

Abstract
Over the past years, monoclonal antibodies have attracted enormous interest as targeted therapeutics, and a number of such reagents are in clinical use. However, responses could not be achieved in all patients with tumors expressing high levels of the respective target antigens, suggesting that other factors such as limited recruitment of endogenous immune effector mechanisms can also influence treatment outcome. This justifies the search for alternative, potentially more effective reagents. Antibody-toxins and cytolytic effector cells genetically modified to carry antibody-based receptors on the surface, represent such tailor-made targeting vehicles with the potential of improved tumor localization and enhanced efficacy. In this way, advances in recombinant antibody technology have made it possible to circumvent problems inherent in chemical coupling of antibodies and toxins, and have allowed construction via gene fusion of recombinant molecules which combine antibody-mediated recognition of tumor cells with specific delivery of potent protein toxins of bacterial or plant origin. Likewise, recombinant antibody fragments provide the basis for the construction of chimeric antigen receptors that, upon expression in cytotoxic T lymphocytes (CTLs) or natural killer (NK) cells, link antibody-mediated recognition of tumor antigens with these effector cells’ potent cytolytic activities, thereby making them promising cellular therapeutics for adoptive cancer therapy. Here, general principles for the derivation of cytotoxic proteins and effector cells with antibody-dependent tumor specificity are summarized, and current strategies to employ these molecules and cells for directed cancer therapy are discussed, focusing mainly on the tumor-associated antigens epidermal growth factor receptor (EGFR) and the closely related ErbB2 (HER2) as targets.
Keywords: Targeted therapy, Single chain Fv antibody fragment, Immunotoxin, Pseudomonal exotoxin A, Cyotoxic T lymphocytes, Natural killer cells, Chimeric antigen receptor, Epidermal growth factor receptor, ErbB2/HER2
Introduction
A major goal of cancer research has been to define consistent alterations in malignant cells which may aid in the development of therapies tailored to a particular tumor. In the past years, monoclonal antibodies have attracted enormous interest as targeted therapeutics and a number of such reagents are already in clinical use. To be superior to conventional treatments, these antibodies must be directed toward antigens which are exclusively or at least preferentially expressed on tumor cells compared to normal tissues. Suitable targets include lineage-specific antigens such as CD19, CD20, or CD22 expressed on certain malignancies of hematologic origin, as well as surface antigens with enhanced expression on carcinoma cells like the epithelial cell adhesion molecule (EpCAM) or carcinoembryonic antigen (CEA).
Of particular interest as targets are cell surface receptors such as the epidermal growth factor receptor (EGFR/ErbB) and the closely related ErbB2 (HER2) receptor tyrosine kinase which are overexpressed on many human tumors of epithelial origin and transmit important growth and survival signals [23, 33]. Antibodies that block ligand binding or interfere with the function of these target receptors can directly inhibit the growth of cancer cells, in addition to their potential to direct effector cells of the immune system to the tumor [29]. Monotherapy with the humanized, ErbB2-specific antibody Herceptin, or a combination of antibody with chemotherapy protocols, for example, resulted in increased clinical benefit for a significant proportion of patients with ErbB2-overexpressing metastatic breast cancers [53]. However, responses could not be achieved in all patients with tumors expressing high ErbB2 levels, suggesting that in addition to enhanced expression of the target receptor, other factors, such as limited recruitment of endogenous immune effector mechanisms or presence of alternative signaling pathways in tumor cells, can also influence treatment outcome. This justifies the search for alternative, potentially more effective reagents.
Antibody-toxins and cytolytic effector cells genetically modified to carry antibody-based receptors on the surface represent such tailor-made targeting vehicles with the potential of improved tumor localization and enhanced efficacy. Advances in recombinant antibody technology have thus made it possible to circumvent problems inherent in chemical coupling of antibodies and toxins, and have allowed construction via gene fusion of recombinant molecules which combine antibody-mediated recognition of tumor cells with specific delivery of potent protein toxins of bacterial or plant origin [24]. Likewise, recombinant antibody fragments provide the basis for the construction of chimeric antigen receptors that, upon expression in cytotoxic T lymphocytes (CTLs) or natural killer (NK) cells, link antibody-mediated recognition of tumor antigens with these effector cells’ potent cytolytic activities, thereby making them promising cellular therapeutics for adoptive cancer therapy [14, 51].
Here, general principles for the derivation of cytotoxic proteins and effector cells with antibody-dependent tumor specificity are summarized, and current strategies to employ these molecules and cells for directed cancer therapy are discussed, focusing mainly on the tumor-associated antigens EGFR and ErbB2 as targets.
Recombinant antibody fragments for tumor targeting
Most therapeutic antibodies currently under clinical evaluation were initially derived by immunization of mice and classical hybridoma technology, followed by partial or complete humanization of murine sequences using recombinant DNA technology. Advances in the rapidly developing field of antibody engineering have resulted in the construction of large combinatorial antibody libraries of human or synthetic origin, and effective in vitro screening systems now allow bypassing immunization and selecting recombinant antibodies of defined specificity without the need for hybridoma production [7, 20]. Over the past decade, a variety of different recombinant antibody formats have been engineered that are suitable for diverse therapeutic applications and include monovalent, bivalent, and multivalent derivatives, single- or double-chain formats, and covalently or noncovalently linked assemblies of antibody heavy (VH) and light chain variable domains (VL) [35, 38, 49].
Among these, single-chain Fv (scFv) antibody fragments appear best suited for their use as targeting domains in chimeric fusion proteins. They consist only of antibody VH and VL sequences genetically linked via a flexible linker, but lack constant regions and Fc domain, thereby preventing possible binding to normal tissues and cells via interaction with Fc receptors. Omitting the constant region sequences from such antibody fragments also allows the use of murine Fv domains without inducing strong human antimouse antibody responses typically associated with application of murine IgG in cancer patients. More importantly, antibody fusion proteins such as recombinant antibody-toxins and chimeric antigen receptors can easily be derived from scFv antibody fragments since this format allows assembly of sequence information from the two antibody chains, and from heterologous effector molecules into an expression unit encoding only a single polypeptide.
