

Abstract
The major barrier for xenotransplantation in humans is the presence of α(1–3) Galactosyl epitopes (αGal) in xenogeneic tissue and the vast quantities of natural antibodies (Ab) produced by humans against this epitope. The binding of anti-αGal Ab to cells expressing αGal triggers a complement-mediated hyperacute rejection of target cells. The hyperacute rejection of whole cancer cells, modified to express αGal epitopes, could be exploited as a new cancer vaccine to treat human cancers. We tested this hypothesis in αGalactosyltransferase knockout (αGT KO) mice which, like humans, do not express αGal on their cell surfaces and can produce anti-αGal Ab. Forty-five percent of mice with preexisting anti-αGal Ab rejected αGal positive melanoma cells (B16αGal). These mice remained tumor-free for more than 90 days. The majority of control mice injected with B16Null, αGal negative cells succumbed to melanoma. The rejection of B16αGal induced strong long-lasting antitumor immunity against B16Null measured by the expansion of cytotoxic T lymphocytes. In addition, mice rejecting B16αGal were protected against melanoma since they survived a second rechallenge with B16Null. Protected mice developed antitumor immunity in the absence of autoimmune depigmentation (vitiligo). These results show that rejection of αGal positive melanoma cells can efficiently boost the immune response to other tumor associated antigens present in αGal negative melanoma cells. This study supports the concept of a novel anticancer vaccine to treat human malignancies.
Keywords: Cancer vaccine; Alpha(1, 3)Galactosyl epitope; Adjuvant; Xenotransplant; Melanoma
Introduction
Analysis of αGal epitope expression and of N-acetyllactosaminide 3-α-galactosyltransferase (αGT, EC 2.4.1.87) activity revealed that the enzyme and its product are abundant in nonprimate mammals, prosimians and New World monkeys [9]. In contrast, nonmammalian vertebrates, Old World monkeys, apes and humans lack αGT activity and the expression of αGal epitopes. In humans, lack of αGT expression is due to the presence of two base pair frameshift mutations and three nonsense mutations within this disrupted reading frame that truncated the human polypeptide inactivating the gene [19]. Whereas humans, due to lack of αGT activity and expression of αGal epitopes, produce large amounts of natural antibodies (Ab) against αGal epitopes as a result of continuous antigenic stimulation by gastrointestinal bacteria that express this epitope [8, 13]. These anti-αGal Ab constitute as much as 1% of total circulating antibodies and represent a major barrier for the xenotransplantation of nonprimate mammalian organs (i.e., pigs) into humans due to the hyperacute organ rejection reaction [6].
The hyperacute rejection of a xenotransplant is characterized by a rapid and acute organ destruction occurring within minutes to hours after transplantation. This reaction is mediated by the binding of anti-αGal Ab from the recipient to αGal epitopes expressed on the xenograft, and complement activation through the classic pathway [16]. Also, noncomplement fixing natural anti-αGal Ab induce Ab-dependent cell-mediated cytotoxicity (ADCC) that initiates tissue damage in xenotransplants by NK cells-mediated mechanism [1, 30, 37, 38].
Our laboratory and others have proposed that human cancer cells modified to express αGal epitopes could be used as a new strategy to generate an anticancer vaccine [5, 7, 23, 28].
This hypothesis was studied using the αGT KO mouse strain [32]. These mice provide an ideal small-animal model to study in vivo the immune response against αGal epitopes, because cells from αGT KO mice, like humans, do not express detectable αGal epitopes on their surface. The αGT KO mice can produce low titers of natural anti-αGal Ab [33]. However, high titers of anti-αGal Ab are produced after immunization with rabbit red blood cell (RRBC) membranes [21, 34] and by oral immunizations with bacteria [27].
Data from our laboratory [34] and others [20] demonstrates that immunization with tumor cells expressing the αGal epitope provides protection in mice challenged with tumor cells that do not express the αGT gene, while no protection was observed after immunization with the αGal negative cells. However, convincing evidence of T-cell mediated immunity induced after rejection of αGal expressing cells recognizing αGal negative target cells has not yet been demonstrated.
In the present report, we show that the presentation of αGal epitopes on cancer cell glycoproteins and glycolipids leads to their direct in vivo hyperacute rejection by anti-αGal Ab and complement activation in presensitized subjects. This reaction induces a strong immune response with specificity extended to other tumor associated antigens present in the native αGal negative tumor, thus conferring complete protection against lethal challenge.
Materials and methods
Cell lines
The following tumor cell lines derived from C57Bl/6 mice were used in this study: B16Bl6 melanoma cell line is a metastatic derivative of the B16.F10 cell line. B16Bl6 cells lack the expression of αGal epitopes due to downregulation of the αGT gene expression [10, 18]. EL-4 (lymphoma, ATCC number TIB-39), MC38 (colon carcinoma), LLC (Lewis lung carcinoma, ATTC number CRL-1642) and GK1.5 (hybridoma ATTC number TIB-207) cells are αGT positive mouse lines.
CA320M is a colon carcinoma, chemically induced in αGT KO mouse, generated and generously provided by Daniel Hellrung from the Iowa Cancer Research Foundation. S91M3 (ATCC number CCL-53.1) is a melanoma cell line lacking the expression of αGT gene [39].
All cells were maintained at 37°C in a 5% CO2 incubator in complete medium consisting of Dulbecco’s Modified Eagles Medium (DMEM) supplemented with 10% FBS, L glutamine, sodium pyruvate, penicillin, streptomycin, HEPES, 2-ME, nonessential amino acids (CM).
B16Bl6 cells were transduced with the retroviral vector pLNCKG encoding αGT under the control of the CMV promoter (LTR-NeoR -CMV-αGT-LTR) or pLNL (LTR-NeoR -LTR) in the presence of protamine sulfate 10 μg/ml. The pLNCKG vector encoding αGT with the N-terminus Golgi localization signal was constructed and kindly provided by Dr. Won Bin Young from NewLink Genetics Corp. After transduction, cells were selected in 1 mg/ml G418 (Geneticin, Invitrogen) for 15 days. The expression of αGT in Neomycin-resistant clones was determined by fluorescent activated cell sorting (FACS.)
