

Abstract.
Engineering antibodies with reduced immunogenicity and enhanced effector functions, and selecting antigen targets with the appropriate specificity, density, and/or functionality, have contributed to the recent clinical successes in using unconjugated "naked" antibody therapies of B-cell lymphoma (rituximab) and breast carcinoma (Herceptin). The non-overlapping toxicities of naked antibodies and chemotherapy, together with their potential synergy, which is based on unique and complementary mechanisms of action, have contributed to the creation of new standards of care in cancer therapy and management. Clinical trial results supporting these concepts are presented. Furthermore, the exquisite specificity of antibodies renders them ideal vehicles for selective delivery of toxic payloads such as drugs or radionuclides. Although successful in therapy of hematological cancers (Zevalin, Mylotarg), the broader application of these technologies to carcinomas still remains to be proven in clinical testing. Engineering of antibody constructs with optimal blood clearance and tumor-targeting kinetics, and selecting the radionuclide that may deliver sufficient radiation energy to kill the more radio-resistant carcinomas, are discussed. With the advent of genomics and proteomics, new membrane-associated tumor antigens are being discovered and will provide novel targets for future antibody therapy of cancer.
Keywords: Antibody therapy, Cancer antigens, Clinical trials, Lymphoma, Radiolabeled antibodies
Introduction
Twenty years passed between the discovery of monoclonal antibody (MAb) technology by Koehler and Milstein [103] and the approval of the first MAb therapy for cancer (MabThera, Roche, Basel, Switzerland), rituximab (Rituxan, Genentech, San Francisco, Calif.), in 1997. Since the approval of ritxuimab for treatment of patients with relapsed low-grade or follicular B-cell non-Hodgkin's lymphoma (NHL), two unconjugated MAb therapies, Herceptin and Campath, and two conjugated MAb therapies, Zevalin and Mylotarg, have also been approved (Table 1).
Table 1.
Therapeutic monoclonal antibodies. FDA U.S. Food and Drug Administration
| Generic name | Trade name | Company | Antibody type | Target | Status |
|---|---|---|---|---|---|
| Anti-tumor MAbs | |||||
| Rituximab | Rituxan | IDEC/Genentech | Chimeric | CD20 | FDA approval 1997 |
| Trastuzumab | Herceptin | Genentech | Humanized | Her2R | FDA approval 1998 |
| Gemtuzumab ozogamicin | Mylotarg | Wyerth-Ayerst | Humanized conjugated to calicheamicin | CD33 | FDA approval 2000 |
| Alemtuzumab | Campath | Millenium/ILEX | Humanized | CD52 | FDA approval 2001 |
| Ibritumomab tiuxetan | Zevalin | IDEC | Murine (90Y labeled) | CD20 | FDA approval 2002 |
The long delay between the discovery and therapeutic applications of monoclonal antibodies can be attributed to limitations of the hybridoma technology and failure of antibody therapy in early clinical trials. Key considerations for rituximab development included:
Immunogenicity of murine antibodies limits repeated administration. Newer recombinant technologies for engineering antibodies have significantly reduced the human immune response to the inoculated MAb (HAMA).
Antibody selection for optimal human effector cell function.
Cost of goods of monoclonal antibodies produced by hybridoma cell lines which rendered the commercial application of antibody therapy non-viable. Development of new gene expression systems and high-production cell lines provided the antibody quantities needed for chronic therapy at an acceptable cost.
Selection of target antigens capable of inducing negative growth signals in neoplastic cells when activated by MAb.
Selection of target indications with accessibility and susceptibility to antibody-mediated effects.
This review outlines some of the important issues associated with identifying and developing unconjugated and radiolabeled antibodies for cancer therapy. The discussion focuses on the recent advances in defining target antigens and therapeutic antibodies, and summarizes clinical trial results which led to the approval of both rituximab and Zevalin.
General considerations for antibody therapy
Immunogenicity
One of the early problems identified with the use of potentially therapeutic murine MAbs was the rapid formation of human antibody responses (HAMA) to the inoculated rodent protein. The HAMA response dramatically altered the pharmacokinetic and pharmacodynamic profiles of the MAb and virtually prevented repeat dosing needed for cancer therapy. Host responses were primarily directed to the murine Fc portion of the antibody, although anti-idiotypic responses have been reported [84]. Recombinant techniques to reduce the immunogenicity of MAbs have been developed, including the engineering of chimeric, humanized, Primatized and human antibodies [124, 127, 136].
Rituximab is a recombinant chimeric MAb directed against the human B-cell differentiation antigen, CD20. The murine anti-CD20 MAb was generated by immunizing mice with human lymphoblastoid CD20+ B cells [84, 124]. Rituximab was genetically engineered by combining the variable region domains of the heavy and light chains of the murine antibody and the constant regions of human IgG1. The immunocompromised nature of NHL patients, together with the remarkable ability of rituximab for depleting normal B cells, resulted in a very low HAMA response.
Antibody selection
Unconjugated MAbs require three key features necessary for success: (a) high-affinity binding to target antigen; (b) ability to activate host effector functions such as complement dependent cytotoxicity (CDC), opsonization, and antibody-dependent cell cytotoxicity (ADCC); and (c) ability to induce apoptosis or inhibit survival signals in the targeted neoplastic cells. The relative significance of each of these mechanisms for therapy of malignant disease is difficult to address, but collectively they all participate in eliminating neoplastic cells in vivo.
The importance of antigen-binding affinity for the target antigen is critical when selecting and/or engineering therapeutic MAbs. Antibody affinity impacts the dose and efficacy of the antibody. Low-affinity antibodies may require large doses to achieve antigen saturation and to activate host-effector functions needed for optimal efficacy.
Engineering effective antibodies requires careful consideration for selecting the most appropriate human constant region used to develop the chimeric or humanized construct. Human-constant-region domains contain sites that interact with complement and Fc gamma receptors present on various host-effector cells. Fc-receptor-positive cell binding to tumor-bound MAb in vivo may lead to ADCC, phagocytosis, and apoptosis. Additionally, a unique class of Fc receptor, FcRn, is involved in antibody catabolism and the maintenance of serum IgG levels in the blood [92].
The selection of human IgG1 construct regions for engineered rituximab was done for optimal effector functions and killing mechanisms. The four human IgG isotypes vary in their ability to bind C1q component of complement and mediate CDC with IgG1=IgG3>IgG2. IgG4 does not activate complement. The critical difference is related to only four residues in the carboxyl portion of the CH2 domain of the human constant region [165]. Recently, the C1q binding sites in the rituximab CH2 domain have been mapped [15, 88]; thus, engineering chimeric or humanized therapeutic MAbs using human IgG1 and IgG3 Fc regions maximize complement-mediated cell killing.
Similarly, the binding of the four human IgG isotypes to Fc receptors has been defined. In humans, there are three identified classes of Fc receptors that participate in mediating host-effector functions: FcR1, FcR2, and FcR3. FcR1 (CD64) has high-affinity binding for monomeric antibody and is expressed on neutrophils, monocytes, and macrophages. FcR2 (CD32) has a high affinity for antibody-antigen complexes and is widely expressed on hematological cells including B cells, neutophils, monocytes, and macrophages, whereas FcR3 is highly expressed on NK cells. Human gamma-1 and gamma-3 constant regions have high affinity to all three Fc receptors. In contrast, human gamma 4 has weak binding to Fc receptor and exhibits weak effector function. In vivo experiments comparing the potency of gamma-4 and gamma-1 versions of anti-CD20 antibodies demonstrated higher effectiveness of the gamma-1 construct in the clearance of normal B cells from the circulation.
While both IgG1 and IgG3 are highly effective molecules in activating host-effector mechanisms, they differ in their FcRn binding, resulting in a longer half-life of the gamma-1 construct. This difference in the pharmacokinetic profile makes the selection of human gamma-1 constant region for engineering antibodies more appropriate for unconjugated MAbs where long circulating half-life is important.
However, Fc receptor binding and activation of host defense mechanisms is not the only mechanism for eliminating neoplastic cells in vivo. Anti-receptor antibodies, including rituximab, have been demonstrated to also induce negative growth signals and tumor cell apoptosis. This can be demonstrated using a tetravalent version of rituximab devoid of the CH2 domain, and thereby lacking C1q and Fc binding [139]. The molecule was effective in inducing apoptosis in vitro without further cross-linking and active in human tumor xenograft models. Growth inhibition of lymphoid cell lines in vitro was first demonstrated using antibody cross-linking of surface IgM molecules [173] and MHC class II [126].
MAb binding to other B-cell markers, CD19, CD22, and CD20, was not sufficient for mediating either growth inhibition or apoptosis. More recently, growth inhibition and induction of apoptosis in vitro was demonstrated by anti-CD20 antibodies under conditions leading to hyper-cross-linking of the CD20 antigen [152]. Similar results have now been published for both CD19 and CD22 when cross-linking of membrane-bound MAb was amplified with anti-mouse IgG antibody [28]. In an in vivo setting, binding to Fc receptor provides the cross-linking of membrane-bound rituximab needed for effective induction of apoptosis. Signaling mechanisms involved in rituximab-mediated apoptosis are not fully elucidated but may involve upregulation of the pro-apoptotic Bax molecule in lymphoma cells [151] or down-regulation of anti-apoptotic Bcl-xl molecule [115].
Application of genetic engineering techniques to reduce immunogenicity, increase binding affinity, and alter biological half-life and antibody effector function will improve the efficacy and safety of antibodies. For example, improved affinity has been achieved via directed mutagenesis of individual amino acids in the variable region [33] and point mutations in the CH2 domain altered Fc receptor binding [154, 191], effector function, and half-life of antibodies. Similarly, it already been established that aglycosylated forms of both gamma-1 and gamma-3 constructs have poorer CD64 interactions [19], C1q binding, and CDC activity than their fully glycosylated forms [164]. Alternatively, introducing a single additional sugar (bisecting GlcNAc) to the biantenary sugar complex enhanced Fc receptor binding and ADCC [46].
Antibody production and cost of goods
An often overlooked issue is the repeated high antibody doses required to achieve a therapeutic response. Hybridoma cell lines are low-MAb producers and are genetically unstable, leading to even further reduction in production levels during scale-up and manufacturing. The engineering of genetically stable production cell lines, capable of producing large quantities of antibody, is essential for the successful commercialization of antibody therapies.
Developing highly productive cells lines and large-scale production methods to economically produce the quantities of MAb required to treat the NHL patient population was a major milestone in the development and commercialization of rituximab. High-production cell lines were achieved by transfection of antibody genes into Chinese hamster ovary (CHO) cells followed by genomic amplification. Vectors used for expression of rituximab were designed for modular expression of Ig genes[138], allowing the insertion of the murine variable region DNA into a vector containing the appropriate human constant regions. High production levels (50 pg/cell day−1) were then achieved by gene amplification [11]. In contrast, traditional hybridoma production levels range from 2 to 5 pg/cell day−1.
Selection of target antigen
Antigen characteristics and level of expression on the cell surface are of paramount importance for consideration as a target for antibody therapy. Blocking of survival signals by antibody binding to growth factor receptors is expected to sensitize cells to killing by chemotherapy drugs and other apoptosis-inducing therapies. Alternatively, antibody binding to pro-apoptotic receptors delivers death signals to the malignant cell and would be expected to show efficacy as a single-agent therapy or in combination with other apoptosis-inducing agents. In most cases, cross-linking of cell-bound antibodies via Fc receptor binding is required for optimal signaling leading to apoptosis. This requires the engineering of the antibodies that contain human gamma-1 or gamma-3 constant regions. These same antibody constructs are expected to exhibit optimal effector functions, such as CDC and ADCC, if supported by acceptable antigen density on the cell surface. Indeed, low-level expression of target antigen may not support antigen cross-linking by antibodies needed for effective complement activation and Fc receptor binding leading to CDC and ADCC, respectively. Effective antibody-mediated cytotoxicity is achieved if the target antigen is not internalized or shed following antibody binding. This should lead to the formation of stable antibody-antigen complexes on the cell membrane capable of mediating CDC, ADCC, and apoptosis, while preventing antigen-negative escape. An antigen which meets the criteria of B-cell-lineage specificity, high density with limited or no modulation or shedding from the cell surface, and, when cross-linked, has the potential for apoptosis induction, is CD20, the target antigen for rituximab.
Selection of target indication
Previous clinical trials failed to demonstrate the efficacy of antibody therapy in cancer with a primary focus on solid tumors. These pioneering studies failed to take into consideration key limitations such as antibody accessibility to solid tumors, heterogeneity of antigen expression, the functionality of the antibody and the target antigen, immunogenicity of murine antibodies, and tumor resistance to antibody-mediated effector mechanisms.
