

Abstract
Antibody-based targeted immunotherapy has shown promise as an approach to treat cancer. However, many known tumor-associated antigens are not expressed as integral membrane proteins and cannot be utilized as targets for antibody-based therapeutics. In order to expand the limited target range of antibodies, we have constructed a soluble single-chain T-cell receptor (TCR) fusion protein designated 264scTCR/IL-2. This fusion protein is comprised of a three-domain HLA-A2-restricted TCR specific for a peptide epitope of the human p53 tumor suppressor protein, which is overexpressed in a broad range of human malignancies. The 264scTCR/IL-2 fusion protein has been expressed at high levels in mammalian cells, and milligram quantities have been purified. MHC-restricted antigen-specific binding properties are maintained in the single-chain, three-domain TCR portion of the fusion protein, and the IL-2 portion retains bioactivity similar to that of free recombinant IL-2. Moreover, this fusion protein is capable of conjugating target and effector cells, remains intact in the blood and substantially increases the half life of the IL-2 portion of the molecule. Finally, the 264scTCR/IL-2 fusion protein can be used to stain tumor cells and is capable of reducing lung metastases in an experimental model of metastasis. Thus, TCR-based fusion proteins may provide a novel class of targeted immunotherapeutics for cancer.
Keywords: T cell receptors, Tumor immunity, Molecular biology, Cytokines
Introduction
There is increasing evidence that immunotherapy is a promising approach to treat cancer. One strategy that has received attention is treatment with cytokines such as IL-2 to enhance anti-tumor immunity. IL-2 has stimulatory effects on a number of immune cell types including T and B cells, monocytes, macrophages, lymphokine activated killer cells (LAK) and NK cells [10, 41]. Thus, systemic administration of IL-2 has been used to treat patients with metastatic melanoma and renal cell carcinoma [42]. Although treatment with cytokines has shown some promise, the toxicity associated with these treatments makes it difficult to achieve an effective dose at the site of the tumor [31, 33, 40, 43, 44, 52]. For example, systemic treatment with IL-2 at tolerated doses induces lymphoid activation in virtually all treated patients, but only a minority of these achieves antitumor responses [42, 46, 47, 55]. Thus, there is some interest in devising strategies for concentrating cytokines at the site of the tumor to increase efficacy and reduce toxicity.
Strategies that have been used to concentrate cytokines at the tumor site include direct injection of cytokine into the tumor, transfer of the cytokine gene into the tumor cells and targeted delivery of the cytokine by fusing it to a tumor antigen-specific antibody [20]. Direct injection and transfer of cytokine genes may be effective on a small scale, but have major drawbacks in that direct injection into micrometastases is impossible and gene transfer is labor-intensive with limited success, making treatment of large patient populations impractical. Antibodies directed against tumor-specific antigens have been fused to cytokines and used as anticancer therapies in animal models. For example, fusion of IL-2 to antibodies that recognize ganglioside GD2 or the Id of a murine lymphoma has shown some promise for stimulating specific tumor responses in animal models [4, 16, 17, 27, 28, 36]. Further, fusion proteins consisting of IL-12 or IL-2 fused to anti-HER2/neu antibodies can mediate killing of HER2/neu positive tumor cells [29, 35]. One potential problem with antibody-based tumor targeting is that antibodies only recognize antigens expressed on the cell surface as integral membrane protein; however, there are many known tumor antigens that are not displayed in this way. Moreover, since many tumor-specific antigens are derived from aberrant expression of cell type specific proteins, they may exist in only a small number of tumor types. Thus, they have limited value as targets for antibody-based therapy. Therefore, it is highly desirable to identify a tumor antigen that is expressed in a broad range of tumors that may be recognized by molecules other than antibodies.
p53 is an intracellular tumor suppressor protein that acts to arrest abnormal cells at the G1/S phase of the cell cycle. When p53 is mutated, it loses its function as a tumor suppressor, thus allowing abnormal cells to continue to proliferate [49]. Mutated p53 has been shown to have a longer half-life than the wild type protein, and this longer half-life allows for accumulation and thus overexpression in a wide range of human malignancies [19, 22, 49]. Furthermore, overexpression of p53 in breast cancer cells has been correlated with aggressiveness of the tumor, such that individuals bearing p53 overexpressing tumors have shorter disease-free time and lower overall survival rates [30, 48]. Hence, p53 overexpression appears to be a significant tumor marker for a large number of human malignancies and represents a good target for broad spectrum targeted tumor immunotherapy. However, p53 cannot be used as a target for antibody-based therapies because it is not displayed independently on the cell surface. Instead the p53 gene product is processed and certain peptide epitopes are displayed on the cell surface in the context of MHC, which are recognized by TCRs [49]. As a result, we are investigating the use of TCRs rather than antibodies in targeting p53 for the creation of tumor immunotherapies.
In this report we describe the construction and characterization of a novel fusion protein comprising a soluble single chain HLA-A2.1-restricted TCR that recognizes an unmutated p53 peptide spanning p53 amino acid residues 264–272 genetically linked to human IL-2. We investigate the peptide-specific binding of the TCR portion of the molecule to peptide-loaded HLA-A2 as well as the specific IL-2 receptor binding capability and bioactivity of the IL-2 portion of the molecule. Our results suggest that these types of TCR-based fusion proteins could serve as an alternative to antibody-based targeted tumor therapies or as an addition to other targeted tumor therapies such as antibody based immunocytokines. Separate and distinct approaches to targeting a tumor may demonstrate additive or synergistic antitumor effects.
Materials and methods
Materials
A2.1 264 CTL clone no. 5 was derived by limiting dilution cloning [50] from a CTL line specific for the human p53 264–272 peptide generated in HLA-A2.1 transgenic mice [49]. The expression vectors for producing peptide-loaded HLA-A2 tetramers were a generous gift from Dr. John Altman at Emery University in Atlanta, Ga. CHO.K1 Chinese hamster ovary, Jurkat human T lymphocyte, CTLL-2 mouse cytotoxic T lymphocyte, T2 human lymphoblast, A375 human melanoma, H57–597 hybridoma and BB7.2 hybridoma cell lines were obtained from American Type Culture Collection (Rockville, Md.). The T2 human lymphoblast cells are positive for HLA-A2.1, but deficient in TAP 1 and 2 proteins, which allows them to display empty MHC molecules that can then be loaded with exogenous peptide [2]. The A375 human melanoma cell line was tested in our laboratory for both HLA-A2.1 and p53 and was found to be positive for both antigens (data not shown). The H57–597 hybridoma produces a monoclonal antibody that recognizes an epitope in the murine TCR β constant region, and the BB7.2 hybridoma produces the BB7.2 monoclonal antibody that specifically recognizes an epitope on the alpha 2 domain of HLA-A2. The highly metastatic subclone of the human melanoma cell line A375, A375-C15N, which was used only for in vivo metastasis studies, was maintained by Dr. John Francis at the Florida Hospital Cancer Research Institute [53]. Recombinant human IL-2 and biotinylated anti-human IL-2 polyclonal antibodies used for the ELISA in the pharmacokinetic study were purchased from R&D Systems, Inc. (Minneapolis, Minn.). Anti-TCR Cβ mAb H57–597, anti-murine TCR Vβ3 mAb, anti-murine CD3ε mAb, anti-human IL-2 mAb, anti-human CD25 blocking antibody and isotype control antibody, and FITC labeled goat anti-mouse IgG were obtained from Pharmingen (San Diego, Calif.). All cell culture media and additives were purchased from CellGro (Herndon, Va.), and all cell culture materials were purchased from Nunc (Rochester, N.Y.) unless otherwise noted. All mice were purchased from Harlan Labs (Indianapolis, Ind.).
