

Abstract
The extracellular domain of the receptor tyrosine kinase Tie2/TEK (exTEK) has been used as an angiopoietin decoy to study the role of angiopoietins in the tumor–host interactions, using a syngeneic model of experimental metastases and subcutaneous tumor. Soluble exTEK secreted by transfected tumor cells inhibited HUVECs from forming tubes in Matrigel. ExTEK-transfected C26 colon carcinoma and TS/A mammary tumor cells displayed reduced growth rate when injected subcutaneously, and reduced ability to form experimental metastases when injected intravenously. Immunohistochemical analysis of tumors and metastases showed increased leukocytes infiltration and signs of inflammation in exTEK-secreting compared to parental tumor, as well as impairment in neo-vessel growth and organization. However, while neoangiogenesis eventually rescued in the subcutis, it failed to organize in the experimental metastases of exTEK-secreting tumor, contributing to the hampering of metastatic growth and to increased mice survival. The reactive infiltrate of C26TEK contained a different percentage of leukocytes and was responsible for the tumor inhibition. In fact, leukopenia induced by γ-irradiation of recipient mice or injection into interferon gamma (IFN-γ) gene knockout (GKO) mice resulted in reduced mouse survival and an increased number of lung metastases. On the other hand, interleukin (IL)-12 treatment prolonged the survival of mice bearing subcutaneous C26TEK but not of those bearing lung metastases, suggesting that IL-12 could exert further antiangiogenic effects at the site where the tumor can restore neoangiogenesis. These results show in vivo that reduced angiopoietin availability at the tumor site induces a local inflammatory response and impairment of neoangiogenesis which act synergistically to limit tumor growth and metastasis.
Keywords: Angiopoietins, Decoy receptor, Leukocyte infiltration, Tumor angiogenesis, Tumor inflammation
Introduction
The growth of tumor metastases in distant organs requires the organization of a new vessel network. It has been shown that angiopoietins play a very early role in the neoangiogenesis related to tumor growth. The early co-option of preexisting vessels that occurs at the beginning of the angiogenic switch is associated with the expression of angiopoietin-2 at the rim between tumor cells and normal tissue, while angiopoietin-1 expression increases later when these vessels regress and new vessels start to sprout [15].
A tight relation between vascular endothelial growth factor (VEGF) and angiopoietin-2 expression has been demonstrated at the beginning of neoangiogenesis, and the balance between the two factors decides the regression or the survival of new vessels, while the presence of angiopoietin-1 is necessary to further organize the network in a functional structured tree [16]. The Tie2/TEK receptor for angiopoietins is expressed on endothelial cells and transduces activating and inhibitory signals in response to angiopoietin-1 and angiopoietin-2, respectively [9, 20].
In vitro experiments have shown that angiopoietin-1 transduces important signals for the stabilization of vessels and contributes to maintaining endothelial cells in a quiescent status, by increasing platelet endothelial cell adhesion molecule-1 (PECAM-1) and vascular endothelial-cadherin expression and by inhibiting vascular cell adhesion molecule-1 (VCAM-1), intracellular adhesion molecule (ICAM-1), and endothelial leukocyte adhesion molecule 1 (ELAM-1) expression [11, 17]. Therefore, angiopoietin-1 has an anti-inflammatory function and counteracts VEGF-induced activation of endothelial cells.
It has been shown that soluble Tie2/TEK extracellular domain is a functional decoy receptor that, when transduced into tumor cells in vitro or in vivo inhibits neoangiogenesis through reduction of angiopoietin availability, and abrogates tumorigenicity of a human melanoma cell line and of a murine mammary carcinoma cell line in xenografts and immunodeficient mouse models, respectively [18, 19, 25, 28]. Angiopoietin withdrawal was intended as a tool for hampering neo-vessel organization, but its relationship with inflammation or immune response could not be studied in absence of a syngeneic tumor model growing in immunocompetent mice.
To analyze the anti-inflammatory effect of angiopoietin-1 in vivo, the local angiopoietin availability should be reduced at sites where VEGF is released, such as at the tumor site, in order to alter the angiopoietin-VEGF balance.
We transduced the soluble extracellular domain of the Tie2/TEK (exTEK) receptor into the C26 colon carcinoma and TS/A mammary carcinoma cell lines and studied how the secretion of such an “angiopoietin decoy” affects the tumor’s growth and its metastases in syngeneic BALB/c mice. Our data show that reduced angiopoietin availability at tumor site affected tumor growth and metastasis and increased mouse survival. Focusing on one of the two models, the C26 colon carcinoma, we showed by immunohistologic analysis that reduced angiopoietin availability at tumor site impairs neo-vessel organization, induces local inflammation, with endothelial cell activation, leukocyte infiltration, and interferon gamma (IFN-γ) release, and synergizes with interleukin (IL)-12 to control subcutaneous tumor outgrowth.
Materials and methods
Mice and tumors
BALB/c and Nu/Nu mice, 8–10 weeks old, were purchased from Charles River (Calco, Italy). IFN-γ-gene knockout (GKO) mice [8] on BALB/c background (BALB/cIfngtm1Ts) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Mice were maintained under standard conditions according to institutional guidelines.
