

Abstract
Major mediators of anti-tumor immunity are CD4+ Th1 cells and CD8+ cytotoxic T lymphocytes (CTLs). In tumor-bearing animals, the Th1- and CTL-mediated anti-tumor immunity is down-regulated in multiple ways. Better understanding of negative regulatory pathways of tumor immunity is crucial for the development of anti-tumor vaccines and immunotherapies. Since immune deviation toward Th2 suppresses Th1 development, it has been thought that induction affecting a Th2 immune response is one of the mechanisms that down-regulate effective tumor immune responses. Recent studies using Th2-deficient signal transducer and activator (Stat6) KO mice demonstrated that this hypothesis was the case. IL-13 is one of the Th2 cytokines that has very similar features to IL-4 through sharing some receptor components and Stat6 signal transduction. It has been thought that IL-13 is not as critical for immune deviation as IL-4 since it cannot directly act on T cells. However, recent studies of IL-13 reveal that this cytokine plays a critical role in many aspects of immune regulation. Studies from our lab and others indicate that IL-13 is central to a novel immunoregulatory pathway in which NKT cells suppress tumor immunosurveillance. Here we will describe biological properties and functions of IL-13, its role in the negative regulation of anti-tumor immunity, and effects of IL-13 on tumor cells themselves.
Keywords: Cerebral Malaria, Tumor Rejection, Immune Deviation, Tumor Immunosurveillance, Negative Regulatory Pathway
Introduction
During the last decade much effort has been made to understand mechanisms by which cancer evades the immune system. It is widely accepted that immune deviation toward Th1 response, including induction of CD8+ cytotoxic T cells (CTLs), results in tumor rejection, whereas deviation toward Th2 response prevents tumor rejection [37]. Among the Th2 cytokines, IL-4 has been demonstrated the most critical cytokine to induce Th2 cells [15]. However, a related cytokine, IL-13, has recently been found to be critical for a number of pathologic processes, from asthma to parasite-induced fibrosis. Recent studies have revealed mechanisms by which the host immune system down-regulates anti-tumor responses to allow tumor to grow in the host. Work from our own lab and others suggests that IL-13, more than IL-4 itself, plays a critical role in this down-regulation of tumor immunosurveillance and is central to a novel immunoregulatory pathway in which NKT cells are induced by tumors to secrete IL-13, which acts through intermediate cells to suppress CTL responses against the tumor.
Biological properties and functions of IL-13
IL-13, originally named P600, was discovered in 1989 [5] by cloning of induction-specific mRNA from a mouse Th2 cell line, and this led to the discovery of human homologues by three independent groups [45, 48, 49]. In both the human and the mouse, the IL-13 gene is very closely located (12 kbp upstream in human) to the IL-4 gene on the chromosome 5q31 and 11 respectively [46, 62]. Although IL-13 has only ~30% homology with IL-4 in amino acid sequence, it shares a conserved hydrophobic structural core with IL-4 [46]. Both IL-13 and IL-4 are made by T cells [5, 45], B cells [36, 40], mast cells [6], and basophils [19]. However, NK cells and dendritic cells produce only IL-13 [12, 25, 54].
IL-13 shares its receptor components with the IL-4 receptor, which explains why it has biological features very similar to those of IL-4, as described below. The IL-13 receptor, which is also called the type II IL-4 receptor, is composed of the IL-4Rα chain and IL-13Rα1 chain [2, 23, 51] (Fig. 1). This type of the receptor is expressed on a broad range of cell types, including hematopoietic and nonhematopoietic cells [4, 18, 72], except for T cells, and can bind both IL-13 and IL-4 [51, 78]. IL-13Rα1 can bind IL-13 but not IL-4 [23]. IL-4Rα can bind only IL-4 [77]. IL-13 binds with low affinity to the IL-13α1 chain (Kd ~2–10 nM), but by IL-4Rα recruitment forms the type II IL-4 receptor that can bind IL-13 with high affinity (Kd ~300–400 pM) [2, 23]. On the other hand, IL-4 first binds to IL-4Rα (Kd ~40–100 pM) [77], which then recruits either the IL-13Rα1 or the IL-2Rγc chain, which increases the binding affinity approximately twofold [47, 51]. With IL-2Rγc, IL-4Rα forms type I IL-4R, which is expressed on hematopoietic cells. Since IL-13 can bind neither IL-4Rα nor IL-2Rγc chains, the type I IL-4R can bind only IL-4. For IL-13, there is another receptor called IL-13Rα2 [7, 14, 75]. This receptor binds only IL-13 with relatively high affinity (Kd ~50–500 pM), and seems to be a decoy [11, 71]. A fusion protein of IL-13Rα2 with the Fc portion of immunoglobulin is used as inhibitor of IL-13 in vitro and in vivo [14]. Since there is no good antibody which is able to inhibit IL-13, this is the only IL-13 antagonist known for in vivo use.