Cell culture
All cell lines were maintained in complete culture medium comprised of IMDM supplemented with 10% heat inactivated FBS, 2 mM L-glutamine and 1 mg/ml G418 (for transfected CHO cells only) at 37°C and 5% CO2. CTLL-2 cells were maintained in the same medium with the addition of 9 U/ml recombinant human IL-2. A375-C15N cells were maintained in RPMI-1640 with 10% heat inactivated FBS, penicillin and streptomycin (Life Technologies).
Mouse splenocytes were isolated by pressing spleens aseptically dissected from BALB/c mice through a nylon mesh screen and washing with culture medium. Red blood cells were lysed with Gey’s solution for 2 min followed by addition of culture medium to stop the lysis. Single cell pellets were washed twice, resuspended at 2.5×106 cells per ml in culture medium and cultured in complete culture medium containing 50 μM 2-ME, 100 IU/ ml recombinant human IL-2, and 50 ng/ml anti-murine CD3ε mAb.
Primers
Oligonucleotide primers were synthesized from sequences matching or complementing the mouse T cell receptor and human IL-2 genes: KC228: 5’-GAGGTGGCCCAGCCGGCCATGGCCCAGTCAGTGACGCAGC-3’; KC229: 5’-GAGGTGACTAGTGTCTGGCTTTATAATTAG-3’; PRIB4: 5’-GGGGGGCTCGAGCAATTCAAAAGTCATTCAGACTC-3’;KC176: 5’-GAGGTGGAGCCCGGGGTCTGCTCGGCCCCAGGC-3’; ET-TCRF1: 5’-CCCACCGGTCAGTCAGTGACGCAGCCC-3’; KC-170: 5’-GTGGAGTTCGAAAAGGTGACTTACGTTTGTCTGCTCGGCCCCAG-3’; KC231: 5’-CGATAAGTGTACTTACGTTTTCATTATTCCATCGGCATGTACTCTTCTTCCTCTCG-3’; KC208: 5’-GTGGAGATCGATAAGTGTACTTACGTTTTCATTATCGCGATCCGGAGTTAACGTCTGCTCGGCCCCAG-3’; KC327B: 5’-TAGGTGTCCGGAGCACCTACTTCAAGTTCTAC-3’; KC328B: 5’-TAGGTGTCGCGAAGTTAGTGTTGAGATGATG-3’; AP2: 5’-ACTCACTATAGGGCTCGAGCGGC-3’; Cα HYB: 5’-GCTGTCCTGAGACCGAGGATCTTTTAACTG-3’; Cβ HYB: 5’-TTGTTTGTTTGCAATCTGTGCTTTTGATGG-3’.
Constructs
The TCR gene was cloned from the T cell clone A2.1 264#5. We designate the single-chain TCR derived from this T cell clone 264scTCR. Poly(A)+ RNA was extracted from the cells using a MicroFast Track kit (Invitrogen, Carlsbad, Calif.), and double-stranded cDNA was prepared and ligated to a double-stranded adaptor oligonucleotide using the Marathon cDNA Amplification Kit (Clontech, Palo Alto, Calif.). To identify the Vα and Vβ segments, 5’-RACE PCR was performed using the A2.1 264#5 cDNA preparation and primers AP2 (specific for the adaptor DNA) and Cα HYB (specific for the constant domain of the α chain) or Cβ HYB (specific for the constant domain of the β chain). PCR fragments were cloned into the pCR2.1 vector using the TA cloning kit (Invitrogen), and the sequence was determined using M13 forward and reverse primers. The T cell receptor Vα chain was amplified using primers KC228 and KC229 to produce an SfiI/SpeI fragment, and the VβCβ chain was amplified using primers PRIB4 and KC176 to generate an XhoI/XmaI fragment. The Cβ chain was truncated just before the cysteine residue at amino acid 127 of the full length Cβ chain. The SfiI/SpeI Vα chain fragment was subcloned into SfiI/SpeI digested pKC60, an E. coli expression vector that encodes an irrelevant TCR, replacing the original TCR insert. The XhoI/XmaI VβCβ fragment was then ligated into an XhoI/XmaI digest of this vector, yielding a vector encoding a soluble three-domain 264scTCR. The three-domain T cell receptor from this construct was amplified using primers ET-TCRF1 and KC170 to generate an AgeI/ClaI DNA fragment, which was then used as a template for PCR with primers KC231 and KC208 to produce an AgeI/HpaI fragment.
The human IL-2 coding sequence was cloned by RT-PCR from total RNA isolated from Jurkat cells using a Mini Total RNA Kit (Qiagen, Valencia, Calif.) and Qiashredder (Qiagen, Valencia, Calif.). Reverse transcription was carried out using primer KC328B, and PCR was carried out using primers KC327B and KC328B to produce a BspEI/NruI human IL-2 fragment. The BspEI/NruI IL-2 fragment was cloned into BspEI/NruI digested p149B1SP, a cloning vector encoding an irrelevant TCR/antibody fusion protein, replacing the antibody portion of the fusion protein. The IL-2 modified vector was digested with AgeI and HpaI, and the AgeI/HpaI 264scTCR fragment described above was ligated into it. Finally, an AgeI/ClaI 264scTCR/IL-2 fusion protein fragment was cloned into AgeI/BstBI digested pSUN27, a scTCR/mouse kappa fusion vector, replacing the irrelevant TCR originally cloned in the vector, yielding the 264scTCR/IL-2 fusion protein expression vector, pSUN38. The 264scTCR/kappa fusion used as a negative control for some of the flow cytometry analyses was generated by cloning an AgeI/BstBI 264scTCR fragment into AgeI/BstBI digested pSUN27, replacing the original TCR.
For production of fusion protein in mammalian cells, CHO.K1 cells were electroporated using a Bio-Rad Gene Pulser, followed by limiting dilution cloning and selection in medium containing 1 mg/ml G418.
Protein purification
264scTCR/IL-2 was purified from cell culture supernatant fluid by immunoaffinity chromatography using the monoclonal anti-murine TCR antibody H57–597, which recognizes an epitope in the constant region of the TCR β chain, coupled to a Sepharose 4B column (Amersham Pharmacia, Piscataway, N.J.). The purified sample was then concentrated and buffer-exchanged into PBS using an Ultrafree-15 centrifugal filter with a 30 kDa molecular weight cutoff membrane (Millipore, Bedford, Mass.). The TCR fusion protein samples were stored at 2–8°C (short term) or at −80°C (long term) for biochemical and functional analysis.
SDS-PAGE
SDS-PAGE was performed under either reducing or non-reducing conditions using 4–12% Nu-PAGE polyacrylamide gels (Novex, San Diego, Calif.) and the Novex EX-Cell II system. SDS-PAGE gels were stained with Coomassie blue.
ELISA
All ELISAs were performed using Maxisorb 96-well plates (Nunc, Rochester, N.Y.) coated with 100–200 ng/well anti-human IL-2 mAb or anti-murine TCR Vβ3 mAb. Fusion protein was detected with biotinylated anti-murine TCR H57 mAb, anti-murine TCR Vβ3 mAb or anti-IL-2 polyclonal Ab followed by streptavidin-HRP (Kirkegaard and Perry Laboratories, Gaithersburg, Md.), TMB substrate, and 0.18 M H2SO4 to quench the reaction (BioFX, Owings Mills, Md.). Absorbance was measured at 450 nm using a 96-well plate reader (Bio-Tek Instruments, Inc., Winooski, Vt.).
Cell staining with TCR fusion proteins
T2 cells pulsed with either p53 (aa 149–157) or p53 (aa 264–272) peptide were incubated with 0.5 μg of 264scTCR/IL-2 fusion protein in 1% FBS in PBS for 30 min at room temperature. The cells were then incubated with 0.5 μg anti-IL-2 Ab or 0.5 μg biotinylated anti-TCR H57–597 mAb for 30 min at room temperature followed by 1 μg anti-murine kappa-PE or 5 ng streptavidin-PE, respectively (both from Becton Dickenson, Franklin Lakes, N.J.). Samples were washed with 1% FBS in PBS before FACScan analysis (Becton Dickenson, Franklin Lakes, N.J.). To determine if both p53 peptides bound to HLA-A2 similarly, peptide-loaded cells were stained with BB7.2 for 30 min at room temperature followed by FITC-labeled goat anti-mouse IgG and analyzed on a FACScan instrument.