Animal treatments
Females and males 8–14-week-old αGT KO mice were used in this study. Several founders of the original colony αGT KO mice were purchased from Dr. J.B. Lowe (University of Michigan) [32]. The original αGT KO mouse expressed both alleles H-2 b and d haplotypes. These animals were generated by crossing C57BL/6×DBA/2J×129sv mice (H-2b × H-2rmd). In an effort to obtain homozygous colonies, by breeding and selection, we generated two αGT KO mouse colonies homozygous for both H-2 b/b and H-2 d/d haplotypes. Animals used in this study express H-2 Kb (Fig. 1) and H-2Db haplotypes (not shown) and they do not express H-2Dd (Fig. 1) or H-2Kd (not shown).
Fig. 1.

a Expression of αGal epitopes in mouse and human cells. Cells were stained with IB4-FITC lectin and analyzed by FACS. LLC (mouse lung carcinoma), GK1.5 (mouse hybridoma), B16F0 (mouse melanoma), B16Bl6 (metastasis derived from B16F10 melanoma, referred in this manuscript as B16Null), B16NeoR (B16Bl6 cells transduced wit control vector encoding Neomycin Resistance gene), peripheral blood lymphocytes from αGT KO mouse (αGT KO PBL), B16αGal (B16Bl6 cells stably transduced with retroviral vector pLNCKG encoding NeoR and αGT genes), A549αGal cells (A549 human lung carcinoma cells transduced with pLNCKG vector), CA320M (carcinoma cell line chemically induced in aGT KO mouse), S91M3 (mouse melanoma), A549 and human PBL. b Complement-mediated lysis of αGal expressing cells. Mouse cell lines were incubated with a 1:10 dilution of heat inactivated mouse pooled serum containing high titer of anti-αGal Ab in Opti-MEM media. Rabbit complement (1:2 dilution of reconstituted commercial product) was added to indicated wells. After 30 min cells were washed and stained with IB4-FITC and 7-AAD to discriminate between live and dead cells. The percentage of dead cells (7-AAD+ cells) was determined in gated αGal-expressing cells by FACS. c Mice used in this study are homozygous for the H-2Kb haplotype. Peripheral blood lymphocytes were double stained with anti-H-2Kb FITC and anti-H-2Dd PE Ab. Panels show stained lymphocytes obtained from αGT KO H-2Kb homozygous haplotype mice (Upper Left); from C57Bl/6 mice H-2Kb haplotype (Upper Right) and from homozygous H-2Dd haplotype mice (Lower Right). The lower left panel shows background staining with PE and FITC isotypes control Ab (negative control)
Mice were immunized i.p. with 1×108 RRBC twice, 2 or 3 weeks apart to increase the anti-αGal antibody titers as previously described [34]. All mice developed high titers of anti-αGal Ab measured by ELISA (not shown). Five days after the last immunization, mice received a lethal subcutaneous (s.c.) challenge of 1×105 of either one of these three cell lines: (a) wild type B16Bl6 (H-2 b/b) melanoma cell line (referred to in this manuscript as “B16Null‘’, αGal negative), (b) B16Bl6 cell transduced with a retroviral vector expressing NeoR gene (referred to as B16NeoR, mock control αGal negative) or (c) B16Bl6 cells transduced with a retroviral vector expressing both NeoR gene and αGT (referred to as B16αGal).
For the preventive vaccination protocol, mice were vaccinated subcutaneously with three doses of 5×105 irradiated whole cell vaccines (B16Null, B16NeoR or B16αGal) 2 weeks apart and subsequently challenged with1×105 B16Null. Developing tumors were measured twice weekly in a blinded manner.
To establish pulmonary melanoma tumors, animals were intravenously injected with 1×105 B16Null cells. Four weeks after metastasis implantation, mice were euthanized and lung burden was determined by counting tumors in a blinded manner. To ensure minimal distress, pain or discomfort, mice were humanely euthanized according to the Institutional Animal Care and Use Committee (IACUC) approved protocol when their tumors reached 1,000 mm3 or before if tumors became hemorrhagic.
Staining for detection of αGal epitopes and fluorescence activated cell sorting (FACS)
To detect the expression of αGal epitopes, cells were stained as previously described [34]. Briefly, the fluorescein isothiocyanate (FITC)-labeled Griffonia simplicifolia isolectin B4 (IB4, Vector Laboratories Inc., Burlingame, CA, USA), was diluted 1:50 in Opti-MEM (Invitrogen-Life Technologies, Carlsbad, CA, USA) and added to the cells. Cells were stained for 30 min, washed and analyzed by FACS. This lectin has previously been shown to bind specifically to αGal epitopes [31]. For haplotype determination, the following antibodies were used, all purchased from BD Pharmingen: PE-labeled antimouse H-2Db (clone KH95), FITC-labeled antimouse H-2Kd (clone SF1-1.1), PE-labeled antimouse H-2Dd (clone 34-2-12) and FITC-labeled antimouse H-2Kb (clone AF6-88.5).
Complement-mediated lysis of αGal expressing cells
Mouse cell lines were incubated with heat inactivated mouse pooled serum containing high titer of anti-αGal Ab (titer IgG greater than 1: 200 and IgM greater than 1:1,000) in presence or absence of rabbit complement. Pooled mouse serum was obtained from 20 RRBC immunized αGT KO mice. After 30 min of incubation, cells were washed and stained with IB4-FITC and 7-AAD (BD Pharmingen). The percentage of dead cells (7-AAD+ cells) was determined in gated αGal expressing cells by FACS.