Non-Hodgkin's B-cell lymphoma provides an ideal indication for antibody therapy because of its accessibility to the therapeutic antibodies and sensitivity to antibody-mediated killing mechanisms including CDC, ADCC, and apoptosis. The NHL is also sensitive to radiation therapy, making it an ideal candidate for radioimmunotherapy as well. The expression of lineage-specific differentiation antigens, such as CD20, provides a unique target for antibody therapy with limited heterogeneity of expression. At the same time, the absence of antigen expression on stem cells allows for the recovery of normal B cells following antibody treatment which leads to the destruction of both malignant and normal B cells. The effectiveness of antibody therapy in NHL was demonstrated by studies using customized anti-idiotype antibodies [123]. Such studies provided proof of concept supporting the utility of "naked" antibodies in NHL therapy.
Radiolabeled antibodies
The clinical efficacy of a radiolabeled antibody for cancer is influenced by the following factors which should be evaluated pre-clinically:
Selection of antigen which does not shed from the cell surface following antibody binding
Selection of an antibody having high specificity for the target antigen
Selection of a suitable radioisotope
Formulation of a radiolabeled antibody maintaining antigen binding and affinity
Assessment of potency
Achievement of acceptable in vitro and in vivo stability
Consideration of specific tumor and normal tissue uptake in animal models
Estimation of human radiation doses based on animal biodistribution data
Antigen and antibody selection
The basic concept of an antibody-targeted radioimmunotherapy (RIT) relies on the ability of the antibody to selectively bind to the tumor target antigen and thereby deliver radiation preferentially to the tumors. All antibody-targeting approaches must, therefore, rely on differential or selective expression of the cell surface target antigen on neoplastic vs normal cells. The usefulness of any target antigen for radioimmunotherapy of cancer is based on antigen density, specificity, heterogeneity of expression, and lack of membrane antigen shedding after antibody binding. Ideally, the targeted antigen should be selectively and abundantly expressed on tumor cells and with limited heterogeneity of expression or shedding from the cell surface. Numerous antigen targets have been considered as targets for a variety of cancers which meet such criteria to differing degrees [24, 26, 67, 147].
The most successful strategies to date have been those using antibodies directed against differentiation antigens expressed on hematological malignancies. While such antigens lack specificity for tumors, they are lineage specific. As normal hematopoetic stem cells differentiate and mature, they express distinct cell surface antigens not present on the immature stem cell. These become attractive targets for RIT, since the antigen-negative stem cell, with its capacity to divide and differentiate to mature cells, rapidly restores the normal cell population.
The CD20 antigen is a B-cell-lineage-specific antigen which is the target for Zevalin (90Y-labeled murine MAb). The CD20 molecule is expressed at high density on normal and malignant B cells and exhibits limited, if any, shedding following antibody binding. It is not expressed either on the immature stem cell or on the fully differentiated plasma cell. The clinical trials results demonstrated that the yttrium-90-labeled anti-CD20 antibody Zevalin is highly effective in therapy of refractory or relapsed low-grade NHL, leading to its approval in 2002.
As discussed previously, the use of murine monoclonal antibodies as targeting molecules may severely limit their effectiveness in humans because of the generation of a HAMA response; however, in limited selective instances, the use of low doses of a murine monoclonal antibody, such as Zevalin (approximately 2 mg/dose), is acceptable for relapsed NHL patients who are typically immunocompromised. In addition, the regimen includes a combination with rituximab, which depletes B cells and further reduces the ability of the host to generate antibody against the murine antibody component of Zevalin therapeutic regimen.
The selection of a murine antibody construct for RIT of NHL was based on its short half-life as compared with chimeric or immunized antibodies. The disadvantage of such engineered constructs is their long half-life in serum, which may lead to increased bone marrow toxicity [121, 122].
Most clinical trials to date have evaluated the use of intact radiolabeled IgG. Despite the clinical success seen in the treatment of hematological malignancies, results of RIT in carcinomas have been less impressive. This may be attributable to the greater radioresistance and limited accessibility of carcinomas as compared with hematologous cancers. Marrow toxicity remains dose limiting, with the circulating radiolabeled antibody providing the majority of this toxicity. Antibody fragments, such as F(ab)2, have been shown to clear much faster from the blood, thereby reducing marrow toxicity, and enabling larger doses to be administered; however, these fragments are not stable and are poorly retained in the tumor, thus delivering much reduced radiation doses.
Improved efficacy of RIT in solid tumors may be attained if larger doses of the radiolabeled antibody could be administered, while maintaining accumulation in the tumor at levels comparable to those of IgG. It has been observed in murine tumor xenograft models that antibodies lacking the CH2 domain have a half-life in blood comparable to that of F(ab)2 but are retained in the tumor to the same extent as IgG [155]; therefore, such antibody constructs may provide effective radiation doses to the tumor with acceptable bone marrow toxicity. Ongoing clinical trials using CH2-domain-deleted antibody may provide support to this concept [97].
Isotope selection
We cannot discuss RIT without addressing the relative advantages of different radioisotopes for this application. Several characteristics that need to be considered are the type of particle and particle energy emitted by the isotope (alpha, beta, gamma, and auger), the physical half-life of the isotope, and the distance traveled by the particle where 90–95% of its energy will be transferred to the cell (linear energy transfer, LET; Table 2). Although several radioisotopes have been considered for RIT, most attention has focused on the use of iodine-131 and yttrium-90 (Table 2). The relative merits of using alpha, gamma, or beta emitters has been extensively discussed in several excellent articles [69, 179, 183]; therefore, this discussion is limited to iodine-131 and yttrium-90.
Table 2.
Properties of α- and β-emitting isotopes for radioimmunotherapy
| Isotope | Half-life (days) | Emission | Emax(MeV) | Max. tissue penetration (mm) | Radiopharmaceutical availability/expense |
|---|---|---|---|---|---|
| 90Y | 2.7 | β | 2.27 | 11.9 | Available/expensive |
| 131I | 8.0 | β | 0.61 | 2.4 | Available/inexpensive |
| 188Re | 0.7 | β | 2.12 | 11.0 | Unavailablea |
| 186Re | 3.8 | β | 1.07 | 5.0 | Unavailablea |
| 177Lu | 6.7 | β | 0.50 | 2.0 | Unavailablea |
| 67Cu | 2.6 | β | 0.58 | 2.1 | Unavailablea |
| 213Bi | 0.03 | α | 5.9 | 0.1 | Unavailablea |
| 225Ac | 10.0 | α | 5.7 | 0.1 | Unavailablea |
| 211At | 0.3 | α | 5.9 | 0.1 | Unavailablea |
aRadioisotope available as radiochemical grade
Unlike drug conjugates that require internalization into the cell for efficacy, both iodine-131 and yttrium-90 possess a beta emission that can travel several millimeters. These relatively long path lengths are typically associated with beta emitters, and enable killing of bystander cells, including antigen negative cells, in the immediate environment of cell-bound radiolabeled antibody. This effect, referred to as "crossfire," is unique for RIT, and not other MAb-targeted approaches such as unconjugated or drug-conjugated therapies. This should allow the consideration of antigens having a good specificity profile but high expression on only a portion of the tumor population for RIT [128]. In contrast, for alpha emitters, although having a higher energy emission spectrum than beta emitters, the energy is absorbed over a very short distance, typically one or two cell diameters; therefore, alpha radioisotopes are considered more appropriate for leukemia and microscopically disseminated cancers having little antigen heterogeneity [105].
Iodine-131 provides a relatively inexpensive and readily available radiopharmaceutical agent. It has a long-lived (8.1 days) beta emission of 0.69 MeV and a gamma emission. The gamma component makes it amenable to imaging using conventional gamma cameras. Key disadvantages of iodine-131 are: (a) the long path length of the gamma component, which can result in increased exposure to hospital staff during treatment administration and follow-up [80, 140]; (b) retention of isotope in the tumor is lower due to dehalogenation and release of the iodine from the targeted antibody [101, 122]; and (c) large variability among patients in radioisotope excretion, thus requiring dosimetry for customized patient dosing [178].
For yttrium-90, the beta energy emission is considerably higher (2.2 MeV) and the maximum distance penetrable by the beta particle is 5 mm as compared with 2 mm for iodine-131 [8, 13, 55]; therefore, yttrium-90 can deposit a greater radiation dose over a much larger distance, which translates into potentially higher efficacy in larger tumors [153, 162, 172]. Since yttrium-90 has no gamma component, shielding of hospital personnel or using high patient doses are easily managed and do not require hospital stay after administration; however, the lack of a gamma component requires the use of a surrogate isotope if an imaging component is desired. Typically, indium-111 is used since binding to the targeting antibody conjugate is similar to that of yttrium-90. This use has been documented in numerous animal and human clinical trials, and although direct correlation between the two isotopes is not absolute, biodistribution profiles are very similar to enable imaging and dosimetric data to be obtained [31, 63]. The gamma component energy of indium-111 is one-half lower than that of iodine-131, and typical imaging doses are much smaller than the high doses needed to achieve a therapeutic response; therefore, exposure of hospital or other personnel after indium-111-labeled antibody administration is minimal.
Preparation and characterization of radiolabeled antibody
Regardless of the type of radioisotope selected, it must be attached to the targeting antibody to provide a stable conjugate without compromising the affinity and specificity of the antibody for the target antigen. Since efficacy is dependent on localization and retention of the isotope at the tumor site, the linkage between the isotope and antibody is critical. For example, using traditional conjugation strategies, dehalogenation of iodine constructs results in a lower retention of isotope in the tumor [101, 122].
A second consideration is the process of conjugating the isotope to the antibody which should result in over 95% radioincorporation. As non-antibody-bound radioisotopes may localize in normal organs leading to toxicity, free iodine or iodine released after dehalogenation will selectively localized in the thyroid. Similarly, free yttrium-90 will accumulate in bone [91], requiring not only having >95% radioincorporation but also special consideration in formulation to prevent bone accumulation.
For yttrium-90, direct attachment to the antibody is not feasible; therefore, a bifunctional chelating agent is first attached to the antibody. Early chelators were not capable of effectively retaining yttrium-90 under physiological conditions due to low affinity [79, 182]. This is particularly important with yttrium-90, given its propensity to localize to bone, causing unacceptable bone marrow toxicity [182]. It was not until the mid-1980s that newer high-affinity chelators became available which enabled easy insertion of the metal and provided enhanced in vivo stability [14, 143, 182]. Several of these improved chelators included derivatives of diethylenetriaminepentaacetic acid (DTPA), such as benzyl DTPA [104, 114] and MX-DTPA [143, 182]. More recently, further modifications to DTPA have provided chelators with even higher affinities for yttrium-90, most notably the cyclohexyl-DTPA derivative [102, 119]. Macrocyclic chelators, such as DOTA, have been increasingly used for attachment of yttrium-90 to antibodies [54, 108]. Although the DOTA analogs conjugated to antibodies exhibit improved in vivo stability for yttrium-90, the radioincorporation process requires harsher conditions, such as elevated temperature, to attain suitable incorporation levels of the radioisotope [107].
Conditions for insertion of the metal into the chelator must be optimized for each antibody conjugate. These conditions are primarily dictated by the amount of chelator attached to the antibody, the pH of labeling, temperature, and incubation time [107, 119]. The ideal chelator enables easy insertion of the yttrium-90 without compromising the integrity of the antibody. The MX-DTPA chelator used in Zevalin provides a highly stable chelation with yttrium-90 and indium-111, as demonstrated by in vitro and in vivo studies [14, 104, 143]. The chelator was attached using isothiocyanate chemistry via lysine amino groups to form a stable, covalent thioether bond. Subsequent insertion of ionic yttrium-90 into the chelator provides the radiolabeled therapeutic antibody.
The relatively short radioactive half-life of yttrium-90 (decays at a rate of 25% per day) makes centralized bulk production difficult. Due to this radioactivity decay and the time required to perform QC testing on a bulk manufactured product, radiolabeling of the antibody with yttrium-90 is best done at the clinical site or regional radiopharmacy. Also, the high-energy beta emission may lead to radiolysis during shipment, requiring the inclusion of a radioprotective formulation to reduce radiolysis. Given the logistical constraints of using yttrium-90, a non-radioactive "cold" kit for on-site preparation of Zevalin was developed [32].
Clinical formulation
Of primary importance in establishing a clinical formulation for radiolabeled antibodies is the minimization of the deleterious effects attributable to radiolysis by iodine-131 and ytrrium-90 [27, 31, 53, 144]. Radiolysis can manifest as a problem during the labeling process and during the storage of the radiolabeled antibody until administered to the patient. It is critical that the integrity of the product be maintained after dose preparation. This potential problem is particularly prevalent with beta emitters, since the radiation emitted is capable of destroying or modifying the affinity of the antibody to the target antigen [144].