CTLL-2 cells were incubated with 0.5 μg of fusion protein for 30 min at room temperature. To detect the bound fusion protein, 0.5 μg biotinylated anti-TCR Vβ3 mAb was added and incubated for 30 min at room temperature followed by incubation with 5 ng streptavidin-PE, or the protein was detected using 0.5 μg PE-labeled HLA-A2.1 p53 (aa 264–272) tetramer for 30 min. Conjugated HLA-A2 tetramers loaded with p53 peptides were produced as described previously [1]. Samples were washed with 1% FBS in PBS before FACScan analysis. For IL-2 receptor blocking experiments, CTLL-2 cells were incubated with α-human CD25 blocking antibody or isotype control antibody for 30 min before incubation with 264scTCR/IL-2 or 264scTCR/kappa fusion protein. For staining of BALB/c mouse splenocytes, staining was carried out as described for the CTLL-2 cells using HLA-A2.1 p53 (aa 264–272) tetramers to detect bound fusion protein.
A375 cells were harvested with enzyme-free cell dissociation buffer (Sigma, St. Louis, Mo.). Samples of 5×105 cells were washed with 1% FBS in PBS and incubated with no fusion protein, 5 μg 3C8 (an irrelevant TCR/IL-2 fusion protein), or 5 μg 264scTCR/IL-2 for 30 min at room temperature followed by incubation with 1 μg biotinylated H57–597 mAb. Cells were then incubated with PE-labeled streptavidin for 15 min at room temperature, washed, and analyzed by FACScan.
Cell conjugation
T2 cells pulsed with either p53 (aa 264–272) peptide or p53 (aa 149–157) peptide were labeled with 7.88 ng/ml dihydroethidium (HE) (Molecular Probes, Inc., Eugene, Ore.), and CTLL-2 cells were labeled with 50 ng/ml calcein AM (Molecular Probes, Inc., Eugene, Ore.). After washing, the two populations of labeled cells were mixed together at a 1:1 ratio in the presence or absence of 2 μg 264scTCR/IL-2 fusion protein for 20 min at room temperature. Cells were then analyzed by FACScan.
Bioassay
CTLL-2 cells were seeded at 4×103 cells/well in 100 μl culture medium containing various concentrations of either recombinant IL-2 or 264scTCR/IL-2 and incubated for 21 h at 37°C and 5% CO2. As a control for specificity CTLL-2 cells were incubated with 264scTCR/IL-2 in the presence or absence of 5 or 50 μg anti-human CD25 blocking antibody or isotype control antibody and incubated for 21 h at 37°C and 5% CO2. Cell proliferation reagent WST-1 (Roche Inc., Indianapolis, Ind.) was added at 20 μl/well and incubated for 4 h at 37°C and 5% CO2. Absorbance was read at 450 nm on a 96-well plate reader.
Pharmacokinetics in mice
For all experiments involving animals, principles of laboratory animal care (NIH publication no. 85-23, revised 1985) were followed, as well as specific national laws where applicable. Female BALB/c mice were injected intravenously via the lateral tail vein with 32 μg 264scTCR/IL-2 fusion protein diluted with PBS to a total volume of 100 μl. Serum was collected from one group of mice not injected with 264scTCR/IL-2 to establish background levels. Serum was collected by tail bleed from the injected groups at 15 and 30 min, 1, 2, 4, 8 and 24 h. Blood samples were centrifuged at 14,000 × g at 4°C for 10 min, and serum was collected and stored at −80°C until use. 264scTCR/IL-2 concentrations were determined by ELISA using anti-TCR Vβ3 or anti-IL-2 monoclonal antibodies for capture and either biotinylated anti-TCR H57 monoclonal or anti-IL-2 polyclonal antibodies followed by streptavidin HRP for detection.
In vivo studies
Female athymic nude mice (nu/nu) were injected with 5.0×105 A375-C15N cells via the lateral tail vein. Animals were injected with varying doses of either 264scTCR/IL-2 (32, 10, 3, 1 or 0.1 μg in 100 μl total volume) or recombinant human IL-2 (8, 2.5, 0.75, 0.25, or 0.025 μg in 100 μl total volume) on days 1, 2, 3, 4, 7, 10, 14, 17, 21, 28 and 35 post-tumor cell injection. Forty-two days after tumor cell injection, all animals were humanely killed, the lungs were removed and fixed in Bouin’s solution, and surface pulmonary tumor nodules were counted. Tumor nodules on each lung were counted by two observers and the average counts were recorded.
Results
Generation of TCR fusion protein constructs
We have constructed a fusion protein comprising a three-domain, HLA-A2.1 restricted mouse TCR specific for a p53 peptide antigen fused to human IL-2. For this TCR fusion protein construct, the Vα and Vβ/Cβ regions were generated by RT-PCR from RNA isolated from a mouse T cell clone that produces TCRs specific for human p53 (aa 264–272) peptide. The carboxyl-terminal end of the variable region of the TCRα chain (Vα3) was fused via a flexible linker (G4S)4 [21] to the N-terminus of the Vβ (Vβ3) to generate the antigen binding portion of the TCR. The Cβ domain, which is directly linked to the Vβ domain, was truncated at the amino acid residue just prior to the final cysteine, removing the transmembrane and cytoplasmic domains, to generate a soluble single-chain TCR molecule (Fig. 1A and B). Human IL-2 was fused to the TCR portion via a short linker (amino acid sequence VNAKTTAPSVYPLAPV). An EE tag (amino acid sequence EEEEYMPME) [11] was inserted just downstream of the IL-2 portion of the fusion molecule to allow for detection of the TCR/IL-2 fusion protein by an anti-EE tag mAb [11] if desired. Mammalian cell expression is driven by a CMV promoter, secretion is directed by an antibody light chain leader, and selection is carried out by G418 resistance.
Fig. 1.

The 264scTCR/IL-2 fusion protein. A Schematic representation of the domain structure of the 264scTCR/IL-2 fusion protein. B Amino acid sequence of 264scTCR/IL-2 fusion protein. Amino acid numbers for each domain of the fusion protein are indicated in the figure
Expression of TCR/IL-2 fusion protein in mammalian cells
To characterize the 264scTCR/IL-2 fusion protein, the 264scTCR/IL-2 construct was stably transfected into CHO-K1 cells. Stable transfectants secreting 264scTCR/IL-2 fusion protein were selected using ELISA assays as described in Materials and Methods. Positive signals for these ELISAs indicate that the transfected cells secrete 264scTCR/IL-2 fusion protein that is recognized by both anti-murine TCR and anti-human IL-2 antibodies, suggesting that the secreted 264scTCR/IL-2 is properly assembled and folded in the transfected cells and that it remains intact when it is secreted from the cells.
264scTCR/IL-2 fusion protein was purified from cell supernates by immunoaffinity chromatography with a yield of approximately 1.8 mg/l of supernate. Purified fusion protein was subjected to SDS-PAGE and Coomassie staining. Under either reducing or non-reducing conditions, the predominant stained band migrated at approximately 60 kDa (Fig. 2), which is consistent with the predicted molecular mass for this protein and indicates that the fusion protein remains intact with no unexpected intramolecular disulfide bonds when it is secreted from the cells. The larger band in the nonreducing gel may be a dimer form of the fusion protein. This conclusion is based on the observation that the larger band has the apparent molecular mass approximately twice that of the fusion protein and this band is reduced to the size of the fusion protein under reducing conditions. Taken together, these data indicate that the transfected CHO cells produce 264scTCR/IL-2 fusion protein of the expected molecular mass and that it is properly folded, assembled and secreted as a soluble fusion protein.
Fig. 2.