Fig. 1.
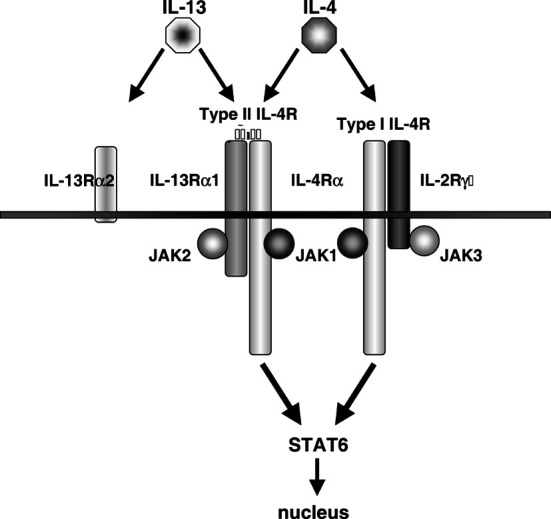
Receptors for IL-13 and IL-4. IL-4 and IL-13 share the type II IL-4R, which shares the IL-4Rα chain with the type I IL-4R as well. Both receptors use JAK1 and Stat6 for signal transduction, but differ in JAK2 and JAK3. The receptors also have different distributions among cell and tissue types. An additional high affinity receptor for IL-13, IL-13Rα2, has no known function at this time, but has served as a component of a soluble IL-13 inhibitor. See [16, 43] for more detailed treatment
Since the IL-4Rα chain is the only component which has kinase-sensitive tyrosine residues in the cytoplasmic domain, signals from both type I and type II IL-4R are transduced by the IL-4Rα chain [34]. Therefore, IL-13 and IL-4 primarily use the same Janus kinase (JAK)-signal transducer and activator of transcription (Stat6) pathway, although each receptor chain associates with different JAKs (IL-4Rα, IL-13Rα1, and IL-2Rγc associate with JAK1, JAK2 or TYK2, and JAK3, respectively) [35]. When the type I or type II IL-4R is dimerized, JAKs associated with the receptor components phosphorylate the second, third, or fourth tyrosine residues of the IL-4Rα cytoplasmic domain [50]. This phosphorylation recruits Stat6, which is then also phosphorylated. Phospho-Stat6 molecules dimerize and migrate to the nucleus to bind to certain promoters.
Accordingly, IL-13 has biological activity very similar to that of IL-4 [76]. Both cytokines induce IgE class switching in B cells [56], enhancement of monocyte/macrophage antigen presentation ability, down-regulation of inflammatory cytokine production by monocytes/macrophages, production of anti-inflammatory molecules [13], chemokine production [55], production of adhesion molecules such as vascular cell adhesion molecule (VCAM)-1 [4], and suppression of apoptosis [42]. However, there are significant differences in certain functions such as induction of Th2 development, since T cells do not express the type II IL-4R and therefore do not respond to IL-13 [73]. IL-4 also has been shown to enhance cytotoxic T cell activity [24, 57, 58, 64]. On dendritic cells, although both IL-4 and IL-13 induce maturation through the type II IL-4R, IL-4 enhances IL-12 p70 production whereas IL-13 does not [41]. Moreover, recently many IL-13-dependent, but IL-4-independent or less dependent, phenomena have been reported, such as expulsion of nematode parasites [68], induction of oxazolone colitis (Th2-induced ulcerative colitis) [22], induction of asthma [20, 69], parasite-induced liver and lung fibrosis [9, 10], and other cases of lung fibrosis [39].
In this review we will focus on the role of IL-13 in regulation of the immune response against tumors and the direct effect of IL-13 on tumor cells.