A number of radioprotective reagents have been used to minimize radiolysis [27], including protein excipients such as human serum albumin [100, 144]. This protein has a long history of parenteral use as a drug, and despite increasing concerns about the use of a human blood-derived product, it still remains a very effective radioprotectant. Other excipients proven effective as radioprotectants include ascorbate [27, 99] and cysteine [112].
Of key importance is the establishment of excipient concentrations enabling adequate protection until patient dosing which for Zevalin was set at 8 h. A final HSA concentration of 5–6% (w/v) provided adequate stability for Zevalin as measured by potency parameters such as radiochemical purity (measuring loss of radioisotope) and retention of CD20 binding for the radiolabeled product. In this case, retention of yttrium-90 was conserved and loss of CD20 binding was negligible (<3%) over 8 h at 2–8°C.
Assessment of potency
The efficacy of a radioimmunotherapeutic is defined by localization of a radioisotope at the tumor site in sufficient amounts and maintained for sufficient time to sterilize the tumor. More specifically, this function is dependent on four critical parameters: (a) accessibility of the tumor; (b) specificity of the targeting antibody; (c) affinity of the targeting antibody; and (d) stable in vivo retention of the radioisotope at the target site. Using these parameters, the potency of a radioimmunotherapeutic can be defined as being dependent on maximal incorporation of the radioisotope into the targeting antibody in conjunction with maximal retention of specific antigen binding.
For a radioimmunotherapeutic, it is conventional to define the immunoreactivity as the fraction of radiolabeled antibodies in a preparation that binds to the antigen. A particularly popular method is that of Lindmo et al. [106] in which antigen is titrated against a constant amount of radiolabeled antibody. By mathematical extrapolation, the binding at a theoretically infinite amount of antibody enables determination of percent immunoreactivity.
Animal studies
The in vivo specific and non-specific targeting attributes of the antibody must be evaluated as a prelude to clinical studies. Historically, this has been most conveniently carried out using rodent models [95, 111, 168, 180]. The objective of such studies is to evaluate biodistribution, specific organ accumulation and toxicity, stability of the radiolabeled antibody, pharmacokinetics, tumor localization, and dosimetric estimations for humans. Extrapolation of results obtained in rodents to humans requires caution. The ratio of transplanted tumors to mouse body size is very large compared with that observed in the clinical setting; therefore, percolation through the tumor is greater in a mouse, relative to a human, resulting in much greater uptake of the antibody in the mouse tumor [16, 17]. Also, the issue of tissue cross-reactivity in which the targeting antibody may react with normal tissue in humans could not be addressed using mouse models.
Despite the limitations of animal studies, conducting pre-clinical tissue localization and dosimetry studies is required to assess potential efficacy and safety of any radioimmunotherapeutic [2]. In general, several techniques have been reported for estimating dosimetry in animal, and extrapolation to human [9, 12, 109, 116, 157]. The use of animal biodistribution data to predict the radiation doses to selected human organs provides information that can identify potential organ toxicity before clinical trials are initiated.
The most frequently used protocols are based on the methods defined by the Medical Internal Radiation Dose (MIRD) committee [109, 157]. Several assumptions are made in using the MIRD methods and include: (a) homogeneous distribution of the radiolabeled antibody in the tumor; (b) radiation deposition does exceed the size of the tumor; and (c) spherical tumors. Very often these assumptions are incorrect and result in erroneous predictions for human radiation doses. Nevertheless, there is widespread use of these methods to extrapolate animal biodistribution data [12, 31, 120, 141, 146] as a prelude to clinical trials. With the exception of overestimating the radiation dose to kidneys, human radiation dose estimates for Zevalin extrapolated from mouse data were comparable to those ultimately obtained in clinical trials [31].
The success of RIT for lymphoma has not yet been attained with solid tumors. As already mentioned, the key reason is the greater insensitivity of carcinomas to radiation and the limitations of targeting radioisotopes using IgG. Several areas of research are being evaluated to improve the chances of success with carcinomas. These areas include: (a) use of engineered antibody constructs such as domain-deleted antibodies; (b) fractionated dose regimens; (c) combination with chemotherapeutic drugs that may provide synergy; and (d) the use of radiosensitizers and/or radioprotectants.
As described above, the use of CH2 domain-deleted constructs may enable larger doses of radiation to be delivered to tumors [94, 155, 156]. Similar results have been noted for other constructs, such as sc-Fv-CH3 antibodies [86, 132, 148]. In both cases the blood clearance is dramatically enhanced, as compared with that of intact IgG, while maintaining antigen binding and tumor residence time comparable to that of the parent antibodies.
Fractionating the radiolabeled antibody dose has improved the efficacy in carcinoma animal models by delivering cumulatively higher radiation doses than that achieved in a single dose [52, 55, 142]. Unfortunately, these results have not translated into significant clinical improvement; however, the potential advantages of using newer antibody constructs in fractionated dosing schedules remains to be explored.
Studies in animals have demonstrated improved efficacy of RIT when combined with conventional chemotherapy [18, 23, 34, 98, 130, 133]. In some cases the different mechanism of anti-tumor activity compliments the effects of radiation, and provides synergistic effects. Bone marrow toxicity remains a limiting factor for adopting such a strategy. The development of a less marrow-toxic RIT may allow a more effective combination without added toxicity.
Preclinical research using murine models has also demonstrated that therapeutic antibodies, such as rituximab, can also effectively synergize with other therapeutic antibodies [139]. The lack of toxicity associated with unconjugated MAb therapy may allow the potential for increased clinical activity when combined with other radiolabeled antibodies.
An intriguing area of research is the use of radiosensitizers [23, 98], or alternatively, the use of drugs which provide protection for normal tissues from the effects or radiation. These reagents include anti-oxidant compounds such as thiols and aminothiols that act as free-radical scavengers [25, 150, 159]. Several of these compounds are preferentially taken up by normal tissues, and may allow larger doses of the radiolabeled antibody to be administered [137]. The ultimate success of RIT for carcinomas therapy may depend on a combination of several of these approaches.
Clinical development of rituximab (Rituxan, MabThera)
In November 1997 the first monoclonal antibody therapy for cancer, rituximab, was approved by the United States Food and Drug Administration (U.S. FDA) for the treatment of low-grade or follicular, relapsed or refractory, B-cell NHL. Less than 1 year later, rituximab was approved for use in the European Union under the indication of stage III/IV, follicular, chemoresistant, or relapsed (two or more relapses) NHL. Approval of rituximab was based on several studies. These studies included phase-I and phase-I/II trials of single-dose and four weekly doses of rituximab; phase-II single-agent studies evaluating retreatment, extended dosing (eight infusions), and treatment of patients with bulky disease (>10 cm); phase-II combination trials with interferon alpha, and with CHOP (cyclophosphamide/doxorubicin/vincristine/prednisone); and a single-agent pivotal trial.
The pivotal trial was a single-arm, multicenter study conducted in 166 patients with relapsed or refractory, low-grade, or follicular NHL. Patients received rituximab at a dose of 375 mg/m2 four times weekly. The population was heavily pretreated and had significant disease burden. The overall response rate (ORR) was 48%, including 6% complete responses (CR) and 42% partial responses (PR). The median time to progression in responders was 13.1 months [71]. In this study rituximab demonstrated activity in chemoresistant disease (29%), and in patients relapsing after anthracycline therapy (51%) [117].
The FDA-approved label was expanded in May 2001 to include information on the treatment of patients with bulky disease, on retreatment of responders to rituximab, and on an extended treatment schedule that includes eight infusions rather than four.
Single-agent rituximab treatment of indolent lymphoma
Since the approval of rituximab in 1997, the antibody has been evaluated in more than 100 trials in other indications and in combination with different agents. As shown in Table 3, overall response rates to single-agent rituximab in patients with relapsed or refractory, indolent NHL have ranged from approximately 36 to 76%. The lower response rates were observed in patients with relapsed or refractory bulky disease or retreated disease [47, 49], the higher ORR of 76% was seen when rituximab was administered six times weekly to patients with recurrent follicular NHL [10].
Table 3.
Single-agent use of rituximab. NHL non-Hodgkin's lymphoma, CLL chronic lymphocytic leukemia, SLL small lymphocytic lymphoma, MALT mucosa-associated lymphoid tissue, PTLD post-transplant lymphoproliferative disorders, MCL mantle-cell lymphoma, DLCL diffuse large cell lymphoma, CR complete response; PR partial response, ORR overall response rate
| Reference | Indication | Dose and schedule | No. of patientsa | Response rate | ||
|---|---|---|---|---|---|---|
| Indolent lymphoma: relapsed or refractory patients | ||||||
| [117 , 118] | Relapsed or refractory low-grade or follicular B-cell NHL | 375 mg/m2, 4× weekly | 166 | ORR=48% (6% CR, 42% PR) | ||
| Median DR=11.2 months | ||||||
| [61] | Relapsed or refractory follicular NHL | 375 mg/m2, 4× weekly | 70 | ORR=46% (3% CR, 43% PR) | ||
| Median DR=11 months | ||||||
| [181] | Recurrent indolent lymphoma | 375 mg/m2, 4× weekly | 34b | ORR=59% (24% CR, 35% PR) | ||
| Median DR=not available | ||||||
| Median TTP=16 months | ||||||
| [134] | Relapsed or refractory follicular, low-grade NHL | 375 mg/m2, 8× weekly | 37 | ORR=57% (14% CR, 43% PR) | ||
| Median DR and TTP not reached at 13.4+ and 19.4+ months, respectively | ||||||
| [10] | Relapsed or refractory follicular NHL | 375 mg/m2, 6× weekly | 17 | ORR=76% (47% CR, 29% PR) | ||
| Median DR=not available | ||||||
| [47, 48] | Rituximab-relapsed low-grade or follicular NHL | 375 mg/m2, 4× weekly | 60 | ORR=38% (10% CR, 28% PR) | ||
| Median DR=15.7+ months | ||||||
| Median TTP in responders=17.3+ months | ||||||
| [1]; pooled data from multiple studies | Relapsed or refractory, bulky disease, low-grade NHL | 375 mg/m2, 4× weekly | 39 | ORR=36% (3% CR, 33% PR) | ||
| Median DR=6.9 months | ||||||
| Indolent lymphoma: front-line patients | ||||||
| [38] | Front-line follicular NHL with low tumor burden | 375 mg/m2, 4× weekly | 49b | ORR=73% (26% CR, 47% PR) | ||
| Median DR=not available | ||||||
| [74] | Front-line low-grade or follicular NHL | 375 mg/m2, 4× weekly | 20 | ORR=50% | ||
| CR and PR not available | ||||||
| Median DR=not available | ||||||
| [75] | Front line in low-grade follicular NHL and CLL; followed by maintenance | 375 mg/m2, 4× weekly; at 6 months, if not PD, repeat courses every 6 months (not to exceed four repeats) | 60b | After first course: | ||
| ORR=47% (7% CR, 40% PR) | ||||||
| [77] | ≥First course (n=46): | |||||
| ORR=65% (27% CR, 38% PR) | ||||||
| Median DR=not available | ||||||
| Median progression-free survival >2 years | ||||||
| [66] | Front-line or relapsed/refractory patients with follicular NHL; followed by maintenance | Randomized, two cohorts: | 202 (58 front line) | In relapsed/refractory patients: ORR=46% | ||
| 375 mg/m2, 4× weekly | ||||||
| Followed by: | In front-line patients: ORR=66% | |||||
| Observation OR | Median event-free survival: | |||||
| Single infusion, months 3, 5, 7, and 9 | 22.4 months if maintenance | |||||
| 13.4 months if observation | ||||||
| Indolent lymphoma subsets | ||||||
| [56] | Front-line or relapsed/refractory Waldenstrom's macroglobulinemia | 375 mg/m2, 4× weekly; after 3 months if not PD, repeat course | 27 | ORR=44% (44% PR) | ||
| Median TTP in all patients: 16 months | ||||||
| [171] | Front-line or relapsed/refractory | 375 mg/m2, 4× weekly; after 3 months if not PD, repeat course | 22b | ORR=72.7% (50% PR, 22.7% minor response) | ||
| Waldenstrom's macroglobulinemia | ||||||
| Median time to treatment failure in responding patients: not reached | ||||||
| [169] | Front-line or relapsed/refractory | Retrospective analysis of several trials | 30b | ORR=60% (27% PR, 33% minor response) | ||
| Waldenstrom's macroglobulinemia | 375 mg/m2, 4× weekly; median of four infusions (range 1–11.3) | Median time to treatment failure in responders: 8 months | ||||
