1 cells (5×04/well respectively) and incubated for 24 h at 37°C. Cells were then removed and a biotinylated IFN-γ detection antibody was added for 2 h. Free antibody was washed out, and the plates were incubated with streptavidin–alkaline phosphatase for 1 h at 37°C, following extensive washing with PBST, and with PBS. Spots were visualized by the addition of the alkaline phosphatase substrate BCIP/NBT. The number of dots in each well was counted by two separate investigators in a blinded manner using a dissecting microscopy. The precursor frequency of MAGE-1–specific T cells was calculated as the number of IFN-γ–secreting cells from splenocytes versus B16-MAGE-1 after subtraction of the background (splenocytes versus B16) per number of splenocytes seeded.
Cytokine detections
Splenocytes (4×106) were harvested 2 weeks after the last vaccination and cocultured with 5×105 irradiated B16-MAGE-1 cells in a total volume of 2 ml of DMEM, supplemented with 10% (v/v) fetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin in 24-well tissue culture plates for 72 h. The supernatants were harvested and assayed for the presence of IL-2 using murine IL-2 ELISA kits (Diaclone, Besancone, France) according to the manufacturer’s protocol. The concentrations of IL-2 in sera of vaccinated mice were calculated by using the curves generated with the standard samples in kits.
Cytotoxicity assays
The CytoTox 96 nonradioactive cytotoxicity assay (Promega, Madison, WI, USA) was performed to measure the cytotoxic activity of the splenocytes in mice vaccinated with various DNA plasmids against B16 and B16-MAGE-1 tumor cells, according to the manufacturer’s protocol with minor modification. Briefly, splenocytes of vaccinated mice were cultured in the presence of human IL-2 (40 U/ml) and irradiated B16-MAGE-1 cells. After 3 days, B16 and B16-MAGE-1 target cells were plated at 1×104 cells/well on 96-well U-bottomed plates (Costar), then the splenocytes (effector cells) were added in a final volume of 100 μl at 1:2.5, 1:5, and 1:10 radios, respectively. The plates were incubated for 45 min in a humidified chamber at 37°C, 5% CO2, and centrifuged at 500 g for 5 min. Aliquots (50 μl) were transferred from all wells to fresh 96-well flat-bottom plates, and an equal volume of reconstituted substrate mix was added to each well. The plates were incubated at room temperature for 30 min and protected from light. Then 50-μl stop solution was added, and the absorbance values were measured at 492 nm. The percentages of cytotoxicity for each effector to target cell ratio was calculated as {[A (experimental) − A (effector spontaneous) − A (target spontaneous)]×100} / [A (target maximum) − A (target spontaneous)]. Percentage of MAGE-1–specific lysis was calculated by subtracting the lysis percentage of splenocytes on B16 from that on B16-MAGE-1 target cells.
Determination of anti-MAGE-1 antibody
The anti-MAGE-1 antibodies in the sera of vaccinated mice were determined by a direct ELISA. Briefly, a 96-well flat-bottom ELISA plate was coated overnight at room temperature with 50 μl of 2.5 μg/ml MAGE-1 protein (amino acid 97–309). The plate was rinsed with PBS, incubated with blocking buffer (5% nonfat dry milk powder and 0.2% Tween 20 in PBS) for 2 h at 37°C, and again rinsed with PBS. Mouse serum samples were 1:50 diluted in blocking buffer, added to the plate, and incubated for 2 h at 37°C. The anti-MAGE-1 sera from mice immunized i.p. at 10-day intervals with recombinant MAGE-1 protein, one in the presence of complete Freund’s adjuvant and three times in the presence of incomplete Freund’s adjuvant, were used as positive controls. After rinsing with PBS, the plate was incubated with horseradish peroxidase–conjugated antimouse IgG (Santa Cruz Biotech, Santa Cruz, CA, USA) for 1 h at 37°C. After extensive washing, 3,3′,5,5′ tetramethyl-benzidine substrate was added, followed by incubation for 20 min at room temperature. The reaction was stopped with 2-M H2SO4, and absorbance was read at 450 nm.
In vivo tumor treatment experiments
Mice (six per group) were s.c. inoculated with lethal doses of B16-MAGE-1 or B16 tumor cells (2×106 cells/mouse, respectively) in the right legs. After 3 days, 100 μg/mouse various plasmids were intramuscularly vaccinated on the left leg, and a boost on day 10. Tumor volumes (length × width2 × π/6) were measured for each individual mouse and were plotted as the mean tumor volume of the group (±standard error, SE) versus days post-tumor challenge. Once tumors became palpable, measurements were taken twice a week. The survival times of mice were recorded, and Kaplan-Meier curves were generated.