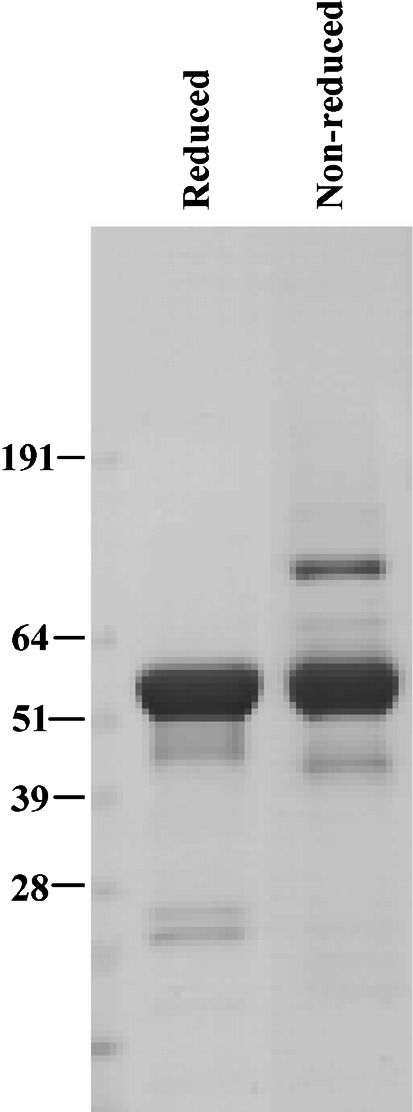
Production of 264scTCR/IL-2 fusion protein in transfected CHO cells. CHO cells were stably transfected with the 264scTCR/IL-2 expression vector. The secreted fusion protein was purified by immunoaffinity chromatography and subjected to SDS-PAGE under either reducing or non-reducing conditions as indicated at the top of the figure. SDS-PAGE gels were stained with Coomassie brilliant blue
MHC/peptide binding ability of the TCR portion of the 264scTCR/IL-2 fusion protein
The ability of the 264scTCR/IL-2 fusion protein to bind to peptide-loaded MHC was determined by flow cytometry. T2 cells were loaded with p53 (aa 264–272) or p53 (aa 149–157) (control) peptide and stained with 264scTCR/IL-2 fusion protein. Cells loaded with p53 (aa 264–272) stained positively with 264scTCR/IL-2 when detected with either the anti-TCR Cβ mAb or the anti-IL-2 detection antibody (Fig. 3A and B). Cells loaded with p53 (aa 149–157) control peptide did not stain with either the anti-TCR Cβ mAb or the anti-IL-2 detection antibodies. To demonstrate that the lack of staining of p53 (aa 149–157) loaded T2 cells is not due to an inability to of the p53 (aa 149–157) peptide to bind to HLA-A2, T2 cells loaded with no peptide, p53 (149–157), or p53 (264–272) peptide were stained with BB7.2 α-HLA-A2 monoclonal antibody. Cells loaded with either p53 peptide stained stronger than cells loaded with no peptide, suggesting that both peptides are capable of binding to HLA-A2 molecules (Fig. 3C). T2 cells were also stained for IL-2 receptor and were found not to express IL-2 receptor (data not shown); thus, these data indicate that binding of the 264scTCR/IL-2 fusion protein is mediated by the TCR component of the fusion protein. The lack of staining by the fusion protein when T2 cells were loaded with the control peptide also indicates that staining is mediated by the TCR component and that the staining is specific for the appropriate peptide. These data indicate that the TCR portion of the 264scTCR/IL-2 fusion protein is capable of recognizing its specific peptide in the context of HLA-A2.
Fig. 3.
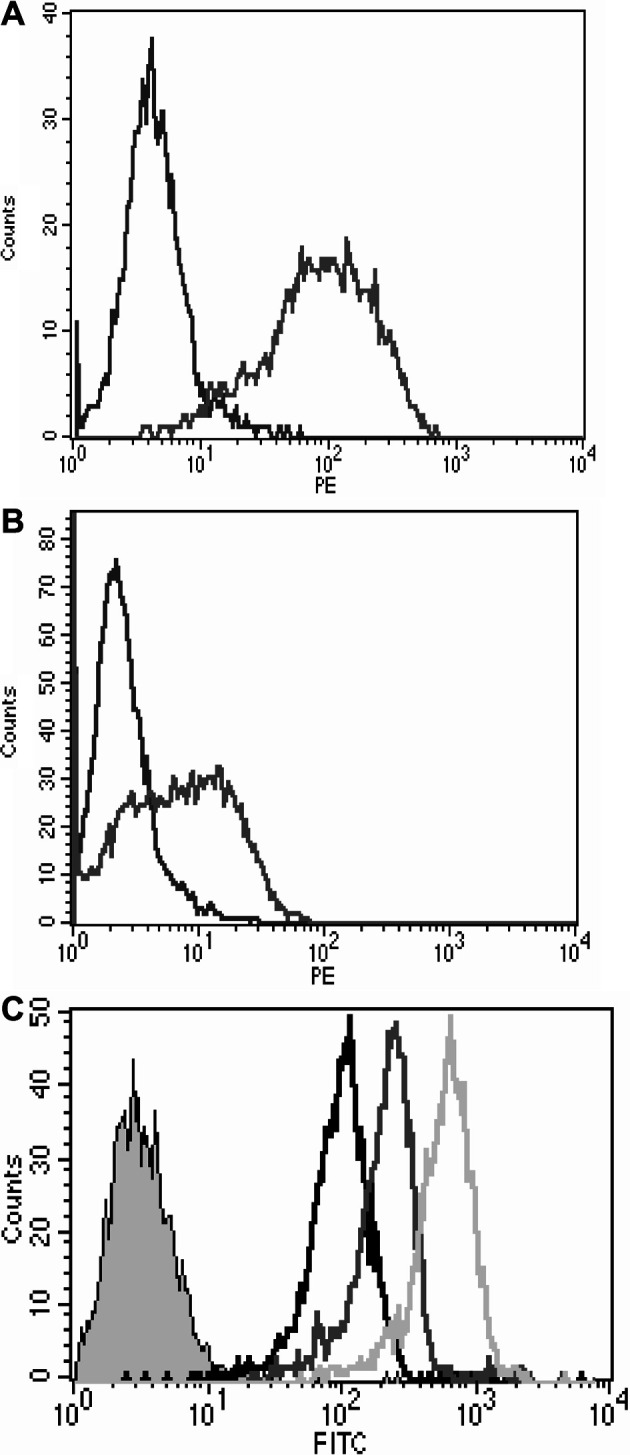
MHC/peptide binding ability of the TCR portion of 264scTCR/IL-2 fusion protein. T2 cells were loaded with p53 (aa 264–272) peptide (gray line) or p53 (aa 149–157) peptide (black line), and stained with either (A) 264scTCR/IL-2 fusion protein and anti-TCR Cβ mAb or (B) 264scTCR/IL-2 fusion protein and anti-IL-2 mAb. (C) T2 cells loaded with p53 (aa 264–272) peptide (dark grey line), p53 (aa 149–157) peptide (light grey line), or no peptide (black line) were stained with anti-HLA-A2 BB7.2 mAb followed by FITC labeled goat anti-mouse IgG. The shaded peak is unstained T2 cells
IL-2 receptor binding ability of the IL-2 portion of the 264scTCR/IL-2 fusion protein
The IL-2 receptor binding capability of the IL-2 portion of the 264scTCR/IL-2 fusion protein was studied by flow cytometry. Primary mouse splenocytes were isolated and stimulated with rIL-2 and anti-CD3 to generate T cell blasts. Stimulated splenocytes that express IL-2 receptor stained positively with p53 (aa 264–272) loaded HLA-A2 tetramers only in the presence of 264scTCR/IL-2 fusion protein (Fig. 4A). Likewise, CTLL-2 mouse cytotoxic T lymphocytes, which constitutively express IL-2 receptor, stained positively with the 264scTCR/IL-2 fusion protein, but not with a 264scTCR/kappa fusion protein (Fig. 4B). When CTLL-2 cells were incubated with α-human CD25 blocking antibody or isotype control antibody followed by 264scTCR/IL-2, staining was substantially reduced when the cells were incubated with the blocking antibody, but not with isotype control antibody. The lack of signal from the CTLL-2 cells incubated with a 264scTCR/mouse kappa chain fusion protein or with IL-2 receptor blocking antibody indicates that staining of these cells is mediated by the IL-2 portion of the 264scTCR/IL-2 fusion protein. These data suggest that the IL-2 portion of the 264scTCR/IL-2 fusion protein is capable of binding to the IL-2 receptor.
