

Abstract
Based on several previous studies indicating that transfection of genomic DNA can stably alter the character of the cells that take up the exogenous DNA, we investigated antitumor immunity conferred by fusions of syngeneic dendritic cells (DCs) and allogeneic fibroblasts (NIH3T3) transfected with genomic DNA from B16 tumor cells. Fusion cells (FCs) composed of dendritic and genetically engineered NIH3T3 cells were prepared with polyethylene glycol, and fusion efficiency was 30.3%. Prior immunization with FCs prevented tumor formation upon challenge with B16 tumor cells. Efficacy was reduced when studies were performed in mice depleted of NK cells. Vaccination with FCs containing DCs and fibroblasts transfected with denatured DNA did not inhibit tumor growth. Cytotoxic T cell (CTL) activity of spleen cells from immunized mice against both Yac-1 and tumor cells was also stimulated by administration of FCs compared with the activity observed for cells obtained from naïve mice. These data demonstrate the therapeutic efficacy of fusion cell–based vaccine therapy using syngeneic DCs and allogeneic fibroblasts transfected with tumor-derived genomic DNA.
Keywords: Cancer, Dendritic cells, Fibroblasts, Fusions, Tumor immunity
Introduction
Dendritic cells (DCs) are professional antigen-presenting cells (APCs) that have a unique potency for activating T cells. DCs express high levels of major histocompatibility complex (MHC) and adhesion and costimulatory molecules [1]. The efficient isolation and preparation of both human and murine DCs are now possible [2, 3]. Therefore, a DC-based vaccine could potentially be used for the treatment of malignant tumors.
Since mature DCs lose the ability to take up antigens, use of mature DCs requires efficient methods for incorporating tumor-associated antigens (TAAs) into DCs. To date, several methods using DCs for the induction of antitumor immunity have been investigated: DCs pulsed with proteins or peptides extracted from tumor cells [4], DCs transfected with genes encoding TAAs [5], DCs cultured with tumor cells [6], and DCs fused with tumor cells (fusion cells) [7, 8, 9]. As we reported previously, systemic vaccination with recombinant interleukin 12 (IL-12) and fusion cells (FCs) containing dendritic and tumor cells prolonged the survival of tumor-bearing mice [7]. Based on these experimental findings, clinical trials of vaccine therapy using FCs and recombinant human IL-12 against recurrent malignant tumors have begun. The advantages of this vaccination are that (1) FCs can be used to induce antitumor immunity against unknown TAAs and (2) the induction of autoimmune responses against normal cells can be avoided. On the other hand, the disadvantages are that (1) cultured tumor cells are needed and (2) irradiated tumor cells may still exhibit tumorigenicity in vivo.
Classic studies indicate that transfection of genomic DNA can stably alter both the genotype and the phenotype of the cells that take up the exogenous DNA [10]. In addition, it has been reported that immunotherapy using fibroblasts transfected with genomic DNA from tumor cells prolonged the survival of tumor-bearing mice [11, 12]. Based on these reports, we investigated antitumor effects of fusions containing autologous dendritic cells and allogeneic fibroblasts transfected with genomic DNA from tumor cells. The use of fibroblasts transfected with tumor cell genomic DNA overcomes the disadvantages of fusion cell therapy. That is, cultured tumor cells are not needed and, even if fibroblasts acquire tumorigenicity, allogeneic fibroblasts are rejected by the host thereby avoiding tumor formation. The present study demonstrates the antitumor effect and therapeutic efficacy of immunotherapy using FCs containing DCs and fibroblasts transfected with tumor-derived genomic DNA.
Materials and methods
Cell lines and animals
Mouse fibroblast cell line NIH3T3 and mouse malignant tumor cell lines B16 and MC38 were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). CT-2A glioma cells were kindly provided by Dr Seyfried [13]. These cell lines were maintained as monolayer cultures in Dulbecco’s Modified Eagle Medium (DMEM; Cosmo Bio, Tokyo, Japan) containing 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 10% heat-inactivated fetal bovine serum (FBS; BRL, Gaithersburg, MD, USA). Yac-1 cells, obtained from Riken Cell Bank (Tsukuba, Japan), were maintained in RPMI 1640 (BRL) supplemented with 10% FBS. Male C57BL/6 J mice were purchased from Sankyo Laboratory, Shizuoka, Japan. Six-week-old mice (body weight 25 ± 2 g) were used in the experiments.