Role of IL-13 in negative regulation of immunosurveillance
In the tumor immunology field, it is widely accepted that induction of a Th1 and/or CTL immune response is critical for tumor rejection. It has been reported that in tumor-bearing animals and cancer patients, typical Th1 responses such as DTH or CD8+ CTL activity were suppressed. One possible explanation of this immunosuppression is that skewing the immune response against tumor toward Th2 suppresses Th1 immune responses. To test this hypothesis, studies using Stat6-deficient mice have recently been carried out in multiple mouse transplantable tumor systems [27, 52, 66], discussed elsewhere in this issue. In these studies, Stat6 knockout (KO) mice, which are deficient for IL-4Rα-mediated signaling, rejected tumors through the action of tumor-specific CD8+ CTLs, implicating a role of IL-13 and/or IL-4 in negative regulation of anti-tumor immunity.
In the case of a murine fibrosarcoma expressing a viral tumor antigen, tumors spontaneously regress by 10 to 15 days after initial growth. However, the tumors recur between 20 and 40 days. We originally found that in this tumor system, CD4+ T cells repress tumor-specific CD8+ CTLs and their IFN-γ production in vivo [43]. IL-4Rα KO and Stat6 KO mice but not IL-4 KO could reject the tumors, implicating IL-13 rather than IL-4 as the inhibitor of tumor immunosurveillance [66]. When IL-13 was inhibited by soluble IL-13Rα2-Fc, not only IL-4 KO mice but also wild-type mice could reject the tumors, similar to IL-4Rα KO and Stat6 KO mice. Therefore, we concluded that IL-13 was the primary cytokine which down-regulated CTL-mediated tumor immunosurveillance [66]. Moreover, NKT cell–deficient CD1d KO mice were highly resistant to the tumor, and IL-13 production by T cells was significantly reduced in this mouse after tumor inoculation. This observation was consistent with the result that CD4+ NKT cells from tumor-bearing mice manifested up-regulated IL-13 production. The results clearly indicated that CD4+ NKT cells were the major source of IL-13 in tumor-bearing mice since CD4+ T cells from NKT cell–deficient mice produced very low levels of IL-13. These findings define a novel immunoregulatory pathway in which IL-13 produced by CD4+ NKT cells down-regulates CD8+ CTL–mediated tumor immunosurveillance through the IL-4Rα–Stat6 pathway [66] (Fig. 2a). However, a remaining question was how IL-13 represses CTLs, since T cells do not express type II IL-4 (IL-13) receptors although they express the type I IL-4R. Recently we found that in tumor-bearing mice, IL-13 made by NKT cells activated CD11b+Gr-1+ myeloid cells to produce TGF-β which acts directly on CD8+ T cells to down-regulate tumor-specific CTL induction [67]. Such myeloid cell populations are discussed elsewhere in this issue.
Fig. 2a, b.
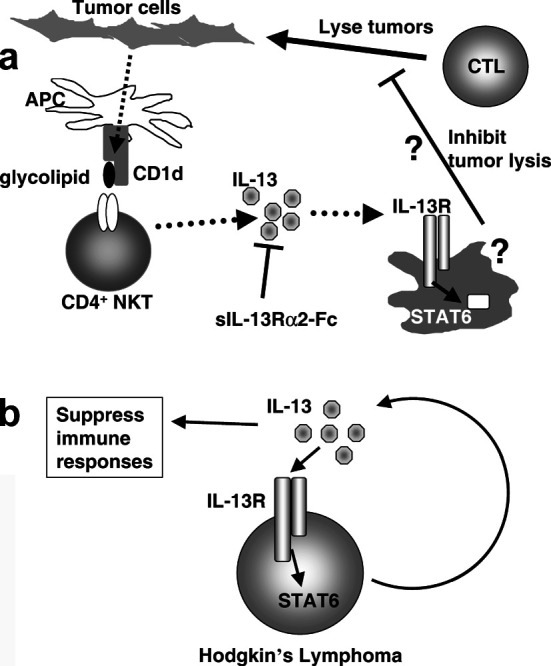
IL-13 can inhibit anti-tumor immune response and enhance tumor growth. a Inhibitory role of IL-13 in anti-tumor immunity. Tumor antigen (glycolipid) presented by antigen-presenting cells via the CD1d molecule is recognized by and activates CD4+ NKT cells. The activated CD4+ NKT cell produces IL-13 to suppress CD8+ cytotoxic T cells (CTLs) that kill tumor cells. However, since T cells do not express the IL-13 receptors, we postulated intermediate cells that express the IL-13 receptors, that are activated by IL-13 to directly suppress CTL. (Based on Terabe et al. [66, 67] with permission.) b IL-13 is a growth factor for Hodgkin’s lymphoma. Hodgkin’s lymphoma makes IL-13. This IL-13 activates growth of Hodgkin’s lymphoma through the IL-13 receptor–Stat6 pathway in an autocrine fashion [61]. The IL-13 made by Hodgkin’s lymphoma can also suppress anti-tumor immune responses by the mechanism shown in a