Fig. 4.
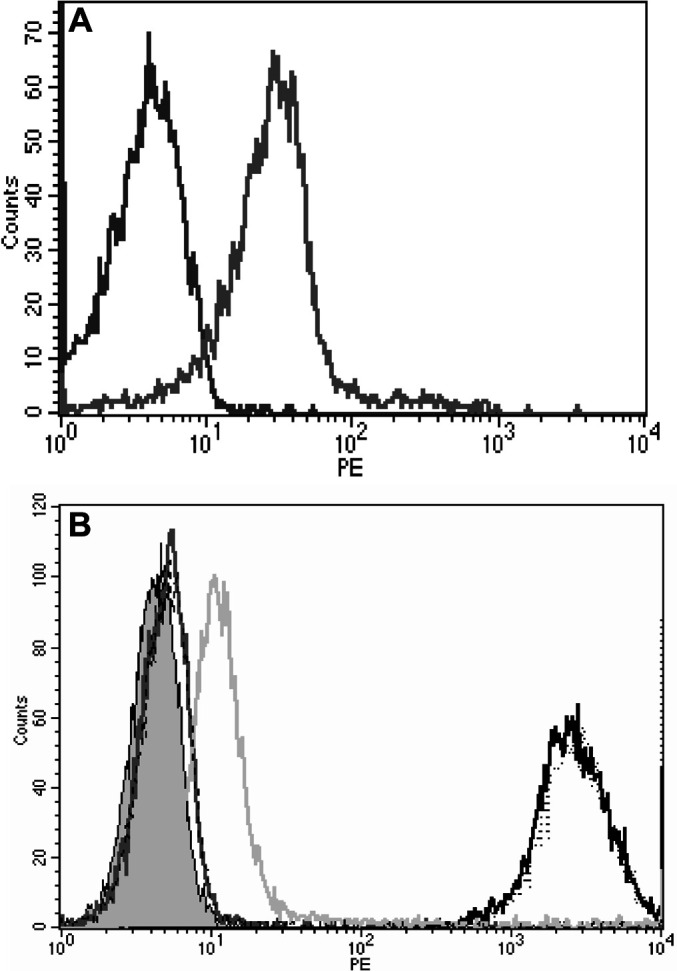
IL-2 receptor binding ability of the IL-2 portion of 264scTCR/IL-2 fusion protein. A Mouse splenocytes were stimulated with IL-2 and anti-CD3ε mAb and then incubated in the presence (gray line) or absence (black line) of 264scTCR/IL-2 fusion protein. Bound fusion protein was detected with PE labeled HLA-A2 p53 (aa 264–272) tetramers. B CTLL-2 cells were incubated with α-human CD25 blocking antibody or isotype control antibody followed by 264scTCR/IL-2 or 264scTCR/kappa fusion protein. Bound fusion protein was detected with PE labeled α-TCR-Vβ3 antibody. The shaded peak is unstained CTLL-2 cells. Black line: CTLL-2 cells stained with 264scTCR/IL-2 only. Gray dotted line: CTLL-2 cells incubated with control antibody followed by 264scTCR/IL-2. Light gray line: CTLL-2 cells incubated with α-human CD25 blocking antibody followed by 264scTCR/IL-2. Dark gray line: CTLL-2 cells stained with 264scTCR/kappa fusion protein. Black dashed line: CTLL-2 cells stained with α-TCR-Vβ
Biological activity of 264scTCR/IL-2 fusion protein
To demonstrate biological activity of the IL-2 portion of the 264scTCR/IL-2 fusion protein, IL-2 dependent CTLL-2 cells were cultured with either 264scTCR/IL-2 or recombinant IL-2 at various concentrations, and cell viability was assessed using WST-1. As shown in Fig. 5A, the ability of rIL-2 or 264scTCR/IL-2 to support the growth of CTLL-2 cells was dose dependent, wherein there was more cell proliferation at higher doses of either recombinant IL-2 or 264scTCR/IL-2. Further, there were similar levels of cell proliferation when equivalent molar amounts of either recombinant IL-2 or 264scTCR/IL-2 were used. As a further control for specificity, CTLL-2 cells were incubated with 264scTCR/IL-2 with α-human CD25 blocking antibody or isotype control. When the blocking antibody was included in the culture, proliferation was substantially decreased with both concentrations of blocking antibody, but proliferation of the CTLL-2 cells was unaffected by either concentration of control antibody (Fig. 5B). Taken together these data indicate that the IL-2 portion of 264scTCR/IL-2 has similar biological activity to recombinant IL-2 in vitro and that the proliferation activity of the fusion protein is dependent on the IL-2 portion of the molecule.
Fig. 5.
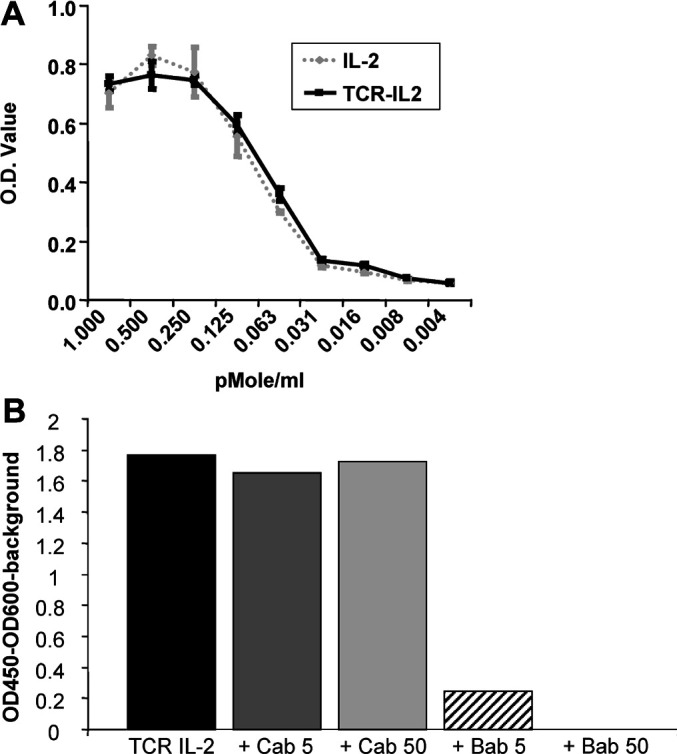
Biological activity of 264scTCR/IL-2 fusion protein. A CTLL-2 cells were cultured with 264scTCR/IL-2 (solid line) or recombinant IL-2 (dotted line) at various concentrations as indicated at the bottom of the figure. B CTLL-2 cells were incubated with 264scTCR/IL-2 and α-human CD25 blocking antibody or isotype control antibody as indicated at the bottom of the figure. Cell viability was measured by incubation with WST-1 and absorbance was read at 450 nm. Cab+5: 5 μg control antibody; Cab+50: 50 μg control antibody; Bab+5: 5 μg blocking antibody; Bab+50: 50 μg blocking antibody
Conjugation of cells mediated by 264scTCR/IL-2 fusion protein
In order for the 264scTCR/IL-2 fusion protein to be useful as a therapeutic agent, it must be able to bring together target and effector cells through its TCR and cytokine portions, respectively. To demonstrate that the 264scTCR/IL-2 fusion protein can effectively conjugate cells, T2 cells were loaded with either p53 (aa 264–272) or p53 (aa 149–157) peptides and then labeled with dihydroethidium (HE). CTLL-2 cells were labeled with calcein AM, and the two labeled cell populations were mixed and incubated in the presence or absence of 264scTCR/IL-2 fusion protein. Samples were analyzed by flow cytometry. When the two cell populations were incubated in the absence of the 264scTCR/IL-2 fusion protein (Fig. 6A and B) or when the T2 cells were loaded with control peptide and incubated with the CTLL-2 cells in the presence of 264scTCR/IL-2 fusion protein (Fig. 6C), the cells remained as two distinct populations on the flow cytometry histograms representing approximately 45% of the total population each (Fig. 6A, B and C, regions 1 and 3), with only approximately 0.46% of the total population falling in the double-stained cell window (Fig. 6A, B and C, region 2). However, when the T2 cells were loaded with p53 (aa 264–272) peptide and incubated with the CTLL-2 cells in the presence of the 264scTCR/IL-2 fusion protein (Fig. 6D), a double staining population of cells appears, representing 4.1% of the total population (Fig. 6D, region 2, conjugated cells), suggesting that T2 cells were conjugated to CTLL-2 cells via the 264scTCR/IL-2 fusion protein.