C26 is a murine colon adenocarcinoma cell line derived from BALB/c mice treated with N-nitroso-N-methylurethane. TS/A is a mammary carcinoma cell line established from a spontaneous carcinoma arising in a BALB/c mouse [23]. F1 fibrosarcoma cell line was obtained in our laboratory from spontaneously transformed embryonic fibroblasts. Tumor cells were cultured in DMEM (GIBCO BRL; Life Technologies Italia, Milan, Italy) supplemented with 10% fetal calf serum (FCS) (GIBCO BRL).
Expression vector construction and tumor cell transfection
The extracellular domain of murine Tie2/TEK (2.3 Kb) was amplified by reverse transcriptase polymerase chain reaction (RT-PCR) from murine lung RNA in two fragments, taking advantage of the unique SpeI site present at base 1,258 of the coding sequence. The following primers were used to amplify respectively the 5’ BamHI-SpeI fragment (A+B) and the 3’ SpeI-EcoRI fragment (C+D): (A) 5’-GGG GAT CCG GGG AAG TAT GGA CTC-3’; (B) 5’-GGA ATT CAC TAG TAG GTA GTG GCC ACC C-3’; (C) 5’-GGG ATC CTA CTA GTG AAG AAA TGA CCC TAG-3’; (D) 5’-CGG AAT TCT CCG AGG TCT GCA GAG CCT GGG GA-3’.
The two fragments were cloned into pGEM7Zf plasmid (Promega, Madison, WI, USA) and sequenced, before being assembled into the BamHI-EcoRI sites of the pCDNA3 expression vector (Invitrogen, Groningen, The Netherlands). In order to introduce a stop codon at the end of the extracellular domain of Tie2/TEK receptor, the XbaI site at 3’ of the EcoRI cloning site was cleaved, filled in with T4 DNA polymerases and re-ligated, thus changing the reading frame and obtaining a TAG stop codon.
The exTEK/pCDNA vector was transiently transfected into COS-7 cells by the DEAE-Dextran procedure and 24 h later the supernatant was tested for the presence of the exTEK by ELISA and immunoblotting. The murine C26 and TS/A tumor cell lines were transfected with 10 μg of exTEK/pCDNA vector by calcium-phosphate precipitation, and selected for G418 resistance. Transduced cells were tested for exTEK secretion by ELISA. The growth rate of exTEK-transfected and parental tumor cell lines was evaluated in vitro at 24, 48, and 72 h, by a spectrophotometric assay, using 1% methylen blue dye in 0.01-M borate buffer, pH 8.5, according to Colombo et al. [5].
Assays for expression of exTEK and other angiogenic molecules
To test the tumor cell supernatant for the presence of exTEK, an ELISA was arranged using the mouse antihuman Tie2/TEK monoclonal antibody Ab33 (kindly provided by Dr P. Lin) [19] for capturing and the rat antimouse mAb TEK4 (kindly provided by Dr Suda) [31] for detection, both recognizing the extracellular domain of the receptor. Parental and exTEK-transfected cell lines were analyzed for the expression of endogenous Tie2/TEK, angiopoietin-1, angiopoietin-2, and VEGF by RT-PCR using the following primers: Tie2/TEK (1,085 bp amplified band): DIR: 5’-CTA GTG AAG AAA TGA CCC TAG-3’; REV: 5’-CCG AGG TCT GCA GAG CCT GGG GA-3’; angiopoietin-1 (764 bp amplified fragment): DIR: 5’-CTT ATG ACA GTT TTC CTT TCC TTT G-3’; REV: 5’-CAT GAG CTC CAG TTG TTG CTT CTG-3’; angiopoietin-2 (1,040 bp amplified fragment): DIR: 5’-CGA CAA GAC TCG AGC TGC AGC-3’; REV: 5’-CCT CTG GTA GTG TAG GCA GGC-3’; VEGF (340 bp amplified fragment): DIR: 5’-CAC GAC AGA AGG AGA GCA GAA GTC-3’; REV: 5’-ACC AAA AGT TTC CCA GGC AGA GG-3’. The amount of secreted VEGF was measured in the supernatant of both cell lines by ELISA (Quantikine M, R&D Systems, Minneapolis, MN, USA) following the manufacturer’s instructions.
In vitro biological assays of ExTEK activity on HUVECs
To test the biological effect of secreted exTEK, we modified the in vitro assay for tube formation on Matrigel by HUVECs. HUVECs were seeded in a transwell coated with Matrigel (Collaborative Research). The transwell was inserted into 24-well plate where 8×104 tumor cells either nontransfected, or transfected with exTEK or with an irrelevant gene (human alpha-folate receptor gene) [22] were previously plated. After 24–48 h of coculture, the slides were removed, fixed, and analyzed on a microscope. To supplement the coculture with an excess of angiopoietin to compete with the soluble decoy, the F1 spontaneous BALB/c fibrosarcoma cell line, constitutively expressing angiopoietin-1 was added to the coculture. In this experiment 4×104 of either parental or exTEK-transfected cells plus 4×104 F1 cells were cocultured with the HUVECs.