Structure of recombinant antibody-toxins
Antibody-toxins, also termed immunotoxins, were initially derived by chemically coupling bacterial or plant toxins to monoclonal antibodies specific for molecules on the surface of tumor cells. The elucidation of the molecular structure of bacterial toxins such as Pseudomonas aeruginosa exotoxin A (ETA, PE), and the development of recombinant antibody formats, have allowed the miniaturizing of these molecules via recombinant DNA techniques and their production as single polypeptides in large quantities, and of consistent quality, in bacteria [24]. Pseudomonal exotoxin A consists of an N-terminal cell recognition domain (Ia), an internal translocation domain (II), and a C-terminal enzymatic domain (III) [55]. Upon cell binding and internalization, ETA is cleaved by the cellular protease furin within domain II and an N-terminal 28-kDa and a C-terminal 37-kDa fragment are generated. Following reduction of a disulfide bond, the C-terminal enzymatically active fragment translocates to the cytoplasm where it gains access to the protein synthesis machinery. The catalytic domain then ADP-ribosylates eukaryotic elongation factor 2 (EF-2) resulting in the inhibition of protein synthesis and subsequent target cell death by apoptosis.
Similar to recombinant toxins employing peptide ligands for targeting, the first ETA-based single-chain antibody-toxin was derived by replacing the toxin’s N-terminal cell-binding domain with an anti-Tac scFv antibody fragment [12]. Upon bacterial expression, the resulting molecule displayed specific binding to tumor cells expressing high levels of the CD25 IL-2 receptor subunit and was selectively cytotoxic for such cells. The same basic principle has been applied in past years for the construction of antibody-toxins targeted to a larger variety of tumor-associated antigens [24] (Fig. 1A). While the combination of antibody domain and toxin fragment in a single polypeptide chain in most cases yielded functional protein, for some antibody-toxins antitumoral activity could be improved by modification of the protein design. Expression of antibody VL and toxin-linked VH as separate chains followed by subsequent connection of VH and VL via an artificial interdomain disulfide bond (dsFv), enhanced thermal stability and binding affinity for some of the resulting dsFv-toxins [37, 38].
Fig. 1A.
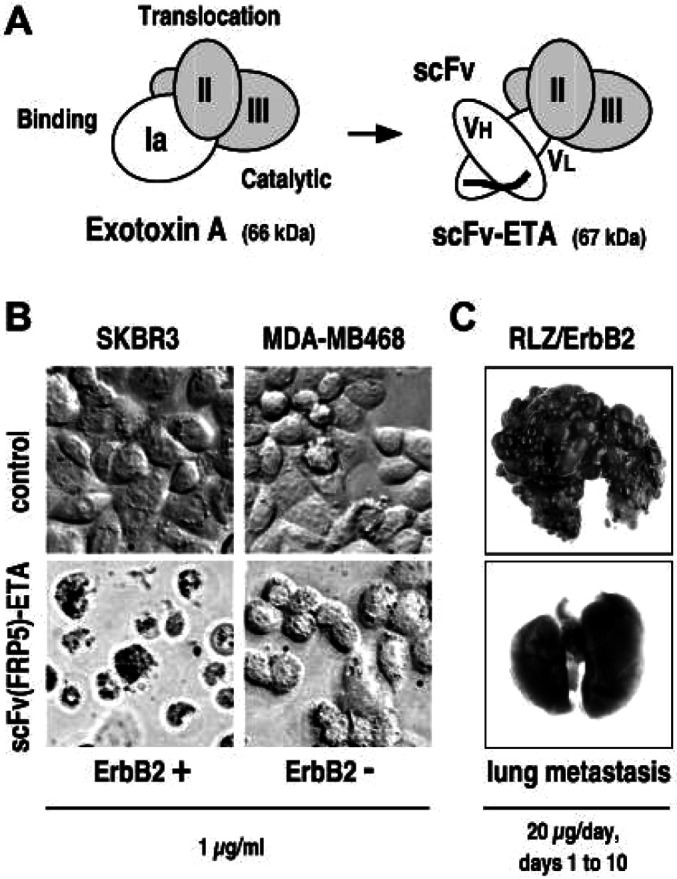
Schematic representation of the structures of Pseudomonas aeruginosa exotoxin A (ETA) and an ETA-based single-chain antibody-toxin. ETA consists of an N-terminal cell binding domain (Ia), an internal translocation domain (II), and a C-terminal enzymatic domain (III) facilitating ADP-ribosylation of eukaryotic elongation factor 2. By replacement of domain Ia with a scFv antibody fragment, chimeric antibody-toxins with novel target cell specificity are derived. B Specificity of scFv(FRP5)-ETA antibody-toxin. ErbB2-overexpressing SKBR3 and ErbB2-negative MDA-MB468 human breast carcinoma cells were incubated for 24 h with 1 μg/ml ErbB2-specific scFv(FRP5)-ETA (lower panels) or PBS (upper panels) before analysis by light microscopy. C Antimetastatic activity of scFv(FRP5)-ETA in vivo. Balb/c mice were injected intravenously with 105 murine renal carcinoma (Renca) cells stably transfected with lacZ and human c-erbB2 constructs (RLZ/ErbB2) at day 0. Animals were treated i.v. with 20 µg/dose of scFv(FRP5)-ETA from days 1–10. Control animals received PBS. Mice were sacrificed at day 28, lungs were excised and analyzed for pulmonary metastasis upon X-gal staining [27]
Successful application of pseudomonal exotoxin A–based antibody-toxins in recent clinical studies has increased interest in these targeted therapeutics for cancer treatment [25, 26]. However, while clinical development of antibody-toxins for the treatment of hematologic malignancies has progressed, reports of a successful clinical application of such molecules in patients suffering from cancers of epithelial origin are still rare. This might in part be due to the type of target antigens available. Normal expression of target receptors such as CD22 and CD25 is restricted to a defined population of differentiated cells limiting potential adverse effects [25, 26], whereas epithelial antigens targeted for therapy usually display significantly enhanced expression in tumors, but might also be present at varying levels on different normal tissues. Therefore, to avoid potential side effects toward vital organs and tissues, recombinant antibody-toxins must be highly selective for tumor cells overexpressing such antigens.