DNA extraction and transfection
Tumor cell genomic DNA was extracted from B16, CT-2A, or NIH3T3 cells using a DNA extraction kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. In some cases, genomic DNA was denatured by heating at 95°C for 5 min and icing at 5°C for 5 min or digested with DNase enzyme (TOYOBO, Tokyo, Japan; 1 U enzyme into 1 μg genomic DNA).
Transfection of genomic DNA into mouse fibroblasts
NIH3T3 cells were transfected with both genomic DNA and pSVNeo (kindly provided by Dr Y. Manome) using LipofectAMINE (BRL) according to the manufacturer’s instructions. Briefly, 2-μg genomic DNA was mixed with 2-μg pSV2neo. The mixture was then mixed with LipofectAMINE and added to 1×104 NIH3T3 cells. After 48 h, selection medium containing 800 μg/ml G418 (BRL) was added. Surviving colonies of transduced NIH-3T3 cells were expended and used for fusion. The transfectants were named NIH/B16, NIH/CT-2A, and NIH/NIH, respectively.
Retroviral transfection of tumor cells
Green fluorescence protein gene plasmid pCMV-GFP [14] was kindly provided by Dr Y. Manome. PAMP51 retroviral producer cells [15] (kindly provided by Dr Yoshimatsu) were transfected with pCMV-GFP (PAMP51/pCMV-GFP). The supernatant from PAMP51/pCMV-GFP was used to transfect MC38 target cells. After infection, MC38 cells were selected by using 800 μg/ml geneticine sulfate. Stable selection was completed after 14 days, and expression of the GFP was monitored by fluorescent microscopy.
Preparation of DCs
Separation of DCs from mouse bone marrow was performed as described previously [7]. Briefly, the bone marrow was flushed from long bones of mice, and red cells were lysed with ammonium chloride (Sigma, St Louis, MO, USA). Lymphocytes and granulocytes were depleted from the bone marrow cells, and the cells were plated on 24-well culture plates (1×106 cells/well) in RPMI 1640 medium supplemented with 5% heat-inactivated FBS, 50-μM 2-mercaptoethanol, 2-mM glutamate, 100 U/ml penicillin, 100 μg/ml streptomycin (all from Sigma), 10 ng/ml recombinant murine granulocyte-macrophage colony-stimulating factor (GM-CSF; Becton Dickinson, San Jose, CA, USA), and 10 ng/ml recombinant mouse interleukin 4 (IL-4; Becton Dickinson). On day 5 of culture, nonadherent and loosely adherent cells were collected as DCs.
Fusions of dendritic and genetically engineered NIH3T3 cells
DCs were fused with genetically engineered NIH3T3 cells as described previously [7]. Briefly, 1×106 DCs were mixed with 1×106 genetically engineered NIH3T3 cells (NIH/B16, NIH/CT-2A, or NIH/NIH). Then, fusion was started by adding 500 μl of a 50% solution of polyethylene glycol (PEG; Sigma) dropwise for 60 s. The fusion was stopped by stepwise addition of serum-free RPMI medium. After washing three times with phosphate-buffered saline (PBS; Cosmo Bio), fusion cells (FCs) were plated on 100-mm petri dishes in the presence of GM-CSF and IL-4 in RPMI medium for 24 h. The various FCs were identified as FC/B16, FC/CT-2A, and FC/NIH, respectively.
Analysis of fusion efficiency
Fusion efficiency was investigated as follows. DCs and NIH3T3 cells were stained with fluorescein isothiocyanate–labeled antimouse CD80 monoclonal antibody (Pharmingen, San Diego, CA, USA) and PKH-26 (Sigma), respectively, according to the manufacturer’s instructions. Immediately, those cells were fused as described above. FCs were resuspended in a buffer (1% BSA, 0.1% Sodium azide in PBS) and analyzed using a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA). Double-positive cells were determined to be fusion cells. Fusion efficiency was calculated as follows: Fusion efficiency = (double positive cells / total cells)x100 (%).
Animal models
FCs were washed twice with PBS, then suspended in PBS at a density of 1×106 cells/ml. FCs (3 x105) were subcutaneously (s.c.) inoculated into the flank of C57/6 mice on days 0 and 7. Subsequently, B16 tumor cells (1×106) were inoculated s.c. into the flank on day 14.