This negative regulatory pathway of tumor immunosurveillance seems to be active also in a colon carcinoma lung metastasis model, in which tumor growth is partially inhibited in CD1d KO mice or mice treated with IL-13 inhibitor, but not in IL-4 KO mice (Park et al., manuscript in preparation). In an orthotopic breast tumor model in which lung metastasis will cause the death of mice after primary resection, Stat6 KO mice and NKT cell–deficient CD1d KO mice were highly resistant against lung metastasis [53], consistent with our observation in the fibrosarcoma model. However, knocking out the IL-4Rα gene and inhibiting IL-13 in IL-4-deficient mice did not induce protection against lung metastasis. Although the explanation for the discrepancy between the results of Stat6 KO and IL-4Rα KO mice remains to be elucidated, it must be noted that CD1d KO mice are also highly resistant to the tumors tested. None of these tumors express CD1d, so the tumor rejection in the CD1d KO mice in all three of these models cannot be due to CD1d itself acting as a tumor rejection antigen. Finally, we have found that IL-13 inhibitor treatment of HER-2/neu transgenic mice can delay the development of spontaneous autologous mammary carcinomas that arise in these mice, whereas blockade of IL-4 had no effect (Park et al., manuscript in preparation). These data collectively support the notion that this negative regulatory pathway involving NKT cells and IL-13 inhibits immunosurveillance against several types of tumors, and its blockade at different steps can reveal spontaneous immunosurveillance that delays or prevents tumor growth. In addition, blockade of this negative regulatory pathway may enhance the efficacy of anti-tumor vaccines analogous to our antiviral vaccine studies in which we found that elimination of NKT cells or blockade of IL-13 enhanced vaccine-induced CTL responses and protection against viral challenge [1, 3].
A critical role of NKT cells in induction of Th2 immune responses through IL-4 or in negative regulation through IL-13 is consistent with many studies in T cell-mediated (Th1-mediated) autoimmune diseases such as type I diabetes [70], multiple sclerosis (EAE in mouse) [65], systematic lupus erythematosus [74], and ulcerative colitis [22]. On the other hand, there are other studies that demonstrated a crucial role of NKT cells in induction of natural immunosurveillance in mouse spontaneous tumor models [63]. These opposing roles of NKT cells may be explained by a recent study demonstrating that NKT cell activity was different between BALB/c and C57BL/6 mice in a mouse cerebral malaria [21]. In this model, using NK gene cluster congenic mice, it was shown that the origin of the NK gene cluster correlates with the activity of NKT cells, which plays a critical role in immune deviation. The hypothesis that this NK gene locus may explain some of the different effects of NKT cells in tumor immunosurveillance is under investigation in our laboratory. Other explanations include differences in the tumors themselves, such as their expression or lack of expression of CD1d, and the glycolipids they produce that could be presented by CD1d on dendritic or other antigen-presenting cells to activate NKT cells. NKT cell cytokine production is sensitive to the type and strength of the stimulus acting on these cells.
Although IL-13 is considered to be a cytokine that modulates adaptive immune responses as described above, it should be noted that there are some reports showing that local delivery of IL-13 at the tumor site induces tumor regression or rejection mediated by granulocytes and macrophages [33, 38]. Thus, IL-13 may be used to activate innate immunity to reduce tumorigenicity.