In vivo tumor protection experiments
For the tumor protection experiment, mice (five per group) were vaccinated i.m. with 100-μg MAGE-1 DNA, HSP70 DNA, MAGE-1-HSP70 DNA, or the empty plasmid. After 1 week, the mice were boosted with the same regimen as the first vaccination. After 2 weeks, for the last vaccination, mice were s.c. inoculated with 2×105 B16 or B16-MAGE-1 tumor cells per mouse in the right leg and then monitored twice a week.
Results
Vaccination with the MAGE-1-HSP70 DNA enhances MAGE-1–specific T-cell–mediated immune responses
CD8+ CTLs are one of the most crucial components among antitumor effectors [21]. To determine the MAGE-1–specific CD8+ T-cell precursor frequencies generated by MAGE-1-HSP70 fusion DNA vaccines, ELISpot, cytokine assays, and cytotoxicity assays were performed. ELISpot is a sensitive functional assay used to measure INF-γ production at the single cell level. For ELISpot, mice received vaccinations on days 0 and 14. After 2 weeks, splenocytes were harvested and coincubated with irradiated B16-MAGE-1 expressing the human MAGE-1 antigen and B16 cells. Following 24-h coculture, cells were removed and IFN-γ production was determined for each type of DNA vaccinations. The number of MAGE-1–specific T-cell precursors in splenocytes were calculated by subtracting the IFN-γ+ spots of splenocytes on B16-stimulating cells from that on B16-MAGE-1 cells. As shown in Fig. 2, the number of spot-forming T-cell precursors specific for MAGE-1 in the splenocytes from mice vaccinated with MAGE-1-HSP70 fusion DNA was three to four times greater than that from mice with MAGE-1 DNA alone or a mixture of MAGE-1 and HSP70 expressing plasmids. The level of MAGE-1–specific IFN-γ–producing T cells/106 splenocytes derived from mice vaccinated with MAGE-1 DNA alone or MAGE-1 DNA mixed with HSP70 DNA was slightly higher than that from mice vaccinated with pcDNA3, HSP70, or PBS. The results suggest that the MAGE-1-HSP70 fusion DNA can enhance the generation of IFN-γ–producing MAGE-1–specific T-cell precursors in vivo.
Fig. 2.
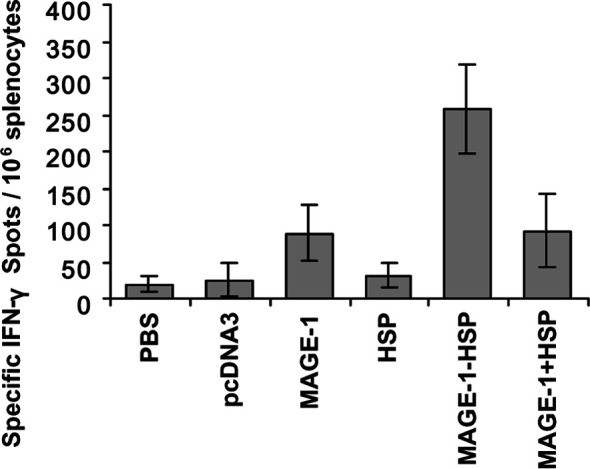
ELISpot assays of MAGE-1–specific T-cell precursors from the splenocytes of vaccinated mice restimulated with irradiated B16-MAGE-1. C57BL/6 mice were intramuscularly vaccinated with empty plasmid (pcDNA3), pcDNA3-MAGE-1 DNA (MAGE-1), pcDNA3-HSP70 DNA (HSP), pcDNA3-MAGE-1-HSP70 fusion DNA (MAGE-1-HSP), or pcDNA3-MAGE-1 DNA mixed with pcDNA3-HSP70 DNA (MAGE-1 + HSP). The control group received only PBS. Vaccinations were performed twice with 100 μg of the above gene construction. The number of INF-γ–producing MAGE-1–specific T-cell precursors was determined by using the ELISpot assay. The spot numbers were the mean ± SE in each group. Statistical analysis by a paired Student’s t-test revealed that mice vaccinated with MAGE-1-HSP70 fusion DNA generated the higher IFN-γ+ spot number than MAGE-1 DNA (p<0.05, n=6), or a mixture of MAGE-1 DNA and HSP70 DNA (p<0.05, n=6)
We then analyzed IL-2 secretion of splenocytes in mice treated with various DNA vaccines by using a sandwich ELISA, to determine the T-cell–mediated antitumor immune responses in vaccinated mice. As shown in Fig. 3, the IL-2 levels in the supernatant of coculture with irradiated B16-MAGE-1 and splenocytes from mice vaccinated with MAGE-1-HSP70 fusion DNA were higher than those from mice vaccinated with other DNA vaccines. The IL-2 concentration in the supernatants from mice vaccinated with MAGE-1 DNA alone was similar to that from mice vaccinated with MAGE-1 mixed with HSP70 DNA, which only showed slightly higher levels above the vector-vaccinated mice or controls (PBS). The results were consistent with the data from ELISpost, suggesting that the MAGE-1-HSP70 can enhance the generation of MAGE-specific T-cell–mediated immune responses.