Generation of melanoma specific cytotoxic T lymphocytes (CTL)
Spleens from two mice protected from melanoma were pooled and single-cell suspensions of total mononuclear cells were used for the generation of melanoma specific effector cells as described [15]. Spleen mononuclear cells from naïve mice were used as effector control cells. One million splenocytes were cultured in the presence or absence of 1×105 irradiated B16Null cells for 5 days in the absence of IL-2 to generate effector cells. After culture, resultant effector cells (CTL) were harvested and tested for specific cytotoxicity with different target cells. B16Null, MC38 and EL-4 cells were used as target cells at two different effector target ratios. Cytotoxicity was measured after 4 h of culturing CTL with specific and nonspecific targets measuring LDH release in the culture supernatant (Promega, Madison, WI, USA). Specific cytotoxicity was calculated using the following formula for each value and the mean and SEM of triplicates was subsequently calculated:
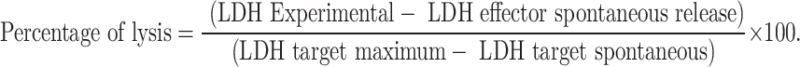 |
Statistical analysis
The software GraphPad Prism was used for the statistical analysis. Kaplan-Meier survival analysis and logrank test were used for curves comparison. ANOVA or t-test were used when appropriate.
Results
Transduction of B16Bl6 melanoma cells with retroviral vectors
All mouse cell lines tested in our laboratory expressed αGal epitopes except for melanoma cell lines. Figure 1a shows the expression of αGal epitopes on the cell surface of LLC (mouse lung carcinoma) and GK1.5 (mouse hybridoma). Mouse cell lines MC38 (colon carcinoma), EMT-6 (mammary carcinoma, ATTC# CRL-2755) and EL-4 (lymphoma) also tested positive for αGal epitope expression (not shown). Confirming previous results, melanoma cell lines such as B16 (B16F0 and B16Bl6) and S91M3 [10, 39] lack the expression of αGT and αGal epitopes (Fig. 1a). No expression of αGal epitopes was detected in B16Bl6 cells transduced with a retroviral vector encoding the Neomycin-resistant gene (pLNL, B16NeoR, Fig. 1a). About 50–80% of B16Bl6 cells transduced with a retroviral vector encoding both the Neomycin-resistant gene and the αGT gene express αGal epitopes (pLNCKG, B16αGal, Fig. 1a). Additionally, αGal epitopes were not detected in peripheral blood lymphocytes (PBL) obtained from αGT KO mouse and human PBL. In addition, αGal epitopes were not detected in A549 cells (human lung carcinoma ATTC# CCL-185) and in the CA320M cell line generated from the αGT KO mouse. A549 cells transduced with pLNCKG vector expressed αGal epitopes at levels similar to mouse αGal positive cells (Fig. 1a).
To corroborate the presence or absence of αGal epitopes in B16 cell lines used in this study, we performed a functional assay based on complement-mediated cell destruction of αGal-expressing cells (Fig 1b). Cells were incubated in the presence or absence of pooled serum from mice with high titers of anti-αGal Ab. Pooled mouse serum was heat inactivated to prevent endogenous complement activation. To induce cell lysis of αGal-expressing cells, an exogenous source of complement was included in the reaction mixture. As shown in Figure 1, LLC (αGal positive cells) are efficiently killed by anti-αGal Ab and complement. Similarly, B16αGal are efficiently killed by the presence of anti-αGal Ab and complement. Cytotoxicity of LLC and B16αGal was not observed in absence of mouse serum and/or complement which indicates that the presence of both anti-αGal Ab and complement is required to induce cell lysis of αGal-expressing cells. No cytotoxicity was observed in the presence of complement and anti-αGal Ab in B16Null, B16NeoR, S91M3 and CA320M cells (Fig. 1b). This result confirmed the presence of αGal epitopes in B16αGal cells and the absence of these epitopes in B16Null and B16NeoR cells.
Animals used in this study were generated by selection of homozygous H-2Kb and Db haplotype, and expanded by breeding brother-sister pairs exclusively (inbreeding). Figure 1c shows the homozygous status of mice used in this study expressing only H-2Kb molecules and not H-2Dd molecules in the cell surface of peripheral blood lymphocytes. B16.Bl6 cells were originally generated from a C57Bl6 mouse of H-2 b/b haplotype. Therefore, mice and cells used in this study are haploidentical for the major histocompatibility locus H-2b.
Reduced lung-tumor burden after in vivo rejection of melanoma cells expressing αGal epitopes
We tested whether the presence of αGal epitopes and preexisting immunity against this epitope would lead to the in vivo destruction of melanoma cells that reach the vascular system. Thus RRBC-primed mice were injected i.v. with B16NeoR, αGal negative (n=7), or with B16αGal (n=11). Thirty days after metastasis implantation, lungs were obtained and tumor burden quantified in a blinded manner. As shown in Fig. 2, a significant reduction in lung melanoma metastasis was observed in mice challenged with αGal expressing cells (Unpaired t test P<0.05). Moreover, not only the average number of pulmonary metastases was significantly reduced, but also, we observed increased number of tumor-free mice. From 11 mice receiving B16αGal, 4 of them had no detectable pulmonary tumors. On the contrary, all mice receiving B16NeoR developed pulmonary melanoma tumors. This result indicates that melanoma cells expressing αGal epitopes are effectively rejected in vivo when administered intravenously.
Fig. 2.

Reduced disseminated melanoma metastases in mice injected intravenously with B16αGal cells. Female and male αGT KO mice were immunized with RRBC to increase the anti-αGal Ab titers. One week after the last immunization, mice were intravenously injected with B16NeoR (n=7) or B16αGal (n=11) in the tail vein. Three weeks after the injection, lungs were harvested and lung melanoma metastases were enumerated in a blinded manner. Results indicate that the difference in lung burden is statistically significant (t test P<0.05. Bars show the average and standard error of the mean (SEM)
Kinetics of tumor development after challenge with αGal-positive B16 cells
Next, we studied whether the expression of αGal epitopes in melanoma cells would lead to their in vivo rejection in presensitized hosts when implanted subcutaneously and whether this rejection would lead to tumor growth retardation and antitumor immunity. Mice were injected with RRBC and then s.c challenged with 105 B16αGal, B16Null or B16NeoR (Fig. 3).