In vivo experiments
Mice were injected s.c. with 5×104 C26 or 105 TS/A transfected or not with exTEK or with an irrelevant gene, in 0.2 ml of saline: tumor growth was monitored twice a week by measuring the two diameters with a caliper, and tumor volume was calculated as: short diameter2 x long diameter. To establish experimental lung metastases, 104 exTEK-transfected or parental tumor cells in 0.4 ml saline were injected i.v. in the tail vein of either BALB/c, Nu/Nu, sublethally irradiated (400 rads), or GKO mice. Mice were sacrificed when displaying respiratory symptoms, and lung metastases were counted after lung insuflation with 15% India ink and bleaching in Fekete solution. To establish liver metastases, BALB/c females were anesthesized, spleen were isolated surgically, and injected with different doses of parental or transfected tumor cells in 0.1 ml of saline. After 30 s the spleen was removed and the wound sutured. Mice were sacrificed after 11 or 14 days, the liver removed and fixed in buffered formalin.
Groups of seven mice injected s.c. with tumor cells were given 1 μg of recombinant murine IL-12 (Genetics Institute, Cambridge, MA, USA) i.p. on days 6, 13, 21, and 28; mice injected i.v. with tumor cells received IL-12 on days 1, 4, 6, 8, 11, and 14 at the same dose.
Morphological analysis and immunohistochemistry
For histologic evaluation, tissue samples were fixed in 10% neutral-buffered formalin, embedded in paraffin, sectioned at 4 μm, and stained with hematoxylin and eosin (lungs) or PAS (livers). Subcutaneous tumors and lungs with metastases were embedded in OCT compound (Miles Laboratories, Elkhart, IN, USA), snap-frozen in liquid nitrogen, and stored at –80°C until sectioning. Immunochemical analysis was performed using the biotin streptavidin/HRP method. In brief, 5-μm cryostat sections were fixed in acetone and immunostained with rat or hamster antimouse mAb against CD45 (M1/9.3.4.HL2 hybridoma, T200), CD8 (53.6.72 hybridoma, Lyt2), CD4 (GK1.5 hybridoma, L3T4), Mac-3 (M37/84 hybridoma, TIB168), all from American Type Culture Collection; DEC-205 (NLDC-145, provided by Dr Ralph Steinman, Rockefeller University, New York); Ly6G (RB6-8C5 hybridoma), CD31/PECAM-1 (Mec 13.3 hybridoma), CD3ε (145-2C11 hybridoma), CD31 (MEC13.3 hybridoma), CD34(49E8 hybridoma), VCAM-1/CD106 (429 MVCAM.A hybridoma) and ELAM-1/CD62E (10E9.6 hybridoma) all from Pharmingen (San Diego, CA, USA). Sections were preincubated with biotin quenching kit (DAKO, Glostrup, Denmark) for 10 min, then with 1% BSA in TBS for 20 min and sequentially incubated with optimal dilutions of primary antibodies, biotinylated-rabbit anti-rat IgG (DAKO) or antihamster IgG (PharMingen), followed by streptavidin/HRP (DAKO). Each incubation step lasted 30 min and was followed by a 10-min wash in TBS. Sections were then incubated with 0.03% H2O2 and 0.06% 3,3’-diaminobenzidine (DAKO) or AEC (DAKO) for 2–5 min, washed in tap water, and counterstained with hematoxylin. The number of immunostained cells was determined by light microscopy at ×400 magnification in five fields on a 1-mm2 grid, and given as cells/mm2 (mean ± SD). The percentage of ELAM-1 and VCAM-1 positive vessels was calculated by counting positive vessels on sequential sections stained with CD31 Ab.
Results
Expression and functional activity of soluble exTEK released by tumor transfected cells
The exTEK cloned into the pCDNA3 expression vector was properly secreted upon transfection into COS-7 cells and detectable as a single specific band of 105 kDa in the supernatant and cell lysate analyzed by Western blot (not shown).
To test whether tumor-secreted exTEK mediated a biological effect, we analyzed in vitro HUVECs for tube formation in Matrigel when cocultured with C26 colon carcinoma and TS/A mammary carcinoma transfected or not with exTEK: if soluble exTEK reduced angiopoietin availability, then tube formation in Matrigel should be impaired. HUVECs formed well-organized tubes when cocultured with C26 (Fig. 1A), but not with C26TEK (Fig. 1B) as they did or did not in the presence of TS/A or TS/A-TEK, respectively (data not shown). We then added a source of angiopoietin-1 to the tumor–HUVEC coculture by mixing the F1 cell line spontaneously expressing angiopoietin-1 at a 1:1 ratio. HUVEC tube formation was restored, thus suggesting that an excess of angiopoietin-1 counteracts the exTEK-mediated effect (Fig. 1D). No modification of tube formation was observed when F1 cells were mixed to C26 or C26 transfected with irrelevant gene and cocultured with HUVECs (Fig. 1C).
Fig. 1A–D.