Fig. 3.

IL-2 secretions of splenocytes restimulated with irradiated B16-MAGE-1 were measured using ELISA. C57BL/6 mice were vaccinated as described in Fig. 1. The splenocytes were pooled 2 weeks after the booster. Splenocytes (4×106) were cocultured with 5×105 irradiated B16-MAGE-1 cells for 3 days in a final volume of 2 ml, and the supernatants were obtained and the concentrations of IL-2 were determined using murine IL-2 ELISA kits. Statistical analysis by a paired Student’s t-test demonstrated that the IL-2 concentration in supernatants of splenocytes from mice vaccinated with MAGE-1-HSP70 fusion DNA was higher than that from mice with MAGE-1 DNA (p<0.05, n=6), or with a mixture of MAGE-1 DNA and HSP70 DNA (p<0.05, n=6)
To determine the MAGE-1–specific lysis on MAGE-1–expressing cells of CTLs induced by vaccination with MAGE-1-HSP70 DNA, the cytotoxicity assay was performed. The splenocytes were pooled 2 weeks after booster with various DNA or PBS, and the cytotoxicity on MAGE-1–expressing tumor cells was measured after coculture of the splenocytes with irradiated B16-MAGE-1 cells for 3 days. Percentage of MAGE-1–specific lysis on MAGE-1–expressing tumor cells was calculated by subtracting the lysis percentage of splenocytes on B16 from that on B16-MAGE-1 target cells. The MAGE-1–specific lysis of CTLs from mice vaccinated with MAGE-1-HSP70 DNA was greater than that from mice vaccinated with other DNA or PBS, whereas mice vaccinated with MAGE-1 DNA or MAGE-1 mixed with HSP70 DNA showed slightly higher levels above the vector-vaccinated mice or controls (PBS). Using these three methods, we have demonstrated that MAGE-1-HSP70 DNA vaccination enhanced the MAGE-1–specific T-cell–mediated immune response in vivo (Fig. 4).
Fig. 4.

MAGE-1–specific lysis against B16-MAGE-1 cells by CTLs induced by vaccination with various DNA. Mice were vaccinated as described in the Fig. 1. The splenocytes of mice vaccinated with various DNA were harvested and restimulated with irradiated B16-MAGE-1 cells for 3 days. The percentage of specific lysis of splenocytes on B16-MAGE-1 target cells was determined by a nonradioactive cytotoxicity assay. Percentage of MAGE-1–specific lysis was calculated by subtracting the percentage lysis of splenocytes on B16 from that on B16-MAGE-1 target cells. The MAGE-1–specific lysis of splenocytes from mice vaccinated with MAGE-1-HSP70 fusion DNA showed a higher level than the other group
MAGE-1-HSP70 fusion DNA vaccine does not generate the MAGE-1–specific antibody
The quantity of anti-MAGE-1 antibodies in the sera of the vaccinated mice was determined by a direct ELISA 2 weeks after the last vaccination. No anti-MAGE-1 antibodies could be detected in the sera of mice of any vaccinated group (Fig. 5). The anti-MAGE-1 sera from mice vaccinated with MAGE-1 protein were used as positive controls. The results show that the MAGE-1-HSP70 DNA does not generate MAGE-1–specific humoral immune responses.
Fig. 5.