Fig. 6.

Conjugation of CTLL-2 cells with peptide-loaded T2 cells mediated by 264scTCR/IL-2 fusion protein. T2 cells were loaded with either p53 (aa 264–272) (B and D) or p53 (aa 149–157) (control) peptides (A and C) and then labeled with HE. CTLL-2 cells were labeled with calcein AM. Labeled cells were mixed and incubated in the presence (C and D) or absence (A and B) of 264scTCR/IL-2 fusion protein, and the samples were analyzed by flow cytometry. Assay conditions including loading peptide used and presence or absence of fusion protein are indicated beneath each histogram. Single-stained regions are marked 1 and 3 and the double-stained cell population is marked 2
Pharmacokinetics of 264scTCR/IL-2 in mice
The pharmacokinetics of the 264scTCR/IL-2 fusion protein was measured in BALB/c mice. Mice were injected intravenously, and serum samples were collected at various time points. The serum levels of 264scTCR/IL-2 fusion protein were measured using ELISA. The ELISA detection was performed using anti-TCR mAb capture/anti-IL-2 Ab detection (Fig. 7A), anti-TCR mAb capture/anti-TCR mAb detection (Fig. 7B) or anti-IL-2 mAb capture/anti-IL-2 polyclonal Ab detection (Fig. 7C) to determine whether the fusion protein is modified or cleaved in vivo. Mice injected with 264scTCR/IL-2 fusion protein showed no obvious signs of toxicity. In these assays we detected a maximum concentration of 0.75 to 2.5 μg/ml of 264scTCR/IL-2 with an apparent serum half-life of 1.6 to 3.0 h, depending on the ELISA format used (Fig. 7). Since the reported serum half-life of free IL-2 is only ~5 min [6], these data suggest that the fusion protein is not cleaved in vivo, but instead remains intact for a relatively long period of time in the blood. The small variability in the half-life of 264scTCR/IL-2 determined in these studies is most likely due to the differences in the sensitivity of the ELISA assays.
Fig. 7.

Serum half-life of 264scTCR/IL-2 fusion protein. BALB/c mice were injected with 264scTCR/IL-2 fusion protein, and serum samples were collected at 15 and 30 min, 1, 4, 8 and 24 h post injection. Serum concentrations of 264scTCR/IL-2 were determined by ELISA using the following formats: (A) anti-TCR mAb capture/anti-IL-2 Ab detection, (B) anti-TCR mAb capture/anti-TCR mAb detection and (C) anti-IL-2 mAb capture/anti-IL-2 Ab detection
Tumor cell staining with 264scTCR/IL-2
In order for the 264scTCR/IL-2 fusion protein to be useful as a therapeutic agent, it must be able to recognize and bind to its target tumor cells. To test whether the 264scTCR/IL-2 is capable of binding to tumor cells, A375 human melanoma cells, which express both HLA-A2.1 and p53, were stained with either 264scTCR/IL-2 or 3C8, an irrelevant TCR/IL-2 fusion protein. Cells not incubated with fusion protein and cells incubated with 3C8 did not stain with the H57–597 detection antibody, while cells incubated with 264scTCR/IL-2 stained positively with the detection antibody (Fig. 8). This result suggests that the 264scTCR/IL-2 fusion protein is capable of recognizing and binding to its target tumor cell and may be useful as an anti-cancer therapeutic in vivo.
Fig. 8.
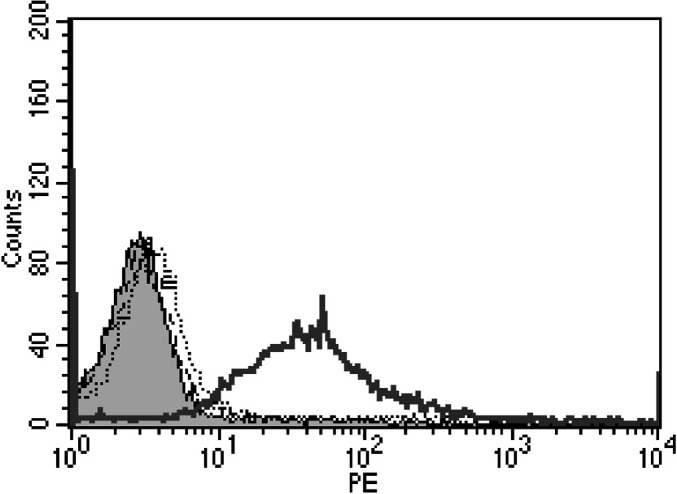
Tumor cell staining with 264scTCR/IL-2. A375 human melanoma cells were incubated with no fusion protein (dashed black line), 5 μg 3C8 TCR/IL-2 fusion protein (control) (dotted line), or 5 μg 264scTCR/IL-2 fusion protein (solid black line) followed by staining with H57–597 mAb. Unstained cells are represented by the shaded area
Anti-tumor effects of 264scTCR/IL-2 fusion protein
To determine if the 264scTCR/IL-2 fusion protein has anti-tumor activity in vivo, an experimental metastasis assay was performed. Female athymic nude mice were injected with the highly metastatic A375 human melanoma subclone, A375-C15N and treated with varying doses of either 264scTCR/IL-2 or recombinant IL-2. Forty-two days after tumor cell injection, lung nodules were counted. Both 264scTCR/IL-2 and recombinant IL-2 reduced lung metastasis in a dose-dependent manner (Fig. 9). However, at all doses lung metastasis was reduced to a greater degree with the 264scTCR/IL-2 fusion protein, suggesting that targeting the cytokine to the tumor may provide greater efficacy as a cancer therapeutic.
Fig. 9.