Inhibition of HUVEC tube formation on Matrigel by tumor-secreted soluble exTEK. HUVECs seeded on Matrigel in a transwell, were cocultured for 24 h with C26 (A, C) or C26TEK (B, D). Spontaneous HUVEC tube formation was inhibited by coculture with C26TEK (B) but not with C26 (A); the addition of F1 tumor cells expressing angiopoietin-1, restored HUVEC tube formation in the presence of soluble exTEK (D) and did not affect HUVECs in coculture with C26 (C)
Soluble exTEK affects subcutaneous tumor growth by inducing inflammation and leukocyte infiltration
In vitro, the growth rate of exTEK-transfected and parental cells was similar (ANOVA on the OD620 values demonstrates no statistical significant difference: at 24 h p>0.4, at 48 h p>0.2, at 72 h p>0.8). Moreover, tumor cell lines were screened by RT-PCR and tested negative for the expression of angiopoietin-1, angiopoietin-2, and Tie2/TEK, and positive for VEGF. The amount of VEGF secreted by 5×106 cells was measured by ELISA and ranged between 8.25 and 9.4 ng/ml/48 h.
When C26TEK and parental C26 cells or C26 transfected with an irrelevant gene was injected s.c. into BALB/c mice, 100% of take was observed, but C26TEK grew slowly in comparison with controls, resulting in reduced tumor volume (Fig. 2A), and in slightly increased survival (not shown). Similar results were obtained with TS/A and TS/A-TEK (Fig. 2B). Immunohistochemical analysis was performed to characterize C26 and C26TEK. Subcutaneous C26TEK showed a higher percentage of infiltrating CD45+ cells than C26 (Fig. 3), particularly of macrophages and granulocytes, while the number of lymphocytes and dendritic cells was similar in both tumors (Table 1). The percentage of CD31+ cells was slightly reduced in C26TEK tumors, but a higher percentage of vessels were positive for ELAM-1 and VCAM-1 compared with C26 (Table 1), indicating endothelial cell activation. These data suggest that reduced angiopoietin availability at the site of a subcutaneous tumor induces inflammatory infiltration and vessel activation, but impairment of neoangiogenesis is inadequate thus resulting in limited control of tumor growth.
Fig. 2A,B.

Secreted soluble exTEK affects subcutaneous tumor growth. A Mean tumor volume of C26TEK (open circles) (n=26) and of parental C26 (filled circles) (n=18). B Mean tumor volume of TS/A-TEK (open squares) and of parental TS/A (filled squares) (n=7)
Fig. 3A,B.

Soluble exTEK secreted at tumor site induces leukocytes infiltration. CD45 immunostaining of subcutaneous tumors. A C26TEK, B C26
Table 1.
Immunohistologic analysis of subcutaneous tumors and lung metastasis
| CD45 | CD3 | CD4 | CD8 | Mac3 | Ly6G | DEC205 | CD31 | CD34 | VCAM-1 | ELAM-1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Subcutaneous | |||||||||||
| C26 | 167±20a | 30±11 | 37±10 | 1±1 | 85±7 | 36±21 | 1±1 | 21±6 | 19±4 | 10%b | 7% |
| C26TEK | 291±43 | 29±5 | 27±7 | 4±2 | 195±15 | 73±31 | 2±1 | 16±2 | 12±4 | 67% | 61% |
| Lung metastasis | |||||||||||
| C26 | 150±51 | 11±3 | 5± 3 | 7±5 | 81±10 | 59±18 | <1 | 33±2 | 31±3 | 6% | 5% |
| C26TEK | 325±61 | 39±15 | 31±18 | 21±9 | 240±7 | 18±7 | 13±11 | 6±3 | 8±4 | 65% | 70% |
aPositive cells/mm2 ± SD
bPercentage of positive vessels identified in the same fields on sequential section
Soluble exTEK hampers experimental metastases by impairing tumor angiogenesis and inducing inflammatory infiltration
The effect of exTEK was also tested in a model of experimental lung and liver metastases. The survival of mice injected i.v. with exTEK-secreting tumors was significantly longer than that of mice injected with parental tumor cell lines (Fig. 4A, B), or with tumor cells transduced with an irrelevant gene (not shown). The occurrence and growth of exTEK-secreting lung metastases was delayed, as shown by comparing the number of lung metastases in mice injected with C26TEK and C26 (Fig. 4C). However, mice injected i.v. with exTEK-transduced tumor cells eventually developed a few large lung metastases, thus allowing the examination of their histologic features at later time points. Similarly, C26TEK liver metastases established through spleen injection were largely impaired in comparison to parental C26: differences were recorded in both liver to body weight ratio and number of metastases (Fig. 4D).
Fig. 4A–E.

