

Abstract

Understanding how signaling proteins like G proteins are allosterically activated is a long-standing challenge with significant biological and medical implications. Because it is difficult to directly observe such dynamic processes, much of our understanding is based on inferences from a limited number of static snapshots of relevant protein structures, mutagenesis data, and patterns of sequence conservation. Here, we use computer simulations to directly interrogate allosteric coupling in six G protein α-subunit isoforms covering all four G protein families. To analyze this data, we introduce automated methods for inferring allosteric networks from simulation data and assessing how allostery is conserved or diverged among related protein isoforms. We find that the allosteric networks in these six G protein α subunits are largely conserved and consist of two pathways, which we call pathway-I and pathway-II. This analysis predicts that pathway-I is generally dominant over pathway-II, which we experimentally corroborate by showing that mutations to pathway-I perturb nucleotide exchange more than mutations to pathway-II. In the future, insights into unique elements of each G protein family could inform the design of isoform-specific drugs. More broadly, our tools should also be useful for studying allostery in other proteins and assessing the extent to which this allostery is conserved in related proteins.
Introduction
Understanding how signaling proteins like G proteins are allosterically activated is a long-standing challenge with significant biological and medical implications. Insight into G protein activation is of great biological significance because these proteins play key roles in regulating a wide range of biological processes including vision, olfaction, taste, neurotransmission, cell proliferation, and motility.1,2 These processes are mediated by 16 Gα protein isoforms, which are categorized into four families based on downstream signaling modality and sequence homology: Gαi/o, Gαs, Gαq/11, and Gα12/13.3,4 Dysfunction of these different isoforms results in diseases ranging from cancer to pseudohypoparathyroidism.5−7 One strategy to treat these diseases is to target the relevant G protein isoform without eliciting off-target effects by simultaneously impacting other isoforms. Achieving this end would be facilitated by understanding how each isoform is activated and identifying structural elements that are shared or unique to each isoform. Such structural elements could be unique either because they play a more important role for some isoforms than others or because they are important in multiple isoforms but have unique sequences/chemistry in some. Ideally, these subtle differences could then be utilized to yield therapeutics with the desired specificity and minimize off-target impacts by uniquely targeting shared, but sequence-diverged, structural elements or structural elements with disparate isoform-specific effects.
Interrogating dynamic processes like the allosteric coupling responsible for G protein activation remains difficult despite significant progress in understanding the structure and function of these molecular switches. G proteins are heterotrimeric complexes composed of an α, β, and γ subunit.8 The α subunit (abbreviated Gα and shown in Figure 1) is the key switch, which in the inactive state has GDP bound between its two domains, the ras-like G domain and the α-helical H domain. G-protein-coupled receptors (GPCRs) activate G proteins by binding the Gα subunit and triggering exchange of GDP for GTP with subsequent separation from Gβγ to yield the active form of Gα. However, many questions remain about how the GPCR- and nucleotide-binding sites communicate given that they are over 30 Å apart. Analysis of sequence conservation and a wealth of biochemical data suggested all G proteins use a universal activation mechanism.9 However, it is necessary to test this hypothesis by directly observing the activation process of every G protein subtype. Molecular dynamics simulations provide a means to study with atomistic detail how similar the allosteric coupling responsible for G protein activation is across families, but so far publications on this subject have focused on single G proteins.10,11
Figure 1.

Structure of a Gα subunit and sequence variation across the 16 Gα isoforms. (A) Structure of Gq (PDB: 3ah8) highlighting GDP (gray spheres) bound to the active site, which is between the G domain (light brown) and H domain (cyan). Key secondary structure elements are labeled according to the CGN system. GPCR-binding regions are shown in orange. (B) Amino acid conservation score computed on the Consurf server using the multiple sequence alignment of the 16 Gα isoforms mapped onto the structure of Gq.
Here, we use molecular dynamics simulations to assess whether the four G protein families use a universal allosteric network to become activated. Specifically, we ran an aggregate of over 210 μs of simulation of six different Gα isoforms: Gi1, Gt, and Go from the Gi/o family; Gq from the Gq/11 family; G13 from the G12/13 family; and Gs from the Gs family. For each isoform, we ran tens of μs of molecular dynamics simulations, quantified the correlations between the structure and dynamics of every pair of amino acids, and identified networks of correlated residues (which we refer to as an allosteric network). While our simulations do not capture the entire activation process or explicitly include perturbations like GPCR binding, we have previously demonstrated that our allosteric networks recapitulate regions that undergo allosteric conformational/dynamical changes in response to perturbations elsewhere in a protein.10,12 We focus on the inactive (i.e., GDP-bound) state of each G protein as this is the biochemical state where activation occurs. We also present new approaches for automating the detection of allosteric networks and assessing the conservation of these networks between multiple proteins. We focus on assessing the extent to which the different G proteins share a universal activation mechanism. To discuss comparisons between isoforms, we use the common Gα numbering (CGN) system.9 In this system, residues are named based on their position in a secondary structure element. For example, PheG.H5.8 refers to the Phe at the eighth residue of the fifth helix of the G domain and CysG.s6h5.2 refers to the Cys at the second residue of the loop between β strand 6 and helix 5 of the G domain. In the future, drawing on our work to delve deeper into the differences between subfamilies/isoforms will likely be of great value for informing the development of inhibitors that specifically target different subsets of G proteins.