Antibody-toxins targeted to members of the ErbB receptor family
Due to their preferential expression in many tumors of epithelial origin, their accessibility from the extracellular space, and their involvement in the transformation process, EGFR and the closely related ErbB2 receptor tyrosine kinase are being considered as suitable targets for directed cancer therapy, and different groups have developed recombinant antibody-toxins with specificity for EGFR or ErbB2 [37, 44, 57, 58]. The first scFv antibody-toxin binding to human EGFR was derived from antagonistic monoclonal antibody 225 [57]. Like the partly humanized antibody derivative C225 which is now in advanced clinical studies [29], bacterially expressed scFv(225)-ETA antibody-toxin retained the ability to block ligand-dependent activation of EGFR. Consequently, targeted delivery of its cytotoxic domain to EGFR overexpressing tumor cells and inhibition of EGFR signaling could both contribute to the therapeutic efficacy of this molecule.
Based on 14E1, another antagonistic antibody blocking the ligand-binding site of EGFR, the scFv(14E1)-ETA antibody-toxin, was derived displaying approximately tenfold higher affinity for EGFR than scFv(225)-ETA, which resulted in enhanced cytotoxic activity toward EGFR-expressing tumor cells in vitro and in vivo [6, 44]. In contrast to a growth factor toxin employing the natural EGFR ligand TGF-α for targeting, both antibody-toxins showed strict selectivity for cells expressing high levels of the target antigen [43, 44]. ScFv(14E1)-ETA and scFv(225)-ETA also displayed strong cytotoxic activity toward cells expressing the ligand-independent, constitutively active EGFR variant EGFRvIII found in brain tumors such as glioblastoma and in various other malignancies [59], suggesting that the antibody-toxins could be particularly useful for the treatment of tumors where coexpression of high levels of full-length EGFR is accompanied by expression of EGFR variants [41, 43].
Recombinant antibody-toxins might be most beneficial for systemic treatment in an adjuvant setting, aimed at eradication of minimal residual disease and the prevention of metastasis formation at distant sites. For scFv(14E1)-ETA, potent activity against disseminated cancer cells expressing human EGFR or EGFRvIII and suppression of organ metastasis could be demonstrated in a murine in vivo model system [41]. Alternatively, direct intratumoral application of antibody-toxins might be a suitable treatment option for localized disease such as glioblastoma and squamous cell carcinoma of the head and neck (SCC-HN). In a murine model system for SCC-HN, marked reduction in tumor size and cures in some of the animals were observed upon intratumoral application of EGFR-specific scFv antibody-toxins [6].
Antitumoral activity of the ErbB2-specific antibody toxin scFv(FRP5)-ETA
While recombinant scFv antibody-toxins targeted to EGFR proved effective in preclinical models, most progress toward clinical application has been made with reagents targeted to ErbB2. Among the different members of the ErbB family of receptor tyrosine kinases, ErbB2 appears to be the most selective in distinguishing tumor cells from normal tissues. Epithelial cells of most organs typically express ErbB2 at low levels. However, in several types of carcinomas, ErbB2 expression is strongly enhanced, often as a result of gene amplification [23].
The bacterially expressed 67-kDa scFv(FRP5)-ETA protein is a recombinant single-chain antibody-toxin with binding specificity for ErbB2-overexpressing tumor cells [57, 58]. In in vitro cell-killing experiments, scFv(FRP5)-ETA displayed potent antitumoral activity against a wide range of established and primary human tumor cells including breast and ovarian carcinomas [42, 48, 58] (Fig. 1B), squamous cell carcinomas [6, 57] and prostate carcinomas [54]. In experimental animals, scFv(FRP5)-ETA effectively inhibited growth of established human tumor xenografts [6, 42, 57, 58] and murine and rat tumor cells stably transfected with human c-erbB2 constructs [3, 27]. Thus both direct intratumoral injection and systemic application of the toxin were effective, resulting in complete elimination of subcutaneously growing tumors [6] and prevention of metastasis formation by disseminated cancer cells [27] (Fig. 1C).
In a first clinical application of scFv(FRP5)-ETA, cancer patients with ErbB2-expressing tumors were treated on a compassionate-use basis by intratumoral injection of scFv(FRP5)-ETA into cutaneous lesions [5]. Eleven patients suffering from metastatic breast and colorectal cancers and from malignant melanoma were treated. Patients received daily intratumoral injections of scFv(FRP5)-ETA for 7 to 10 days. Total daily doses ranged from 60 to 900 μg, and total doses per treatment cycle ranged from 0.6 to 6.0 mg. Treatment caused tumors to shrink in 60% of the patients evaluated. Complete regression of injected tumor nodules was achieved in 40% and partial reduction in the size of injected tumors in another 20% of patients. While no response could be achieved upon treatment of a tumor with moderate ErbB2 expression (1+), partial reduction in the size of the injected tumor nodule was seen in one of two patients with intermediate ErbB2 overexpression (2+) and complete regression or partial reduction in tumor size was noted in five of seven patients whose tumors showed high (3+) ErbB2 expression [5]. This indicates that the selectivity of scFv(FRP5)-ETA for ErbB2-overexpressing target cells seen in preclinical models can also be reproduced in cancer patients. Similar to studies with other ETA-based antibody-toxins, neutralizing antibodies directed against the bacterial toxin domain, developed during scFv(FRP5)-ETA treatment in two of three patients analyzed in detail.