Secreted soluble exTEK affects tumor metastasis. A Survival of BALB/c mice injected i.v. with C26TEK (n=14) (empty symbols) or parental C26 (n=14) (filled symbols). B Survival of BALB/c mice injected i.v. with TS/A-TEK (n=14) (empty symbols) or parental TS/A (n=14) (filled symbols). C Mean number of lung metastases in mice injected i.v. with C26TEK (empty bars) or C26 (filled bars) (n=7). D Mean number of liver metastases in mice injected intrasplenically with C26TEK (empty bars) or C26 (filled bars) (n=7). E Survival of nude mice (squares) or sublethally irradiated BALB/c mice (diamonds) injected i.v. with C26TEK (n=7) (empty symbols) or C26 (n=7) (filled symbols)
We focused on the C26TEK model to analyze the mechanisms of tumor impairment by histology and immunohistochemistry. Interestingly, C26TEK lung and liver metastases differed in size and localization from C26 metastases. The few C26TEK lung metastases were large and localized mainly on the lung surface, with rare micrometastases within the organ parenchyma, and their growth was slow and progressed constraining the lung parenchyma (Fig. 5A). In contrast, C26 metastases were numerous, disseminated throughout the lung (Fig. 5B), and grew by actively infiltrating the alveolar spaces. Similar to lung metastases, C26TEK liver metastases were few, isolated, larger than C26 metastases (not shown), and had a tendency to grow into the vessel spaces.
Fig. 5A,B.
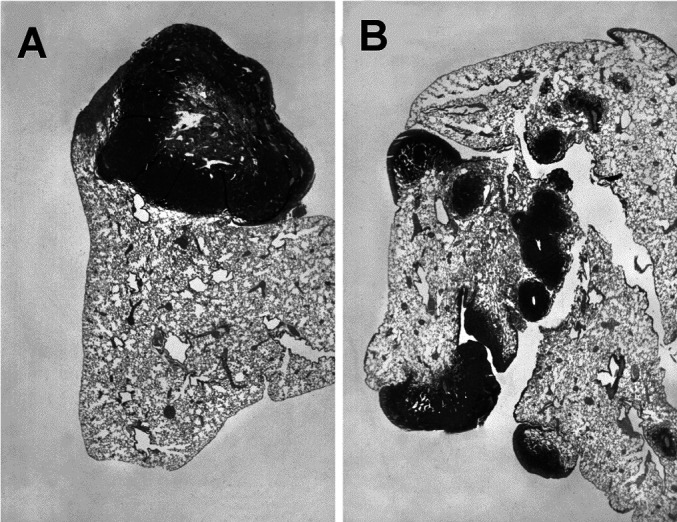
Histologic analysis of lung metastases in BALB/c mice. A Single, isolated, large C26TEK metastasis displaying central necrosis, at day 31. B Multiple C26 metastases spread all over the lung lobe at day 18. Hematoxylin & eosin staining (×60)
C26TEK lung metastases were largely infiltrated by CD45+ reactive cells (Table 1), with a high percentage of T lymphocytes, macrophages, and DEC205+ dendritic cells compared with C26 (Table 1). Independently from metastasis size, C26TEK contained much fewer CD31+/CD34+ endothelial cells than C26. Moreover, sparse CD31+/CD34+ cells not organized into vessels were frequently detected in C26TEK metastases, even at day 31, and many of them were also positive for VCAM-1 (Table 1; Fig. 6A). Isolated CD31+/CD34+ cells were rare in C26 and located at the periphery of the metastases. The hallmarks of C26 metastases were well-organized vessels negative for ELAM-1 and VCAM-1 (Fig. 6B), while in C26TEK metastases, the vessels were few and highly positive for ELAM-1 and VCAM-1 (Table 1; Fig. 6). Outside the tumor, the nearby lung parenchyma of mice injected with either tumors, displayed a limited and comparable infiltration in terms of cell type and number.
Fig. 6A,B.
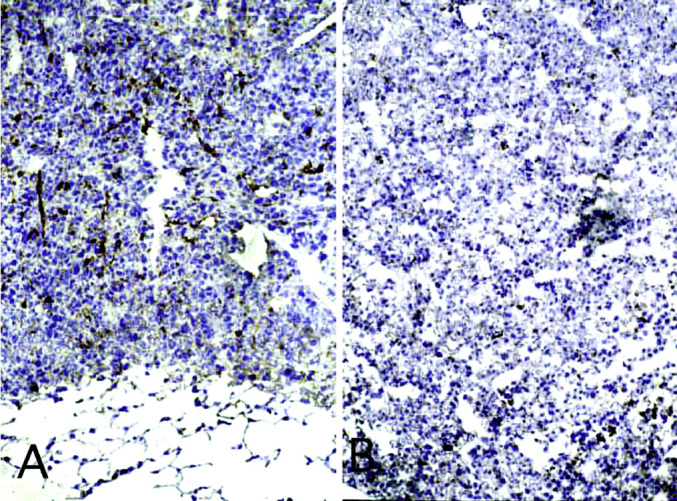
Immunohistochemical staining of lung metastases with Ab to VCAM-1. Positive immunostaining reveals endothelial cell activation in C26TEK (A) but not in C26 (B). (×350)
To test whether leukocytes might take part in C26TEK metastases inhibition, mice were sublethally irradiated prior to i.v. tumor injection. The survival of mice injected with C26TEK was reduced (Fig. 4E). However, no differences in survival were observed between BALB/c Nu/Nu mice and euthymic counterparts injected i.v. with C26TEK (compare Fig. 4A and E), suggesting that leukocytes other than T lymphocytes, such as NK cells, were responsible for delaying metastasis growth in these mice.