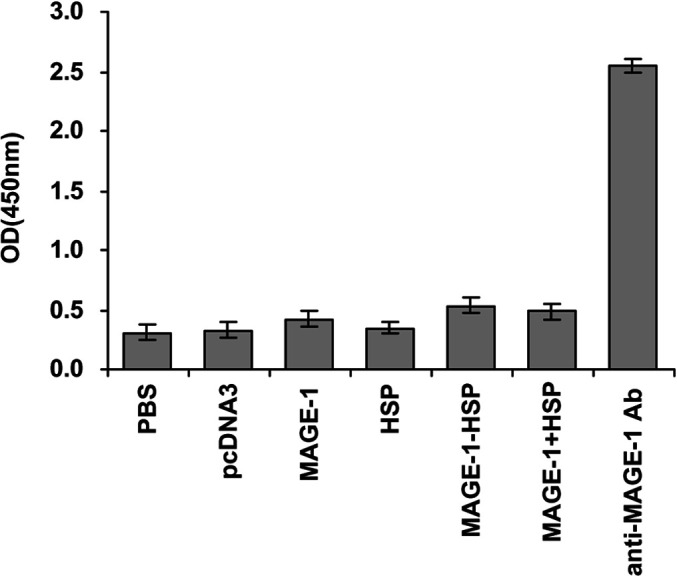
MAGE-1–specifc antibody titers in C57BL/6 mice vaccinated with various recombinant DNA vaccines. C57BL/6 mice were vaccinated as described in Fig. 1. Serum samples were obtained from vaccinated mice 2 weeks after the boost vaccination. The presence of MAGE-1–specific antibody was examined by ELISA. The anti-MAGE-1 sera from mice vaccinated with MAGE-1 protein were used as positive controls. The results of the 1:50 dilution are presented showing the mean absorbance (A 450nm)±SE. No anti-MAGE-1 antibodies could be detected in the sera of mice of any vaccinated group
Vaccination with MAGE-1-HSP70 fusion DNA delays tumor growth and prolongs survival of mice challenged with B16-MAGE-1 tumor cells
We had found that MAGE-1-HSP70 DNA vaccinations could generate the CTLs against the MAGE-1–expressing tumor cell B16-MAGE-1. We sought to test the efficacy of MAGE-1-HSP70 fusion DNA vaccines against the MAGE-1–expressing tumor model in order to demonstrate a relevant and universal clinical value. In these experiments, lethal doses (1×106 cells/mouse) of B16-MAGE-1 melanoma cells were inoculated in mice s.c. on day 0, and 100 μg of various DNA vaccines was administered on day 3, followed by a booster on day 10. As demonstrated in Fig. 6a, vaccination with MAGE-1-HSP70 fusion DNA significantly delayed tumor growth in the B16-MAGE-1 tumor model compared with vaccination with PBS and other plasmids. Survival of mice with a preexisting B16-MAGE-1 tumor model was also significantly prolonged in the mice treated with MAGE-1-HSP70 fusion DNA vaccine, as shown in the Kaplan-Meier plots of Fig. 6c.
Fig. 6a–d.

The immunotherapy of preestablished B16-MAGE-1 melanoma (a and c) and B16 melanoma (b and d) with DNA vaccination. Groups of mice were s.c. inoculated with a lethal dose of B16-MAGE-1 and B16 tumor cells on day 0 (1×106 cells/mouse, respectively). Mice were immunized with 100 μg of various DNA vaccines (or PBS) on days 3 and 10. Tumor growth kinetics (a and b) were followed over time by caliper measurements, and mean tumor volumes (in mm3) were calculated. Error bars depict SE (n=6 mice/group). For a, MAGE-1-HSP70 fusion DNA vaccines give in statistically significantly better results than all other vaccines from day 24 (p<0.05, n=6), and MAGE-1 DNA and a mixture of MAGE-1 DNA and HSP70 DNA were significantly better than PBS on day 28 (p<0.05, n=6). For b, the difference between MAGE-1-HSP70 versus B16 and MAGE-1-HSP70 versus B16-MAGE-1 was significant from day 21 (p<0.05, n=6). Kaplan-Meier curves (c and d) were generated from survival data (n=6 mice/group). For c, MAGE-1-HSP70 vaccination versus PBS mock vaccination (p<0.01); versus MAGE-1 (p<0.05); versus HSP70 (p<0.05); versus MAGE-1 mixed with HSP70 (p<0.05; while all other comparisons were n.s. Median survival times for each treatment group: PBS, 36 days; pcDNA3, 38 days; MAGE-1, 47 days; HSP70, 41 days; MAGE-1-HSP70, 60 days; MAGE-1 mixed with HSP70, 44 days. For d, MAGE-1-HSP70 vaccination against B16-MAGE-1 tumor cells was significantly better than against B16 (p<0.01). Median survival times were 40 days for MAGE-1-HSP70 against B16, and 36 days for PBS against B16
To test whether or not MAGE-1 is the main contributor to the antitumor response, we inoculated mice with either B16 parental tumor cells on day 0, and vaccinated those mice with MAGE-1-HSP70 fusion DNA plasmid on days 3 and 10. As shown in Fig. 4b, d, the MAGE-1-HSP70 fusion DNA vaccination had minimal effect in retarding the tumor growth and prolonging the survival times in the B16 tumor model group. The results demonstrated that the MAGE-1 substantially contributed to the main antitumor immune response, and MAGE-1-HSP70 was a potent therapeutic vaccine against MAGE-1–expressing tumor cells.