Assay of cytolytic activity
The cytolytic activity of spleen cells (SPCs) was tested in vitro using a standard 51Cr-release assay [16]. Single-cell suspensions of SPCs from individual mice were washed and resuspended in 10% FCS-RPMI at a density of 1x107 cells/ml in six-well plates (Falcon Labware, Lincoln Park, NJ, USA). After removing adherent cells, 10 U/ml of recombinant human IL-2 was added to the cultures every other day. Four days after culture initiation, cells were harvested and cytotoxic T cell (CTL) activity was determined. Target cells were labeled by incubation with 51Cr for 90 min at 37°C and then cocultured with effector lymphocytes for 4 h. The effector to target ratio ranged from 10:1 to 80:1. All determinations were made in triplicate and percentage lysis was calculated using the formula: [(experimental cpm − spontaneous cpm) / (maximum cpm − spontaneous cpm)]x100%.
Antibody ablation studies
In vivo ablation of lymphocyte subsets was accomplished as previously described [16]. Briefly, 3x105 FCs were inoculated subcutaneously into the flank of C57/6 mice on days 0 and 7. B16 tumor cells (1×106) were inoculated into the flank on day 14. Antiasialo GM1 (Wako Pure Chemicals, Tokyo, Japan) was injected i.p. (0.5 mg/injection/mouse) on days –1, 3, 7, and 10.
Results
Transfer of tumor DNA leads to expression of encoded genes by MC38 cells
MC38 cells were first stably transfected with a retroviral construct containing a gene for GFP. Genomic DNA isolated from MC38/GFP cells was transferred to NIH3T3 cells, and after 48 h in culture, the transduced cells were tested for expression of GFP by fluorescence microscopy. Clusters of cells expressing GFP were present among transduced cells (Fig. 1A). Flow cytometry confirmed that about 8% of the recipient cells expressed GFP (data not shown). Most of the MC38/GFP cells were positive for GFP (Fig. 1B) and parental NIH3T3 cells were negative (Fig. 1C). These data indicated that the phenotype of the recipient cells was altered by transfer of genomic DNA from the genetically modified MC38 cells.
Fig. 1A–C.
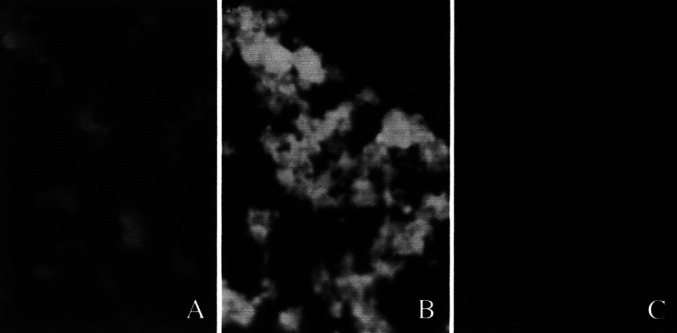
Expression of GFP. Clusters of cells expressing GFP were present among transduced cells (A). Most MC38/GFP cells were positive for GFP (B), and parental NIH3T3 cells were negative (C)
Fusion efficiency
DCs and genetically engineered fibroblasts were stained with antimouse CD80 monoclonal antibody and PKH-26, respectively, and fused by using PEG. Double-positive cells were determined to be fusion cells. Figure 2A shows that 85% of DCs were positive for anti-CD80 monoclonal antibody. More than 97% of NIH/B16 cells were positive for PKH26 (Fig. 2B). The percentage of double-positive cells was 30.3% (Fig. 2C). These experiments were repeated three times and the representative data were shown.
Fig. 2A–C.

Fusion efficiency. DCs and genetically engineered fibroblasts were stained with antimouse CD80 monoclonal antibody and PKH-26, respectively, and fused using PEG. Double-positive cells were determined to be fusion cells. A 85% of DCs were positive for anti-CD80 monoclonal antibody. B More than 97% of NIH/B16 cells were positive for PKH26. C The percentage of double-positive cells was 30.3%
Immunization with FCs followed by tumor inoculation
We examined the antitumor effects of prior immunization with FCs on subcutaneous tumors. FCs (3x105 cells) containing DCs and NIH/B16 (FC/B16), 3x105 NIH/B16 cells (not fused with DCs), 3x105 FCs containing DCs and NIH/CT-2A (FC/CT-2A), or 3x105 NIH3T3 cells as a control were injected s.c. into the flank of C57/6 mice on days 0 and 7 (n=5 in each group). On day 14, 1x106 B16 cells were inoculated s.c. into the flank. The administration of FC/B16 prolonged the latency period before tumor appearance, while the administration of FC/CT-2A, NIH/B16, or NIH3T3 cells did not shorten the latency period before tumor appearance (p<0.05) (Fig. 3A). Vaccination with DCs alone, PBS alone, and a mixture of DCs and NIH/B16 cells had no antitumor effect (data not shown).