Methods
Molecular Dynamics Simulations of Gα Isoforms
Crystal structures of Gαi1 (PDB id: 3QE0),13 Gαt (PDB id: 1GOT),14 Gαo (PDB id: 3C7K),15 Gα13 (PDB id: 1ZCB),16 and Gαs (PDB id: 6EG8)17 were used to set up simulations of GDP-Gα for these isoforms. A Mg2+ ion coordinating to GDP at the active site was also included in each system. Helix HN at the N-terminal and residues after position G.H5.21 at the C-terminal were removed from these structures to make the protein lengths uniform across isoforms including Gαq system prepared in our previous study.10
Each protein structure was solvated in a dodecahedron box of TIP3P water model18 that extended 10 Å from the protein in every dimension. Na+ and Cl– were added to prepare a neutral system with 0.15 M NaCl. AMBER03 force field was used for protein.19 We chose AMBER03 in part to be consistent with our past work on G proteins.10 We have also found that it gives comparable performance to other force fields,20 explains published experimental data,21 and predicts the outcome of new experiments.22 The force field parameters of GDP were obtained from the AMBER Parameter Database23 as described in our earlier work on Gαq.10 Virtual sites were used to allow for a 4 fs time step24 during the production runs of molecular dynamics.
Solvated protein systems were energy minimized using the steepest descent algorithm for 50000 steps until the maximum force reached below 100 kJ/mol/nm. A step size of 0.02 nm and a cutoff distance of 0.9 nm for the neighbor list, electrostatic interactions, and van der Waals interactions were used during the energy minimization step. Subsequently, NVT simulations were run with the constraint of 1000 kJ/mol/nm applied to the protein heavy atoms using a time step of 2 fs. During these simulations, the temperature of the system was constrained to 300 K using a V-rescale thermostat with a time constant of 0.1 ps.25 A cutoff distance of 1 nm was used for the van der Waals, short-range electrostatic interactions, and periodic boundary conditions that were applied to the x, y, and z directions. The particle-mesh Ewald method was employed to recover the long-range electrostatic interactions with 0.16 nm as the grid spacing and with a fourth-order spline.26 All the bonds connecting to hydrogens are constrained using the LINCS algorithm.27 Following the NVT simulations, the systems were equilibrated using NPT simulations for 1 ns using the integration time step of 2 fs. All simulation parameters were the same as those used during NVT equilibration. In addition, the Parrinello–Rahman barostat for pressure coupling28 was applied to constrain the pressure of the system to 1 bar. The position constraints on heavy atoms were removed during the subsequent production runs set up on Folding@home.
We performed 100 parallel simulations of the GDP-bound state of Gα isoforms on the Folding@home29 distributed computing platform. For each isoform, aggregate simulations of more than 35 μs were performed.
Correlation of All Rotameric and Dynamical States (CARDS) Analysis
We applied the CARDS10 framework to derive inter-residue structural and dynamical correlations to understand allosteric communications in Gα isoforms. The CARDS method measures correlations between every pair of dihedral angles (backbone and side chain) by quantifying correlated changes in their rotameric (structural) states and dynamical behavior. Structural states of backbone dihedrals are determined by categorizing Φ and Ψ dihedrals into rotameric states cis and trans, respectively. The side-chain dihedrals are categorized into three states, gauche+, gauche-, and trans. Every dihedral is also discretized into dynamical states based on whether the dihedral is stable in a single rotameric state (ordered) or rapidly transitioning between multiple states (disordered) as described in earlier studies.10,12
From the probability distribution of the structural and dynamical states of every dihedral angle, a holistic communication for every pair of dihedrals is computed as,
where 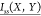 is the normalized mutual information
between
the rotameric states of dihedral X and dihedral Y,
is the normalized mutual information
between
the rotameric states of dihedral X and dihedral Y, 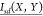 is the normalized mutual information
between
the rotameric state of dihedral X and the dynamical
state of dihedral Y,
is the normalized mutual information
between
the rotameric state of dihedral X and the dynamical
state of dihedral Y,  is the normalized mutual information
between
the dynamical state of dihedral X and the rotameric
state of dihedral Y, and
is the normalized mutual information
between
the dynamical state of dihedral X and the rotameric
state of dihedral Y, and 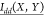 is the normalized mutual information
between
the dynamical state of dihedral X and the dynamical
state of dihedral Y. The mutual information (I) is computed as,
is the normalized mutual information
between
the dynamical state of dihedral X and the dynamical
state of dihedral Y. The mutual information (I) is computed as,
Community Detection
To detect clusters of strongly intercommunicating residues from the CARDS allosteric network, we applied Louvain community detection method.30 Louvain algorithm identifies clusters by maximizing the modularity function defined as follows
Here, Aij is the edge weight connecting two nodes i and j; ki and kj are the sum of all the edge weights connected to nodes i and j, respectively; m is the sum of all the edge weights in the network; ci and cj are the clusters of nodes i and j, respectively; and δ(cicj) is a Kronecker delta function such that δ(cicj) = 1 if two nodes i and j are in the same cluster, 0 otherwise.
Plasmid Construction
pBAD 6xHis-Gαi1 was a gift from Dr. Sergej Djuranovic, Washington University in St. Louis, St. Louis, MO.31 Further mutations were generated by site-directed mutagenesis using specifically designed and synthesized primers for each mutation (Integrated DNA Technologies). All plasmids were verified by Sanger sequencing (GeneWiz).