In contrast to treatment with erb-38, a similar ErbB2-specific, pseudomonal exotoxin–based dsFv-toxin investigated previously [34, 37], treatment with the scFv(FRP5)-ETA molecule resulted in clinical responses and was well tolerated by the majority of patients, with adverse reactions mainly restricted to fully reversible symptoms such as pain and inflammation at the injection sites. In the case of erb-38, intravenous injection of 1 or 2 µg/kg caused grade 2 or 3 hepatotoxicity in all patients [34]. Intratumoral injection of scFv(FRP5)-ETA only resulted in liver toxicity in one patient after treatment with high daily doses of 600 to 900 μg. Another patient treated with 600 μg, completed a 10-day treatment cycle without any systemic adverse effects. While these differences might be partly due to the different routes of toxin administration, it appears likely that other factors also contribute to toxicity and treatment outcome. For erb-38, an LD50 in mice of 450 μg/kg was reported [34], whereas scFv(FRP5)-ETA could be applied at very high doses of 1,000 and 2,000 μg/kg without signs of toxicity [27, 42]. The efficacy of local application of scFv(FRP5)-ETA and the low degree of toxicities observed in most patients suggest that therapeutically effective doses of the molecule could also be reached upon intravenous administration. Therefore, a phase I clinical study to investigate the effects of intravenous application of scFv(FRP5)-ETA in patients suffering from ErbB2-overexpressing tumors has now been initiated.
T cells and chimeric antigen receptors
Recombinant antibody-toxins represent one approach to covalently link antibody-dependent recognition of specific antigens on the tumor cell surface with a potent cytotoxic activity, thereby bypassing the requirement for natural antibodies to induce complement fixation or, upon binding to Fc receptors, to redirect killer cells to the tumor in order to elicit antitumoral activity. Providing cytolytic effector cells such as cytotoxic T lymphocytes (CTLs) or natural killer (NK) cells with chimeric antigen receptors constitutes an alternative strategy to harness potent cytotoxic effectors of natural origin for a specific attack on tumor cells.
Cytotoxic T lymphocytes are important effector cells operative in the elimination of tumors. They have the intrinsic potential to extravasate from the circulation and to reach their target cells in almost all body tissues. At the site of cytolytic action they secrete cytokines and chemoattractants thereby activating additional nonspecific effector cells. A prerequisite for a T-cell response, however, is the activation of the cells by suitable antigens. CTLs recognize short peptide sequences presented by HLA class I molecules on the target cell surface. Interaction of these antigens with the T-cell receptor (TCR) leads to proliferation and clonal expansion. While there is increasing evidence that a large proportion of human tumors might express antigens suitable for recognition by T cells, strategies for the development of effective cancer vaccines are still hampered by our limited understanding of the complex interactions between tumor cells and immune effector cells in vivo [47]. Tumors might escape immune surveillance by a variety of mechanisms such as the prevention of efficient antigen presentation or the absence of costimulatory signals during initial T-cell activation. Endowing T cells with chimeric antigen receptors was therefore developed as an alternative strategy to circumvent the dependence on professional antigen-presenting cells for activation of naïve T cells, and to bypass MHC-restricted recognition of peptide antigens on target cells as a requirement for the initiation of cytolytic effector functions.
Strategies to genetically manipulate T lymphocytes and exploit them for directed tumor therapy were made possible by insights into the structure and function of the T-cell receptor complex [22]. The TCR consists of multiple polypeptide chains that can be separated into functional subunits facilitating ligand binding and signal transduction. Dimers of α and β (or γ and δ) chains confer antigen specificity to the receptor and recognize antigenic peptides presented by MHC molecules. Complexed with these dimers are the invariant chains of the CD3 complex CD3γ, CD3δ, and CD3ε, and the ζ chains that couple the antigen-recognizing TCR proteins to the intracellular signaling machinery (Fig. 2A). The initial signaling event following antigen recognition is the activation of Src family protein tyrosine kinases (PTKs) that in turn phosphorylate immunoreceptor tyrosine-based activation motifs (ITAMs) within the cytoplasmic tails of the CD3 complex proteins. The phosphorylated ITAMs serve as docking sites for PTKs of the ZAP-70 family that further activate downstream signaling pathways ultimately resulting in cellular responses such as proliferation, differentiation, and initiation of effector functions.
Fig. 2A.
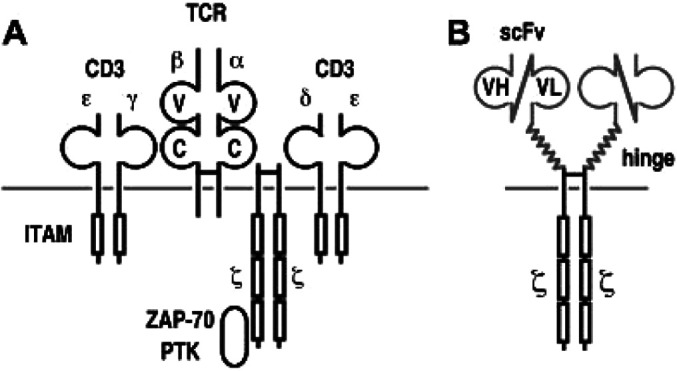
Schematic representation of the T-cell receptor (TCR) complex. The TCR consists of the α/β dimer recognizing antigenic peptides presented by MHC molecules. Complexed with the TCR are the invariant chains of the CD3 complex CD3γ, CD3δ, CD3ε, and the ζ chains. The invariant chains couple the antigen-recognizing TCR proteins to intracellular signaling molecules such as the ZAP-70 protein tyrosine kinase (PTK) that binds to phosphorylated immunoreceptor tyrosine–based activation motifs (boxed). B Single-chain chimeric antigen receptor consisting of a target-cell specific scFv antibody fragment comprising antibody heavy (VH) and light chain (VL) variable domains fused via a flexible hinge region to transmembrane and intracellular regions of the TCR-associated ζ chain. Alternative designs for chimeric antigen receptors are mentioned in the text
Specificity of chimeric antigen receptors is provided by the incorporation of heterologous binding domains derived from natural ligands or antibodies for direct, MHC-independent recognition of antigens expressed on the surface of target cells. The binding domains are genetically fused to effector molecules derived from proteins that are naturally involved in T-cell signaling such as the T-cell receptor α and β chains [18], or components of the TCR-associated CD3 complex [51]. Upon antigen binding, the chimeric receptors link to endogenous signaling pathways and generate activating signals similar to those initiated by the T-cell receptor complex.