MAGE-1-HSP70 is a potent protective vaccine against MAGE-1–expressing tumors
To determine the protection effect of MAGE-1-HSP70 fusion DNA vaccines, vaccinated mice were challenged with 1×105 B16-MAGE-1 or B16 melanoma cells. Groups of mice were vaccinated with various DNA or PBS 3 weeks and 1 week before the tumor challenged. Mice were monitored for evidence of tumor growth by palpation and inspection twice a week. As shown in Fig. 7, all of the mice vaccinated with pcDNA3 DNA, HSP70 DNA, or PBS grew tumors within 21 days. Four out of five mice vaccinated with MAGE-1 DNA or MAGE-1 DNA mixed with HSP70 DNA grew tumors. In contrast, all of mice vaccinated with MAGE-1-HSP70 fusion DNA remained tumor-free 50 days after challenge with tumor cells. To determine whether MAGE-1 was the main contributor in the protective effect, the mice were s.c. challenged with the B16 melanoma cells after vaccinated with MAGE-1-HSP70 fusion DNA. Within 17 days, five mice except one grew tumors. These results suggested that MAGE-1-HSP70 fusion DNA vaccine enhanced the MAGE-1–specific cellular immune response, and generated the protective effect against MAGE-1–expressing tumors. However, the results also show that a mixture of MAGE-1 and HSP70 genes was relatively ineffective in generating the protective effect.
Fig. 7a, b.
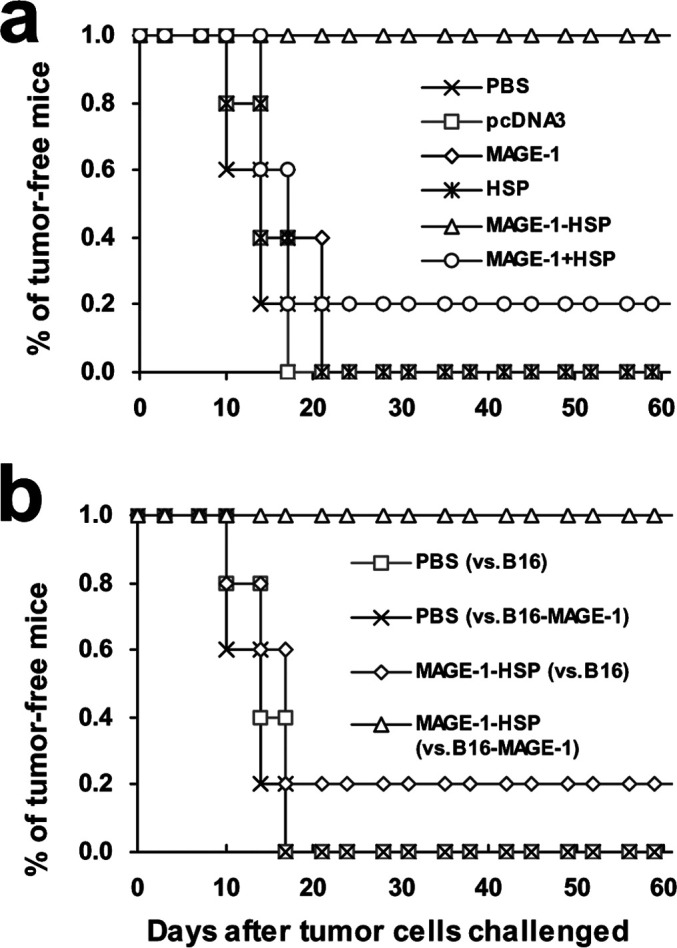
Vaccination with MAGE-1-HSP70 DNA protects mice against the growth of MAGE-1–expressing B16 tumor cells. C57BL/6 mice were immunized with empty plasmid pcDNA3, MAGE-1 DNA, HSP70 DNA, MAGE-1-HSP70 DNA and MAGE-1 mixed with HSP70 DNA, or saline (PBS), 21 and 7 days before challenge with melanoma cells. Mice were inoculated with 2×105 B16-MAGE-1 or B16 melanoma cells per mouse subcutaneously in the right leg. The mice were monitored for evidence of tumor growth by palpation and inspection twice a week. a B16-MAGE-1 melanoma cells were s.c. inoculated. All mice vaccinated with pcDNA3, HSP70, or PBS grew tumors within 21 days. Four out of five mice vaccinated with MAGE-1 or MAGE-1 mixed with HSP70 grew tumors. In contrast, all of mice vaccinated with MAGE-1-HSP70 fusion DNA remained tumor-free 50 days after the tumor challenged. b Mice vaccinated with PBS or MAGE-1-HSP70 fusion DNA were challenged with B16 melanoma cells on day 0, and four of five mice treated with MAGE-1-HSP70 fusion DNA and all five mice treated with PBS grew tumors within 17 days
Discussion
In this study, we demonstrated that M. tuberculosis HSP70 could dramatically enhance the potency of MAGE-1–expressing DNA vaccines. DNA vaccines with HSP70 fused to MAGE-1 elicited strong MAGE-1–specific cellular immunity and generated significant T-cell–dependent preventive and therapeutic effects against MAGE-1–expressing murine tumors.