Direct effect of IL-13 on tumors
Since the type II IL-4R is expressed on a wide range of cells, it is not surprising that some types of tumor cells express the type II IL-4R. Indeed, there are many reports of expression of the type II IL-4R on tumor cells including lymphoma [8], renal cell carcinoma, glioblastoma [30], head and neck cancer [26, 30], hepatoma [17], pancreatic cancer, breast cancer [33], prostate cancer [32], and colon cancer [28]. However, except for lymphoma, few studies have been conducted to address the function of this receptor in the tumors. Among the tumors expressing the type II IL-4R, the role of the receptor and IL-13 itself is well studied only in Hodgkin’s lymphoma/Reed-Sternberg cells (H/RS). Expression of IL-13 by H/RS was found by using gene-chip technology to identify a gene uniquely expressed in an H/RS cell line compared with an EBV-immortalized B-lymphoblastoid cell line [29]. Interestingly, the IL-13 was used by the H/RS cells as an autocrine growth factor in vitro, since blocking of IL-13 induced apoptosis in a dose-dependent manner [60]. The H/RS lines were also observed to have activated Stat6, implying signal transduction through the receptor [59]. It should be noted that these observations were made not only in vitro in tumor cell lines but also in vivo by examining biopsies of H/SR [59]. Testing lymphoma biopsy samples for expression of IL-13, the IL-13Rα1, and activated Stat6 also demonstrated that H/RS express IL-13 and use it as a growth factor in an autocrine manner [61] (Fig. 2b). The expression of IL-13 and phospho-Stat6 is unique to H/RS, and was not observed in other types of lymphoma. In addition to the autocrine effect of IL-13, the IL-13 made by H/RS may also affect the immune response to this tumor [61]. One of the mechanisms of enhancing tumor metastasis is thought to be reducing cell-cell adhesion. In a colon cancer cell line which expresses the type II IL-4R, both IL-4 and IL-13 down-regulate expression of E-cadherin and carcinoembryonic antigen that are responsible for cell adhesion [28]. In glioma cell lines, which express IL-4Rα, IL-13Rα1, and IL-13Rα2, it has been shown that IL-13 up-regulates VCAM-1 in a dose- and time-dependent manner [31]. IL-13 also directly acts on B chronic lymphocytic leukemia (B-CLL) [8]. In B-CLL, in contrast to the situation in H/RS, however, IL-13 does not act as a growth factor, but rather it inhibits IL-2-induced proliferation and spontaneous apoptosis. Thus, by several different mechanisms, IL-13 can promote growth or survival of certain types of tumors through direct action on the tumor, rather than or in addition to acting through suppression of immunosurveillance.
Conclusions
The ultimate goal of cancer immunology is to develop vaccines or immunotherapy. Understanding of mechanisms of negative regulatory pathways that down-regulate anti-tumor immune responses is critical, since most cancer vaccine and immunotherapy trials have had little success so far. Such negative regulatory pathways, rather than weakness of the vaccines per se, may be thwarting those efforts. Studies using both spontaneous and transplantable mouse tumor models indicate that there is a novel negative regulatory pathway inhibiting CD8+ CTL-mediated tumor immunosurveillance in which IL-13 and NKT cells, as a key source of the cytokine, play a critical role. Interrupting this pathway has been shown to prevent or delay tumor growth or metastasis in several types of murine tumors, revealing an underlying immunosurveillance capacity of the host that had been suppressed by this regulatory pathway. Blockade of this pathway also has been shown to enhance vaccine efficacy, amplifying the CTL response that is otherwise dampened by this IL-13-dependent mechanism. IL-13 also has direct effects on tumor growth, metastasis, and escape from apoptosis for certain types of tumors. Since IL-13 overlaps in its features with IL-4 by virtue of sharing a receptor, many of the other biological activities of IL-13 may be compensated by IL-4 when IL-13 is inhibited, avoiding serious adverse effects of IL-13 blockade. Thus, blockade of IL-13 can be an attractive target for anti-tumor immunotherapy either alone to unmask immunosurveillance or in combination with other immunotherapy, and an IL-13 inhibitor is likely to be synergistic with cancer vaccines.
Footnotes
This article forms part of the Symposium in Writing “Inhibitors of immunosurveillance and anti-tumor immunity,” published in Vol. 53.