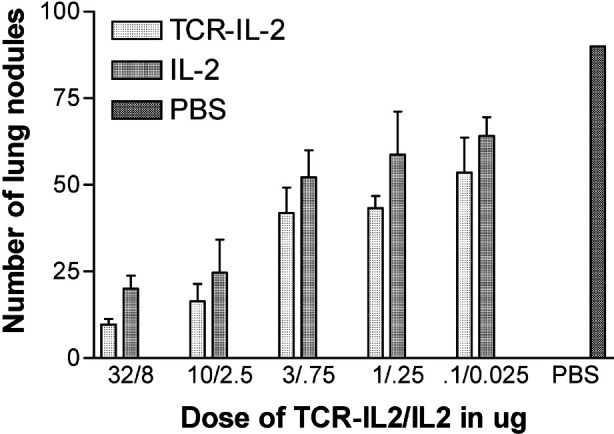
Anti-tumor effect of 264scTCR/IL-2 fusion protein. Female athymic nude mice were injected with highly metastatic A375-C15N cells and treated with 264scTCR/IL-2, recombinant IL-2 or PBS. Forty-two days after tumor cell injection, the lungs were removed, lung nodules were counted, and the mean number of lung nodules relative to the PBS-treated control group was plotted
Discussion
In these studies we describe the construction and expression of a soluble three-domain mouse scTCR that recognizes human p53 peptide (aa 264–272) in the context of HLA-A2.1. The three-domain scTCR is fused to human IL-2 yielding a soluble 264scTCR/IL-2 fusion protein that is expressed at high levels and secreted from mammalian cells. The TCR portion of the 264scTCR/IL-2 fusion protein retains its MHC-restricted, peptide-specific antigen-binding properties, and the IL-2 portion binds to IL-2 receptor and is biologically active. Moreover, these studies further demonstrate that this fusion protein is capable of conjugating target and effector cells, exhibits favorable pharmacokinetics in mice, can bind to target tumor cells and has anti-tumor effects. Therefore, soluble scTCR fusion proteins may provide access to another repertoire of antigens for targeted immunotherapy that are not recognizable by antibody-based immunotherapies. TCR-based therapies may serve as an alternative to antibody-based treatments or as a useful addition to other targeted tumor therapies. Treating tumors using multiple distinct approaches may have additive or synergistic antitumor effects and hence greater success in the clinical setting.
There has been some difficulty expressing soluble recombinant TCRs that are stable and retain antigen binding properties. Several attempts have been made at expressing recombinant TCRs as membrane-associated fusion proteins or as inclusion bodies in E. coli. Each of these methods was hindered by low or unstable expression or dimerization and also by the need for proteolytic cleavage and/or refolding to generate soluble TCRs [5, 8, 9, 18, 24, 26, 54]. Further, the activity of soluble TCRs made by these methods tends to be low. Our scTCRs are produced in mammalian cells as soluble fusion proteins without any membrane association, eliminating the extra step of enzymatic cleavage or refolding, which could affect fusion protein function. As the data presented here demonstrate, these TCRs fold correctly and are secreted intact from transfected eukaryotic cells. Moreover, fusion of a TCR in this format to a cytokine via a short linker does not interfere with MHC-restricted, peptide-specific antigen-binding or specific cytokine activity. No enzymatic modification or protein refolding is necessary to produce soluble active TCRs, and the effector molecule, in this case IL-2, is covalently bound, eliminating the need for chemical crosslinking, which can be unstable. Thus, this is the first report of the production of a truly soluble recombinant TCR that retains its MHC-restricted, peptide-specific antigen-binding properties.
One potential drawback of using recombinant TCR fusion proteins as targeted immunotherapies is the weak binding affinity of the TCR for its cognate MHC/peptide. We have determined the dissociation constant of the 264scTCR for its cognate MHC/peptide to be approximately 10-7 M at physiological conditions using surface plasmon resonance detection (unpublished results). This high affinity for TCR interaction with MHC/peptide complex is likely due to its CD8 independence as the TCR was generated in HLA-A2.1 transgenic mice [49] and mouse CD8 has been shown not to interact with human HLA [45]. The affinity of the 264scTCR is sufficient to bind to unmanipulated tumor cells and effectively conjugate target and effector cells, both of which we have shown in these studies. Another potential problem with this type of therapy is that the use of TCR-based cancer therapies may be limited by the HLA type of the patient, which may be confounded by the downregulation of HLA expression in some tumors. Therefore, it will be necessary to screen patients before treatment with a TCR-based therapy to determine if they would benefit from this type of therapy in much the same way that breast cancer patients are screened for ErbB-2 expression before treatment with herceptin. We are currently developing tumor screening methods using another 264scTCR fusion protein so that tumor histoarrays may be stained to determine the proportion and types of tumors that may be treated with TCR-based therapies. Development of TCR-based fusion proteins targeting other HLA types should broaden the treatable patient population.
With these data in mind, we performed preliminary studies using the 264scTCR/IL-2 fusion protein as an anti-tumor agent against A375-C15N human melanoma cells in an experimental lung metastasis model in nude mice. Mice treated with either 264scTCR/IL-2 or recombinant IL-2 showed no obvious signs of toxicity. Both treatments resulted in reduction of lung metastasis; however, at all doses treatment with 264scTCR/IL-2 was more effective than recombinant IL-2. At this point it is unclear whether the improved anti-tumor efficacy of the 264scTCR/IL-2 fusion protein is due to the increase in the half-life of IL-2 or to the concentration of the cytokine at the site of the tumor. These studies were performed in athymic nude mice in the absence of T-cells; thus, the effector cells in these studies were those of the innate system such as natural killer cells and macrophages that can be stimulated by IL-2 [10, 41]. IL-2 has a profound effect on T-cells, which this model system does not address. Thus, we are currently performing T-cell reconstitution studies in this model to determine if the fusion protein has a stronger effect than free IL-2 in the presence of T-cells. In addition, we are performing imaging studies to determine the degree of 264scTCR/IL-2 fusion protein localization at the site of the tumor.
The main problems with systemic administration of cytokines to treat tumors are their short serum half lives and toxicity. To circumvent this problem cytokines have been delivered over a period of time as infusions; however, high systemic doses of cytokines have widely reported toxic side effects, and delivery by infusion is time consuming and inconvenient for the patient and has its own undesirable side effects [7, 31, 34, 44]. Delivery of cytokines such as interferon alpha-2a in pegylated forms is being tested, and although the pegylated cytokine has a longer half life, the side effects are similar to that of the unpegylated form [32, 39]. Targeting delivery via antibody-cytokine fusion proteins is thought to take advantage of the paracrine nature of IL-2, and because these molecules concentrate cytokine at the site of the tumor, they may allow the cytokine to be used at lower, less toxic doses. Antibody-cytokine fusion proteins such as the anti-disialoganglioside ch14.18/IL-2 fusion protein (ch14.18-IL-2) have been shown to have serum half lives of ~4 h, which significantly increases the serum half life of IL-2 [23]. However, it was reported that this fusion protein is cleaved in vivo and that the IL-2 portion is then cleared with kinetics similar to those of free IL-2 [23]. Our 264scTCR/IL-2 fusion protein has an apparent serum half-life of ~3 h and appears to remain intact in the blood. Thus, the 264scTCR/IL-2 fusion protein effectively increases the half-life of IL-2 and survives intact in the blood, suggesting that it has potential as a new agent for immunomodulatory cancer therapy. As reported with other biologics, at higher doses the serum half life of our fusion protein may increase [3, 25, 37, 38]. If this is the case for 264scTCR/IL-2, it should further improve the efficacy of the molecule against tumors. Escalating dose studies to further investigate toxicity and pharmacokinetic parameters are underway.
Besides T-cells, IL-2 has been shown to have stimulatory effects on a broad range of immune cell types including B cells, monocytes, macrophages, LAK cells and NK cells [10, 41]. For this reason, IL-2 concentrated at the tumor site should activate local T-cells as well as other IL-2 responsive cells, thereby recruiting effector cells to the site of the tumor. In addition, recirculation of these effector cells may be useful for eradication of distant micrometastases, which may be heterogeneous with respect to expression of the targeted tumor antigen. Straten et al. reported that targeted delivery of IL-2 via anti-ganglioside ch14.18-IL-2 fusion protein to murine melanoma resulted in clonal T-cell expansion [51]. Moreover, both target antigen positive and negative tumors implanted in the same mouse regressed as a result of this treatment, indicating that T cell clones activated locally by targeted therapy recirculated and mediated eradication of the distant tumor that was not targeted by the therapy [51]. The anti-GD2 ch14.18-IL-2 fusion protein has also been shown to be effective in the eradication of liver and bone marrow metastases in a murine neuroblastoma model. However, in this case the anti-tumor effect was shown to be NK cell mediated [28]. The activation of other types of immune effector cells by IL-2 for cancer therapy may be useful because many cancer patients have severe T-cell dysfunction either from the cancer itself or from the damaging effects of the radiotherapy or chemotherapy used to treat the cancer [14, 56]. However, most patients retain the ability to expand and activate NK cells after IL-2 treatment [12, 13, 15]. Thus, by concentrating IL-2 at the site of a tumor our 264scTCR/IL-2 fusion protein may potentiate a wide ranging immune response including activation and proliferation of a variety of T-cell clones as well as activation of NK cells or other members of the innate immune system. Such a multifaceted anti-tumor response may be more effective for the eradication of primary tumors as well as distant metastases.