Protein Expression and Purification
All G proteins were expressed recombinantly in TOP10 cells (ThermoFisher). 0.5 L of cultures was autoinduced for 72 h at room temperature in 2.4 L Erlenmeyer flasks with media containing 2% tryptone, 1% yeast extract, 1% glycerol, 0.5% NaCl, 50 mM KH2PO4, and 25 mM (NH4)2SO4 buffered to pH 8.0 and supplemented with 100 μg/mL ampicillin, 2 mM MgSO4, 0.4x Trace Metals, 5 mM d-glucose, and 0.1 mM l-arabinose at time of induction.32,33 The induced cells were collected by centrifugation at 4 °C (4000g for 15 min), the supernatant was discarded, and the pellets were snap-frozen in liquid nitrogen and stored at −80 °C until purification. The harvested cells were thawed on ice, suspended in lysis buffer (50 mM Tris, pH 8, 150 mM NaCl, 150 mM KCl, 25 mM (NH4)2SO4, 10% glycerol, 10 mM BME, 1× cOmplete EDTA-free Protease Inhibitor Cocktail (Roche)), and then lysed by sonication on ice. The supernatant was cleared by centrifugation (100,000g for 30 min) and then bound to Ni-NTA Agarose (Qiagen) for 1 h at 4 °C with shaking. Agarose resin was collected by centrifugation at 4 °C (4000g for 5 min), and the supernatant was discarded. Beads were washed twice with 50 mL of wash buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 150 mM KCl, 25 mM (NH4)2SO4, 10 mM BME, 10% glycerol) and shaking for 5 min at 4 °C, transferred to a disposable gravity flow column, and washed once with 25 mL of wash buffer. Protein was eluted with elution buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 150 mM KCl, 25 mM (NH4)2SO4, 10% glycerol, 500 mM imidazole), and a sample was taken for activity assays. The eluted protein was then dialyzed in dialysis buffer (50 mM HEPES, 10 mM MgCl2, 1 mM EDTA, 1 mM DTT, 10% glycerol, pH 8.0) starting with dialysis in 500 mL for at least 4 h, then 500 mL overnight, and finally 1 L for at least 4 h. Protein concentration was determined spectroscopically in dialysis buffer using an extinction coefficient of 35 870 M–1 cm–1. The eluted protein was then aliquoted, snap-frozen in liquid nitrogen, and stored at −80 °C until use.
Nucleotide Exchange Assays
Nucleotide loading activity of purified Gα subunits was detected with BODIPY-FL-GTPγS, a nonhydrolyzable fluorescent analog of GTP, and a Synergy H4 plate reader26. For association assays, BODIPY-FL-GTPγS (25 nM final concentration) was aliquoted into the wells of a black-wall clear-bottom 96-well plate (catalog number 3603, Costar). Nucleotide loading was initiated by injecting Gα subunits (1 μM final concentration) in reaction buffer (50 mM Tris–HCl, pH 8.0, 1 mM EDTA, 10 mM MgCl2). For dissociation assays, BODIPY-FL-GTPγS (25 nM final concentration) and Gα subunits (1 μM final concentration) were preincubated for 1 h in the wells of a black-wall clear-bottom 96-well plate (catalog number 3603, Costar) before injecting GDP (20 μM final concentration) in reaction buffer +25 nM BODIPY-FL- GTPγS to initiate dissociation. Fluorescence intensity was monitored in well mode at 30 °C for 15 min with 1s intervals for association or in plate mode for 180 min with 5s intervals for dissociation with excitation and emission bandpass filters with wavelengths of 485/20 nm and 528/20 nm, respectively, and a 510 nm dichroic mirror. The resulting curves were fit to one-phase association models for kobs and one-phase decay models for kd using GraphPad Prism v6.01. From these values, ka was determined from the relationship kobs = [L]·ka + kd which, when rearranged, yields ka = (kobs – kd)/[L]. Kd values were subsequently calculated using the relationship Kd = kd/ka.
Results and Discussion
Automated Identification of the Allosteric Network in Gq Recapitulates Prior Results
To assess how well the allosteric network responsible for G protein activation is conserved, we first sought to develop an automated method for detecting allostery that would ensure robust and reproducible results. Many methods have been developed to dissect allosteric networks.34−38 Most focus on concerted structural changes. For this work, we chose to use CARDS (Correlation of All Rotameric and Dynamical States, see the Supporting Information for details, Figure S1).12 CARDS is unique in its ability to account for coupling between both the structure and dynamics of pairs of dihedral angles. We previously showed that the dynamical insight it provides is important for understanding allostery in G proteins.10 While determining the coupling between pairs of dihedrals and coarse-graining this information to determine the coupling between pairs of residues is automated, further interpretation of the allosteric network previously required manual input. For instance, in our past work, we manually identified sets of residues with strong coupling to one another that spanned the GPCR- and nucleotide-binding sites.10 While this analysis provided significant insight, performing the same analysis on multiple isoforms is labor-intensive and potentially introduces an unwanted element of subjectivity into any claims about conservation. Therefore, we sought to develop an automated process of identifying allosteric networks based on CARDS.
To automate the process of finding allosteric networks, we applied a community detection method called the Louvain algorithm to the matrix of all pairwise correlations between the structure and dynamics of residues that we calculated with our CARDS methods. The Louvain algorithm identifies clusters of strongly interconnected nodes within a network.30 Like many clustering algorithms, the Louvain algorithm seeks to group together nodes (in this case, residues) that have strong links to each other while separating weakly connecting nodes into separate clusters. This method has a number of desirable capabilities, particularly the ability to determine the number of clusters automatically. At the start of this method, each node in a network is assigned to its own cluster. In subsequent steps, these nodes are iteratively grouped together into larger clusters to maximize a quantity called modularity, which is a measure of the density and strength of connections among nodes within a cluster compared to connections between clusters.
Applying Louvain clustering to CARDS data from simulations of Gαq recapitulated insights from our past work, as expected.10 Louvain clustering identified six clusters of residues. We will refer to the three in the G domain as GDC1–3, the one in the H domain as HdC1, and the three at the interdomain interface IDC1–3 (Figure 2). GDC1 encompasses the GPCR-binding interface (the C-terminal segment of H5 and the hNs1 loop), β strands S1–S4, residues from helix H1, and the P-loop that engages the nucleotide phosphates, implying that these regions have strong structural and dynamical correlations. GDC1 also has strong coupling to GDC3 (Figure 2B), which includes other key components of the nucleotide-binding site, such as the switch-I and switch-II motifs which contain the active site residues responsible for GTP hydrolysis and undergo important nucleotide-dependent structural changes that distinguish the active and inactive states.39−41 We refer to GDC1 and GDC3 as pathway-I for communication between the GPCR- and nucleotide-binding sites. GDC2 constitutes a second pathway, which we refer to as pathway-II, as it encompasses GPCR-binding elements like β strand S6 and nucleotide-binding elements like helix HG and the s6h5 loop. These pathways are consistent with previous computational studies of allostery in Gq and other G protein isoforms.9,10,42
Figure 2.