Tumor cells express defined antigens that can be recognized by tumor-destroying (CD8+) CTLs. Recently, several genes or gene families encoding tumor-associated antigens have been isolated by analyzing autologous serologic or cytotoxic T-cell responses [3, 22, 23]. Cancer-testis antigens, a subgroup of these tumor-associated antigens, are expressed in a variety of malignant neoplasms, and in the testis as the only normal tissue [3]. The MAGE gene family was the first cancer-testis antigen isolated, and 17 different MAGE-encoding genes have now been identified [24], of which MAGE-1 ought to be strictly tumor-specific because the only normal cell type that expresses the encoding genes, the spermatogonia, does not bear HLA molecules on its surface. The antigen encoded by the MAGE-1 gene can be recognized and processed as a foreign antigen by the immune system, and the MAGE-1–specific immune response can be generated in vivo. For these reasons, MAGE-1 can be used as an attractive gene for tumor immunotherapy. It will be important to increase the therapeutic potential in favoring T-cell survival and specificity since clinical responsiveness will depend on the enhancement of T-cell immunity [25].
Nucleic acid vaccines represent an attractive approach to generating antigen-specific immunity because of their stability and simplicity of delivery. However, there is a need to find effective tumor-specific antigens and to increase the potency of DNA vaccines. As most hosts obviously do not mount efficient T-cell responses against their tumors, the task is clear: immunotherapies must induce effective cancer-destroying T cells in patients [26]. HSPs are among the most highly conserved molecules of the biosphere. They are widely found in eukaryotes and in prokaryotes, and their functions are essential for life since they serve as molecular chaperones [27]. Under normal conditions, they help to achieve suitable protein folding and subunit assembly. In stress situations, HSP synthesis is increased to protect cells; they prevent the aggregation of partially denatured proteins and initiate their refolding or their proteolytic degradation if the unfolding is irreversible. HSPs are very important in the control of protective immunity [28]. Thanks to their chaperone function they participate in the assembly of antibody molecules, and they are involved in the stabilization of MHC class I and class II molecules and they can stimulate the synthesis of cytokines [29]. It has been even proposed to name one of them “chaperokine” [30]. Expression of HSP70 influences the presentation and processing of antigen to CD4+ T cells by antigen-presenting cells (APCs) [31], and increasing the expression of HSP70 in tumor cells promotes the presentation of peptides on the tumor cell surface via an enhancement of MHC class I expression [32]. HSP70 expression in tumor cells also enhances their recognition by T cells via pathways other than MHC up-regulation, such as by influencing antigen processing [33]. As intracellular chaperones, HSPs bind a large number of peptides derived from the cells from which they are isolated [34, 35], the so-called antigenic fingerprint of that cell [36]. HSPs such as HSP70, HSP90, gp96, calreticulin, HSP110, and grp170 can act as carriers of protein antigens from the cell from which they are derived. Studies have demonstrated that immunization of animals with these HSPs purified from tumor cells elicits effective antitumor immunity and protects the animals from subsequent challenge with the same, but not different tumor cells [37–39]. Such an approach can also be used to generate effective immunity against established tumors [17]. It is now known that immunization leads to the generation of tumor-specific CTLs, the specificity of which is defined by the peptides associated with the HSP rather than by the HSP itself [40].
Evidences suggest that HSP70 may work through its effects on APCs, which play a key role in induction of immune response. The implication of the original studies was that HSP immunization was able to generate antitumor CTLs by transferring the antigenic peptides with which they were associated into the MHC class I antigen presentation pathway within APCs, a pathway usually restricted to antigens derived from the cytoplasm of APCs. Such antigen cross-presentation is now considered to be a key process in the action of HSPs [41]. For DNA vaccinations, APCs can directly uptake the DNA vaccine and present the expressed antigen. Alternatively, the DNA vaccine is expressed by somatic cells, and the released antigen is picked up by APCs. Dendritic cells are considered one of the major APCs involved in DNA vaccines [42].