In summary, we demonstrate that it is possible to construct a biologically active bi-functional molecule comprised of a TCR and a cytokine. This fusion protein is capable of binding to tumor cells, mediating the conjugation of target and effector cells, and appears to have reasonable pharmacokinetic properties in mice. Although this report restricts its discussion to p53 as a target, other gene products that are upregulated and presented in the context of MHC on tumor or virally infected cells may be used as targets for TCR-based immunotherapies. In addition, other immunomodulatory molecules such as GM-CSF, IFNγ or IFN-α could be linked to the TCR to activate other effector cells for an anti-tumor or anti-viral response. Thus, we are also constructing and characterizing additional TCR fusion proteins that target other cancer and viral antigens containing different effector molecules. These types of novel molecules have the potential of forming a new class of immunotherapeutics for the treatment of cancer and viral infection.
Acknowledgements
This work was supported in part by grant no. 1R43CA88615-01 from the National Institutes of Health.
References
- 1.Altman Science. 1996;274:94. [PubMed] [Google Scholar]
- 2.Anderson J Immunol. 1993;151:3407. [PubMed] [Google Scholar]
- 3.Bauer J Pharmacokinet Biopharm. 1999;27:397. doi: 10.1023/A:1020917122093. [DOI] [PubMed] [Google Scholar]
- 4.Becker J Clin Invest. 1996;98:2801. doi: 10.1172/JCI119107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung Proc Natl Acad Sci USA. 1994;91:12654. [Google Scholar]
- 6.Donohue J Immunol. 1983;130:2203. [PubMed] [Google Scholar]
- 7.Dummer Cancer. 1995;75:1038. [Google Scholar]
- 8.Engel Science. 1992;256:1318. doi: 10.1126/science.1598575. [DOI] [PubMed] [Google Scholar]
- 9.Gregoire Proc Natl Acad Sci USA. 1991;88:8077. [Google Scholar]
- 10.Grimm J Exp Med. 1982;155:1823. doi: 10.1084/jem.155.6.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grussenmeyer Proc Natl Acad Sci USA. 1985;82:7952. [Google Scholar]
- 12.Hank J Immunother. 1993;14:329. doi: 10.1097/00002371-199311000-00013. [DOI] [PubMed] [Google Scholar]
- 13.Hank Cancer Res. 1990;50:5234. [PubMed] [Google Scholar]
- 14.Hank J Biol Response Mod. 1990;9:5. [PubMed] [Google Scholar]
- 15.Hank J Immunother. 1994;15:29. [Google Scholar]
- 16.Harvill Immunotechnology. 1995;1:95. doi: 10.1016/1380-2933(95)00009-7. [DOI] [PubMed] [Google Scholar]
- 17.Harvill J Immunol. 1996;157:3165. [PubMed] [Google Scholar]
- 18.Hilyard Proc Natl Acad Sci USA. 1994;91:9057. [Google Scholar]
- 19.Hinds Cell Growth Differ. 1990;1:571. [PubMed] [Google Scholar]
- 20.Hurford Nat Genet. 1995;10:430. doi: 10.1038/ng0895-430. [DOI] [PubMed] [Google Scholar]
- 21.Huston Proc Natl Acad Sci USA. 1988;85:5879. [Google Scholar]
- 22.Iggo Lancet. 1990;335:675. doi: 10.1016/0140-6736(90)90801-b. [DOI] [PubMed] [Google Scholar]
- 23.Kendra Cancer Immunol Immunother. 1999;48:219. doi: 10.1007/s002620050569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klausner Annu Rev Cell Biol. 1990;6:403. doi: 10.1146/annurev.cb.06.110190.002155. [DOI] [PubMed] [Google Scholar]
- 25.Lewis J Immunol Methods. 2001;248:149. doi: 10.1016/s0022-1759(00)00355-0. [DOI] [PubMed] [Google Scholar]
- 26.Lin Science. 1990;249:677. [Google Scholar]
- 27.Lode J Natl Cancer Inst. 1997;89:1586. doi: 10.1093/jnci/89.21.1586. [DOI] [PubMed] [Google Scholar]
- 28.Lode Blood. 1998;91:1706. [PubMed] [Google Scholar]
- 29.Lustgarten J Immunol. 1999;162:359. [PubMed] [Google Scholar]
- 30.McLaughlin Ir J Med Sci. 2001;170:11. doi: 10.1007/BF03167712. [DOI] [PubMed] [Google Scholar]
- 31.Motzer Clin Cancer Res. 1998;4:1183. [PubMed] [Google Scholar]
- 32.Motzer J Clin Oncol. 2001;19:1312. doi: 10.1200/JCO.2001.19.5.1312. [DOI] [PubMed] [Google Scholar]
- 33.Nastala J Immunol. 1994;153:1697. [PubMed] [Google Scholar]
- 34.Pardoll Annu Rev Immunol. 1995;13:399. doi: 10.1146/annurev.iy.13.040195.002151. [DOI] [PubMed] [Google Scholar]
- 35.Peng J Immunol. 1999;163:250. [Google Scholar]
- 36.Penichet Hum Antibodies. 1997;8:106. [PubMed] [Google Scholar]
- 37.Posey J Immunother. 1999;22:371. [Google Scholar]
- 38.Pullarkat Cancer Immunol Immunother. 1999;48:9. doi: 10.1007/s002620050543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reddy Hepatology. 2001;33:433. doi: 10.1053/jhep.2001.21747. [DOI] [PubMed] [Google Scholar]
- 40.Rosenberg Transplantation. 1983;35:631. [Google Scholar]
- 41.Rosenberg N Engl J Med. 1987;316:889. doi: 10.1056/NEJM198704093161501. [DOI] [PubMed] [Google Scholar]
- 42.Rosenberg Ann Surg. 1989;210:474. doi: 10.1097/00000658-198910000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenberg Ann Surg. 1998;228:307. doi: 10.1097/00000658-199809000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Royal Cancer J Sci Am. 1996;2:91. [Google Scholar]
- 45.Sherman Science. 1992;258:815. doi: 10.1126/science.1439792. [DOI] [PubMed] [Google Scholar]
- 46.Sondel Cancer Res. 1988;48:2561. [PubMed] [Google Scholar]
- 47.Sosman Cancer Invest. 1991;9:35. doi: 10.3109/07357909109032798. [DOI] [PubMed] [Google Scholar]
- 48.Temmim Anticancer Res. 2001;21:743. [PubMed] [Google Scholar]
- 49.Theobald Proc Natl Acad Sci USA. 1995;92:11993. [Google Scholar]
- 50.Theobald J Exp Med. 1997;185:833. doi: 10.1084/jem.185.5.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thor Eur J Immunol. 2001;31:250. doi: 10.1002/1521-4141(200101)31:1<250::AID-IMMU250>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 52.Tsung J Immunol. 1997;158:3359. [PubMed] [Google Scholar]
- 53.van Clin Exp Metastasis. 1996;14:95. doi: 10.1007/BF00121206. [DOI] [PubMed] [Google Scholar]
- 54.Weber Nature. 1992;356:793. doi: 10.1038/356793a0. [DOI] [PubMed] [Google Scholar]
- 55.Weil-Hillman Cancer Res. 1990;50:2683. [PubMed] [Google Scholar]
- 56.Wiebke J Clin Oncol. 1988;6:1440. doi: 10.1200/JCO.1988.6.9.1440. [DOI] [PubMed] [Google Scholar]