References
- 1.Ahlers Proc Natl Acad Sci USA. 2002;99:13020. doi: 10.1073/pnas.192251199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aman J Biol Chem. 1996;271:29265. doi: 10.1074/jbc.271.46.29265. [DOI] [PubMed] [Google Scholar]
- 3.Berzofsky Nat Rev Immunol. 2001;1:209. doi: 10.1038/35105075. [DOI] [PubMed] [Google Scholar]
- 4.Bochner J Immunol. 1995;154:799. [PubMed] [Google Scholar]
- 5.Brown J Immunol. 1989;142:679. [Google Scholar]
- 6.Burd J Exp Med. 1995;181:1373. doi: 10.1084/jem.181.4.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caput J Biol Chem. 1996;271:16921. doi: 10.1074/jbc.271.28.16921. [DOI] [PubMed] [Google Scholar]
- 8.Chaouchi Blood. 1996;87:1022. [PubMed] [Google Scholar]
- 9.Chiaramonte J Clin Invest. 1999;104:777. doi: 10.1172/JCI7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiaramonte J Immunol. 1999;162:920. [PubMed] [Google Scholar]
- 11.Chiaramonte J Exp Med. 2003;197:687. doi: 10.1084/jem.20020903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de J Immunol. 1998;160:1666. [PubMed] [Google Scholar]
- 13.de J Immunol. 1993;151:6370. [PubMed] [Google Scholar]
- 14.Donaldson J Immunol. 1998;161:2317. [PubMed] [Google Scholar]
- 15.Fallon Immunity. 2002;17:7. [Google Scholar]
- 16.Finkelman Curr Opin Immunol. 1999;11:420. doi: 10.1016/S0952-7915(99)80070-3. [DOI] [PubMed] [Google Scholar]
- 17.Gabay Blood. 1999;93:1299. [PubMed] [Google Scholar]
- 18.Gauchat Eur J Immunol. 1997;27:971. doi: 10.1002/eji.1830270425. [DOI] [PubMed] [Google Scholar]
- 19.Gibbs Eur J Immunol. 1996;26:2493. doi: 10.1002/eji.1830261033. [DOI] [PubMed] [Google Scholar]
- 20.Grunig Science. 1998;282:2261. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen Immunity. 2003;18:391. [Google Scholar]
- 22.Heller Immunity. 2002;17:629. [Google Scholar]
- 23.Hilton Proc Natl Acad Sci USA. 1996;93:497. doi: 10.1073/pnas.93.1.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horohov J Immunol. 1988;141:4217. [PubMed] [Google Scholar]
- 25.Hoshino J Immunol. 1999;162:51. [Google Scholar]
- 26.Joshi Clin Cancer Res. 2002;8:1948. [Google Scholar]
- 27.Kacha J Immunol. 2000;165:6024. doi: 10.4049/jimmunol.165.11.6024. [DOI] [PubMed] [Google Scholar]
- 28.Kanai Br J Cancer. 2000;82:1717. doi: 10.1054/bjoc.2000.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kapp J Exp Med. 1999;189:1939. doi: 10.1084/jem.189.12.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawakami Hum Gene Ther. 2000;11:1829. doi: 10.1089/10430340050129459. [DOI] [PubMed] [Google Scholar]
- 31.Kawakami Oncol Res. 2000;12:459. [Google Scholar]
- 32.Kawakami Cancer Gene Ther. 2001;8:861. doi: 10.1038/sj.cgt.7700373. [DOI] [PubMed] [Google Scholar]
- 33.Kawakami J Exp Med. 2001;194:1743. doi: 10.1084/jem.194.12.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keegan Cell. 1994;76:811. doi: 10.1016/0092-8674(94)90356-5. [DOI] [PubMed] [Google Scholar]
- 35.Kelly-Welch Science. 2003;300:1527. doi: 10.1126/science.1085458. [DOI] [PubMed] [Google Scholar]
- 36.Kindler Eur J Immunol. 1995;25:1239. doi: 10.1002/eji.1830250516. [DOI] [PubMed] [Google Scholar]
- 37.Kobayashi J Immunol. 1998;160:5869. [PubMed] [Google Scholar]
- 38.Lebel-Binay Eur J Immunol. 1995;25:2340. doi: 10.1002/eji.1830250833. [DOI] [PubMed] [Google Scholar]
- 39.Lee J Exp Med. 2001;194:809. doi: 10.1084/jem.194.6.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J Immunol. 1996;156:4833. [PubMed] [Google Scholar]
- 41.Lutz J Immunol. 2002;169:3574. doi: 10.4049/jimmunol.169.7.3574. [DOI] [PubMed] [Google Scholar]
- 42.Manna J Immunol. 1998;161:2863. [PubMed] [Google Scholar]
- 43.Matsui J Immunol. 1999;163:184. [Google Scholar]