An important improvement in the design of chimeric antigen receptors was the combining of antigen recognition and signaling capability in a single-chain configuration. For the construction of such chimeric antigen receptors, scFv antibody fragments were genetically fused to different receptor subunits that serve as signaling molecules in lymphocytes (Fig. 2B). Among the effector proteins tested, the most commonly used are the γ and ζ chains of the Fc receptor (FcR) and TCR, respectively [14], since these effectors are sufficient for coupling extracellular recognition events to receptor-associated signal transduction pathways. When expressed in T cells, the chimeric receptors form both homodimers and heterodimers with endogenous signaling chains. Concerning T-cell activation, cytokine production, and target cell killing, single-chain chimeric receptors showed characteristics similar to those observed for chimeric double-chain TCRs, but in contrast to the latter, were not dependent on endogenous CD3 complex for surface expression and transduction of stimulatory signals [14]. This expanded the spectrum of cell types suitable for retargeting from T cells to NK cells and neutrophils [39].
Factors influencing the activity of retargeted T cells
A common aim of the different designs of chimeric antigen receptors is to mimic, upon contact with the selected antigen, the signaling events that usually follow activation of a cytotoxic T cell. For complete activation, naïve T cells must encounter an antigen-specific signal provided through the TCR-CD3 complex. In addition, costimulatory activity, generated after interaction of the CD28 receptor on the T cell with its cognate ligands of the B7 protein family expressed on professional APCs, is required. TCR activation in the absence of costimulation usually results in unresponsiveness or T-cell anergy. Consequently, for resting T cells expressing a chimeric receptor, the first signal is not sufficient for activation [10].
Since most protocols for the generation of retargeted T cells are based on ex vivo transduction with retroviral vectors after stimulation with anti-CD3 antibody or phytohemagglutinin in the presence of IL-2, the absence of a specific costimulatory stimulus might not present a problem [14]. This could be different if retargeted effector cells are to be generated by in vivo transfection of naïve T cells, or upon transplantation of hematopoietic stem cells transduced with the chimeric construct. The potential problem of anergy induction in the absence of costimulation has been addressed by modifying the structure of chimeric receptors. Fusion of a scFv antibody fragment to the intracellular signal-transducing part of CD28 resulted in a receptor that provides a costimulatory signal upon contact with target antigen and, when combined with activation of the TCR-CD3 complex, initiated signals similar to those elicited by cross-linking of unmodified CD28 [4]. To circumvent the requirement for coexpression of two chimeric receptors, signaling components providing primary and costimulatory signals were also combined in a single gene product comprising a scFv antibody fragment for target recognition fused to the intracellular domain of CD28 and the signaling region of the TCR ζ chain [15]. T cells expressing this scFv-CD28-ζ construct produced up to 20 times more IL-2 upon stimulation with solid phase antigen when compared with transfectants expressing chimeric receptors that only carried the signaling domain of the ζ chain.
Effector molecules, such as the ζ chain, depend on close interaction with other components of the TCR complex for activation. In T cells isolated from tumor patients, this might present a problem since these cells often display defects in TCR-mediated signaling and exhibit impaired immune responses. In most cases studied, an altered composition of the TCR-CD3 complex or impaired phosphorylation and kinase activity were identified as the underlying molecular mechanisms [14]. To address this problem, chimeric receptors were constructed that are able to bypass defective TCR proximal signaling and directly access the T cell’s effector mechanisms by employing a protein tyrosine kinase of the ZAP-70 family as an effector domain [16]. Activation of such PTKs normally follows downstream of TCR-CD3 initiated signaling. When intracellular signaling after antigenic stimulation was analyzed, a construct harboring the PTK Syk was far superior to a ZAP-70–containing construct and T cells expressing this construct were able to mediate efficient lysis of antigen-expressing cells.
T lymphocytes with grafted recognition specificity for tumor cells
Recombinant antibody technology allowed the construction of chimeric receptors that employ scFv fragments derived from a variety of previously characterized antibodies with specificity for disease-associated antigens. Therefore, the majority of chimeric antigen receptors have so far been designed to target relevant cancer antigens such as the pan-carcinoma antigen TAG-72 expressed by most human adenocarcinomas [28] and the carcinoembryonic antigen [13]. To target these antigens, fully humanized constructs suitable for clinical studies were developed. In addition, chimeric receptors were constructed that recognize carbohydrate antigens such as Lewis Y [30] or antigens such as the high molecular weight melanoma associated antigen (HMW-MAA) only expressed by a subset of tumors [36]. Approaches for the treatment of hematopoietic malignancies include the use of chimeric receptors that target CD30 expressed on Hodgkin/Reed-Sternberg cells [19], or the B cell malignancy associated antigens CD19 [9] and CD20 [21].
As in the case of recombinant antibody-toxins, growth factor receptors, such as EGFR and ErbB2 that are aberrantly expressed on the tumor cell surface, present another group of interesting therapeutic targets. A chimeric T-cell receptor for targeting to ErbB2-expressing cells was constructed by assembling DNA fragments encoding an immunoglobulin heavy-chain leader peptide, the ErbB2-specific scFv(FRP5) antibody fragment previously used as a targeting domain for antibody-toxins, a Myc tag for immunological detection, a flexible hinge region derived from CD8α (amino acids 105–165) and the TCR ζ chain transmembrane and intracellular domains into a single open reading frame in the retroviral vector pLXSN [1]. In initial studies a similar construct was introduced into the murine CTL line Cl96 by retroviral gene transfer. Upon coincubation with ErbB2-expressing cells, the transduced T cells secreted IFN-γ and specifically lysed the target cells [32]. Interestingly, alternative chimeric receptors without a spacer or hinge region were not functional [31]. When Cl96-scFv(FRP5)-ζ cells were adoptively transferred into nude mice, the growth of subcutaneously implanted ErbB2-expressing tumor cells could be retarded. T cells bearing the chimeric receptor in contrast to parental Cl96 cells could thus be shown to home to the tumor site [32].