These results suggest that angiopoietin withdrawal at metastatic sites impairs the correct organization of endothelial cells into vessels and induces local inflammation with endothelial cell activation and leukocyte recruitment, and these two components are both necessary to control metastasis outgrowth.
Role of IFN-γ in the control of C26TEK lung metastasis
The strong leukocytic infiltration of C26TEK tumors versus C26 and the inflammatory features of tumor-associated vessels prompted us to test whether secondary cytokines were locally released and were responsible for the control of lung metastasis. We analyzed the role of IFN-γ which is known to mediate the release of antiangiogenic molecules, such as IP-10 and MIG [6], and to increase the NO-dependent macrophage tumor cell killing. Toward this aim we injected C26TEK i.v. into GKO mice: the survival of GKO mice was reduced and the number of lung metastases increased significantly (Fig. 7), thus proving that IFN-γ takes part in the inhibition of lung metastasis.
Fig. 7A,B.

Role of IFN-γ in the control of exTEK-secreting tumor metastases. A Survival of GKO mice (filled symbols) and BALB/c mice (empty symbols) injected with C26TEK (squares) (n=9) or C26 (circles) (n=7). B Mean number of C26TEK lung metastases in GKO mice (empty bars) and BALB/c mice (filled bars) at two different time points (n=5)
Effect of recombinant IL-12 on C26TEK tumor growth
In an attempt to boost the antiangiogenic effect of secreted exTEK, we treated BALB/c mice injected i.v. or s.c. with C26 or C26TEK, with repeated doses of recombinant IL-12, and monitored tumor progression and mice survival. The survival of mice bearing s.c. C26TEK was greatly prolonged by IL-12, and in two out of seven mice the tumor completely regressed after an initial growth (Fig. 8A). However, IL-12 treatment did not affect the survival of mice bearing C26TEK lung metastases (Fig. 8B).
Fig. 8A,B.

Effect of recombinant IL-12 on survival of mice bearing C26TEK or C26. A Survival of mice bearing s.c. C26TEK (squares) or C26 (circles) treated (filled symbols) or not (empty symbols) with recombinant IL-12. B Survival of mice bearing C26TEK (squares) or C26 (circles) lung metastases treated (filled symbols) or not (empty symbols) with recombinant IL-12
Discussion
The establishment and remodeling of blood vessels in physiologic, as well as in pathologic conditions, is controlled by paracrine signals, most of which are ligands of transmembrane receptor tyrosine kinases [13]. The Tie2/TEK receptor and its ligands, angiopoietin-1 and angiopoietin-2, create a paracrine circuit responsible for vessel maturation and maintenance of vessel integrity. Angiopoietin expression has been detected focally, at the front of vascular sprouts invading ovary follicles, and developing corpus luteum [12, 20] and tumor masses [16, 27].
In vitro, angiopoietin-1 has been demonstrated to counter the action of VEGF on endothelial cells, by down-regulating VCAM-1 and ELAM-1 expression, therefore reducing vessel permeability and leukocytes extravasation, and acting as an anti-inflammatory agent [11, 17]. Moreover, in vivo, cutaneous vessels overexpressing angiopoietin-1 are resistant to leaks caused by inflammatory agents and VEGF [29]. In vivo, the delivery of soluble extracellular domain of Tie2/TEK or its transduction into tumors by viral vectors was shown to impair tumor growth in xenografts or Nu/Nu mice, by disrupting the neoangiogenesis[18, 19, 25, 28].
The aim of this study was to test if reduced angiopoietin availability at the tumor site would activate an inflammatory/immune response that, together with the impairment of neoangiogenesis, would affect tumor growth and metastasis in a syngeneic immunocompetent mouse model. We transfected the murine colon carcinoma C26 and the mammary carcinoma TS/A with an expression vector encoding the extracellular domain of the Tie2/TEK receptor, acting as a receptor decoy, and tested their tumorigenicity and ability to form experimental metastases in lung and liver.
To test the biologic activity of soluble exTEK, we reasoned that depletion of angiopoietins would affect the morphogenic ability of endothelial cells cultured in matrices such as Matrigel. HUVEC tube formation in Matrigel was effectively inhibited by coculture with exTEK-secreting tumor cells, while tubes were restored when an excess of angiopoietin-1, produced by the F1 fibrosarcoma cell line, was added to the coculture. Though this is not a formal proof that soluble exTEK released by tumor cells prevent the angiopoietin-1 and angiopoietin-2 present in the Matrigel from binding to the Tie2/TEK receptor on HUVECs, this result indicates that the soluble exTEK secreted by transfected tumor cells is active on endothelial cells. It also confirms that the integrity of the angiopoietins–Tie2/TEK pathway is necessary for tube formation in Matrigel as well as in collagen matrix [14, 24].
In our study, the in vivo growth of exTEK-secreting tumors, either colon or mammary carcinoma cell lines, was reduced compared with controls, and sensibly affected by the environment in which tumor cells were seeded. C26TEK grown s.c. was highly infiltrated by leukocytes, and vessels displayed signs of activation; however, the percentage of CD31+ and CD34+ endothelial cells in C26TEK tumors was slightly reduced compared with that of control C26, suggesting that angiopoietin withdrawal induced tumor inflammation but neoangiogenesis impairment was promptly rescued in the subcutis. Both the initial reduction in tumor growth and the late angiogenic rescue of C26TEK in the subcutis may be due to infiltrating macrophages and granulocytes. In fact, macrophages are capable of both protumor and antitumor activity [21], and activated inflammatory and stromal cells have been shown to secrete proangiogenic molecules like VEGF, bFGF, IL-8, as well as diverse enzymatic activities involved in extracellular matrix remodeling [2, 7, 10].