Allosteric network in Gq that was automatically identified by Louvain clustering. (A) A structure of Gq with residues colored based on which of the six clusters they are grouped into. (B) A representation of the allosteric network showing the color coding of the G domain clusters (GDC1–3), the H domain cluster (HDC1), and the interdomain clusters (IDC1–3). Each node represents a cluster, the size of the node is proportional to the average strength of CARDS communication between amino acids within the cluster, and the width of the edges between nodes is proportional to the average strength of CARDS communication between residues in the two clusters.
Automated Analysis of Other Isoforms Suggests Similar Allosteric Networks
Encouraged by the success of Louvain clustering in finding important allosteric pathways in Gq, we extended this approach to other Gα isoforms to understand and compare their allosteric dynamics. We selected at least one representative isoform from each of the other families besides Gq/11, specifically Gi1, Gt, and Go from the Gi family, G13 from the G12/13 family, and Gs from the Gs family. We collected ∼35 μs of molecular dynamics simulation data for each isoform and applied CARDS to quantify the coupling between every pair of dihedral angles. Then, we applied Louvain clustering to coarse-grain these networks and begin understanding how similar they are.
Examining the allosteric networks identified by Louvain clustering suggests that the different isoforms share many common features. For example, several residues from the C-terminal of helix H5, β strands S1–S3, and helix H1 are in a common cluster in multiple isoforms (Figure 3), suggesting that these regions are strongly coupled across subfamilies. These regions are an important part of pathway-I, suggesting that this path may be shared by many isoforms. Similarly, parts of activation pathway-II, such as residues from β strand S6 and the s6h5 loop, are grouped together in all the isoforms. There are also similarities outside the activation pathways. For example, a large part of the H domain is grouped into a single cluster in multiple isoforms.
Figure 3.

Allosteric networks for six different G proteins covering all four subfamilies. (A) Louvain clustering of CARDS communication networks for G protein isoforms (Gq, Gi1, Gt, Go, G13, and Gs) mapped on their structures. (B) Network diagrams showing coupling between clusters in each G protein isoform shown in panel (A). Each node is a cluster with a size proportional to the average strength of coupling between residues in that cluster. Edge widths are proportional to the strength of communication between clusters.
Despite the common allosteric aspects discussed above, we observed significant differences in the number and composition of clusters among the different isoforms that could be useful for targeting specific subsets of them. For example, the residues in GDC2 of Gq are split between two clusters in Gt, suggesting that pathway-II may be weaker in Gt than it is in Gq. Parts of switch-II also were assigned to different clusters in different isoforms. These differences are noteworthy because switch-II is important for nucleotide binding and is part of the binding interface that G proteins use to engage effector molecules. Therefore, differences between the allosteric coupling that controls the conformation of switch-II in various isoforms could have important functional consequences.
Quantifying the Conservation of Allostery between Isoforms Identifies a Universal Activation Mechanism
While there are similarities between the networks depicted in Figure 3, it is not clear how to interrogate these networks more deeply to quantify the extent to which the allosteric network is conserved or to identify specific residues that behave the same or differently. Obtaining such insight is crucial for using our models to guide future experiments, such as the selection of residues to mutate to test a proposed allosteric network or the design of allosteric modulators.
To quantify the extent of conservation at the residue level, we adapted a network alignment method originally developed to study how protein–protein interaction networks are conserved between organisms.43 The first step was to create an alignment network from two parent networks (A and B) one wishes to compare. Each node in the alignment network represents a pair of equivalent nodes from the parent networks. In this case, a node in the alignment network refers to equivalent nodes from two Gα isoforms, where equivalency is determined by the CGN numbering scheme. Edges between nodes in the alignment network quantify how strongly conserved the allosteric coupling between a pair of residues is. This conservation score (Δs(a,b)) is defined as
where a is the strength of
coupling between the two residues in parent network A and b is the strength of coupling between the two
residues in parent network B. 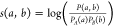 , where P(a,b) is the joint probability
that allosteric correlations
between equivalent residues in two isoforms A and B have strengths a and b, respectively, PA (a) is the probability that an allosteric correlation has
strengths a in the isoform A, ⟨s(a,b′)⟩b′ is the
average over all aligned inter-residue allosteric correlations with
strength a fixed, and ⟨s(a′,b)⟩a′ is the average over all aligned
inter-residue allosteric correlations with strength b fixed. We used Gq as a reference and compared each isoform to it.
Further details are given in the Supporting Information.
, where P(a,b) is the joint probability
that allosteric correlations
between equivalent residues in two isoforms A and B have strengths a and b, respectively, PA (a) is the probability that an allosteric correlation has
strengths a in the isoform A, ⟨s(a,b′)⟩b′ is the
average over all aligned inter-residue allosteric correlations with
strength a fixed, and ⟨s(a′,b)⟩a′ is the average over all aligned
inter-residue allosteric correlations with strength b fixed. We used Gq as a reference and compared each isoform to it.
Further details are given in the Supporting Information.
This approach confirmed that the different Gα isoforms share a largely conserved allosteric network and highlighted specific residues of importance (Figures 4 and 5). Many of the inter-residue correlations that constitute activation pathway-I in Gq are conserved in multiple isoforms. For example, allosteric coupling between residue PheG.H5.8 from helix H5, several residues from β strands S1–S3, LysG.H1.1, MetG.H1.8, IleG.H1.11, and HisG.H1.12 from helix H1, and the P-loop were found to be conserved in all isoforms. Similarly, key parts of activation pathway-II are conserved, such as TyrG.S6.2, HisG.S6.4, and PheG.S6.5 from β strand S6 that engages GPCRs and residues ThrG.s6h5.1 and CysG.s6h5.2 from the s6h5 loop that engages the nucleotide. The binding site of a known Gq inhibitor, called YM-254890 (YM), is also part of this conserved allosteric network, suggesting that molecules like YM might be designed to inhibit other Gα isoforms by targeting this region. This largely universal allosteric network is depicted in Figure 5. Interestingly, the conservation of allosteric coupling does not necessitate the conservation of the sequence. Several of the key residues just highlighted are of different amino acid types in different isoforms.