Upon optimization of transduction protocols, efficient retroviral transfer of the chimeric receptor construct into primary T cells from rats and mice could be achieved [1, 2]. In vivo antitumoral activity of the modified cells was demonstrated in immunocompetent mice. Syngeneic T cells transduced with the scFv(FRP5)-ζ vector by cocultivation with the retroviral producer cell line were injected into established subcutaneous tumors of Balb/c-derived HC11 R2 cells which express an oncogenically activated human ErbB2. This resulted in complete tumor regression [2].
Natural killer cells as alternative effectors
So far, only a limited number of clinical trials with retargeted T cells have been initiated, partly due to the requirement for efficient transduction of patient-derived effector cells and expansion in quantities sufficient for therapy. Employing retargeted cytotoxic cell lines for adoptive transfer in an allogeneic setting might help to overcome some of the current limitations and could result in the development of more generally applicable cell therapeutics.
As a first step in this direction, a genetically modified variant of the continuously growing cytotoxic natural killer cell line NK-92 was recently generated. NK cells are a subgroup of lymphocytes that play an essential role in the cellular immune defense against virus-infected and malignant cells. NK cells do not rearrange their immune receptor genes, and the cytotoxicity toward tumor and virus-infected cells is not MHC-restricted [11]. In patients with malignant disorders, NK-cell function can be impaired resulting in a reduced proliferative response and reduced cytotoxic activity [46]. The human NK-92 cell line was originally established from peripheral blood lymphocytes of a patient with large granular lymphoma [17]. NK-92 cells are similar to activated primary NK cells with respect to the expression of typical NK-cell surface receptors and functional characteristics, but do not harbor Fc-γRIII (CD16) and display a much higher cytolytic activity against a broad spectrum of tumor targets, in particular against malignant cells of hematologic origin. They do not affect normal human cells and, based on this selectivity, the potential utility and application of NK-92 cells for adoptive therapy is currently being investigated in phase I clinical studies [50].
In contrast to malignant cells of hematologic origin, the proportion of NK-92–sensitive cancer cells derived from solid tumors appears to be significantly lower. In a recent study, six out of seven established tumor cell lines and primary cancer cells originating from human breast, ovarian, and squamous cell carcinomas, and expressing elevated levels of receptor tyrosine kinases such as ErbB2 or EGFR, were completely resistant to NK-92–mediated lysis, with the remaining cell line being only weakly sensitive [52]. To extend NK-92 cytotoxicity to these cancer cell types and to enhance tumor-specific targeting, NK-92 cells were transduced with a retroviral vector encoding the ErbB2-specific scFv(FRP5)-ζ antigen receptor previously used for retargeting of T cells [1, 52]. An NK-92 cell population was selected that displayed homogeneous and high-level surface expression of the chimeric receptor. The high cytotoxic potential of the parental cell line against malignant cells of hematologic origin was retained by these NK-92-scFv(FRP5)-ζ cells, indicated by their continued ability to lyse typical NK cell targets such as erythroleukemic K562 cells. Likewise, expression of the chimeric antigen receptor did not change the activity of NK-92 against ErbB2-negative tumor cells of epithelial origin. In contrast, all ErbB2-expressing tumor cell lines and primary cancer cells that were insensitive to NK-92–mediated killing, were now efficiently lysed by NK-92-scFv(FRP5)-ζ [52]. This demonstrates that NK-cell resistance could be overcome alone by expression of the chimeric antigen receptor. Specific antitumoral effects of NK-92-scFv(FRP5)-ζ were also observed in vivo when these cells, in the absence of exogenous IL-2, were implanted simultaneously with ErbB2-transformed fibroblasts into nude mice. In comparison to animals treated with parental NK-92 cells, tumor growth was markedly delayed.
Even at low effector to target ratios, NK-92-scFv(FRP5)-ζ cells rapidly induced apoptosis in ErbB2-expressing target cells, and upon prolonged incubation, completely eliminated tumor cells from the culture (Fig. 3A–D). In this way, expression of ErbB2 on the tumor cell surface was sufficient to enable recognition and lysis by NK-92-scFv(FRP5)-ζ. Otherwise, NK-92–resistant murine renal carcinoma cells became highly sensitive to the genetically modified variant after stable transfection with a human c-erbB2 cDNA construct [52]. Cytolytic activity of this NK-92-scFv(FRP5)-ζ population expressing high levels of antigen receptor was retained toward target cells expressing varying levels of ErbB2. This corresponds well with recent studies addressing the influence of target antigen and receptor densities on cytotoxic activity. Whereas T cells with large amounts of a chimeric receptor specific for the tumor-associated G250 antigen were able to lyse tumor cells expressing high or low G250 levels, T cells carrying fewer receptors were only triggered for cytolysis and cytokine production if target cells expressed relatively high antigen levels [56]. Similarly, when NK-92-scFv(FRP5)-ζ populations carrying lower numbers of the chimeric antigen receptor were investigated, more moderate killing activity toward ErbB2-expressing targets was found (Fig. 3E, and unpublished data). These results suggest that T and NK cells can be manipulated to discriminate between different antigen densities. In a clinical setting, this might allow the development of retargeted effector cells that specifically lyse tumor cells carrying large amounts of antigen, but spare normal cells harboring the same antigen at physiological levels.
Fig. 3A–D.