Fig. 3.

Mice challenged with B16αGal developed smaller tumors. Female and male αGT KO mice were immunized with RRBC. One week after the last immunization, mice received a lethal subcutaneous (s.c.) challenge of 1×105 of either one of these three cell lines: a B16Bl6 melanoma cell line (αGal negative, B16Null, n=19) (filled triangle), b B16Bl6 cell transduced with a retroviral vector expressing NeoR gene (B16NeoR, control αGal negative n=21) (filled square) or c B16Bl6 cells transduced with a retroviral vector expressing both, NeoR and αGT genes (B16αGal n=20) (filled circle). After challenge, tumors were measured in a blinded manner. Average tumor size and SEM is depicted. A second independent experiment was performed, and similar results were obtained (not shown)
Results demonstrate that fewer animals developed smaller tumors when challenged with B16αGal cells. Only 5 from 20 animals (25%) in this group developed palpable tumors 15 days after tumor implantation. On the contrary, 11 of 19 mice that received B16Null developed measurable tumors 2 weeks after the challenge and 10 out of 20 mice receiving the B16NeoR cells developed tumors (57, 50%, respectively). Tumors were larger in animals challenged with the control and mock transduced B16 cells compared with tumors in mice receiving B16αGal. Both control groups had tumors of 200 mm3 average sizes (t test P>0.05, F test to compare variances P>0.05). On the contrary, a significant reduction in the tumor size was observed in mice receiving B16αGal cells. The mean tumor size of the test group was 36 mm3, which represents about 80% reduction in the average tumor size compared with control animals (Bartlett’s test for equal variances P<0.01, variances are different, Fig. 3).
Comparable results were observed when tumors were measured 23 days after the challenge. Thirteen mice out of 17 mice in the group that received B16Null cells had tumors (76%), and 13 out of 18 mice developed large tumors in the group receiving B16NeoR cells (72%). More importantly, a total of four animals, two in each of the control groups, had to be euthanized because of the development of large tumors. In a striking difference, only 50% of mice in the group receiving the B16αGal cells developed tumors. None of the animals that received B16αGal cells died within the first 3 weeks of tumor implantation.
The kinetics of tumor development after challenge suggests that in mice injected with B16αGal, tumor cells were initially destroyed by anti-αGal Ab and complement activation. This rejection yields to less number of cells capable of developing a tumor. Consequently, the size of tumors generated in this group is smaller than the size of tumors developed in both control groups. In addition, the growth rate of B16Null, B16NeoR and B16αGal differed significantly between groups. Tumors in mice injected with B16Null or B16NeoR grew at the same rate almost doubling the volume every 7 days (preferred model for best fit is straight line, curve comparison B16Null vs. B16NeoR, p>0.05, Fig. 3). On the contrary, tumors from mice challenged with B16αGal cells grew significantly slower at early time points suggesting the development of active immune mechanism(s) that restrain the growth of the tumor and help prolong the survival of tumor-bearing mice (Best fit straight line, curve comparison all data P<0.03, Fig. 3).
Long-term survival of mice after rejection of B16αGal cells
Mice challenged subcutaneously with B16Null, B16NeoR, and B16αGal were observed weekly for 85 days (Fig. 4a). As shown in the figure, 9 out of 20 mice (45%) survived the lethal challenge of B16αGal while only 2 out of 19 (10%) and 4 out of 21 (19%) survived the challenge with B16Null and B16NeoR, respectively. The challenge with B16αGal significantly prolonged the survival of mice since 45% remained tumor-free for more than 80 days (Fig. 4a, logrank test P<0.05). This experiment was repeated in a second independent experiment and similar results were obtained (Fig. 4b). As shown in the figure, 47% of the animals receiving B16αGal cells survived the lethal challenge. Nine out of 19 mice remained tumor-free for 100 days after the challenge. A significant prolonged survival response was observed in this group compared to control group (Kaplan-Meier survival analysis, Logrank Test P<0.02).
Fig. 4.

αGT KO mice challenged with B16αGal showed significant prolonged survival. a Mice treated as described in Fig. 3 were studied for 85 days. b Second experiment of mice receiving B16Null (n=11 filled triangle), B16NeoR (n=12 filled square) and B16αGal (n=19 filled circle) were studied for 80 days. Kaplan–Meier survival analysis indicated significant increased survival in mice injected with B16αGal (logrank test for both experiments P<0.05). c C57Bl/6 mice (Jackson Laboratories) were s.c injected with 1×105 B16Null (n=10 filled triangle), B16NeoR (n=10 filled square) and B16αGal (n=10 filled circle). Logrank test P>0.05
On the contrary, none out of ten animals challenged with B16Null survived and only one out of 11 mice receiving B16NeoR cells survived the challenge. Both control groups showed very similar survival curves, indicating that the NeoR gene product is not inducing a significant change in the rejection and/or immunogenicity of B16 (Logrank Test P=0.5, differences not statistically significant).
It has been shown that alteration in the surface carbohydrates of B16 cells modifies their metastasic potential [10]. Consequently, it could be possible that the prolonged survival of mice injected with B16αGal is due to differences in the tumorigenic properties of these cell lines rather than the presence of anti-αGal Ab. To rule out this possibility, wild type C57Bl/6 mice, which do not have detectable anti-αGal Ab and anti-αGal producing B cells [17], were challenged subcutaneously with 105 B16Null, B16NeoR and B16αGal (Fig. 4c). None of the injected C57Bl/6 mice survived the challenge with B16Null, B16NeoR or B16αGal. This result demonstrates that the presence of anti-αGal Ab in αGT KO mice was responsible for the prolonged survival of mice challenged with B16αGal.
Rejection of B16αGal leads to induction of protective immunity against wild type B16Null
We hypothesized that those mice surviving the initial challenge with B16αGal cells developed cellular immunity extended to the native αGal negative melanoma tumor. To test this hypothesis, all surviving tumor-free mice were re-challenged with wild type B16 (B16Null αGal negative). Twelve additional, age-matched mice were included as controls.