Figure 4.

Conservation of the allosteric networks of each Gα isoform compared to Gq. The upper panel shows a representative structure with residues whose allosteric coupling is conserved color-coded according to the network representation of the conserved allosteric network in the lower panel. Each node represents a group of amino acids with conserved allosteric coupling compared to Gq. Residues and nodes are colored based on the allosteric network identified for Gq. The size of each node is proportional to the strength of coupling within that cluster for the given isoform, and the edge width is proportional to the strength of coupling between clusters. The edge color is proportional to the conservation score, taken with respect to Gq (dark means highly conserved and light means weakly conserved).
Figure 5.

A largely universal allosteric network is not directly linked to sequence conservation. (A) Key residues mapped onto a representative structure and colored according to the allosteric network shown in panel (B). (C) Amino acid conservation scores of residues in the unified allosteric network mapped on a representative structure. The amino acid conservation score is computed on the Consurf server using a multiple sequence alignment of 16 Gα isoforms.
Activation Pathway-I is the Dominant Communication Channel
Examining the allosteric networks for each isoform and the conservation between them suggested that allosteric pathway-I (GDC1 and GDC3) is generally stronger than pathway-II (GDC2). Each of the clusters in pathway-I has stronger coupling between residues within the cluster than is found in pathway-II, as denoted by the larger sizes of GDC1/3 compared to GDC2 in the network diagrams (Figures 3 and 4). The coupling between GDC1 and GDC3 is typically stronger than the coupling of GDC2 to any other cluster, again supporting the greater strength of pathway-I. Within pathway-I, our analysis of the conservation of allosteric coupling suggests that PheG.H5.8 has strongly conserved coupling with the S1–S3 strands and helix H1, notably PheG.S2.6, PheG.S3.3, MetG.H1.8, and HisG.H1.12 (Figures 4 and 5A). The strength of this coupling is consistent with the analysis of amino acid conservation by Flock et al., who highlighted the hydrophobic interactions of PheG.H5.8 with PheG.S2.6 and PheG.S3.3 from the S1–S3 strands and MetG.H1.8 and HisG.H1.12 from the H1 helix.9 Breaking down the allosteric coupling we observe into contributions from the side chains and the backbone suggests that the side chains are more important (Figure S5). Therefore, mutating PheG.H5.8 is likely to disrupt pathway-I. This prediction is also consistent with a previous simulation study.44 Similarly, HisG.S6.4 stood out as having strong and conserved coupling within pathway-II. The side chain of this residue also appears to be more important than the backbone (Figure S6), suggesting mutating HisG.S6.4 is likely to disrupt pathway-II.
To test this hypothesis, we mutated key residues identified in each pathway and experimentally assessed their effect on nucleotide exchange. As in previous work,45,46 we reasoned that mutations that increase the basal rate of nucleotide exchange are disrupting allostery. We focused on Gi because it has the biggest difference between the strength of pathway-I and pathway-II (Figure 3). We focus on Phe336G.H5.8 in pathway-I and His322G.S6.4 in pathway-II. These residues were selected based on their importance to the pathway (i.e., they maximize the sum of the correlations to other residues in the pathway) subject to the constraint that they do not directly interact with the nucleotide and, therefore, any effect is more likely due to allostery than to mitigating binding. To confirm that the effects on nucleotide exchange are due to allostery, we also measured the association rate, dissociation rate, and binding affinity for nucleotide (Table 1). To test the importance of pathway-II, we chose to make the substitution His322G.S6.4Ala and found that this variant behaved reasonably in kinetic assays, yielding single exponential association/dissociation curves. For pathway-I, we chose to mutate Phe336G.H5.8 to a leucine because a well-known mutation at this site (F156L) in the small GTPase Ras leads to a fast exchange variant47 and a clinically relevant mutation in Gα11 at this position (F341L) leads to an autosomal dominant form of hypercalcemia,48 indicating that a leucine substitution at this site would be permitted while demonstrating some perturbative effect. We also tried Phe336G.H5.8Ala but found that this variant had no activity, thereby precluding us from disentangling the effects of this mutation on allostery and binding. We reasoned the volume change from introducing an alanine was too perturbative in the context of the hydrophobic core. Similarly, Met53G.H1.8, a member of pathway-I which projects into the hydrophobic core, could be mutated to leucine to yield single exponential association/dissociation curves, while kinetic data for Met53G.H1.8Ala could not be interpreted via simple kinetic models, so Met53G.H1.8Leu was further characterized. Finally, we chose His57G.H1.12Phe to avoid dramatic volume changes and to manipulate a key π–π stacking interaction with an adjacent phenylalanine at residue 189. The correlations of the structure and dynamics of these residues to others in the protein are stable for simulation lengths over 1 μs (Figure S7).