Fig. 3A–C.

Antitumor effects of immunization with FCs. A We examined the antitumor effects of prior immunization with FCs on subcutaneous tumors. FC/B16 (solid circles), NIH/B16 cells (not fused with DCs; open squares), FC/CT-2A (solid squares), or NIH3T3 cells (open circles) as a control, were injected s.c. into the flank of C57/6 mice on days 0 and 7 (n=5 in each group). On day 14, 1x106 B16 cells were inoculated s.c. into the flank. The administration of FC/B16 prolonged the latency period before tumor appearance, while the administration of FC/CT-2A, NIH/B16, or NIH3T3 cells did not shorten the latency period before tumor appearance. B We used FCs containing DCs and NIH/3T3 transfected with B16 genomic DNA digested with DNase (solid pyramids) or denatured DNA (open triangles) as a negative control. We also used FC/NIH (open squares). Immunization with these FCs did not shorten the latency period before tumor appearance. C NIH/3T3 cells were transfected with 2 μg (solid circles), 0.2 μg (solid squares), or 0.02 μg (open triangles) of genomic DNA from B16 cells. FCs containing DCs and each type of NIH/3T3 were identified as FC/high, FC/mid, and FC/low, respectively. No difference in antitumor effects was observed in response to immunization with FC/low or NIH3T3 (open circles), whereas immunization with FC/high or FC/mid remarkably inhibited the growth of subcutaneous tumors
Subsequently, we investigated whether the antitumor effect was dependent on the quality of genomic DNA transferred into NIH3T3 cells. We used FCs containing DCs and NIH/3T3 transfected with B16 genomic DNA digested with DNase or denatured DNA as a negative control. We also used FC/NIH (FCs containing DCs and NIH3T3 transfected with genomic DNA from NIH3T3). Immunization with these FCs did not shorten the latency period before tumor appearance (p<0.05) (Fig. 3B), indicating that the antitumor effect induced by FC/B16 was dependent on the quality of tumor-derived genomic DNA transferred into NIH3T3 cells.
Furthermore, we also examined whether the antitumor effect was dependent on the dose of genomic DNA transfected into NIH/3T3 cells. NIH/3T3 cells (3×105) were transfected with 2, 0.2, or 0.02 μg of genomic DNA from B16 cells. FCs containing DCs and each type of NIH/3T3 were identified as FC/high, FC/mid, and FC/low, respectively. FCs were injected s.c. into C57/6 mice on days 0 and 7 (n=5 in each group). On day 14, 1x106 B16 cells were inoculated s.c. into the flank. No differences were observed in antitumor effects upon immunization with FC/mid or FC/low (p>0.05), whereas immunization with FC/high remarkably inhibited the growth of subcutaneous tumors (p<0.05), indicating that the antitumor effect induced by FC/B16 was DNA-dose dependent (Fig. 3C).
Induction of CTL activity
CTL activity was analyzed using a 51Cr-release assay. After immunization with FCs (on days 0 and 7), SPCs were separated from untreated mice and the mice were immunized with FCs on day 14. Figure 4 shows that CTL activity on tumor cells from mice immunized with FC/B16 was considerably higher than in the control or other immunizations, and that antitumor activity on Yac-1 cells from mice immunized with FC/B16 increased. Antitumor activity on NIH/3T3 and CT-2A cells from mice immunized with FC/B16 did not increase (data not shown). These results suggest that vaccination with FC/B16 induced systemic antitumor immunity.
Fig. 4.

Cytotoxicity of spleen cells from tumor-bearing mice. SPCs were separated from mice injected with FC/B16 (solid circles, open circles), mice injected with FCs containing DCs and NIH/3T3 transfected with B16 genomic DNA digested with DNase (solid pyramids), mice injected with FC/CT-2A (solid squares), or mice injected with NIH3T3 cells (open squares) on days 0 and 7. SPCs were separated from the mice on day 14. CTL activity on B16 cells from mice immunized with FC/B16 (solid circles) was considerably higher than in the control and other mice, and antitumor activity on Yac-1 cells from mice immunized with FC/B16 increased (open circles)
NK cells are required for antitumor effects of FCs
We examined the role of NK cells in the antitumor response generated by vaccination with FCs. NK cells were depleted by administering antiasialo GM1 into mice given injections of B16 cells and FCs. On days 0 and 7, FC/B16 were subcutaneously inoculated into the flank. Subsequently, on day 14, B16 cells were inoculated into the same flank. Antiasialo GM1 was injected i.p. on days −1, 3, 7, and 10. The antitumor effect was reduced in mice depleted of NK cells compared with the controls (n=5 in each group) (Fig. 5), suggesting that, in these experiments, NK cells are required for the antitumor effect induced by immunization with FC/B16.