As expected, almost all control mice died from subcutaneous melanoma, developing large and pigmented tumors while none of the surviving mice developed tumors and remained tumor-free for more than 70 days (Fig. 5). In a second independent experiment, mice surviving the first encounter with B16αGal (n=9) received a second challenge with B16Null. All mice survived and remained tumor-free for more than 50 days (not shown).
Fig. 5.

Complete protection of mice surviving lethal challenge of B16αGal against B16Null. Eight mice that survived the first challenge with B16αGal cells from Fig. 4, were subsequently rechallenged with 1×105 B16Null cells (n=8 filled circle). Age-matched control mice received same dose of B16Null (n=12 filled triangle). Kaplan–Meier analysis was performed for a period of 60 days after the injection of B16Null (Logrank test P<0.002). Similar results were obtained in a second independent experiment performed with nine tumor-free mice that survived the first B16αGal challenged (not shown)
This result demonstrated that mice that survived the lethal challenge with αGal-expressing cells developed strong long-lasting anti-tumor immunity against the native αGal-negative tumor in all protected mice.
We performed in vitro T-cell cultures to further demonstrate the hypothesis that cytotoxic T cells against B16Null were induced in melanoma protected mice. Splenocytes from mice that effectively rejected B16αGal were harvested and expanded in vitro in the presence or absence of irradiated B16Null cells. Effector cells generated in these cultures were tested against B16Null melanoma and control target cells at two different effector target ratios (Fig. 6).
Fig. 6.

Induction of melanoma specific cytotoxic T cells in mice that survived a lethal injection of B16αGal cells. Spleen mononuclear cells from control mice (naive) and from mice that survived the first challenge with B16αGal (melanoma-protected mice) were used to generate effector cytotoxic T lymphocytes (CTL). Effector CTL were generated by culturing 1×106 splenocytes in presence (stimulated) or absence (non-stimulated) of 1×105 irradiated B16Null cells during 5 days in the absence of IL-2. Effector CTL were harvested and tested in triplicate wells against the specific target B16Null or the nonspecific haploidentical cell lines MC38 and EL-4. Specific cytotoxicity was determined after 4 h of incubation by measuring LDH release in the culture supernatant using formula depicted in Materials and methods. a Effector target ratio 5:1. b Effector:target ratio 10:1. Effector cells: Splenocytes from naïve mice non-stimulated (white bars), Splenocytes from naïve mice cultured with B16Null (dashed bars), Splenocytes from melanoma-protected mice nonstimulated (gray bars) and Splenocytes from melanoma-protected mice cultured with B16Null (black bars). Bars represent the mean and errors the SEM of triplicates. * One way ANOVA P<0.001
As shown in Fig. 6, strong cytotoxic cells recognizing B16Null were expanded from mice challenged with B16αGal that survived tumor development. These cytotoxic lymphocytes are melanoma specific since they were not able to lyse MC38 and EL-4 control target cells.
Altogether, these results demonstrate that in vivo rejection of αGal positive melanoma cells increased the survival in mice with preexisting anti-αGal Ab. Moreover, mice that rejected B16αGal cells and survived the initial lethal challenge were protected against B16Null. This indicated the induction of strong antitumor immunity extended to the native αGal negative tumor cell in these mice.
Prevention of subcutaneous tumor development by αGal expressing melanoma vaccines induced long-lasting memory immunity
We tested whether retrovirally transduced B16 cells expressing αGT were able to induce antitumor immunity against B16Null in a preventive protocol of vaccination as previously described [20]. Thus, RRBC-immunized mice were subcutaneously vaccinated with irradiated B16αGal cells or control cells (B16Null). Twelve out of 20 mice vaccinated with B16αGal cells survived the challenge with B16Null and remained tumor-free for more than 90 days. Some protection was observed in mice that were vaccinated with B16Null or B16NeoR control vaccines (4 out of 14 and 5 out of 12, respectively). None of ten nonvaccinated mice survived the s.c challenge with B16. Vaccination with B16αGal cells effectively prevented the development of s.c melanoma tumors in 60% of vaccinated mice and prolonged the survival of challenged mice (not shown). These results are consistent with previous data using B16 cells transfected with plasmid DNA containing the αGT gene [20]. This experiment confirmed previous evidence indicating that preventive vaccination with B16αGal induced antitumor immunity against B16Null. We asked the question of whether this antitumor immunity was long-lasting as demonstrated by experiments shown in Fig. 5. Ten mice vaccinated with B16αGal that survived the first B16Null challenge, were subsequently rechallenged with B16Null. Only one mice developed melanoma while nine mice remained tumor-free for additional 50 days. This result indicates that long-lasting immune memory was induced in 90% of mice after vaccination with B16αGal (Fig. 7).
Fig. 7.

Preventive vaccination with B16αGal induces long-lasting antitumor immunity. Females from the αGT knockout mice colony, haplotype H-2b/b were injected i.p. RRBC to increase the anti-αGal Ab titers. One week after last RRBC injection, they were s.c injected with irradiated B16Null, B16NeoR or B16αGal (preventive protocol of vaccination). They received two doses of s.c vaccines two weeks apart. One week after the last vaccination, mice received 1×105 live B16Null cells (not shown). Ten mice vaccinated with B16αGal survived remaining tumor-free. These surviving mice were rechallenged with a second dose of B16Null. Mice were observed surviving for additional 50 days (filled circle). Age-matched naïve mice were used as controls (filled triangle). Kaplan–Meier analysis was performed for a period of 53 days after the second injection of B16Null (Logrank test P<0.001)
Lack of autoimmune depigmentation in melanoma protected mice
It is increasingly recognized that effective antitumor immunity is accompanied in many cases with autoimmune reactions [24–26, 36]. For future clinical utilization of vaccines designed to express xenoepitopes such as αGal, it is relevant to give consideration to the possibility of the development of autoimmunity following an effective antitumor therapy [26]. In the present report, none of 17 mice that rejected B16αGal and survived the second rechallenge with B16Null developed clinical signs of autoimmune depigmentation (vitiligo). These melanoma-protected mice were observed for at least 5 months without signs of autoimmune reactions to the skin (Table 1). Moreover, major perfused organs were examined by histopathology, including skin, and no microscopic alterations were found (not shown).