Specific killing of ErbB2-overexpressing tumor cells by NK-92 natural killer cells expressing an ErbB2-specific chimeric antigen receptor. Human SKBR3 breast carcinoma cells were incubated with ErbB2-specific NK-92-scFv(FRP5)-ζ (A, C) or parental NK-92 cells (B, D) at a low effector to target ratio of 1:1. Cells were analyzed by light microscopy after incubation at 37°C for 90 min (A, B) and 29 h (C, D). Adherent cells represent SKBR3. NK-92 and NK-92-scFv(FRP5)-ζ cells are characterized by their smaller size and rounded shape. E Influence of antigen receptor density on cell killing activity. Selected populations of NK-92 cells expressing different levels of ErbB2-specific antigen receptors that either employ murine or human ζ chains as signal-transducing elements were established by FACS and magnetic beads sorting. Cytolytic activity of isolated cell populations was then analyzed in europium (Eu3+) release assays. SKBR3 target cells were labeled with Eu3+ solution by electroporation followed by incubation for 2 h with NK-92-scFv(FRP5)-ζ cells (open symbols) or parental NK-92 transduced with empty vector (mock) at an effector to target (E/T) ratio of 5:1. Release of Eu3+ complexes into the medium was determined by fluorimetric analysis and served as a measure of target cell lysis
Conclusions
Progress in such diverse areas of research as molecular oncology, tumor immunology, molecular biology, biotechnology, and gene therapy converges in the development of new therapeutic modalities. In the near future it will become possible to “individualize” cancer therapy: i.e., to exploit the biochemical characteristics of the cancer cells of individual patients and administer drugs which act specifically on these cells. This might diminish the current adverse effects of cytotoxic drugs and increase the success rate of the treatment.
While differing in the type of cytotoxic effectors employed, both of the targeting strategies discussed above make use of recombinant antibody fragments for specific recognition of tumor-associated cell surface antigens. In the case of antibody-toxins, covalently linking antibody-dependent recognition of the tumor cell with a potent protein toxin allows the resulting molecules to act independently of endogenous immune effector mechanisms, which classical therapeutic antibodies must recruit for antitumoral activity. Refinement of the molecular design of antibody-toxins and more careful selection of target antigens have increased safety and efficacy of such reagents. Successful application of pseudomonal exotoxin A–based antibody-toxins directed against CD22- or CD25-expressing malignancies in recent clinical studies, suggests that such recombinant molecules could indeed become a valuable treatment option even in the case of advanced disease [25, 26]. Also for immunotoxins targeting antigens overexpressed on the surface of tumor cells of epithelial origin, promising results have been obtained and further clinical evaluation is ongoing [5]. However, at present, prolonged treatment of patients with antibody-toxins can still be complicated by the immunogenicity of their bacterial protein domain. Employment of apoptosis-inducing cytokines such as Fas-ligand [40] or other cytotoxic protein domains of human origin instead of a bacterial toxin might result in similarly effective immunotoxin-like molecules that allow continued therapy.
In contrast to recombinant antibody-toxins which can be manufactured in large amounts and to consistent levels of quality by bacterial expression, the production of retargeted, autologous killer cells for clinical application is complicated by the variability of effector cells derived from a patient, and depends on sophisticated vector technology to achieve efficient transduction of the cells with chimeric antigen receptor constructs. This might explain why, despite successful demonstration of the general applicability of this approach in numerous in vitro and in vivo studies, clinical application of retargeted effector cells so far has only rarely been attempted [51]. Improvements in the design of retroviral vectors—i.e., pseudotyping with heterologous viral envelopes more suitable for transduction of T cells or NK cells, or the use of lentiviral vector constructs—might enhance transduction efficiencies and allow gene transfer in selected effector cell subpopulations [45]. Furthermore, suicide gene constructs might be included in such refined strategies to control potential adverse effects of modified CTLs and NK cells toward normal tissues [8]. Also, allogeneic cells or established effector cell lines carrying chimeric antigen receptors similar to donor lymphocytes that induce graft-versus-leukemia effects after allogeneic bone marrow transplantation, might prove beneficial for the control of minimal residual disease or micrometastasis.
Acknowledgements
This work was supported in part by grants from the Deutsche Forschungsgemeinschaft (SFB 364-C1) and from the National Genome Research Network (NGFN) program of the German Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie (BMBF).
Footnotes
This work was presented at the first Cancer Immunology and Immunotherapy Summer School, 8–13 September 2003, Ionian Village, Bartholomeio, Peloponnese, Greece.
References
- 1.Altenschmidt Clin Cancer Res. 1996;2:1001. [PubMed] [Google Scholar]
- 2.Altenschmidt J Immunol. 1997;159:5509. [PubMed] [Google Scholar]
- 3.Altenschmidt Int J Cancer. 1997;73:117. doi: 10.1002/(SICI)1097-0215(19970926)73:1<117::AID-IJC18>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez-Vallina Eur J Immunol. 1996;26:2304. doi: 10.1002/eji.1830261006. [DOI] [PubMed] [Google Scholar]