The identification of HSP receptors on professional APCs explains, at least in part, the ability of HSP-containing formulations (such as HSP fusions) to induce significant immune response to associated antigens. Recently, a specific receptor involved in the capacity of HSP70 to induce peptide-specific immunity has been identified as the α2-microglobulin receptor CD91 [43]. CD91 is expressed by many APCs, including dendritic cells, and it is also a receptor for the other HSPs (calreticulin, HSP90, pg96) that have the capacity to induce tumor-specific immunity by similar approaches. Another molecule also involved in the delivery of HSP-peptide complexes to APCs is CD40 [39, 44]. Scavenger receptors have also been implicated in HSP uptake an cross-presentation, as the receptor for oxidized low density lipoprotein (LDL) and poly(I), namely LOX-1, has been identified as a novel HSP receptor [45].
In this research, we chose the M. tuberculosis HSP70 gene as an enhancer for the construction of a MAGE-1 gene–based DNA vaccine. The observation that the fusion of HSP70 to MAGE-1 enhances MAGE-1–specific CTL-mediated immune responses and antitumor effect is consistent with previous reports [10]. Compared with animals receiving a vector containing the E7 gene alone, E7-HSP70 fusion vector immunized mice exhibited significantly higher levels of IFN-γ secreting and increased numbers of E7 peptide–specific CD8+ T CTLs. In contrast, there was no significant enhancement of IFN-γ–secreting CD4+ T cells or production of anti-E7 antibody in animals receiving the E7-HSP70 vector compared with the E7 vector alone. Immunization with the E7-HSP70 vector contributed to superior protection against challenge with E7-expressing tumor cells.
Although the MAGE-1-HSP70 fusion DNA vaccine holds promise for mass immunization, two safety issues need to be resolved. First, the plasmid DNA may integrate into the host genome, resulting in the inactivation of tumor suppresser genes or the activation of oncogenes, which may lead to a malignant transformation of the host cell. Fortunately, it is estimated that the frequency of integration is much lower than that of spontaneous mutation, and integration should not pose any real risk [42]. The second issue concerns potential risks associated with the presence of MAGE-1 protein in host cells. The MAGE-1 gene encodes a cancer-testis antigen, which is silent in the normal tissues except in testis and in placenta. The expression of MAGE-1 genes in testis is restricted to the germline cells. Because male germline cells lack expression of the MHC molecules, they are not expected to present MAGE-encoded antigens. With regard to the MAGE-1 and HSP70 fusion DNA vaccines, the potential usefulness of the antigens encoded by the MAGE genes largely rests on the tumor-specific expression of these genes.
In summary, our results indicate that the fusion of HSP70 gene to the MAGE-1 gene can generate stronger MAGE-1–specific CTL-mediated immune responses and antitumor effects against MAGE-1–expressing murine tumors than those generated by MAGE-1 DNA vaccines. The present study holds out great promise for the generation of enhanced DNA vaccines by fusing the gene of HSP70 to those of tumor-specific antigens, e.g., MAGE-1.
Acknowledgements
We think Prof. An-Gang Yang for critical review of the manuscript. This work was supported in part by the National Natural Science Foundation and Military Foundation for Medicine and Health of China.