Table 1.
Lack of autoimmune depigmentation (vitiligo) in melanoma protected mice
| Type of experiment | Number of mice protected from melanoma | Duration of experiment (Months) |
|---|---|---|
| Survived rechallenged. EXP#1a | 8 | 6 |
| Survived rechallenged. EXP#2a | 9 | 5 |
| Preventive vaccination. Tumor-free mice rechallenged (Fig. 7) . EXP#3 | 10 | 5 |
| Treatment of s.c. melanoma (not shown)b . EXP#4 | 5 | 2.5 |
| Treatment of s.c. melanoma (not shown)b . EXP#5 | 11 | 2 |
aExperiment showed in Fig. 5 and not shown.
bRRBC primed mice were injected with B16Null and subsequently, they were vaccinated with three doses of B16αGal (treatment protocol, manuscript in preparation). Tumor-free mice are shown
In the preventive vaccination protocol, 10 out of 12 surviving mice were rechallenged s.c with B16Null and only one developed melanoma (Fig. 7). Surviving mice were observed for 5 months. None of them showed detectable clinical signs of vitiligo. Two other experiments were conducted for the treatment of preestablished s.c melanoma tumors (manuscript in preparation). Mice were injected with 1×105 B16Null and 4 days later they were treated with three doses 1 week apart of irradiated B16αGal vaccines. Protected mice (tumor-free) were followed for 2 months and signs of vitiligo were not observed in any of them (Table 1 Experiments 4 and 5, manuscript in preparation). These results indicate that rejection of αGal positive cells induced effective antitumor immunity in absence of detectable signs of autoimmune reactions against the skin.
Discussion
In this report, we demonstrated that rejection of αGal expressing cells leads to the generation of strong long-lasting T-cell mediated immunity against αGal negative targets. With this report, we establish the preclinical basis for the development of a cancer vaccine using the concept of hyperacute rejection to xenoantigens. Recently, we demonstrated that preexisting humoral immunity against αGal epitopes protected mice challenged with αGal positive mouse cells, while mice challenged with αGal negative targets were not protected [34]. In this report, we extended those studies to establish that preexisting humoral immunity against αGal epitopes could be utilized as a potent immune-adjuvant to induce antitumor immunity to αGal negative tumors. Expression of the αGT gene and the change in the glycosylation pattern of B16 induces in vivo destruction of living cancer cells. This resulted in reduction of tumor growth, prolonged survival of tumor-bearing mice, as well as induction of a strong long-lasting antitumor immunity to the αGal negative cells.
Previously, La Temple et al. [20] demonstrated that vaccination with αGal expressing melanoma cells prevented the development of melanoma. Although successful preventive vaccination was demonstrated, no data was available in regard to the induction of melanoma-specific T cells that recognize αGal negative melanoma cells. In this report, we demonstrated that CTL able to recognize and specifically kill B16Null cells were induced after rejection of αGal expressing B16 cells. Moreover, the rejection of B16αGal induced long-lasting antitumor immunity with a specificity that goes beyond the initial triggering response. All mice that survived the first encounter with B16αGal cells survived a second challenge with B16Null.
In our experiments, using αGT KO mice, backcrossed to C57BL/6 mice, we observed a low rate of survival (~10–20%) in the control groups challenged with either B16Null or B16NeoR . This low background of immunogenicity is rarely observed when C57BL/6 mice are challenged with completely syngeneic B16 cells. This suggests that our αGT KO mice colony has some degree of allogeneicity with respect to B16 cells, despite having the same H2 haplotype. In spite of that, our experiments indicate that the survival rate of αGT KO mice challenged with B16αGal is significantly higher than the background survival rate of the control groups. This observation also suggests that αGal vaccination can be effective in allogeneic tumor vaccination scenarios.
Very few strategies have convincingly shown effective treatment of B16 melanoma. B16 whole cell vaccines transduced to express GM–CSF alone or in combination with CTLA-4 have been demonstrated to be an effective treatment for B16 tumors. Also, vaccination with recombinant vaccinia virus expressing TRP-1 has shown some effect to treat preexisting B16 melanoma tumors [24, 35]. In those and other reports [4, 25, 36], effective antitumor immunity was accompanied by autoimmune depigmentation. Similarly, autoimmune reactions were associated with clinical responses in melanoma patients [26, 29]. In our report, we did not observe autoimmune depigmentation in any of 43 mice protected from melanoma. Some mice were observed for a period of 6 months and yet they did not develop any detectable clinical sign of vitiligo. Other reports, using different vaccine strategies, have also shown effective antitumor immunity in absence of autoimmunity. Naked DNA vaccines encoding an alphavirus replicon and the self antigen TRP-1 induced effective antimelanoma immunity without autoimmune depigmentation [22]. Also, transgenic CEA mice vaccinated with recombinant poxviral vectors encoding the CEA transgene as well as costimulatory molecules B7-1, ICAM-1 and LFA-3 were effectively treated from CEA-expressing tumors. Autoimmunity was studied 6 months and 1 year later. No sign of toxicity was reported [11, 14]. Vaccination with cryptic peptides in IFA also induced antitumor immunity in absence of detectable autoimmune reactions [12]. Recently, a genetic trait was described for mice that spontaneously rejected large tumors. These mice showed a powerful resistance against cell lines from multiple types of cancer. Regression was age-dependent and occured without apparent destruction of normal tissues. Cytolytic destruction of cancer cells was exclusively dependent on innate leukocytes (macrophages, NK and leukocytes) and independent of T and B cells [2, 3].