| [39] | Front-line or relapsed/refractory marginal zone (MALT type) | 375 mg/m2, 4× weekly | 35 | ORR=74% (49% CR, 25% PR) | ||
| Median DR=not available | ||||||
| Chronic lymphocytic leukemia | ||||||
| [87] | Relapsed or refractory CLL | 375 mg/m2, 4× weekly | 30 | ORR=23% (23% PR) | ||
| Median DR=20 weeks | ||||||
| [129] | Front-line or relapsed/refractory CLL or other mature B-cell lymphoid leukemias | Four weekly doses, dose 1 375 mg/m2 with dose escalation beginning at dose 2 (500 to 2, 250 mg/m2) | 45b (39b=CLL) | All patients: ORR=40% | ||
| Median TTP in responders: 8 months | ||||||
| In CLL patients: ORR=36% (all PR) | ||||||
| In CLL patients with highest dose, (2250 mg/m2): ORR=75% (all PR) | ||||||
| [20] | Front-line or relapsed SLL/CLL | First dose 100 mg then randomized to two cohorts: 250 (n=3) OR; 375 (n=30) mg/m2 thrice weekly for 4 weeks | 33 | In all patients: ORR=45% (3% CR, 42% PR) | ||
| Median DR: 10 months | ||||||
| [78] | Front-line and maintenance SLL/CLL | 375 mg/m2, 4× weekly | 56b | After first course: ORR=44% (9% CR, 35% PR) | ||
| At 6 months, if not PD, repeat courses every 6 months (not to exceed four repeats) | Median progression free-survival in first 24 patients: 35 months | |||||
| [167] | Front-line, early-stage CLL | 375 mg/m2, 8× weekly | 21b | ORR=86% (19% CR, 19% nodular PR, 48% PR) | ||
| Median DR=not available | ||||||
| Aggressive lymphoma | ||||||
| [36] | Relapsed or refractory aggressive lymphoma | Randomized, two cohorts: | 54 | ORR=31% (9% CR, 22% PR) | ||
| 1. 375 mg/m2, 8× weekly OR 2. 375 mg/m2 on day 1, followed by 500 mg/m2 7× weekly | 30 DLCL | DLCL patients: ORR=37% | ||||
| 13 MCL | MCL patients: ORR=33% | |||||
| 11 other | Median DR ≥246 days | |||||
| [62] | Front-line or relapsed mantle-cell, relapsed immunocytoma and small B-cell lymphocytic lymphoma | 375 mg/m2, 4× weekly | 120b | ORR=30% (8% CR, 22% PR) | ||
| Front-line MCL patients: ORR=38% | ||||||
| Relapsed MCL patients: ORR=37% | ||||||
| Median DR in MCL=1.2 years | ||||||
| [89] | Relapsed or refractory aggressive NHL | 375 mg/m2, 8× weekly | 57b | ORR=37% (12% CR, 25% PR) | ||
| Median DR=not available | ||||||
| Other lymphoproliferative diseases | ||||||
| [170] | Relapsed multiple myeloma | 375 mg/m2, 4× weekly | 19 | PR=5% | ||
| At 3 months, if not PD, second course | SD=27% | |||||
| [192] | Relapsed, classical Hodgkin's disease | 375 mg/m2, 6× weekly | 22b | ORR=23% | ||
| [57] | Lymphocyte-predominant Hodgkin's disease | 375 mg/m2, 4× weekly | 19b | ORR=100% (42% CR, 11% CRu, 47% PR) | ||
| [110] | Lymphocyte-predominant Hodgkin's disease | 375 mg/m2, 4× weekly | 9b | ORR=100% (67% CR, 33% PR) | ||
| [58] | PTLD | 375 mg/m2 weekly (1–9 infusions) | 12 | ORR=66% (66% CR) | ||
| [83] | PTLD | 375 mg/m2, 4× weekly | 13b | ORR=62% (23% CR) | ||
| [125] | PTLD | 375 mg/m2, 4× weekly, every 6 months | 8 | ORR=75% (37.5% CR) | ||
| Median duration of remission 5+ months | ||||||
aPatients in the intent-to-treat population are listed, unless otherwise noted
bEvaluable patients only
Rituximab has been evaluated in previously untreated patients as a single course and also as maintenance therapy. An ORR of 73%, with a complete response rate of 26%, was achieved in 49 evaluable, previously untreated patients with indolent lymphoma and low tumor burden, receiving a single course (375 mg/m2 times four) of rituximab [158].
The efficacy of the front-line use with maintenance rituximab was evaluated in patients with follicular or small lymphocytic lymphoma (SLL). In one study, patients who had not progressed following 375 mg/m2 four times weekly received repeated 4-week maintenance courses of rituximab every 6 months for a maximum of four courses. An initial assessment of 60 treated patients at 6 weeks post-treatment revealed an overall response rate of 47%. Overall response rate increased to 65% following additional courses of maintenance treatment. Complete response rate increased from 7 to 27%. Ultimately, repeated courses of rituximab improved responses in 30% of the patients. After a minimum follow-up of 15 months, median progression-free survival has not been reached [75].
A second trial evaluated maintenance therapy in newly diagnosed and relapsed or refractory patients with follicular NHL. Following the standard regimen of rituximab (375 mg/m2 four times weekly), 151 of 202 treated patients were randomized to receive additional single infusions of rituximab at months 3, 5, 7, and 9, or to observation only. Preliminary analysis shows a median event-free survival (EFS) of 22.4 months for maintenance and 13.4 months for observation (p<0.05). Although circulating B cells returned to normal by 1 year in patients receiving only induction rituximab, B-cell levels remained low in those on maintenance therapy (p<0.05); however, infection rate was not greater with maintenance therapy than with induction treatment only [66].
Rituximab results have been reported in other indolent lymphoma subsets including Waldenstrom's macroglobulinemia (WM) and extranodal marginal zone lymphomas (MALT type), and also in chronic lymphocytic leukemia (CLL). Dimopoulos has reported on 27 patients with WM treated with a single course of rituximab (375 mg/m2 four times weekly). Patients who remained progression free at 3 months received a second course of treatment. Response rate in these 27 patients was 44% (all partial responses) with a median time to progression in all patients of 16 months [56]. A retrospective analysis of 30 patients with WM from several trials reported a partial response rate of 27%, minor response rate of 33%, and no complete responses [169]. Conconi et al. reported a 74% response rate in 35 patients with marginal zone lymphoma with a 49% complete response rate [39].
Initial reports of the use of rituximab in patients with SLL noted a lower response rate (13%) than that seen in follicular NHL [117]; however, pharmacokinetic studies also demonstrated lower serum levels of rituximab monitored at 1–4 weeks and 3 and 6 months. It was not clear at that time whether lower response rates were related to the lower density of CD20 antigen on the surface of SLL and CLL cells, or whether higher doses were necessary due to significantly higher numbers of easily accessible circulating and bone marrow cells.
With this in mind, O'Brien et al. [129] treated CLL patients with escalating doses of rituximab. Patients received the standard dose of 375 mg/m2 for the first infusion and up to 2250 mg/m2 for each of infusions two, three, and four [129]. They demonstrated a dose response with an overall response rate (only partial responses were achieved) of 75% in CLL patients at the highest dose and an ORR of 22% in patients receiving the lowest doses of 500–825 mg/m2. Byrd et al. [20] explored 375 mg/m2 administered three times weekly for 4 weeks with initial doses of 100 mg to lower the risk of tumor lysis syndrome. In their study, ORR was 45%.
Single-agent rituximab in aggressive lymphoma
Median overall response rates ranging from 22 to 38% have been achieved in trials including patients with mantle-cell lymphoma (MCL), diffuse large cell lymphoma (DLCL), and other aggressive lymphomas [35, 60, 65].
In a multicenter study in which patients were treated with the standard rituximab regimen, newly diagnosed and previously treated MCL patients achieved overall response rates of 38 and 37%, respectively [62].
Single-agent rituximab for other lymphoproliferative diseases
Rituximab has demonstrated efficacy in other CD20+ malignancies. Faye et al. [58] reported a 66% complete response rate in patients with post-transplant lymphoproliferative disorders (PTLD) receiving between one and nine weekly rituximab infusions. Rituximab therapy resulted in significant clinical efficacy in CD20-positive, lymphocyte-predominant Hodgkin's disease (ORR of 100%) [57, 110]. Results were even noted in relapsed CD20-negative classical Hodgkin's disease (ORR of 23%) [192]. In multiple myeloma, one partial response was observed in a study of 19 patients with relapsed disease [170]. The observed safety and efficacy in other lymphoproliferative diseases has warranted the proposal for additional studies investigating the longer-term efficacy of rituximab-inclusive combination therapies.
Rituximab–chemo combination therapy in indolent NHL
The differing mechanisms of action and lack of cross resistance between chemotherapy and rituximab make combination therapy an attractive option. In vitro studies demonstrate that rituximab sensitizes lymphoma cell lines to the cytotoxic and apoptotic effects of various therapeutic agents such as cisplatin, fludarabine, vinblastine, doxorubicin, and taxol [6, 7, 51, 90]. Other studies have revealed synergistic apoptotic effects between rituximab and the glucocorticoids dexamethasone or hydrocortisone [174]. Several trials have investigated use of rituximab in combination with chemotherapy. Results of trials combining rituximab with anthracycline-containing or fludarabine-containing chemotherapeutic regimens are summarized in Tables 4, 5, and 6.
Table 4.
Response to treatment with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or CHOP plus rituximab
| Response | CHOP plus rituximab (n=202) | CHOP (n=197) |
|---|---|---|
| N | N | |
| Complete response | 106 (52) | 72 (37) |
| Unconfirmed complete response | 46 (23) | 52 (26) |
| Partial response | 15 (7) | 11 (6) |
| Stable disease | 2 (1) | 1 (1) |
| Progressive disease | 19 (9) | 43 (22) |
| Death without progression | 12 (6) | 11 (6) |
| Could not be assesseda | 2 (1) | 7 (4) |
Numbers in parentheses are percentages
Tumor responses were classified as complete response, unconfirmed complete response, partial response, stable disease, or progressive disease according to the International Workshop criteria [30].
aTreatment was stopped because of toxic effects, the patient's decisions, or the investigator's decision before evaluation of the tumor [37]
Table 5.
Rituximab (R) in combination with anthracycline-based regimens. RCD rituximab/cyclophosphamide/decadron, EPOCH etopside/vincristine/doxorubicin/cyclophosphamide/prednisone, ICE ifosamide/etopside/carboplatin, MCP mitoxantrone/chlorambucil/prednisolone, HCVAD fractionated cyclophopshamide/vincristine/doxorubicin/dexamethasone
| Reference | Indication | Dose and schedulea | No. of patientsb | Response rate |
|---|---|---|---|---|
| Anthracycline-based regimens: indolent lymphoma | ||||
| [41] | Front-line or relapsed/refractory low-grade B-cell lymphoma | (R+CHOP) × 6 | 40 | ORR=95% |
| [44] | In completely treated patients | |||
| ORR=100% (63% CR, 37% PR) | ||||
| Median DR: 63.6+ months | ||||
| [76] | Front-line follicular NHL | Randomized, two cohorts: | 82c | ORR=97% (57% CR, 40% PR) |
| R, 375 mg/m2, 4× weekly then: | After a median of 15 months follow-up, 87% of patients are progression free | |||
| 1. (R+CHOP) × 3 OR | ||||
| 2. (R+CVP) × 3 | ||||
| [113] | Front-line follicular NHL | CHOP × 6 | 84 | ORR=72% |
| Followed by R, 375 mg/m2, 4× weekly | (54% CR and Cru; 18% PR); 2-year progression-free survival=76% | |||
| 2-year overall survival=95% | ||||
| [131] | Relapsed or refractory low-grade NHL | RCD × median of 6 | 10 | ORR=100% (40% CR, 60% PR) |
| Median DR=not reached at 10+ months | ||||
| [81] | Advanced indolent NHL, lymphoplasmacytic and MCL | Randomized, two cohorts: | 106c | ORR=82% (40% CR, 42%PR) |
| 1. (R+MCP) × 8 OR | All patients | Results similar and unblinded | ||
| 2. MCP × 8 | Median DR=not available | |||
| Anthracycline-based regimens: chronic lymphocytic leukemia | ||||
| [135] | Relapsed/refractory CLL and autoimmune hematological disease | RCD × 1 | 4 | All patients responded |
| Median DR=not available | ||||
| [72] | Relapsed/refractory advanced chronic CLL | RCD × median of 4 | 22 | ORR=77% (36% CR, 41% PR) |
| Median survival: not reached at 27+ months | ||||
| Anthracycline-based regimens: aggressive lymphoma | ||||
| [37] | Front-line diffuse large B-cell lymphoma: elderly patients | Randomized, two cohorts: | 202 (R-CHOP) 197 (CHOP) | CHOP+R: ORR=82% (76% CR, 7% PR) |
| 1. (R+CHOP) × 8 OR | At 2 years, 70% survival | |||
| 2. CHOP × 8 | CHOP alone: ORR=66% (63% CR, 3% PR) | |||
| [177] | Front-line advanced aggressive NHL | (R+CHOP) × 6 | 33 | ORR=94% (61% CR, 33% PR) |
| Median DR=not reached after 26+ months | ||||
| [185] | Front-line or relapsed/refractory aggressive NHL | (R+EPOCH) × a minimum of 6 | 20c | Front-line patients: ORR=85% (85% CR) |
| Front line | At median of 12 months, 85% progression-free survival | |||
| 14c Relapsed/refractory | Relapsed/refractory patients: ORR=85% (64% CR, 21% PR) | |||
| [85] | Front-line mantle-cell lymphoma | (R+CHOP) × 6 | 40 | ORR=96% (48% CR+CRu, 48% PR) |
| Median progression-free survival: 16.6 months | ||||
| [96] | Relapsed or refractory diffuse large B-cell lymphoma | (R+ICE) × 3 | 31c | ORR=81% (55% CR, 26% PR) |
| Median DR=not available | ||||
| [175] | Front-line diffuse large-cell lymphoma or MCL | (CHOP+R+GMCSF) × 6 | 14c | ORR=100% (21% CR, 79% PR) |
| Median DR=not available | ||||
| [166] | Front-line Burkitt's, Burkitt's-like leukemia, or lymphoma | [R (375 mg/m2, two doses within 12 days)+HCVAD] × 4 | 15c | CR=93% |
| 1 year estimated disease-free survival 86% | ||||
aUnless otherwise noted, one infusion of rituximab (375 mg/m2) is included in each combination cycle
bPatients in the intent-to-treat population are listed, unless otherwise noted
cEvaluable patients only
Table 6.