When exTEK-secreting tumors were injected i.v., the metastasis was impaired either in the lung or in the liver, with the few metastases occurring later than in control mice.
Since both parental and transfected tumor cells reached the lung through the blood flow, they seem equally able to extravasate and seed the parenchyma. Here the lack of available angiopoietin-1 at the tumor site would be responsible for a reduced chemotactic signal for the endothelial cells [30], while angiopoietin-2 withdrawal could impair the “destabilization” of preexisting vessels and their sprouting in response to VEGF [13, 15]. Because C26 and C26TEK constitutively express VEGF, the effect of locally secreted exTEK on tumor vascularization suggests that, despite a potent angiogenic factor, the neoangiogenesis can be disrupted at a stage later than endothelial cell activation and proliferation, by interrupting the morpho-angiogenic pathway mediated by Tie2/TEK signaling. The finding of isolated CD31+/CD34+ cells within C26TEK lung metastases supports such a hypothesis; some endothelial cells can be recruited but without being able to organize into tubular structures in absence of angiopoietins. Therefore, the peculiar pleural distribution of C26TEK lung metastases is likely to be due to the survival of tumor cells that, by reaching the organ surface, were fed by diffusion of metabolites. Similarly, in liver, C26TEK cells remain close to the vessel they have extravasated, and metastases protrude into the portal spaces. The C26TEK metastases grow slowly and appear to constrain rather than invade the organ parenchyma.
Whether the late recovery of metastatic growth could be due to the loss of transgene expression was ruled out by showing persistent expression of exTEK in four out of four lung metastases analyzed at day 31 (not shown).
C26TEK lung metastases were characterized by a large leukocyte infiltration that is likely to have contributed to inhibit tumor take and progression, since sublethally irradiated mice survived for a shorter time than normal mice. However, Nu/Nu and immunocompetent BALB/c mice injected i.v. with C26TEK cells showed similar survival, suggesting that NK cells take part in metastasis inhibition together with other leukocytes. We had evidence that NK cells infiltrate C26TEK but not C26 lung metastases, and express IFN-γ, as evaluated by in situ hybridization (data not shown). Therefore, NK cells may contribute to metastasis inhibition in nude mice where they represent the main source of IFN-γ.
The C26 tumor has been shown to be marginally infiltrated by host leukocytes unless genetically engineered to secrete selected cytokines [4, 26]. To explain leukocytic infiltration, it can be hypothesized that endothelial cells directly induce inflammation by sensing the drop of angiopoietins caused by the decoy receptor. Angiopoietin-1 has been shown to act as an antipermeability and anti-inflammatory agent [11, 29] which counteracts the VEGF action on vessel permeability and leukocyte transmigration by reducing expression of adhesion molecules and therefore by maintaining endothelial cells in a quiescent status [17]. Our observations are in agreement with these results and show for the first time in vivo that, in the presence of tumor-secreted VEGF, subtraction of angiopoietin-1 by exTEK directly correlates with up-regulation of VCAM-1 and ELAM-1 on tumor vessels, and with increased leukocytic infiltration. The few organized vessels and the isolated CD31+/CD34+ endothelial cells of C26TEK lung metastases and subcutaneous tumors, strongly expressed VCAM-1 and ELAM-1. Activated endothelial cells may contribute to the local inflammatory condition through cytokine production and leukocyte recruitment [3]. Then, sustained recruitment and inappropriate retention of inflammatory infiltrate work together with stromal factors associated with the local microenvironment [2], a feature that could account also for the rescue of tumor angiogenesis occurring in C26TEK growing subcutaneously but not in the lung. This may explain the results obtained with IL-12 treatment that prolonged the survival of BALB/c mice bearing subcutaneous C26TEK tumors, but not lung metastases. Perhaps, IL-12 potentiates the antitumor effect of soluble exTEK in the subcutis by exerting its antiangiogenic activity where tumor neoangiogenesis is rescued [1, 6].
Finally, recruited leukocytes contribute to maintaining an inflammatory environment through the release of secondary cytokines such as IFN-γ [6]. In fact, i.v. injection of C26TEK into GKO mice resulted in an increased number of lung metastases and reduced survival, suggesting that IFN-γ is needed to counter C26TEK lung metastases.