References
- 1.Weiss Infect Immun. 2000;68:5914. doi: 10.1128/IAI.68.10.5914-5919.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Donnelly Annu Rev Immunol. 1997;15:617. doi: 10.1146/annurev.immunol.15.1.617. [DOI] [PubMed] [Google Scholar]
- 3.Van Curr Opin Immunol. 1997;9:684. doi: 10.1016/s0952-7915(97)80050-7. [DOI] [PubMed] [Google Scholar]
- 4.Banchereau Cancer Res. 2001;61:6451. [PubMed] [Google Scholar]
- 5.Gajewski Clin Cancer Res. 2001;7:895s. [PubMed] [Google Scholar]
- 6.Marchand Eur J Cancer. 2003;39:70. doi: 10.1016/S0959-8049(02)00479-3. [DOI] [PubMed] [Google Scholar]
- 7.Bueler Mol Med. 1996;2:545. [Google Scholar]
- 8.Sun Vaccine. 2002;20:1466. doi: 10.1016/S0264-410X(01)00476-5. [DOI] [PubMed] [Google Scholar]
- 9.Kim J Interferon Cytokine Res. 2000;20:487. doi: 10.1089/10799900050023906. [DOI] [PubMed] [Google Scholar]
- 10.Chen Cancer Res. 2000;60:1035. [Google Scholar]
- 11.Hsu Gene Ther. 2001;8:376. doi: 10.1038/sj.gt.3301408. [DOI] [PubMed] [Google Scholar]
- 12.Huang J Exp Med. 2000;191:403. doi: 10.1084/jem.191.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Todryk Immunology. 2003;110:1. doi: 10.1046/j.1365-2567.2003.01725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaufmann Immunol Today. 1990;11:129. doi: 10.1016/0167-5699(90)90050-j. [DOI] [PubMed] [Google Scholar]
- 15.Murray J Bacteriol. 1992;174:4193. doi: 10.1128/jb.174.13.4193-4196.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young Annu Rev Immunol. 1990;8:401. doi: 10.1146/annurev.iy.08.040190.002153. [DOI] [PubMed] [Google Scholar]
- 17.Tamura Science. 1997;278:117. doi: 10.1126/science.278.5335.117. [DOI] [PubMed] [Google Scholar]
- 18.Heikema Immunol Lett. 1997;57:69. doi: 10.1016/S0165-2478(97)00048-5. [DOI] [PubMed] [Google Scholar]
- 19.Blachere J Exp Med. 1997;186:1315. doi: 10.1084/jem.186.8.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzue J Immunol. 1996;156:873. [PubMed] [Google Scholar]
- 21.Melief Immunol Rev. 1995;145:167. doi: 10.1111/j.1600-065x.1995.tb00081.x. [DOI] [PubMed] [Google Scholar]
- 22.Boon Curr Opin Immunol. 1997;9:681. doi: 10.1016/S0952-7915(97)80049-0. [DOI] [PubMed] [Google Scholar]
- 23.Cheville Mod Pathol. 1999;12:974. [PubMed] [Google Scholar]
- 24.De Immunogenetics. 1994;40:360. doi: 10.1007/BF01246677. [DOI] [PubMed] [Google Scholar]
- 25.Offringa Curr Opin Immunol. 2000;12:576. doi: 10.1016/s0952-7915(00)00145-x. [DOI] [PubMed] [Google Scholar]
- 26.Gouttefangeas Nat Biotechnol. 2000;18:491. doi: 10.1038/75343. [DOI] [PubMed] [Google Scholar]
- 27.Smith Pharmacol Rev. 1998;50:493. [PubMed] [Google Scholar]
- 28.Melnick Immunol Today. 1995;16:243. doi: 10.1016/0167-5699(95)80167-7. [DOI] [PubMed] [Google Scholar]
- 29.More Int Immunol. 2001;13:1121. doi: 10.1093/intimm/13.9.1121. [DOI] [PubMed] [Google Scholar]
- 30.Asea Cell Stress Chaperones. 2000;5:425. doi: 10.1379/1466-1268(2000)005<0425:hpbapn>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panjwani J Immunol. 1993;163:1936. [PubMed] [Google Scholar]
- 32.Wells Int Immunol. 1998;10:609. doi: 10.1093/intimm/10.5.609. [DOI] [PubMed] [Google Scholar]
- 33.Dressel Eur J Immunol. 1999;29:3925. doi: 10.1002/(SICI)1521-4141(199912)29:12<3925::AID-IMMU3925>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 34.Gething Nature. 1992;355:33. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- 35.Young Philos Trans R Soc Lond B Biol Sci. 1993;339:363. doi: 10.1098/rstb.1993.0035. [DOI] [PubMed] [Google Scholar]
- 36.Srivastava Curr Opin Immunol. 1994;6:728. doi: 10.1016/0952-7915(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 37.Basu J Exp Med. 1999;189:797. doi: 10.1084/jem.189.5.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chandawarkar J Exp Med. 1999;189:1437. doi: 10.1084/jem.189.9.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Immunity. 2001;15:971. doi: 10.1016/S1074-7613(01)00242-4. [DOI] [PubMed] [Google Scholar]
- 40.Schild Curr Opin Immunol. 1999;11:109. doi: 10.1016/s0952-7915(99)80019-3. [DOI] [PubMed] [Google Scholar]
- 41.Colaco Cell Mol Biol. 1998;44:883. [PubMed] [Google Scholar]
- 42.Gurunathan Annu Rev Immunol. 2000;18:927. doi: 10.1146/annurev.immunol.18.1.927. [DOI] [PubMed] [Google Scholar]
- 43.Basu Immunity. 2001;14:303. [Google Scholar]
- 44.Becker J Cell Biol. 2002;158:1277. doi: 10.1083/jcb.200208083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Delneste Immunity. 2002;17:353. [Google Scholar]