Our data demonstrates that complete protection against melanoma was achieved in mice rejecting αGal expressing cells. The expansion of melanoma specific lymphocytes and microscopic examination of tumors undergoing rejection (not shown) suggest that classical T-cell mediated immunity and nonclassical innate immunity leukocytes mingle together to induce a potent cell-mediated antitumor immunity. Current investigations are underway to determine the mechanism by which antitumor immunity is induced in the context of rejection of cells expressing the xenoepitope αGal.
In summary, in the present report, we demonstrated for the first time the presence of CTL against αGal negative melanoma cells induced after rejection of αGal positive cells. The presence of CTL and the induction of long-lasting immunologic memory indicate that human cancer cells engineered to express the xenoepitope αGal could be efficiently utilized in vaccination strategies to treat human malignancies. Based on this and other studies [34] (manuscript in preparation), two phase I/II clinical trails were FDA approved and are currently open for the treatment of lung and breast cancer patients.
Acknowledgments
Rossi GR was funded by NewLink Genetics Corporation through a Research and Development Agreement. The authors would like to thank Dawn Bertrand, for her outstanding efforts and invaluable help in the management of the Animal Care Facility at the Iowa Cancer Research Foundation. The authors also would like to thank Dr Sergei Kisselev for his assistance in the FACS and Dr Mario R Mautino for his help in revising this manuscript.
References
- 1.Baumann BC, Forte P, Hawley RJ, Rieben R, Schneider MK, Seebach JD. Lack of galactose-alpha-1,3-galactose expression on porcine endothelial cells prevents complement-induced lysis but not direct xenogeneic NK cytotoxicity. J Immunol. 2004;172:6460–6467. doi: 10.4049/jimmunol.172.10.6460. [DOI] [PubMed] [Google Scholar]
- 2.Cui Z, Willingham MC. The effect of aging on cellular immunity against cancer in SR/CR mice. Cancer Immunol Immunother. 2004;53:473–478. doi: 10.1007/s00262-003-0488-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cui Z, Willingham MC, et al. Spontaneous regression of advanced cancer: identification of a unique genetically determined, age-dependent trait in mice. Proc Natl Acad Sci USA. 2003;100:6682–6687. doi: 10.1073/pnas.1031601100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finkelstein SE, Heimann DM, et al. Bedside to bench and back again: how animal models are guiding the development of new immunotherapies for cancer. J Leukoc Biol. 2004;76:333–337. doi: 10.1189/jlb.0304120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galili U. Autologous tumor vaccines processed to express alpha-gal epitopes: a practical approach to immunotherapy in cancer. Cancer Immunol Immunother. 2004;53(11):935–945. doi: 10.1007/s00262-004-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galili U, Anaraki F, Thall A, Hill-Black C, Radic M. One percent of human circulating B lymphocytes are capable of producing the natural anti-Gal antibody. Blood. 1993;82:2485–2493. [PubMed] [Google Scholar]
- 7.Galili U, LaTemple DC. Natural anti-Gal antibody as a universal augmenter of autologous tumor vaccine immunogenicity. Immunol Today. 1997;18:281–285. doi: 10.1016/S0167-5699(97)80024-2. [DOI] [PubMed] [Google Scholar]
- 8.Galili U, Mandrell RE, Hamadeh RM, Shohet SB, Griffiss JM. Interaction between human natural anti-alpha-galactosyl immunoglobulin G and bacteria of the human flora. Infect Immun. 1988;56:1730–1737. doi: 10.1128/iai.56.7.1730-1737.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galili U, Shohet SB, Kobrin E, Stults CL, Macher BA. Man, apes, and Old World monkeys differ from other mammals in the expression of alpha-galactosyl epitopes on nucleated cells. J Biol Chem. 1988;263:17755–17762. [PubMed] [Google Scholar]
- 10.Gorelik E, Duty L, Anaraki F, Galili U. Alterations of cell surface carbohydrates and inhibition of metastatic property of murine melanomas by alpha 1,3 galactosyltransferase gene transfection. Cancer Res. 1995;55:4168–4173. [PubMed] [Google Scholar]
- 11.Greiner JW, Zeytin H, Anver MR, Schlom J. Vaccine-based therapy directed against carcinoembryonic antigen demonstrates antitumor activity on spontaneous intestinal tumors in the absence of autoimmunity. Cancer Res. 2002;62:6944–6951. [PubMed] [Google Scholar]
- 12.Gross DA, Graff-Dubois S, et al. High vaccination efficiency of low-affinity epitopes in antitumor immunotherapy. J Clin Invest. 2004;113:425–433. doi: 10.1172/JCI200419418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamadeh RM, Jarvis GA, Galili U, Mandrell RE, Zhou P, Griffiss JM. Human natural anti-Gal IgG regulates alternative complement pathway activation on bacterial surfaces. J Clin Invest. 1992;89:1223–1235. doi: 10.1172/JCI115706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hodge JW, Grosenbach DW, Aarts WM, Poole DJ, Schlom J. Vaccine therapy of established tumors in the absence of autoimmunity. Clin Cancer Res. 2003;9:1837–1849. [PubMed] [Google Scholar]
- 15.Induction and measurement of cytotoxic t lymphocyte activity (2001) Current protocols in immunology Chapter 3. In: Coico R (ed) Vitro Assays for Mouse Lymphocyte Function. John Wiley& Sons, New York
- 16.Joziasse DH, Oriol R. Xenotransplantation: the importance of the Galalpha1,3Gal epitope in hyperacute vascular rejection. Biochim Biophys Acta. 1999;1455:403–418. doi: 10.1016/s0925-4439(99)00056-3. [DOI] [PubMed] [Google Scholar]