In conclusion, we have shown that reducing angiopoietin availability at the tumor site impairs tumor growth and metastatic potential, not only through a reduced endothelial cell recruitment and ineffective assembly into functional neo-vessels, but also through the reactive leukocytes that infiltrate the tumor in response to endothelial cell activation, likely caused by angiopoietin-1 drop [11, 17]. Local tumor inflammation may result from the imbalance between the proinflammatory/permeability action of tumor-secreted VEGF and the antiinflammatory/antipermeability action of angiopoietin-1. The complex cross-talk among redundant angiogenic factors may interfere with any therapeutic application of single or a few antagonists of such angiogenic agents. Our data showing the proinflammatory effects of local angiopoietin-1 withdrawal and the consequent tumor leukocytic infiltration, suggest the possibility of targeting the Tie2/TEK–angiopoietin pathway not only to alter the tumor neoangiogenesis, but to activate the host antitumor response, and provide new perspectives for combinatorial antiangiogenic and immunotherapeutic approaches.
Acknowledgements
This work was supported by the Italian Association for Cancer Research (AIRC), by the CNR (grant 99.02526.CT04), the Italian Ministry of University and Scientific Research (MURST), and the ISS Italy-USA Program on Tumor Therapy. We thank Mr Ivano Arioli and Ms Daniela Nicosia for their skillful technical assistance, and Mr Mario Azzini for art work. We are indebted to Dr Pengnian Charles Lin (Duke University Medical Center, Durham, NC) and Prof. Toshio Suda (Institute of Molecular Embryology and Genetics, Kumamoto University School of Medicine, Kumamoto, Japan) for providing the anti-Tie2/TEK-specific antibodies.
Abbreviations
- AEC
amino-ethylcarbazole
- ELISA
enzyme-linked immunosorbent assay
- HRP
horseradish peroxidase
- HUVEC
human umbilical vascular endothelial cell
- i.v.
intravenous
- s.c.
subcutaneous
- TBS
Tris-HCl buffered solution
References
- 1.Boggio Cancer Res. 2000;60:359. [PubMed] [Google Scholar]
- 2.Buckeley Trends Immunol. 2001;22:199. doi: 10.1016/S1471-4906(01)01863-4. [DOI] [PubMed] [Google Scholar]
- 3.Butcher Adv Immunol. 1999;72:209. doi: 10.1016/s0065-2776(08)60022-x. [DOI] [PubMed] [Google Scholar]
- 4.Colombo J Exp Med. 1991;173:889. doi: 10.1084/jem.173.4.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colombo J Immunol. 1992;149:113. [PubMed] [Google Scholar]
- 6.Coughlin Immunity. 1998;9:25. [Google Scholar]
- 7.Coussens LM, Werb Z. Inflammatory cells and cancer: think different! J Exp Med. 2001;193:F23. doi: 10.1084/jem.193.6.f23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dalton Science. 1993;259:1739. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- 9.Dumont Oncogene. 1993;8:1293. [PubMed] [Google Scholar]
- 10.Foda Drug Discov Today. 2001;6:478. doi: 10.1016/S1359-6446(01)01752-4. [DOI] [PubMed] [Google Scholar]
- 11.Gamble Circ Res. 2000;87:603. doi: 10.1161/01.res.87.7.603. [DOI] [PubMed] [Google Scholar]
- 12.Goede Lab Invest. 1998;78:1385. [PubMed] [Google Scholar]
- 13.Hanahan Science. 1997;277:48. doi: 10.1126/science.277.5322.48. [DOI] [PubMed] [Google Scholar]
- 14.Hayes Res. 1999;58:224. doi: 10.1006/mvre.1999.2179. [DOI] [PubMed] [Google Scholar]
- 15.Holash Oncogene. 1999;18:5356. doi: 10.1038/sj.onc.1203035. [DOI] [PubMed] [Google Scholar]
- 16.Holash Science. 1999;284:1994. doi: 10.1126/science.284.5422.1994. [DOI] [PubMed] [Google Scholar]
- 17.Kim Circ Res. 2001;89:477. doi: 10.1161/hh1801.097034. [DOI] [PubMed] [Google Scholar]
- 18.Lin J Clin Invest. 1997;100:2072. doi: 10.1172/JCI119740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin Proc Natl Acad Sci U S A. 1998;95:8829. doi: 10.1073/pnas.95.15.8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maisonpierre Science. 1997;277:55. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 21.Mantovani Immunol Today. 1992;13:265. doi: 10.1016/0167-5699(92)90008-U. [DOI] [PubMed] [Google Scholar]
- 22.Melani Cancer Res. 1998;58:4146. [PubMed] [Google Scholar]
- 23.Nanni Clin Exp Metastasis. 1983;1:373. doi: 10.1007/BF00121199. [DOI] [PubMed] [Google Scholar]
- 24.Papapetropoulos Lab Invest. 1999;79:213. [PubMed] [Google Scholar]
- 25.Siemeister Cancer Res. 1999;59:3185. [PubMed] [Google Scholar]
- 26.Stoppacciaro J Exp Med. 1993;178:151. doi: 10.1084/jem.178.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stratmann Am J Pathol. 1999;153:1459. doi: 10.1016/S0002-9440(10)65733-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stratmann Int J Cancer. 2001;91:273. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1054>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 29.Thurston Science. 1999;286:2511. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- 30.Witzenbichler J Biol Chem. 1998;273:18514. doi: 10.1074/jbc.273.29.18514. [DOI] [PubMed] [Google Scholar]
- 31.Yano Blood. 1997;89:4317. [PubMed] [Google Scholar]