Rituximab (R) in combination with fludarabine (F)-based regimens. FND fludarabine/mitoxantrone/dexamethasone, M mitoxantrone, RFC rituximab/fludarabine/cyclophosphamide, FCM fludarabine/cyclophosphamide/mitoxantrone
| Reference | Indication | Dose and schedulea | No. of patientsb | Response rate | ||
|---|---|---|---|---|---|---|
| Fludarabine-based regimens: indolent lymphoma | ||||||
| [176] | Front-line or relapsed follicular or indolent NHL | FND × 4→R, 375 mg/m2, 4× weekly | 16c | After FND alone: ORR=94% (37.5% CR, 19% Cru, 37.5% PR) | ||
| After FND+R: ORR=94% (75% CR, 6% CRu, 13% PR) | ||||||
| Median DR=not available | ||||||
| [22] | Front-line stage-IV indolent lymphoma | Randomized, two cohorts, interferon maintenance for both: | 95c | Molecular responses (12 months) | ||
| 1. Concurrent: (Ra+FND) × 6, FND × 2 OR | Concurrent: 79% blood (n=14); 73% marrow (n=12) | |||||
| 2. Sequential: FND × 8→R, 375 mg/m2, 6× weekly | Sequential: 92% blood (n=13); 67% marrow (n=12) | |||||
| One infusion of Ra, then (R+F) × 6 | At 2 years, projected failure-free survival of 88% for responders in blood at 6 months | |||||
| [42] | Front-line or relapsed/refractory low-grade lymphoma | Randomized, two cohorts: | 40 | ORR=90% (82.5% CR/Cru, 7.5% PR) | ||
| Median DR not reached at 15+ months | ||||||
| [194] | Front-line follicular NHL | 1. (F+M) × 6 OR | 69c | Addition of R improved molecular responses by >30% in both arms | ||
| 2. CHOP × 6 | ||||||
| If CR or PR, then →R, 375 mg/m2, 4× weekly | Median DR=not available | |||||
| (F+M) × 6, →R, 375 mg/m2, 4× weekly | ||||||
| [68] | Front-line advanced low-grade NHL | 19c | ORR=95% (53% CR, 42% PR) | |||
| Median DR=not available | ||||||
| Fludarabine-based regimens: chronic lymphocytic leukemia | Randomized: 2 cohorts | |||||
| [21] | Front-line CLL | 1. Concurrent: (Ra+F) × 6→R, 375 mg/m2, 4× weekly OR | Concurrent: 51c | Concurrent: ORR=90% (47% CR, 43% PR) | ||
| 2. Sequential: F × 6 →R, 375 mg/m2, 4× weekly | ||||||
| (R+F) × 4 | Sequential: 53c | Sequential: ORR=77% (28% CR, 49% PR) | ||||
| Median DR=not available | ||||||
| [149] | Front-line or relapsed/refractory, but anthracycline and fludarabine-naive CLL | FCR × 6 (R at 375 mg/m2 for cycle 1 and 500 mg/m2 for cycles 2–6) | 29c | ORR=90% | ||
| Median TTP not reached at median follow-up of 6 months | ||||||
| [64] | Relapsed or refractory CLL | FCR × 6 (R at 375 mg/m2 for cycle 1 and 500 mg/m2 for cycles 2–6) | 102c | ORR=73% (23% CR, nodular PR 14%, PR 36%) | ||
| Median DR=not available | ||||||
| [184] | Front-line CLL | FCR × 6 (R at 375 mg/m2 for cycle 1 and 500 mg/m2 for cycles 2–6) | 79c | ORR=95% (66% CR, 14% nodular PR, 15% PR) | ||
| Median DR=not available | ||||||
| [93] | Front-line CLL | 35c, six courses | Six courses: ORR=77% (57% CR, 20% nodular PR) | |||
| 21c | Three courses: ORR=43% (14% CR, 29% nodular PR) | |||||
| Three courses | Median DR=not available | |||||
| Fludarabine-based regimens: aggressive lymphoma | Randomized: 2 cohorts | |||||
| [82] | Relapsed or refractory follicular or mantle-cell lymphomas | 1. FCM × 4 OR | 80c | FCM alone: ORR=53% (15% CR, 38% PR) | ||
| 2. RFCM X 4 | ||||||
| RFCM: ORR=89% (36% CR, 53% PR) | ||||||
| Median DR=not available | ||||||
aOne standard infusion of rituximab (375 mg/m2) is included in each combination
bPatients in the intent-to-treat population are listed, unless otherwise noted
cEvaluable patients only
The longest follow-up is available for a trial of rituximab plus CHOP (R-CHOP) in indolent NHL. Thirty-eight patients with either relapsed or previously untreated NHL received rituximab, once weekly for 6 weeks, in combination with six cycles of CHOP. The overall response rate was 100%, consisting of 63% complete responses and 37% partial responses. The median duration of response is 63.6+ months and progression-free survival has not been reached after a median observation of 65.1 months [41, 43].
As summarized in Table 5, addition of rituximab to fludaribine-containing regimens resulted in response rates as high as 90% [45].
Rituximab–chemo combination therapy in aggressive NHL
During the past 3 decades, the four-drug CHOP chemotherapy regimen has been the standard for treatment of aggressive NHL. Second- and third-generation regimens have not proved to be more effective as reported by Fisher et al. in 1994 [59], with the results of a phase-III SWOG (Southwest Oncology Group) trial comparing CHOP with the more intensive regimens, ProMACE–CytaBOM, and MACOP-B. The CHOP was as effective (response rate, time to treatment failure, and overall survival), while less toxic, than these later-generation regimens [59].
Coiffier [37] reports that efficacy can be improved by adding rituximab to the CHOP regimen. A randomized, clinical trial of CHOP plus rituximab in patients 60–80 years old with previously untreated diffuse large-B-cell lymphoma was carried out in 399 patients by the Groupe d'Etude des Lymphomes de l'Adulte (GELA) [37]. Patients were randomized to receive eight cycles of CHOP every 3 weeks or eight cycles of CHOP plus rituximab, with rituximab given on day 1 of every cycle. Patients who received the combination therapy achieved a complete and unconfirmed complete response (CRu) rate of 76% which was significantly greater than the CR+CRu rate (63%) achieved in patients treated with CHOP alone (p=0.005; see Table 4). The event-free survival was also significantly longer (p<0.001; Fig. 1) as was survival (p=0.007; Fig. 2). The number of patients alive at 2 years post-treatment with R-CHOP was 70 vs 57% of those treated with CHOP alone. The incidence of grade-3 and grade-4 adverse events was consistent with the expected toxicity profile of CHOP alone. Patients who received the combination therapy had a higher incidence of grade-1 cardiac events.
Fig. 1.
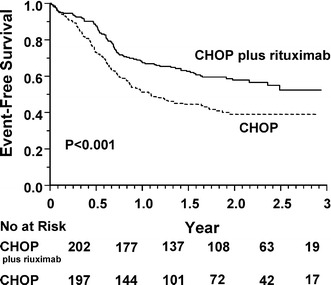
Event-free survival among 399 patients assigned to chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), or with CHOP plus rituximab. (From [37])
Fig. 2.
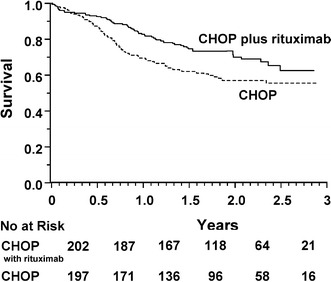
Overall survival among 399 patients assigned to chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), or with CHOP plus rituximab. (From [37])
Rituximab has also been evaluated in combination with EPOCH in patients with newly diagnosed and relapsed aggressive NHL. Response rate was 85% in both subpopulations [185]. The presence of the bcl-2 protein was associated with a significantly shorter progression-free survival in 29 patients treated with EPOCH alone (50% for bcl-2+ patients and 82% for bcl-2− patients at 24 months; p2=0.39). In 23 patients receiving EPOCH+R, no difference in progression-free survival was noted (88% for bcl-2+ patients and 82% for bcl-2− patients at 24 months; p2=0.62) [186].
Rituximab use in nonmalignant disorders
Early anecdotal reports of efficacy of rituximab in immune thrombocytopenic purpura (ITP), autoimmune hemolytic anemia (AIHA), cryoglobulinemia, rheumatoid arthritis (RA), and other autoimmune diseases have led to studies in these areas (Table 7). Clinical efficacy of rituximab in ITP [29, 40, 50, 145, 161], AIHA [73, 193], and RA has been reported [163, 197]. Additional clinical trials in these indications are ongoing along with trials in lupus, graft-vs-host disease, and various neurological disorders including multiple sclerosis and peripheral neuropathies.
Table 7.
Rituximab in non-malignant disorders. ITP immune thrombocytopenic purpura, AIHA autoimmune hemolytic anemia, RA rheumatoid arthritis
| Reference | Indication | Treatment | No. of patientsa | RR (%) |
|---|---|---|---|---|
| [161] | Chronic ITP | 375 mg/m2, 4× weekly | 25 | 52 (20 CR, 20 PR, 12 MR) |
| [50] | Refractory ITP | 375 mg/m2, 4× weekly | 4 | 25 CR |
| [40] | ITP | 375 mg/m2, 4× weekly | 14b | 57 (36 CR, 21 PR) |
| [145] | Refractory ITP | Dose escalation, three arms | 20b | 25 (15 CR, 10 PR) |
| [29] | Severe thrombotic thrombocytopenic purpura | R+steroids+vincristine | 2 | 100 CR |
| [163] | NHL and arthritis in a CLL patient | R 375 mg/m2, 4× weekly+EPOCH | 1 | RA and NHL in remission |
| [73] | AIHA in CLL | R+cyclophosphamide+dexamethasone | 5 | 100 remission |
| [195] | Systemic lupus erythematosus | Dose escalation | 12 | Only low- and medium-dose data: improved arthritis, rash, fatigue but not anti-ds DNA titers or serum complement |
| 100 mg/m2 single infusion | ||||
| 375 mg/m2 single infusion | ||||
| 375 mg/m2 4× weekly |
aPatients in the intent-to-treat population are listed, unless otherwise noted
bEvaluable patients only
Safety
The majority of adverse events associated with rituximab are infusion-related and occur with the first infusion; the most common are fever, chills, nausea, fatigue, headache, angioedema, and pruritus; less common are hypotension and bronchospasm. Although B-cell depletion occurs in most patients, mean serum immunoglobulins remain normal over a 1-year observation period and the incidence of infection does not appear to be increased [117]. Rare severe mucocutaneous reactions, some of which have been fatal, have been observed in association with rituximab. Eight deaths (p<0.01%) and 20 cases occurred in 125,000 treated patients. The incidence of serious skin reactions in lymphoma patients in general is estimated to be 0.07%. Other severe adverse events observed in association with rituximab include tumor lysis syndrome occurring at a rate of 0.1–0.15% and infusion-related deaths at a rate of 0.04–0.07%. For the vast majority of patients receiving rituximab, treatment is well tolerated [70].