Table 1. BODIPY-FL-GTPγS Exchange Rates for Gαi1 WT and Mutants with Standard Error.
| protein | kobs (min–1) | kd (min–1) | ka (M–1·min–1) | Kd (nM) |
|---|---|---|---|---|
| WT | 0.17 ± 0.01 | 0.031 ± 0.001 | 5.5 ± 0.4 × 1006 | 5.7 ± 0.6 |
| Met53Leu | 1.16 ± 0.06 | 0.35 ± 0.01 | 3.2 ± 0.3 × 1007 | 11 ± 1 |
| His57Phe | 0.28 ± 0.01 | 0.073 ± 0.003 | 8.2 ± 0.6 × 1006 | 9 ± 1 |
| His322Ala | 0.15 ± 0.01 | 0.056 ± 0.003 | 3.7 ± 0.5 × 1006 | 15 ± 3 |
| Phe3366Leu | 0.43 ± 0.01 | 0.23 ± 0.02 | 8 ± 1 × 1006 | 29 ± 6 |
Our results from mutagenesis and nucleotide exchange experiments corroborated the prediction that pathway-I is stronger than pathway-II (Figure 6 and Table 1). We found that introducing the mutation Phe336G.H5.8Leu to disrupt pathway-I in Gi resulted in a greater than twofold increase in a nucleotide exchange rate. Previous work also showed that the equivalent mutation in Gt results in enhanced nucleotide exchange,46 consistent with our prediction that this allosteric pathway is conserved across multiple Gα isoforms. Furthermore, the equivalent substitution in Ras has also been observed to accelerate nucleotide exchange,47 suggesting that the conservation of pathway-I extends beyond Gα subunits of heterotrimeric G proteins to the large family of monomeric G proteins. Meanwhile, introducing the His322G.S6.4Ala mutation to disrupt pathway-II did not have a statistically significant effect on nucleotide exchange rate. This result is also consistent with a previous study that found that Tyr320G.S6.2Ala in pathway-II had little effect.49 To further test our hypothesis, we selected two additional mutations to pathway-I, Met53G.H1.8Leu and His57G.H1.12Phe. Both mutations resulted in enhanced nucleotide exchange, again consistent with the importance of pathway-I. In contrast, mutations we and others have previously performed elsewhere in the protein have had a negligible effect on both nucleotide exchange and binding affinity. For example, we made eight mutations in the binding site of an inhibitor called YM, such as Tyr69Phe, and did not observe any significant change in nucleotide exchange or binding affinity.50 Some of these residues were near pathway-I, but none of them was in pathway-I or pathway-II.
Figure 6.

Mutations in pathway-I have a significant impact on nucleotide exchange, while mutations in pathway-II do not. (A) BODIPY-FL-GTPγS loading of each mutant. Curves are the average of 9 independent measurements with fitted curves. (B) BODIPY-FL-GTPγS dissociation of each mutant. Curves are the average of 6 independent measurements with fitted curves.
Conclusions
We have identified a largely universal mechanism of G protein activation by directly observing allosteric coupling in molecular dynamics simulations of representatives of all four subclasses of Gα subunits. Our results are largely consistent with previous inferences from sequence conservation.9 This complementarity is important as our ability to see allostery in action is free of confounding factors based purely on sequence conservation, such as requirements for protein stability. Our models also provide insight into elements of the allosteric coupling that are unique to different isoforms and, therefore, may help inform the design of isoform-specific drugs. The automated tools for inferring allostery and conservation that we have developed suggest that there are two dominant pathways for allosteric coupling in G proteins, which we call pathway-I and pathway-II. Our models suggest that pathway-I is dominant, and this finding is supported by experimental characterization of mutations designed to disrupt the two pathways. We expect our contributions to understanding G protein biology will help inform the design of new inhibitors. More broadly, our tools should also be useful for studying allostery in other proteins and assessing the extent to which this allostery is conserved in related proteins.
Acknowledgments
We are grateful to the Folding@home community for volunteering their personal computing power to run simulations for this work. This work was funded by the NIH R01 GM124093 to KJB and the NIH GM124007 and the NSF MCB-2218156 to GRB. GRB holds a Packard Fellowship from the David and Lucile Packard Foundation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.3c07028.
CARDS analysis for allosteric communication in Gα isoforms (PDF)
The authors declare no competing financial interest.
Special Issue
Published as part of The Journal of Physical Chemistry Bvirtual special issue “Gregory A. Voth Festschrift”.
Supplementary Material
References
- Johnston C. A.; Siderovski D. P. Receptor-Mediated Activation of Heterotrimeric G-Proteins: Current Structural Insights. Mol. Pharmacol. 2007, 72 (2), 219–230. 10.1124/mol.107.034348. [DOI] [PubMed] [Google Scholar]
- Oldham W. M.; Hamm H. E. Structural Basis of Function in Heterotrimeric G Proteins. Q. Rev. Biophys. 2006, 39 (2), 117–166. 10.1017/S0033583506004306. [DOI] [PubMed] [Google Scholar]
- Anantharaman V.; Abhiman S.; de Souza R. F.; Aravind L. Comparative Genomics Uncovers Novel Structural and Functional Features of the Heterotrimeric GTPase Signaling System. Gene 2011, 475 (2), 63–78. 10.1016/j.gene.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathmann M. P.; Simon M. I. G Alpha 12 and G Alpha 13 Subunits Define a Fourth Class of G Protein Alpha Subunits. Proc. Natl. Acad. Sci. U. S. A. 1991, 88 (13), 5582–5586. 10.1073/pnas.88.13.5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lania A. G.; Mantovani G.; Spada A. Mechanisms of Disease: Mutations of G Proteins and G-Protein-Coupled Receptors in Endocrine Diseases. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2 (12), 681–693. 10.1038/ncpendmet0324. [DOI] [PubMed] [Google Scholar]