- 44.McKenzie Pharmacol Ther. 2000;88:143. doi: 10.1016/s0163-7258(00)00088-7. [DOI] [PubMed] [Google Scholar]
- 45.McKenzie Proc Natl Acad Sci USA. 1993;90:3735. [Google Scholar]
- 46.McKenzie J Immunol. 1993;150:5436. [PubMed] [Google Scholar]
- 47.Miloux FEBS Lett. 1997;401:163. doi: 10.1016/s0014-5793(96)01462-7. [DOI] [PubMed] [Google Scholar]
- 48.Minty Nature. 1993;362:248. doi: 10.1038/362248a0. [DOI] [PubMed] [Google Scholar]
- 49.Morgan Nucleic Acids Res. 1992;20:5173. doi: 10.1093/nar/20.19.5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nelms Annu Rev Immunol. 1999;17:701. doi: 10.1146/annurev.immunol.17.1.701. [DOI] [PubMed] [Google Scholar]
- 51.Obiri J Biol Chem. 1995;270:8797. doi: 10.1074/jbc.270.15.8797. [DOI] [PubMed] [Google Scholar]
- 52.Ostrand-Rosenberg J Immunol. 2000;165:6015. doi: 10.4049/jimmunol.165.11.6015. [DOI] [PubMed] [Google Scholar]
- 53.Ostrand-Rosenberg J Immunol. 2002;169:5796. doi: 10.4049/jimmunol.169.10.5796. [DOI] [PubMed] [Google Scholar]
- 54.Peritt J Immunol. 1998;161:5821. [PubMed] [Google Scholar]
- 55.Pope J Allergy Clin Immunol. 2001;108:594. doi: 10.1067/mai.2001.118600. [DOI] [PubMed] [Google Scholar]
- 56.Punnonen Proc Natl Acad Sci USA. 1993;90:3730. [Google Scholar]
- 57.Schuler J Exp Med. 1999;189:803. doi: 10.1084/jem.189.5.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schuler J Exp Med. 2001;194:1767. doi: 10.1084/jem.194.12.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Skinnider Blood. 2001;97:250. doi: 10.1182/blood.V97.1.250. [DOI] [PubMed] [Google Scholar]
- 60.Skinnider Blood. 2002;99:618. doi: 10.1182/blood.v99.2.618. [DOI] [PubMed] [Google Scholar]
- 61.Skinnider Leuk Lymphoma. 2002;43:1203. doi: 10.1080/10428190290026259. [DOI] [PubMed] [Google Scholar]
- 62.Smirnov Gene. 1995;155:277. doi: 10.1016/0378-1119(94)00720-d. [DOI] [PubMed] [Google Scholar]
- 63.Smyth J Exp Med. 2000;191:661. doi: 10.1084/jem.191.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spits J Immunol. 1988;141:29. [Google Scholar]
- 65.Sumida J Exp Med. 1995;182:1163. doi: 10.1084/jem.182.4.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Terabe Nat Immunol. 2000;1:515. doi: 10.1038/82771. [DOI] [PubMed] [Google Scholar]
- 67.Terabe M, Matsui S, Park J-M, Mamura M, Noben-Trauth N, Donaldson DD, Chen W, Wahl SM, Ledbetter S, Pratt B, Letterio JJ, Paul WE, Berzofsky JA (2003) TGF-b productin and myeloid cells are an effector mechanism through which CD1d-restricted T cells block CTL-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med (in press) [DOI] [PMC free article] [PubMed]
- 68.Urban Immunity. 1998;8:255. doi: 10.1016/s1074-7613(00)80477-x. [DOI] [PubMed] [Google Scholar]
- 69.Wills-Karp Science. 1998;282:2258. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 70.Wilson Nature. 1998;391:177. doi: 10.1038/34419. [DOI] [PubMed] [Google Scholar]
- 71.Wood J Exp Med. 2003;197:703. doi: 10.1084/jem.20020906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wynn Annu Rev Immunol. 2003;21:425. doi: 10.1146/annurev.immunol.21.120601.141142. [DOI] [PubMed] [Google Scholar]
- 73.Yokota Proc Natl Acad Sci USA. 1986;83:5894. [Google Scholar]
- 74.Zeng J Immunol. 2000;164:5000. [Google Scholar]
- 75.Zhang J Biol Chem. 1997;272:9474. doi: 10.1074/jbc.272.14.9474. [DOI] [PubMed] [Google Scholar]
- 76.Zurawski Immunol Today. 1994;15:19. doi: 10.1016/0167-5699(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 77.Zurawski Embo J. 1993;12:2663. doi: 10.1002/j.1460-2075.1993.tb05927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zurawski J Biol Chem. 1995;270:13869. doi: 10.1074/jbc.270.23.13869. [DOI] [PubMed] [Google Scholar]