- 5.Azemar M, Djahansouzi S, Jäger E, Solbach C, Schmidt M, Maurer AB, Mross K, Unger C, Minckwitz G, Dall P, Groner B, Wels W. Regression of cutaneous tumor lesions in patients intratumorally injected with a recombinant single-chain antibody-toxin targeted to ErbB2/HER2. Breast Cancer Research and Treatment. 2003;82:155–164. doi: 10.1023/b:brea.0000004371.48757.19. [DOI] [PubMed] [Google Scholar]
- 6.Azemar Int J Cancer. 2000;86:269. doi: 10.1002/(sici)1097-0215(20000415)86:2<269::aid-ijc18>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 7.Benhar J Mol Biol. 2000;301:893. doi: 10.1006/jmbi.2000.4021. [DOI] [PubMed] [Google Scholar]
- 8.Bonini Science. 1997;276:1719. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 9.Brentjens Nat Med. 2003;9:279. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- 10.Brocker J Exp Med. 1995;181:1653. doi: 10.1084/jem.181.5.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bukowski J Exp Med. 1985;161:40. doi: 10.1084/jem.161.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaudhary Nature. 1989;339:394. doi: 10.1038/339394a0. [DOI] [PubMed] [Google Scholar]
- 13.Darcy J Immunol. 2000;164:3705. [Google Scholar]
- 14.Eshhar Cancer Immunol Immunother. 1997;45:131. doi: 10.1007/s002620050415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finney J Immunol. 1998;161:2791. [PubMed] [Google Scholar]
- 16.Fitzer-Attas J Immunol. 1998;160:145. [PubMed] [Google Scholar]
- 17.Gong Leukemia. 1994;8:652. [PubMed] [Google Scholar]
- 18.Gross Proc Natl Acad Sci U S A. 1989;86:10024. [Google Scholar]
- 19.Hombach Cancer Res. 1998;58:1116. [PubMed] [Google Scholar]
- 20.Hoogenboom Immunotechnology. 1998;4:1. doi: 10.1016/s1380-2933(98)00007-4. [DOI] [PubMed] [Google Scholar]
- 21.Jensen Biol Blood Marrow Transplant. 1998;4:75. doi: 10.1053/bbmt.1998.v4.pm9763110. [DOI] [PubMed] [Google Scholar]
- 22.Kane Curr Opin Immunol. 2000;12:242. doi: 10.1016/s0952-7915(00)00083-2. [DOI] [PubMed] [Google Scholar]
- 23.Klapper Adv Cancer Res. 2000;77:25. [PubMed] [Google Scholar]
- 24.Kreitman Curr Opin Mol Ther. 2003;5:44. [PubMed] [Google Scholar]
- 25.Kreitman N Engl J Med. 2001;345:241. doi: 10.1056/NEJM200107263450402. [DOI] [PubMed] [Google Scholar]
- 26.Kreitman J Clin Oncol. 2000;18:1622. [Google Scholar]
- 27.Maurer-Gebhard Cancer Res. 1998;58:2661. [PubMed] [Google Scholar]
- 28.McGuinness Hum Gene Ther. 1999;10:165. doi: 10.1089/10430349950018968. [DOI] [PubMed] [Google Scholar]
- 29.Mendelsohn Oncogene. 2000;19:6550. doi: 10.1038/sj.onc.1204082. [DOI] [PubMed] [Google Scholar]
- 30.Mezzanzanica Cancer Gene Ther. 1998;5:401. [PubMed] [Google Scholar]
- 31.Moritz Gene Ther. 1995;2:539. [PubMed] [Google Scholar]
- 32.Moritz Proc Natl Acad Sci U S A. 1994;91:4318. [Google Scholar]
- 33.Olayioye EMBO J. 2000;19:3159. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pai-Scherf Clin Cancer Res. 1999;5:2311. [PubMed] [Google Scholar]
- 35.Pluckthun Immunotechnology. 1997;3:83. doi: 10.1016/S1380-2933(97)00067-5. [DOI] [PubMed] [Google Scholar]
- 36.Reinhold J Invest Dermatol. 1999;112:744. doi: 10.1046/j.1523-1747.1999.00586.x. [DOI] [PubMed] [Google Scholar]
- 37.Reiter J Biol Chem. 1994;269:18327. [PubMed] [Google Scholar]
- 38.Reiter Clin Cancer Res. 1996;2:245. [PubMed] [Google Scholar]
- 39.Roberts J Immunol. 1998;161:375. [PubMed] [Google Scholar]
- 40.Samel D, Muller D, Gerspach J, Assohou-Luty C, Saas G, Tiegs G, Pfizenmaier K, Wajant H (2003) Generation of a FasL based proapoptotic fusion protein devoid of systemic toxicity due to cell surface antigen-restricted activation. J Biol Chem (in press) [DOI] [PubMed]
- 41.Schmidt Oncogene. 1999;18:1711. doi: 10.1038/sj.onc.1202489. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt Gynecol Oncol. 2001;80:145. doi: 10.1006/gyno.2000.6040. [DOI] [PubMed] [Google Scholar]
- 43.Schmidt Int J Cancer. 1998;75:878. doi: 10.1002/(SICI)1097-0215(19980316)75:6<878::AID-IJC10>3.3.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt Br J Cancer. 1997;75:1575. doi: 10.1038/bjc.1997.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schnierle Proc Natl Acad Sci U S A. 1997;94:8640. doi: 10.1073/pnas.94.16.8640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sedlmayr J Immunother. 1991;10:336. doi: 10.1097/00002371-199110000-00005. [DOI] [PubMed] [Google Scholar]
- 47.Smyth Nat Immunol. 2001;2:293. doi: 10.1038/86297. [DOI] [PubMed] [Google Scholar]
- 48.Spyridonidis Blood. 1998;91:1820. [PubMed] [Google Scholar]
- 49.Todorovska J Immunol Methods. 2001;248:47. doi: 10.1016/S0022-1759(00)00342-2. [DOI] [PubMed] [Google Scholar]
- 50.Tonn J Hematother Stem Cell Res. 2001;10:535. doi: 10.1089/15258160152509145. [DOI] [PubMed] [Google Scholar]
- 51.Uherek J Hematother Stem Cell Res. 2001;10:523. doi: 10.1089/15258160152509136. [DOI] [PubMed] [Google Scholar]
- 52.Uherek Blood. 2002;100:1265. [PubMed] [Google Scholar]
- 53.Vogel J Clin Oncol. 2002;20:719. doi: 10.1200/JCO.20.3.719. [DOI] [Google Scholar]
- 54.Wang Prostate. 2001;47:21. doi: 10.1002/pros.1043.abs. [DOI] [Google Scholar]
- 55.Wedekind J Mol Biol. 2001;314:823. doi: 10.1006/jmbi.2001.5195. [DOI] [PubMed] [Google Scholar]
- 56.Weijtens Gene Ther. 2000;7:35. doi: 10.1038/sj.gt.3301051. [DOI] [PubMed] [Google Scholar]
- 57.Wels Int J Cancer. 1995;60:137. doi: 10.1002/ijc.2910600120. [DOI] [PubMed] [Google Scholar]
- 58.Wels Cancer Res. 1992;52:6310. [PubMed] [Google Scholar]
- 59.Wikstrand Cancer Res. 1995;55:3140. [PubMed] [Google Scholar]