Rituximab as a component of ibritumomab tiuxetan (Zevalin)
In February 2002 ibritumomab tiuxetan (Zevalin) radioimmunotherapy was approved by the U.S. FDA for the treatment of patients with relapsed or refractory low-grade, follicular, or CD20+ transformed B-cell NHL, and rituximab-refractory follicular NHL. As discussed in the preclinical section, the antibody moiety of Zevalin, ibritumomab, is a CHO-expressed, murine IgG1 kappa monoclonal antibody, targeting the same epitope on the CD20 antigen as its chimeric counterpart, rituximab. Tiuxetan (MX-DTPA; 1,4-methyl-benzyl isothiocyanate diethylenetriamine pentaacetic acid), a second-generation radiometal chelator, is covalently bound to ibritumomab, allowing for formation of a stable radioimmunoconjugate by chelating 90Y for therapy and 111In for imaging.
The rituximab–ibritumomab tiuxetan (R-ibritumomab tiuxetan) regimen includes rituximab on day 1 (250 mg/m2) immediately followed by 111In ibritumomab tiuxetan (5 mCi), followed 1 week later by rituximab (250 mg/m2) and an injection of 90Y ibritumomab tiuxetan (0.4 mCi/kg for patients with platelets ≥150,000/mm3 and 0.3 mCi/kg for patients with platelets 100,000–149,000/mm3). Gamma camera scans are required at 2–24 h and 48–72 h to rule out rare instances of altered biodistribution.
Six clinical trials were performed to support approval. The initial phase-I and phase-I/II trials demonstrated that unlabelled pretreatment antibody (initially the murine antibody, ibritumomab, later replaced by the chimeric antibody, rituximab) results in improved biodistribution of the radioimmunoconjugate. The phase-III randomized trial demonstrated statistically superior ORR and CR rate in combination R-ibritumomab tiuxetan-treated patients compared with rituximab-treated controls. A trial in rituximab-refractory patients resulted in a 74% response rate [189]. An additional supportive study evaluated unlabeled rituximab and the radioimmunoconjugate of ibritumomab tiuxetan in patients with mild thrombocytopenia. A final open-label study provided additional safety and efficacy data. The studies are summarized in Table 8.
Table 8.
Ibritumomab tiuxetan clinical studies. LG low grade, IG intermediate grade, MC mantle cell, F follicular
| Reference | Indication | Treatment | No. of patients | ORR (%) | CR+CRu (%) | PR (%) | Estimated median TTP | |
|---|---|---|---|---|---|---|---|---|
| [3] | Relapsed or refractory NHL | Phase-I (pretreatment with unlabeled murine antibody, ibritumomab) | 17 (14 single dose) | 64 | 28 | 36 | 9.3 months in responders (6–13 months) | |
| [188, 190] | Relapsed or refractory, LG/IG/MC | Phase I/II | 51 total: | 67 | 26 | 41 | 15.4 months in responders | |
| Dose finding (pretreatment with unlabeled rituximab)a | 34 LG | 82 | 27 | 56 | ||||
| [189] | Relapsed or refractory, LG/F, or transformed | Phase-III randomized, controlled ibritumomab tiuxetan (0.4 mCi/kg) vs rituximab | 143 total: | 80 ibritumomab tiuxetan | 34 ibritumomab tiuxetan | 45 ibritumomab tiuxetan | In all patients, ibritumomab tiuxetan: 11.2+ months vs rituximab: 10.1+ months | |
| 73 ibritumomab tiuxetan | ||||||||
| 70 rituximab | 56 rituximab | 20 rituximab | 36 rituximab | |||||
| [196] | Rituximab-refractory follicular NHL | Phase II (0.4 mCi/kg) | 54 follicular patients | 74 | 15 | 59 | 8.7 months in responders | |
| [187] | Relapsed or refractory, LG/F, or transformed thrombocytopenic patients | Phase-II reduced dose (0.3 mCi/kg) | 30 | 83 | 43 | 40 | 12.6 months in responders |
aTreatment with 0.2, 0.3, or 0.4 mCi/kg
Studies were limited to patients with adequate bone marrow reserve. This included patients with <25% bone marrow involvement with lymphoma, baseline platelet counts ≥150,000/mm3 for standard dosing of 0.4 mCi/kg and 100,000–149,000/mm3 for reduced dosing of 0.3 mCi/kg, absolute neutrophil count ≥1500/mm3, external beam radiation to <25% of active bone marrow, and no prior myeloablative therapies.
The phase-I/II [190] dose-escalating trial was performed in 51 patients with relapsed or refractory low-grade, intermediate-grade, and mantle-cell NHL to determine the maximum tolerated dose (MTD) of ibritumomab tiuxetan. Nonmyeloablative doses of ibritumomab tiuxetan were evaluated as stem cells and were not harvested. Patients in this study had a poor prognosis in that 43% had bone marrow involvement; 59% had bulky disease ≥5 cm, and 37% ≥7 cm; nearly all had had a prior anthracycline-based chemotherapy regimen; 20% had not responded to at least one prior chemotherapy regimen; and 27% had two or more extranodal disease sites. A response rate of 67% was achieved in all patients, with a higher response rate in the low-grade population (82%). The MTD was established as 0.4 mCi/kg in patients with baseline platelet count ≥150,000/mm3 and 0.3 mCi/kg in patients with pre-existing mild thrombocytopenia.
The randomized, phase-III, multicenter, controlled study compared the efficacy and safety of a single-course of pretreatment rituximab followed by ibritumomab tiuxetan (0.4 mCi/kg; n=73) with standard-dose rituximab (375 mg/m2 four times weekly; n=70) in patients with relapsed or refractory low-grade, follicular, or transformed NHL. Median number of prior therapies was 2 (range 1–6). Nearly half of all patients (45%) had tumors ≥5 cm, 20% had tumors ≥7 cm, 8% ≥10 cm, and 56% had not responded to at least one prior chemotherapy regimen.
The overall response rate was 80% for the rituximab plus 90Y ibritumomab tiuxetan group vs 56% for the rituximab group (p=0.002). Complete response rates were 30 and 16%, respectively (p=0.04). An additional 4% in each group achieved an unconfirmed complete response (CRu). Although the study was not powered for comparison of time to progression (TTP), there was a trend towards longer TTP in rituximab plus ibritumomab tiuxetan-treated follicular patients and nontransformed patients (see Figs. 3, 4). Kaplan-Meier estimated median duration of response was 14.2 months in the R-90Y ibritumomab tiuxetan group vs 12.1 months in the control group (p=0.64) and TTP was 11.2 vs 10.1 months (p=0.173) in all patients. Time to next anticancer therapy was 17.6 months in patients treated with 90Y ibritumomab tiuxetan vs 12.4 months in patients treated with rituximab (p=0.1) In all of the trials, there was no difference in response rate between patients ≥65 years of age and patients <65 years of age. [4, 5, 189].
Fig. 3.

Kaplan-Meier analysis of time to progression in follicular patients
Fig. 4.
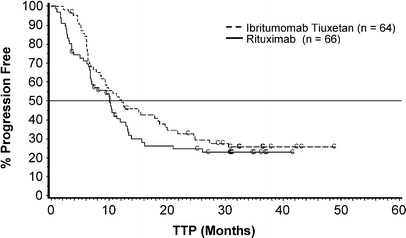
Kaplan-Meier analysis of time to progression in nontransformed patients
A phase-II trial was performed evaluating ibritumomab tiuxetan in 57 patients with NHL refractory to rituximab with refractory defined as disease in which patients achieved no objective response to rituximab (375 mg/m2, four times weekly) or TTP was ≤6 months. The median age was 54 years; 74% had tumors ≥5 cm, 44% had tumors ≥7 cm, 19% ≥10 cm, all were extensively pretreated (median four prior therapies; range 1–9), and 82% had not responded to at least one prior chemotherapy regimen. The ORR for the 54 follicular patients was 74% (15% CR, 59% PR). Kaplan-Meier estimated TTP is 6.8 months (range 1.1–25.9+) for all patients, and 8.7 months (range 1.7–25.9+) for responders [196].
Thrombocytopenic patients have an increased risk of chemotherapy-induced myelosuppression following treatment. The safety and efficacy of radioimmunotherapy with a reduced dose of 90Y ibritumomab tiuxetan (0.3 mCi/kg) was evaluated in a phase-II study in 30 patients with mild thrombocytopenia (platelets 100,000–149,000/mm3) who had advanced, relapsed, or refractory low-grade, follicular, or CD20+ transformed B-cell NHL. Patients had had a median of two prior therapy regimens (range 1–9), 47% had tumors ≥5 cm, 17% had tumors ≥7 cm, 7% ≥10 cm, 63% were resistant to at least one prior chemotherapy regimen, and 67% had bone marrow involvement. The ORR was 83% (37% CR, 6.7% CRu, and 40% PR). Kaplan-Meier estimated median TTP was 9.4 months (range 1.7–24.6+) for all patients and 12.6 months (range 4.9–24.6+ months) in responders [187].
The primary adverse event associated with ibritumomab tiuxetan therapy is transient hematological toxicity. In an integrated analysis of safety in 349 patients, grade-4 neutropenia was observed in 30% of patients, grade-4 thrombocytopenia in 10%, and grade-4 anemia in 4% though only 7% of patients were hospitalized due to infection or febrile neutropenia Nadirs occurred at 7–9 weeks.
The most frequent, nonhematological adverse events (asthenia, chills, fever, nausea, and headache) are those known to be associated with rituximab infusions. Human antimouse antibodies were observed in 1% of patients (3 of 211) and human antichimeric antibodies in 0.05% (1 of 211). No associated toxicity occurred in conjunction with these immune responses. The B-cell depletion recovered by 6–9 months. Serum immunoglobulins were minimally affected and T cells remained normal. Rare cases of myelodysplasia occurred in these trials but were within the expected rate for this heavily pretreated patient population. Patients ≥65 years had no additional safety risk as compared with those <65 years.
Dosimetry studies were performed for 179 patients in these trials. MIRDOSE3 [160] was used to estimate radiation-absorbed dose to uninvolved, major organs and red marrow, and to confirm patient eligibility for radioimmunotherapy. All patients studied with dosimetry met the protocol-defined criteria for proceeding with treatment. Estimated median radiation-absorbed doses were 742 cGy to spleen, 450 cGy to liver, 211 cGy to lungs, 23 cGy to kidneys, and 62 cGy to red marrow. Median effective blood half-life was 27 h and median area under the curve (AUC) was 25 h. Comprehensive statistical analyses revealed that hematological toxicity could not be predicted by any of the dosimetric or pharmacokinetic parameters analyzed, including red marrow dose, total body dose, blood half-life, or blood AUC. This finding was attributed to shortcomings of techniques for estimating marrow dose and the wide spectrum of hematological reserve in these patients with marrow damage from chemotherapy.
Conclusion
The advent of effective immunotherapy and radioimmunotherapy has led to meaningful advances in the treatment of B-cell NHL. The role of rituximab and ibritumomab tiuxetan in the treatment of other CD20+ malignancies and autoimmune diseases will be determined by results of ongoing and future studies.
Footnotes
This article forms part of the Symposium in Writing on "Antibodies in Cancer Immunotherapy", published in this issue (Vol. 52) of the journal.