Fig. 5.

NK cells are required for antitumor effects of FCs. NK cells were depleted by administering antiasialo GM1 into mice given injections of B16 cells and FCs. On days 0 and 7, FC/B16 were subcutaneously inoculated into the flank. Subsequently, on day 14, B16 cells were inoculated into the same flank. Antiasialo GM1 was injected i.p. on days −1, 3, 7, and 10. The antitumor effect was reduced in mice depleted of NK cells compared with the controls (n=5 in each group)
Discussion
The results of the present study demonstrate that vaccination with FCs containing DCs and fibroblasts transduced with tumor-derived genomic DNA elicits antitumor immunity. To date, several methods using DCs for the induction of antitumor immunity have been investigated [4, 5, 6, 7, 8]. The advantages of the present method are that (1) fibroblasts have no tumorigenicity, (2) allogeneic fibroblasts are rejected even if those cells acquire tumorigenicity, (3) cultured tumor cells are not needed, (4) antitumor immunity against unknown TAAs can be induced, and (5) several types of genetically engineered fibroblasts can be prepared in advance (e.g., IL-12–transduced fibroblasts). Although fibroblasts exhibit no tumorigenicity, it remains unclear whether genetically engineered fibroblasts are altered such that they acquire tumorigenicity. In the present study, allogeneic fibroblasts were used, and therefore, the host rejected the fibroblasts even if they had acquired tumorigenicity.
In previous studies, genetically engineered DCs were used to elicit antitumor immunity. Ordinary DCs were adenovirally transduced with genes including those for IL-2, IL-12, GM-CSF, chemokines, and TAAs [5, 17, 18, 19, 20]. The disadvantage of this method was that transfection had to be performed each time DCs were used. On the other hand, although naïve fibroblasts were used in the present study, fibroblasts transfected in advance with specific genes could be used instead of naïve fibroblasts to enhance antitumor effects. The transduction of tumor-derived DNA into genetically engineered fibroblasts such as IL-12 producing fibroblasts or CD40 ligand–expressing fibroblasts, is expected to induce stronger antitumor immunity. We are currently investigating antitumor effects of fusions containing DCs and genetically engineered fibroblasts transfected with both CD40L and tumor-derived genomic DNA.
Studies have reported that genetically engineered fibroblasts alone elicit antitumor immunity [11]. In those studies, fibroblasts were used as APCs. Schoenberger et al. reported that CTL induction required the engineered fibroblasts to express CD80 molecules [21]. These fibroblasts were pulsed with tumor peptides. Therefore, the fibroblasts expressed both TAA and costimulatory molecule. On the other hand, in our study, fibroblasts were used as a transporter of genomic DNA from tumor cells, and both TAA and costimulatory molecules were expressed on DCs. As we reported previously, vaccination with genetically engineered tumor cells that expressed both CD54 and CD80 inhibited the growth of tumors [22], suggesting that a single costimulatory molecule could not induce antitumor effects. DCs express high levels of MHC, adhesion, and costimulatory molecules. Therefore, the FCs used in the present study elicited stronger antitumor immunity than genetically engineered fibroblasts alone. Additionally, injection of allogeneic fibroblasts may induce an allogeneic reaction in the host, resulting in enhanced antitumor effects.