- Schöneberg T.; Liebscher I. Mutations in g Protein–Coupled Receptors: Mechanisms, Pathophysiology and Potential Therapeutic Approaches. Pharmacol. Rev. 2021, 73 (1), 89–119. 10.1124/pharmrev.120.000011. [DOI] [PubMed] [Google Scholar]
- Spiegel A. M.; Weinstein L. S. Inherited Diseases Involving G Proteins and G Protein-Coupled Receptors. Annu. Rev. Med. 2004, 55, 27–39. 10.1146/annurev.med.55.091902.103843. [DOI] [PubMed] [Google Scholar]
- Hurowitz E. H.; Melnyk J. M.; Chen Y. J.; Kouros-Mehr H.; Simon M. I.; Shizuya H. Genomic Characterization of the Human Heterotrimeric G Protein α, β, and γ Subunit Genes. DNA Res. 2000, 7 (2), 111–120. 10.1093/dnares/7.2.111. [DOI] [PubMed] [Google Scholar]
- Flock T.; Ravarani C. N. J.; Sun D.; Venkatakrishnan A. J.; Kayikci M.; Tate C. G.; Veprintsev D. B.; Babu M. M. Universal Allosteric Mechanism for Gα Activation by GPCRs. Nature 2015, 524 (7564), 173–179. 10.1038/nature14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Singh S.; Blumer K. J.; Bowman G. R. Simulation of Spontaneous G Protein Activation Reveals a New Intermediate Driving GDP Unbinding. eLife 2018, 7, e38465 10.7554/eLife.38465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X. Q.; Malik R. U.; Griggs N. W.; Skjærven L.; Traynor J. R.; Sivaramakrishnan S.; Grant B. J. Dynamic Coupling and Allosteric Networks in the α Subunit of Heterotrimeric G Proteins. J. Biol. Chem. 2016, 291 (9), 4742–4753. 10.1074/jbc.M115.702605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.; Bowman G. R. Quantifying Allosteric Communication via Both Concerted Structural Changes and Conformational Disorder with CARDS. J. Chem. Theory Comput. 2017, 13 (4), 1509–1517. 10.1021/acs.jctc.6b01181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch D. E.; Willard F. S.; Ramanujam R.; Kimple A. J.; Willard M. D.; Naqvi N. I.; Siderovski D. P. A P-Loop Mutation in Gα Subunits Prevents Transition to the Active State: Implications for G-Protein Signaling in Fungal Pathogenesis. PLoS Pathog. 2012, 8 (2), e1002553 10.1371/journal.ppat.1002553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambright D. G.; Sondek J.; Bohm A.; Skiba N. P.; Hamm H. E.; Sigler P. B. The 2.0 Å Crystal Structure of a Heterotrimeric G Protein. Nature 1996, 379 (6563), 311–319. 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- Slep K. C.; Kercher M. A.; Wieland T.; Chen C. K.; Simon M. I.; Sigler P. B. Molecular Architecture of Gαo and the Structural Basis for RGS16-Mediated Deactivation. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (17), 6243–6248. 10.1073/pnas.0801569105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutz B.; Yau D. M.; Nance M. R.; Tanabe S.; Tesmer J. J. G.; Kozasa T. A New Approach to Producing Functional Gα Subunits Yields the Activated and Deactivated Structures of Gα12/13 Proteins. Biochemistry 2006, 45 (1), 167–174. 10.1021/bi051729t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Xu X.; Hilger D.; Aschauer P.; Tiemann J. K. S.; Du Y.; Liu H.; Hirata K.; Sun X.; Guixà-González R.; Mathiesen J. M.; Hildebrand P. W.; Kobilka B. K. Structural Insights into the Process of GPCR-G Protein Complex Formation. Cell 2019, 177 (5), 1243–1251.e12. 10.1016/j.cell.2019.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Duan Y.; Wu C.; Chowdhury S.; Lee M. C.; Xiong G.; Zhang W.; Yang R.; Cieplak P.; Luo R.; Lee T.; Caldwell J.; Wang J.; Kollman P. A Point-Charge Force Field for Molecular Mechanics Simulations of Proteins Based on Condensed-Phase Quantum Mechanical Calculations. J. Comput. Chem. 2003, 24 (16), 1999–2012. 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- Bowman G. R. Accurately Modeling Nanosecond Protein Dynamics Requires at Least Microseconds of Simulation. J. Comput. Chem. 2016, 37 (6), 558–566. 10.1002/jcc.23973. [DOI] [PubMed] [Google Scholar]
- Zimmerman M. I.; Hart K. M.; Sibbald C. A.; Frederick T. E.; Jimah J. R.; Knoverek C. R.; Tolia N. H.; Bowman G. R. Prediction of New Stabilizing Mutations Based on Mechanistic Insights from Markov State Models. ACS Cent. Sci. 2017, 3 (12), 1311–1321. 10.1021/acscentsci.7b00465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz M. A.; Frederick T. E.; Mallimadugula U. L.; Singh S.; Vithani N.; Zimmerman M. I.; Porter J. R.; Moeder K. E.; Amarasinghe G. K.; Bowman G. R. A Cryptic Pocket in Ebola VP35 Allosterically Controls RNA Binding. Nat. Commun. 2022, 13 (1), 2269 10.1038/s41467-022-29927-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meagher K. L.; Redman L. T.; Carlson H. A. Development of Polyphosphate Parameters for Use with the AMBER Force Field. J. Comput. Chem. 2003, 24 (9), 1016–1025. 10.1002/jcc.10262. [DOI] [PubMed] [Google Scholar]
- Feenstra K. A.; Hess B.; Berendsen H. J. C. Improving Efficiency of Large Time-Scale Molecular Dynamics Simulations of Hydrogen-Rich Systems. J. Comput. Chem. 1999, 20 (8), 786–798. 10.1002/(SICI)1096-987X(199906)20:8<786::AID-JCC5>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126 (1), 014101 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Kolafa J.; Perram J. W. Cutoff Errors in the Ewald Summation Formulae for Point Charge Systems. Mol. Simul. 1992, 9 (5), 351–368. 10.1080/08927029208049126. [DOI] [Google Scholar]
- Hess B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4 (1), 116–122. 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52 (12), 7182–7190. 10.1063/1.328693. [DOI] [Google Scholar]