References
- 1.IDEC Pharmaceuticals Corporation (2001) Rituxan (rituximab). IDEC Pharmaceuticals Corporation, San Diego, vol 19, p 2
- 2.U.S. J Immunother. 1997;20:214. doi: 10.1097/00002371-199705000-00007. [DOI] [PubMed] [Google Scholar]
- 3.IDEC Pharmaceuticals Corporation (1996) A phase I/II clinical trial of yttrium-[90]-labeled IDEC-2B8 given every 6 to 8 weeks to patients with B-cell lymphoma. BB-IND 4850 and 4851. IDEC Pharmaceuticals Corporation, San Diego, vol 106-01-01
- 4.IDEC Pharmaceuticals Corporation (2000) A phase II, open-label, multicenter trial to evaluate the safety and efficacy of IDEC-Y2B8 radioimmunotherapy of relapsed or refractory low-grade or follicular B-cell non-Hodgkin's lymphoma in patients with mild thrombocytopenia. IDEC Pharmaceuticals Corporation, San Diego, vol 106-05
- 5.IDEC Pharmaceuticals Corporation (2000) A phase III, open-label, nonrandomized controlled, multicenter trial to evaluate the safety and efficacy of IDEC-Y2B8 radioimmunotherapy in patients with B-cell non-Hodgkin's lymphoma who are refractory to prior rituximab therapy. IDEC Pharmaceuticals Corporation, San Diego
- 6.Alas Anticancer Res. 2000;20:2961. [PubMed] [Google Scholar]
- 7.Alas Clin Cancer Res. 2001;7:709. [PubMed] [Google Scholar]
- 8.Anderson J Natl Cancer Inst Monogr. 1987;3:149. [Google Scholar]
- 9.Austin Int J Radiat Biol. 2000;76:101. doi: 10.1080/095530000139069. [DOI] [PubMed] [Google Scholar]
- 10.Aviles Cancer Biother Radiopharm. 2001;16:159. doi: 10.1089/108497801300189245. [DOI] [PubMed] [Google Scholar]
- 11.Barnett RS, Limoli KL, Huynh TB, Ople EA (1995) Antibody production in Chinese hamster ovary cells using an impaired selectable marker. In: Wang HY, Imanaka T (eds) Antibody expression and engineering. American Chemical Society, Washington D.C., pp 27–40
- 12.Beatty Cancer. 1994;73:958. doi: 10.1002/1097-0142(19940201)73:3+<958::aid-cncr2820731331>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 13.Berger J Nucl Med. 1971;13:5. [Google Scholar]
- 14.Brechbiel Bioconjug Chem. 1991;2:187. doi: 10.1021/bc00009a008. [DOI] [PubMed] [Google Scholar]
- 15.Brekke Eur J Immunol. 1994;24:2542. doi: 10.1002/eji.1830241042. [DOI] [PubMed] [Google Scholar]
- 16.Buchsbaum DJ (1995) Experimental radioimmunotherapy and methods to increase therapeutic efficacy. In: Goldenberg DM (ed) Cancer therapy with radiolabelled antibodies. CRC Press, Boca Raton, Florida, p 115
- 17.Buchsbaum Med Phys. 1993;20:551. doi: 10.1118/1.597142. [DOI] [PubMed] [Google Scholar]
- 18.Burke Cancer. 2002;94:1320. doi: 10.1002/cncr.10303. [DOI] [PubMed] [Google Scholar]
- 19.Burton Adv Immunol. 1992;51:1. doi: 10.1016/s0065-2776(08)60486-1. [DOI] [PubMed] [Google Scholar]
- 20.Byrd J Clin Oncol. 2001;19:2153. doi: 10.1200/JCO.2001.19.8.2153. [DOI] [PubMed] [Google Scholar]
- 21.Byrd Blood. 2001;98:772a. [Google Scholar]
- 22.Cabanillas Blood. 2000;96:331a. [Google Scholar]
- 23.Cardillo Int J Cancer. 2002;97:386. doi: 10.1002/ijc.1613. [DOI] [PubMed] [Google Scholar]
- 24.Caron Cancer. 1994;73:1049. doi: 10.1002/1097-0142(19940201)73:3+<1049::aid-cncr2820731344>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 25.Cassatt Semin Radiat Oncol. 2002;121:97. doi: 10.1053/srao.2002.31382. [DOI] [PubMed] [Google Scholar]
- 26.Ceriani Antib Immunoconjug Radiopharm. 1990;3:181. [Google Scholar]
- 27.Chakrabarti J Nucl Med. 1996;37:1384. [PubMed] [Google Scholar]
- 28.Chaouchi J Immunol. 1995;154:3096. [PubMed] [Google Scholar]
- 29.Chemnitz Blood. 2001;98:33a. [Google Scholar]
- 30.Cheson J Clin Oncol. 1999;17:1244. doi: 10.1200/JCO.1999.17.4.1244. [DOI] [PubMed] [Google Scholar]
- 31.Chinn Int J Oncol. 1999;15:1017. doi: 10.3892/ijo.15.5.1017. [DOI] [PubMed] [Google Scholar]
- 32.Chinn J Nucl Med. 2001;42:265P. [Google Scholar]
- 33.Chowdhury Nat Biotechnol. 1999;17:568. doi: 10.1038/9872. [DOI] [PubMed] [Google Scholar]
- 34.Clarke Clin Cancer Res. 2000;6:3621. [Google Scholar]
- 35.Coiffier Ann Hematol. 1998;77:A7. [Google Scholar]
- 36.Coiffier Blood. 1998;92:1927. [PubMed] [Google Scholar]
- 37.Coiffier N Engl J Med. 2002;346:235. [Google Scholar]
- 38.Colombat Blood. 2001;97:101. doi: 10.1182/blood.V97.1.101. [DOI] [PubMed] [Google Scholar]
- 39.Conconi Blood. 2001;98:3354. doi: 10.1182/blood.V98.3.781. [DOI] [Google Scholar]
- 40.Cooper Blood. 2001;98:521a. [Google Scholar]
- 41.Czuczman Blood. 2001;98:601a. [Google Scholar]
- 42.Czuczman Blood. 2001;98:601a. [Google Scholar]
- 43.Czuczman J Clin Oncol. 1999;17:268. [Google Scholar]
- 44.Czuczman J Clin Oncol. 1999;17:268. [Google Scholar]
- 45.Czuczman Blood. 2000;96:729a. [Google Scholar]
- 46.Davies Biotechnol Bioeng. 2001;74:288. doi: 10.1002/bit.1119. [DOI] [PubMed] [Google Scholar]
- 47.Davis Blood. 2000;96:235b. [Google Scholar]
- 48.Davis J Clin Oncol. 2000;18:3135. [Google Scholar]
- 49.Davis J Clin Oncol. 1999;17:1851. [Google Scholar]
- 50.Delgado Blood. 2001;98:59b. [Google Scholar]
- 51.Demidem Cancer Biother Radiopharm. 1997;12:177. doi: 10.1089/cbr.1997.12.177. [DOI] [PubMed] [Google Scholar]
- 52.DeNardo Cancer Biother Radiopharm. 1998;13:239. doi: 10.1089/cbr.1998.13.239. [DOI] [PubMed] [Google Scholar]
- 53.DeNardo Cancer Biother Radiopharm. 2000;15:547. doi: 10.1089/cbr.2000.15.547. [DOI] [PubMed] [Google Scholar]
- 54.DeNardo Clin Cancer Res. 1998;4:2483. [PubMed] [Google Scholar]
- 55.DeNardo Cancer. 2002;94:1332. doi: 10.1002/cncr.10304. [DOI] [PubMed] [Google Scholar]
- 56.Dimopoulos J Clin Oncol. 2002;20:2327. doi: 10.1200/JCO.2002.09.039. [DOI] [PubMed] [Google Scholar]
- 57.Ekstrand Blood. 2002;21:264a. [Google Scholar]
- 58.Faye Br J Haematol. 2001;115:112. doi: 10.1046/j.1365-2141.2001.03041.x. [DOI] [PubMed] [Google Scholar]
- 59.Fisher Ann Oncol. 1994;5:91. [Google Scholar]
- 60.Foran Ann Oncol. 2000;11:S117. doi: 10.1023/A:1008309405678. [DOI] [Google Scholar]
- 61.Foran Br J Haematol. 2000;109:81. doi: 10.1046/j.1365-2141.2000.01965.x. [DOI] [PubMed] [Google Scholar]
- 62.Foran J Clin Oncol. 2000;18:317. doi: 10.1200/JCO.2000.18.2.317. [DOI] [PubMed] [Google Scholar]
- 63.Gansow Nucl Med Biol. 1991;18:369. [Google Scholar]
- 64.Garcia Blood. 2001;98:633a. [Google Scholar]
- 65.Ghielmini Ann Oncol. 1999;10:33. [Google Scholar]
- 66.Ghielmini Ann Oncol. 2002;13:38. doi: 10.1093/annonc/mdf159. [DOI] [Google Scholar]
- 67.Goldenberg Cancer Res. 1981;41:4354. [PubMed] [Google Scholar]
- 68.Gregory Blood. 2001;98:605a. [Google Scholar]
- 69.Griffiths GL (1995) In: Goldenberg DM (ed) Radiochemistry of therapeutic radionuclides. Cancer therapy with radiolabelled antibodies. CRC Press, Boca Raton, Florida, p 47
- 70.Grillo-Lopez Semin Oncol. 2002;29:105. doi: 10.1053/sonc.2002.30145. [DOI] [PubMed] [Google Scholar]
- 71.Grillo-López Blood. 2000;96:238b. [Google Scholar]
- 72.Gupta NK, Kavuru S, Patel D, Driscoll N, Janson D, Prasad B, Rai KR (2002) Long-term results from a monthly regimen of rituximab (R), cyclophosphamide (C), and dexamethosone (D) in advanced chronic lymphocytic leukemia (CLL). The 38th Annual Meeting of the American Society of Clinical Oncology, Orlando, Florida, 18–21 May, 21:275a, no. 1098
- 73.Gupta Blood. 2001;98:363a. [Google Scholar]
- 74.Gutheil JC, Finucane D, Rodriquez F, Saleh F, Stahler S, Royston I (2000) Phase II study of rituximab (Rituxan) in patients with previously untreated low-grade or follicular non-Hodgkin's lymphoma. Proc Am Soc Clin Oncol, New Orleans,19:22a, no. 79
- 75.Hainsworth Semin Oncol. 2002;29:25. doi: 10.1053/sonc.2002.30154. [DOI] [PubMed] [Google Scholar]
- 76.Hainsworth JD, Burris HA, Yardley DA, Litchey S, Morrissey LH, Grimaldi M, McCarthy M, Greco FA (2002) Rituximab plus short duration chemotherapy as first-line treatment for follicular non-Hodgkin's lymphoma (NHL): a Minnie Pearl Cancer Research Network phase II trial. The 38th Annual Meeting of the American Society of Clinical Oncology, Orlando, Florida, 7–11 December, 21:268a, no. 1070
- 77.Hainsworth Blood. 2000;95:3052. [PubMed] [Google Scholar]
- 78.Hainsworth Blood. 2001;98:363a. [Google Scholar]
- 79.Harrison Nucl Med Biol. 1991;18:469. [Google Scholar]
- 80.Harwood Cancer. 2002;94:1358. doi: 10.1002/cncr.10306. [DOI] [PubMed] [Google Scholar]
- 81.Herold Blood. 2001;98:601a. [Google Scholar]
- 82.Hiddemann Blood. 2001;98:844a. doi: 10.1182/blood.v98.12.3500. [DOI] [PubMed] [Google Scholar]
- 83.Horwitz SM, Tsai D, Twist C, Sarwal M, Breslin S, Dahl G, Stadtmauer E, Horning S (2001) Rituximab is effective therapy for post-transplant lymphoproliferative disorder (PTLD) not responding to reduction in immunosuppression: a prospective trial in adults and children. Proc Am Soc Clin Oncol, San Francisco, 20:284a, no. 1134
- 84.Hosono Br J Cancer. 1992;65:197. doi: 10.1038/bjc.1992.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Howard J Clin Oncol. 2002;20:1288. doi: 10.1200/JCO.20.5.1288. [DOI] [Google Scholar]
- 86.Hu Cancer Res. 1996;56:3055. [PubMed] [Google Scholar]
- 87.Huhn Blood. 2001;98:1326. doi: 10.1182/blood.V98.5.1326. [DOI] [PubMed] [Google Scholar]
- 88.Idusogie J Immunol. 2000;164:4178. [Google Scholar]
- 89.Igarashi T, Itoh K, Kobayashi Y, Ogura M, Kinoshita T, Taniwaki M, Hiraoka A, Hotta T, Aikawara K, Tsushita K, Tobinai K (2002) Phase II and pharmacokinetic study of Rituximab with eight weekly infusions in relapsed aggressive B-cell non-Hodgkin's lymphoma (B-NHL). The 38th Annual Meeting of the American Society of Clinical Oncology, Orlando, Florida, 18–21 May, 21:286a, no. 1142
- 90.Jazirehi Blood. 2000;96:131a. [Google Scholar]
- 91.Jowsey Radiat Res. 1958;8:490. [PubMed] [Google Scholar]
- 92.Junghans Immunol Res. 1997;16:29. doi: 10.1007/BF02786322. [DOI] [PubMed] [Google Scholar]
- 93.Keating Blood. 2000;96:514a. [Google Scholar]
- 94.Kennel Cancer Biother Radiopharm. 2002;17:219. doi: 10.1089/108497802753773847. [DOI] [PubMed] [Google Scholar]