- 17.Kawahara T, Ohdan H, Zhao G, Yang YG, Sykes M. Peritoneal cavity B cells are precursors of splenic IgM natural antibody-producing cells. J Immunol. 2003;171:5406–5414. doi: 10.4049/jimmunol.171.10.5406. [DOI] [PubMed] [Google Scholar]
- 18.Kim M, Rao MV, Tweardy DJ, Prakash M, Galili U, Gorelik E. Lectin-induced apoptosis of tumour cells. Glycobiology. 1993;3:447–453. doi: 10.1093/glycob/3.5.447. [DOI] [PubMed] [Google Scholar]
- 19.Larsen RD, Rivera-Marrero CA, Ernst LK, Cummings RD, Lowe JB. Frameshift and nonsense mutations in a human genomic sequence homologous to a murine UDP-Gal:beta-D-Gal(1,4)-D-GlcNAc alpha(1,3)- galactosyltransferase cDNA. J Biol Chem. 1990;265:7055–7061. [PubMed] [Google Scholar]
- 20.LaTemple DC, Abrams JT, Zhang SY, Galili U. Increased immunogenicity of tumor vaccines complexed with anti-Gal: studies in knockout mice for alpha1,3galactosyltransferase. Cancer Res. 1999;59:3417–3423. [PubMed] [Google Scholar]
- 21.LaTemple DC, Galili U. Adult and neonatal anti-Gal response in knock-out mice for alpha1,3galactosyltransferase. Xenotransplantation. 1998;5:191–196. doi: 10.1111/j.1399-3089.1998.tb00027.x. [DOI] [PubMed] [Google Scholar]
- 22.Leitner WW, Hwang LN, deVeer MJ, Zhou A, Silverman RH, Williams BR, Dubensky TW, Ying H, Restifo NP. Alphavirus-based DNA vaccine breaks immunological tolerance by activating innate antiviral pathways. Nat Med. 2003;9:33–39. doi: 10.1038/nm813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Link CJ, Jr, Seregina T, Atchison R, Hall A, Muldoon R, Levy JP. Eliciting hyperacute xenograft response to treat human cancer: alpha(1,3) galactosyltransferase gene therapy. Anticancer Res. 1998;18:2301–2308. [PubMed] [Google Scholar]
- 24.Overwijk WW, Lee DS, et al. Vaccination with a recombinant vaccinia virus encoding a “self” antigen induces autoimmune vitiligo and tumor cell destruction in mice: requirement for CD4(+) T lymphocytes. Proc Natl Acad Sci USA. 1999;96:2982–2987. doi: 10.1073/pnas.96.6.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Overwijk WW, Theoret MR, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phan GQ, Yang JC, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA. 2003;100:8372–8377. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Posekany KJ, Pittman HK, Bradfield JF, Haisch CE, Verbanac KM. Induction of cytolytic anti-Gal antibodies in alpha-1,3-galactosyltransferase gene knockout mice by oral inoculation with Escherichia coli O86:B7 bacteria. Infect Immun. 2002;70:6215–6222. doi: 10.1128/IAI.70.11.6215-6222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Posekany KJ, Pittman HK, Swanson MS, Haisch CE, Verbanac KM. Suppression of Lewis lung tumor development in alpha 1,3 galactosyltransferase knock-out mice. Anticancer Res. 2004;24:605–612. [PubMed] [Google Scholar]
- 29.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411:380–384. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 30.Schaapherder AF, Daha MR, te Bulte MT, van der Woude FJ, Gooszen HG. Antibody-dependent cell-mediated cytotoxicity against porcine endothelium induced by a majority of human sera. Transplantation. 1994;57:1376–1382. doi: 10.1097/00007890-199405150-00016. [DOI] [PubMed] [Google Scholar]
- 31.Spiro RG, Bhoyroo VD. Occurrence of alpha-D-galactosyl residues in the thyroglobulins from several species. Localization in the saccharide chains of the complex carbohydrate units. J Biol Chem. 1984;259:9858–9866. [PubMed] [Google Scholar]
- 32.Thall AD, Maly P, Lowe JB. Oocyte Gal alpha 1,3Gal epitopes implicated in sperm adhesion to the zona pellucida glycoprotein ZP3 are not required for fertilization in the mouse. J Biol Chem. 1995;270:21437–21440. doi: 10.1074/jbc.270.37.21437. [DOI] [PubMed] [Google Scholar]
- 33.Thall AD, Murphy HS, Lowe JB. alpha 1,3-Galactosyltransferase-deficient mice produce naturally occurring cytotoxic anti-Gal antibodies. Transplant Proc. 1996;28:556–557. [PubMed] [Google Scholar]
- 34.Unfer RC, Hellrung D, Link CJ., Jr Immunity to the alpha(1,3)galactosyl epitope provides protection in mice challenged with colon cancer cells expressing alpha(1,3)galactosyl-transferase: a novel suicide gene for cancer gene therapy. Cancer Res. 2003;63:987–993. [PubMed] [Google Scholar]
- 35.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190:355–366. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Elsas A, Sutmuller RP, et al. Elucidating the autoimmune and antitumor effector mechanisms of a treatment based on cytotoxic T lymphocyte antigen-4 blockade in combination with a B16 melanoma vaccine: comparison of prophylaxis and therapy. J Exp Med. 2001;194:481–489. doi: 10.1084/jem.194.4.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watier H, Guillaumin JM, Piller F, Lacord M, Thibault G, Lebranchu Y, Monsigny M, Bardos P. Removal of terminal alpha-galactosyl residues from xenogeneic porcine endothelial cells. Decrease in complement-mediated cytotoxicity but persistence of IgG1-mediated antibody-dependent cell-mediated cytotoxicity. Transplantation. 1996;62:105–113. doi: 10.1097/00007890-199607150-00020. [DOI] [PubMed] [Google Scholar]
- 38.Watier H, Guillaumin JM, Vallee I, Thibault G, Gruel Y, Lebranchu Y, Bardos P. Human NK cell-mediated direct and IgG-dependent cytotoxicity against xenogeneic porcine endothelial cells. Transpl Immunol. 1996;4:293–299. doi: 10.1016/S0966-3274(96)80050-5. [DOI] [PubMed] [Google Scholar]
- 39.Zhu Z, Sanchez-Sweatman O, Huang X, Wiltrout R, Khokha R, Zhao Q, Gorelik E. Anoikis and metastatic potential of cloudman S91 melanoma cells. Cancer Res. 2001;61:1707–1716. [PubMed] [Google Scholar]