- Shirts M.; Pande V. S. Screen Savers of the World Unite. Science 80-. 2000, 290 (5498), 1903–1904. 10.1126/science.290.5498.1903. [DOI] [PubMed] [Google Scholar]
- Blondel V. D.; Guillaume J. L.; Lambiotte R.; Lefebvre E. Fast Unfolding of Communities in Large Networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008 10.1088/1742-5468/2008/10/P10008. [DOI] [Google Scholar]
- Verma M.; Choi J.; Cottrell K. A.; Lavagnino Z.; Thomas E. N.; Pavlovic-Djuranovic S.; Szczesny P.; Piston D. W.; Zaher H. S.; Puglisi J. D.; Djuranovic S. A Short Translational Ramp Determines the Efficiency of Protein Synthesis. Nat. Commun. 2019, 10, 5774 10.1038/s41467-019-13810-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning D. R.G ProteinsTechniques of Analysis; CRC Press, 1999 10.1201/9781420048575. [DOI] [Google Scholar]
- Studier F. W. Protein Production by Auto-Induction in High Density Shaking Cultures. Protein Expr. Purif. 2005, 41 (1), 207–234. 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Feher V. A.; Durrant J. D.; Van Wart A. T.; Amaro R. E. Computational Approaches to Mapping Allosteric Pathways. Curr. Opin. Struct. Biol. 2014, 25, 98–103. 10.1016/j.sbi.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiye T.; Karplus M. Collective Motions in Proteins: A Covariance Analysis of Atomic Fluctuations in Molecular Dynamics and Normal Mode Simulations. Proteins Struct. Funct. Bioinf. 1991, 11 (3), 205–217. 10.1002/prot.340110305. [DOI] [PubMed] [Google Scholar]
- Lange O. F.; Grubmüller H. Generalized Correlation for Biomolecular Dynamics. Proteins Struct. Funct. Genet. 2006, 62 (4), 1053–1061. 10.1002/prot.20784. [DOI] [PubMed] [Google Scholar]
- Weinkam P.; Pons J.; Sali A. Structure-Based Model of Allostery Predicts Coupling between Distant Sites. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (13), 4875–4880. 10.1073/pnas.1116274109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley M.; Lee A. Frameworks for Understanding Long-Range Intra-Protein Communication. Curr. Protein Pept. Sci. 2009, 10 (2), 116–127. 10.2174/138920309787847563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamato D.; Thach L.; Bernard R.; Chan V.; Zheng W.; Kaur H.; Brimble M.; Osman N.; Little P. J. Structure, Function, Pharmacology, and Therapeutic Potential of the G Protein, Gα/q,11. Front. Cardiovasc. Med. 2015, 2, 14. 10.3389/fcvm.2015.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eps N.; Oldham W. M.; Hamm H. E.; Hubbell W. L. Structural and Dynamical Changes in an α-Subunit of a Heterotrimeric G Protein along the Activation Pathway. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (44), 16194–16199. 10.1073/pnas.0607972103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall M. A.; Posner B. A.; Sprang S. R. Structural Basis of Activity and Subunit Recognition in G Protein Heterotrimers. Structure 1998, 6 (9), 1169–1183. 10.1016/S0969-2126(98)00117-8. [DOI] [PubMed] [Google Scholar]
- Dror R. O.; Mildorf T. J.; Hilger D.; Manglik A.; Borhani D. W.; Arlow D. H.; Philippsen A.; Villanueva N.; Yang Z.; Lerch M. T.; Hubbell W. L.; Kobilka B. K.; Sunahara R. K.; Shaw D. E. Structural Basis for Nucleotide Exchange in Heterotrimeric G Proteins. Science 2015, 348 (6241), 1361–1365. 10.1126/science.aaa5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg J.; Lässig M. Cross-Species Analysis of Biological Networks by Bayesian Alignment. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (29), 10967–10972. 10.1073/pnas.0602294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louet M.; Martinez J.; Floquet N. GDP Release Preferentially Occurs on the Phosphate Side in Heterotrimeric G-Proteins. PLoS Comput. Biol. 2012, 8 (7), e1002595 10.1371/journal.pcbi.1002595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya A. I.; Lokits A. D.; Gilbert J. A.; Iverson T. M.; Meiler J.; Hamm H. E. A Conserved Phenylalanine as a Relay between the A5 Helix and the GDP Binding Region of Heterotrimeric Gi Protein α Subunit. J. Biol. Chem. 2014, 289 (35), 24475–24487. 10.1074/jbc.M114.572875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin E. P.; Krishna A. G.; Sakmar T. P. Rapid Activation of Transducin by Mutations Distant from the Nucleotide-Binding Site. J. Biol. Chem. 2001, 276 (29), 27400–27405. 10.1074/jbc.C100198200. [DOI] [PubMed] [Google Scholar]
- Quilliam L. A.; Zhong S.; Rabun K. M.; Carpenter J. W.; South T. L.; Der C. J.; Campbell-Burk S. Biological and Structural Characterization of a Ras Transforming Mutation at the Phenylalanine-156 Residue, Which Is Conserved in All Members of the Ras Superfamily. Proc. Natl. Acad. Sci. U. S. A. 1995, 92 (5), 1272–1276. 10.1073/pnas.92.5.1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbit M. A.; Hannan F. M.; Howles S. A.; Babinsky V. N.; Head R. A.; Cranston T.; Rust N.; Hobbs M. R.; Heath H.; Thakker R. V. Mutations Affecting G-Protein Subunit α 11 in Hypercalcemia and Hypocalcemia. N. Engl. J. Med. 2013, 368 (26), 2476–2486. 10.1056/NEJMoa1300253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D.; Flock T.; Deupi X.; Maeda S.; Matkovic M.; Mendieta S.; Mayer D.; Dawson R. J. P.; Schertler G. F. X.; Babu M. M.; Veprintsev D. B. Probing Gα I1 Protein Activation at Single-Amino Acid Resolution. Nat. Struct. Mol. Biol. 2015, 22 (9), 686–694. 10.1038/nsmb.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken M. D.; Makepeace C. M.; Kaltenbronn K. M.; Kanai S. M.; Todd T. D.; Wang S.; Broekelmann T. J.; Rao P. K.; Cooper J. A.; Blumer K. J. Targeting Nucleotide Exchange to Inhibit Constitutively Active G Protein a Subunits in Cancer Cells. Sci. Signal. 2018, 11 (546), eaao6852 10.1126/scisignal.aao6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.