- 95.Kennel Radiat Res. 2002;157:633. doi: 10.1667/0033-7587(2002)157[0633:vtrwta]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 96.Kewalramani T, Zelenetz A, Bertino J, Donnelly G, Hedrick E, O'Connor O, Portlock C, Straus D, Yahalom J, Gencarelli A, Remy D, Qin J, Nimer S, Moskowitz C (2001) Rituximab significantly increases the complete response rate in patients with relapsed or primary refractory DLBCL receiving ICE as second-line therapy (SLT). The 43rd Annual Meeting of the American Society of Hematology, Orlando, Florida, 7–11 December, 98:346a, no. 1459
- 97.Khazaeli Proc Am Assoc Cancer Res. 2002;43:1015. [Google Scholar]
- 98.Kievit Int J Radiat Oncol Biol Phys. 1997;38:419. doi: 10.1016/S0360-3016(97)82501-1. [DOI] [PubMed] [Google Scholar]
- 99.Kinuya Jpn J Cancer Res. 1998;89:870. doi: 10.1111/j.1349-7006.1998.tb00642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kishore Int J Radiat Appl Instrum B. 1986;13:457. doi: 10.1016/0883-2897(86)90025-5. [DOI] [PubMed] [Google Scholar]
- 101.Knox Cancer Res. 1990;50:4935. [PubMed] [Google Scholar]
- 102.Kobayashi J Nucl Med. 1998;39:829. [PubMed] [Google Scholar]
- 103.Kohler Nature. 1975;256:495. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 104.Kozak Cancer Res. 1989;49:2639. [PubMed] [Google Scholar]
- 105.Kvinnsland Radiat Res. 2001;155:288. doi: 10.1667/0033-7587(2001)155[0288:rwapem]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 106.Lindmo J Immunol Methods. 1984;72:77. doi: 10.1016/0022-1759(84)90435-6. [DOI] [PubMed] [Google Scholar]
- 107.Liu Bioconjug Chem. 2001;12:559. doi: 10.1021/bc000146n. [DOI] [PubMed] [Google Scholar]
- 108.Liu Bioconjug Chem. 2002;13:902. doi: 10.1021/bc010134h. [DOI] [PubMed] [Google Scholar]
- 109.Loeevinger J Nucl. 1968;Med:9. [PubMed] [Google Scholar]
- 110.Lucas Blood. 2000;96:831a. [Google Scholar]
- 111.Ma Leukemia. 2002;16:60. doi: 10.1038/sj.leu.2402320. [DOI] [PubMed] [Google Scholar]
- 112.Maisin Adv Space Res. 1989;9:205. doi: 10.1016/0273-1177(89)90439-0. [DOI] [PubMed] [Google Scholar]
- 113.Maloney Blood. 2001;98:843a. [Google Scholar]
- 114.Mardirossian Nucl Med Biol. 1993;20:65. doi: 10.1016/0969-8051(93)90137-j. [DOI] [PubMed] [Google Scholar]
- 115.Mathas Cancer Res. 2000;60:7170. [PubMed] [Google Scholar]
- 116.Mayer Int J Radiat Oncol Biol Phys. 1994;28:505. [Google Scholar]
- 117.McLaughlin J Clin Oncol. 1998;16:2825. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]
- 118.McLaughlin Semin Oncol. 1999;2:79. [PubMed] [Google Scholar]
- 119.McMurry J Med Chem. 1998;41:3546. doi: 10.1021/jm980152t. [DOI] [PubMed] [Google Scholar]
- 120.Mehta Int J Radiat Oncol Biol Phys. 1990;19:627. [Google Scholar]
- 121.Meredith J Nucl Med. 1992;33:23. [Google Scholar]
- 122.Meredith Antibody Immunoconj Radiopharm. 1992;5:75. [Google Scholar]
- 123.Miller N Engl J Med. 1982;306:517. doi: 10.1056/NEJM198203043060906. [DOI] [PubMed] [Google Scholar]
- 124.Morrison Proc Natl Acad Sci USA. 1984;81:6851. [Google Scholar]
- 125.Morrison VA, Bartlett N, Dunn DL, Peterson A (2001) Rituximab therapy for post-transplant lymphoproliferative disorders (PTLD): preliminary outcome results. Proc Am Soc Clin Oncol, San Francisco, 20:295a, no. 1177
- 126.Newell Proc Natl Acad Sci USA. 1993;90:10459. [Google Scholar]
- 127.Newman Biotechnology. 1992;10:1455. doi: 10.1038/nbt1192-1455. [DOI] [PubMed] [Google Scholar]
- 128.Nourigat J Natl Cancer Inst. 1990;82:47. doi: 10.1093/jnci/82.1.47. [DOI] [PubMed] [Google Scholar]
- 129.O'Brien J Clin Oncol. 2001;19:2165. doi: 10.1200/JCO.2001.19.8.2165. [DOI] [PubMed] [Google Scholar]
- 130.O'Donnell Prostate. 2002;50:27. doi: 10.1002/pros.10029. [DOI] [PubMed] [Google Scholar]
- 131.Patel D, Gupta NK, Mehrotra B, Russo L, Driscoll N, Gor A, Padmanabhan S, Miller N,Rai K R (2001) Rituximab,cyclophosphamide and decadron (RCD) combination is an effective salvage regimen for previously treated low grade lymphoma. Proceedings of the American Society of Clinical Oncology, San Francisco, CA.;20(2):228b, 2665.
- 132.Pavlinkova Clin Cancer Res. 1999;5:2613. [PubMed] [Google Scholar]
- 133.Pedley Int J Cancer. 1994;57:830. doi: 10.1002/ijc.2910570611. [DOI] [PubMed] [Google Scholar]
- 134.Piro Ann Oncol. 1999;10:655. doi: 10.1023/A:1008389119525. [DOI] [PubMed] [Google Scholar]
- 135.Poretta TA, Devereux L, Grana G, Hageboutros A, Sbar E, Patibandla S, O'Brien A, Goldberg J. Rituximab, cyclophosphamide, and dexamethasone provide hematologic and immunologic response in patients with relapsed refractory chronic lymphocytic leukemia and associated autoimmune activity. Proc Am Soc Clin Oncol, San Francisco. 2001;20(2):225b. [Google Scholar]
- 136.Queen Proc Natl Acad Sci USA. 1989;86:10029. [Google Scholar]
- 137.Quinones Clin Cancer Res. 2002;8:1295. [PubMed] [Google Scholar]
- 138.Reff Blood. 1994;83:435. [PubMed] [Google Scholar]
- 139.Reff Cancer Control. 2002;9:152. doi: 10.1177/107327480200900207. [DOI] [PubMed] [Google Scholar]
- 140.Risse Eur J Nucl Med. 2001;28:914. doi: 10.1007/s002590100542. [DOI] [PubMed] [Google Scholar]
- 141.Roberson Cancer Res. 1995;55:5811s. [PubMed] [Google Scholar]
- 142.Roberson Cancer. 1997;80:2567. doi: 10.1002/(SICI)1097-0142(19971215)80:12+<2567::AID-CNCR32>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 143.Roselli Nucl Med Biol. 1991;18:389. [Google Scholar]
- 144.Salako J Nucl Med. 1998;39:667. [PubMed] [Google Scholar]
- 145.Saleh Blood. 2001;98:521a. [Google Scholar]
- 146.Sautter-Bihl Strahlenther Onkol. 1993;169:595. [PubMed] [Google Scholar]
- 147.Schlom Cancer Treat Res. 1990;51:313. doi: 10.1007/978-1-4613-1497-4_16. [DOI] [PubMed] [Google Scholar]
- 148.Schultz Cancer Res. 2000;60:6663. [PubMed] [Google Scholar]
- 149.Schulz Blood. 2001;98:364a. [Google Scholar]
- 150.Schumacher Farmaco. 1997;52:729. [PubMed] [Google Scholar]
- 151.Shan Cancer Immunol Immunother. 2000;48:673. doi: 10.1007/s002620050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shan Blood. 1998;91:1644. [PubMed] [Google Scholar]
- 153.Sharkey Cancer Res. 1990;50:2330. [PubMed] [Google Scholar]
- 154.Shields J Biol Chem. 2001;276:6591. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 155.Slavin-Chiorini Int J Cancer. 1993;53:97. doi: 10.1002/ijc.2910530119. [DOI] [PubMed] [Google Scholar]
- 156.Slavin-Chiorini Cancer Biother Radiopharm. 1997;12:305. doi: 10.1089/cbr.1997.12.305. [DOI] [PubMed] [Google Scholar]
- 157.Snyder WS, Ford MR, Warner GG, Watson SB (1975) "S," absorbed dose per unit cumulated activity for selected radionuclides and organs Medical Internal Radiation Dose (MIRD) pamphlet 11
- 158.Solal-Celigny Anti Cancer Drugs. 2001;12:S11. [PubMed] [Google Scholar]
- 159.Srinivasan Int J Radiat Biol. 2002;78:535. doi: 10.1080/095530002317577358. [DOI] [PubMed] [Google Scholar]
- 160.Stabin J Nucl Med. 1996;37:538. [PubMed] [Google Scholar]
- 161.Stasi Blood. 2001;98:952. doi: 10.1182/blood.V98.4.952. [DOI] [PubMed] [Google Scholar]
- 162.Stein Cancer. 1994;73:816. doi: 10.1002/1097-0142(19940201)73:3+<816::aid-cncr2820731311>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 163.Stewart Ann Rheum Dis. 2001;60:892. [PMC free article] [PubMed] [Google Scholar]
- 164.Tao J Immunol. 1989;143:2595. [PubMed] [Google Scholar]
- 165.Tao J Exp Med. 1993;178:661. doi: 10.1084/jem.178.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Thomas Blood. 2001;98:804a. [Google Scholar]
- 167.Thomas Blood. 2001;98:364a. [Google Scholar]
- 168.Tian Chin Med. 2000;J:53. [Google Scholar]
- 169.Treon J Immunother. 2001;24:272. doi: 10.1097/00002371-200105000-00012. [DOI] [Google Scholar]
- 170.Treon Blood. 2000;11:164a. [Google Scholar]
- 171.Treon SP, Emmanouilides CE, Kimby E, Kelliher A, Preffer F, Freeman A, Mitsiades CS, Mitsiades N, Ainsley S, Fernando D, Anderson K, Frankel SR (2002) Extended rituximab therapy is highly effective and facilitates hematological recovery in patients with lymphoplasmacytic lymphoma (Waldenstrom's macroglobulinemia, WM). The 38th Annual Meeting of the American Society of Clinical Oncology, Orlando, Florida, 18–21 May, 21:268a, no. 1068
- 172.Ugur Nucl Med Biol. 1996;23:1. doi: 10.1016/0969-8051(95)02001-2. [DOI] [PubMed] [Google Scholar]
- 173.Valentine Eur J Immunol. 1992;22:3141. doi: 10.1002/eji.1830221217. [DOI] [PubMed] [Google Scholar]
- 174.Veatch AL, Smith BE, Maloney DG (2001) Synergy between rituximab and glucocorticoids in vitro. Proc Am Soc Clin Oncol, San Francisco, 20:274a, no. 1094
- 175.Venugopal Blood. 2001;98:251b. [Google Scholar]
- 176.Vitolo Blood. 2001;98:251b. [Google Scholar]
- 177.Vose J Clin Oncol. 2001;19:389. doi: 10.1200/JCO.2001.19.2.389. [DOI] [PubMed] [Google Scholar]
- 178.Vose J Clin Oncol. 2000;18:1316. [Google Scholar]
- 179.Vriesendorp Int J Radiat Oncol Biol Phys. 1992;22:37. [Google Scholar]
- 180.Wahl RL (1994) Experimental radioimmunotherapy. A brief overview. Cancer (Suppl) 73:989–992 [DOI] [PubMed]
- 181.Walewski Med Oncol. 2001;18:141. doi: 10.1385/mo:18:2:141. [DOI] [PubMed] [Google Scholar]
- 182.Washburn Nucl Med Biol. 1991;18:313. [Google Scholar]
- 183.Wessels Med Phys. 1984;11:637. doi: 10.1118/1.595559. [DOI] [PubMed] [Google Scholar]
- 184.Wierda Blood. 2001;98:771a. [Google Scholar]
- 185.Wilson Semin Oncol. 2002;29:41. doi: 10.1053/sonc.2002.30151. [DOI] [PubMed] [Google Scholar]
- 186.Wilson Blood. 2001;98:343a. [Google Scholar]
- 187.Wiseman Blood. 2002;99:4336. doi: 10.1182/blood.v99.12.4336. [DOI] [PubMed] [Google Scholar]
- 188.Witzig Semin Oncol. 2000;27:74. [PubMed] [Google Scholar]
- 189.Witzig J Clin Oncol. 2002;20:2453. [Google Scholar]
- 190.Witzig J Clin Oncol. 1999;17:3793. doi: 10.1200/JCO.1999.17.12.3793. [DOI] [PubMed] [Google Scholar]
- 191.Wu Proc Natl Acad Sci USA. 1998;95:6037. doi: 10.1073/pnas.95.11.6037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Younes Blood. 2001;98:132a. doi: 10.1182/blood.v98.9.2877. [DOI] [PubMed] [Google Scholar]
- 193.Zecca Blood. 2001;97:3995. doi: 10.1182/blood.V97.12.3995. [DOI] [PubMed] [Google Scholar]
- 194.Zinzani Blood. 2001;98:842a. [Google Scholar]
- 195.Anolik Arthritis Rheum. 2001;44:S387. [Google Scholar]
- 196.Witzig J Clin Oncol. 2002;20:3262. doi: 10.1200/JCO.2002.11.017. [DOI] [PubMed] [Google Scholar]
- 197.Leandro Ann Rheum Dis. 2002;61:883. doi: 10.1136/ard.61.10.883. [DOI] [PMC free article] [PubMed] [Google Scholar]