DCs can sensitize CD4+ T cells to specific antigens in a MHC-restricted manner. CD4+ T cells are critical in priming both cytotoxic T lymphocytes and antigen nonspecific effector immune responses, implying that both CD4+ and CD8+ T cells are equally important in antitumor immunity. As reported previously, antitumor effects of cells fused with DCs and tumor cells are mediated via CD8+ T cells, although the role of CD4+ T cells and NK cells is less obvious [7]. In the present study, CTL activity on both B16 cells and Yac-1 cells from mice immunized with FCs was considerably higher than in the control and otherwise immunized mice. In addition, the antitumor effect was reduced in mice depleted of NK cells. These results suggest that antitumor effects of FCs were mediated mainly via NK cells. Coculture of the NK cells with DCs resulted in significant enhancement of NK cell cytotoxicity [23], indicating that the FCs, used in the present study, may stimulate NK cells directly. However, mice cured of their subcutaneous tumors by administration of FCs develop long-term systemic immunity against the parental tumor (data not shown). Additionally, vaccination with FC/CT-2A did not inhibit the growth of B16 cells, suggesting that the antitumor effect in this model is both tumor specific and nonspecific and that T lymphocytes also play a role in antitumor effects induced by vaccination with FCs.
In conclusion, our data suggest that vaccination with FCs containing DCs and fibroblasts transfected with tumor-derived DNA can be used to treat malignant tumors in a mouse model. In the present study, allogeneic fibroblasts were used as a fusion partner. However, allogeneic tumor cells derived from the same organ may potentially be used instead of fibroblasts. The advantage of this method is that an antitumor immunity against common tumor antigens may be induced. Future research will focus on investigating antitumor effects of vaccination with fusion cells composed of syngeneic DCs and allogeneic tumor cells transfected with tumor-derived genomic DNA.
Acknowledgements
This study wassupported by the Bio-Venture Research Fund Project Aid, from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1.Steinman Annu Rev Immunol. 1991;9:271. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 2.Nestle Nat Med. 1998;4:328. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 3.Reeves Cancer Res. 1996;56:5672. [PubMed] [Google Scholar]
- 4.Zitvogel J Exp Med. 1996;183:87. doi: 10.1084/jem.183.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuting J Immunol. 1998;160:1139. [PubMed] [Google Scholar]
- 6.Celluzzi J Immunol. 1998;160:3081. [PubMed] [Google Scholar]
- 7.Akasaki J Immunother. 2001;24:106. [PubMed] [Google Scholar]
- 8.Gong Nat Med. 1997;3:558. doi: 10.1038/nm0597-558. [DOI] [PubMed] [Google Scholar]
- 9.Gong Proc Natl Acad Sci U S A. 1998;95:6279. doi: 10.1073/pnas.95.11.6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wigler Cell. 1978;14:725. doi: 10.1016/0092-8674(78)90254-4. [DOI] [PubMed] [Google Scholar]
- 11.de J Immunol. 1999;162:6934. [PubMed] [Google Scholar]
- 12.Whiteside Proc Natl Acad Sci U S A. 2002;99:9415. doi: 10.1073/pnas.142302399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ecsedy Cancer Res. 1997;57:1580. [PubMed] [Google Scholar]
- 14.Housey Cell. 1988;52:343. doi: 10.1016/s0092-8674(88)80027-8. [DOI] [PubMed] [Google Scholar]
- 15.Yoshimatsu Hum Gene Ther. 1998;9:161. doi: 10.1089/hum.1998.9.2-161. [DOI] [PubMed] [Google Scholar]
- 16.Kikuchi Int J Cancer. 1999;80:425. doi: 10.1002/(sici)1097-0215(19990129)80:3<425::aid-ijc15>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 17.Ribas Anticancer Res. 1999;19:1165. [PubMed] [Google Scholar]
- 18.Kaneko Gene Ther. 2003;10:143. doi: 10.1038/sj.gt.3301872. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura Clin Cancer Res. 2002;8:2742. [PubMed] [Google Scholar]
- 20.Nelson WG, Simons JW, Mikhak B, Chang JF, DeMarzo AM, Carducci MA, Kim M, Weber CE, Baccala AA, Goeman MA, Clift SM, Ando DG, Levitsky HI, Cohen LK, Sanda MG, Mulligan RC, Partin AW, Carter HB, Piantadosi S, Marshall FF (2000) Cancer cells engineered to secrete granulocyte-macrophage colony-stimulating factor using ex vivo gene transfer as vaccines for the treatment of genitourinary malignancies. Cancer Chemother Pharmacol 46[Suppl]:S67 [DOI] [PubMed]
- 21.Schoenberger Cancer Res. 1998;58:3094. [PubMed] [Google Scholar]
- 22.Joki Int J Cancer. 1999;82:714. doi: 10.1002/(SICI)1097-0215(19990827)82:5<714::AID-IJC15>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 23.Yu J Immunol. 2001;166:1590. doi: 10.4049/jimmunol.166.3.1590. [DOI] [PubMed] [Google Scholar]