

Parkinson’s disease is a chronic and progressive neurodegenerative disorder characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta, which leads to the featured motor impairment of parkinsonism, including akinesia, bradykinesia, rigidity, and tremor (McGregor and Nelson, 2019). The natural progression of Parkinson’s disease can last for decades in humans, during which dopamine levels in the brain continue to drop as a consequence of the gradual degeneration of midbrain dopaminergic cells and their axonal projections to the striatum. At the same time, other parts of the brain adapt to the immediate consequences of the decreased dopamine levels. Some adaptations can act as compensatory mechanisms, delaying the onset of motor impairments. However, motor deficits become clinically noticeable when such adaptations fail to counteract the damages associated with the loss of dopamine. Over the last two decades, a series of adaptative changes at the molecular, cellular, and synaptic levels throughout the basal ganglia circuits have been reported (McGregor and Nelson, 2019; Chu, 2020). However, it remains undefined whether loss of dopamine also triggers similar adaptative changes in brain regions downstream of the basal ganglia.
Involvement of the motor cortex in Parkinson’s disease has been proposed as a direct consequence of the elevated basal ganglia inhibition, leading to decreased excitatory drive from the thalamus to the cerebral cortex as well as altered cortical outputs to the subcortical motor regions and the spinal cord (McGregor and Nelson, 2019). The reduced cortical output has been hypothesized as the pathophysiology basis underlying the hypokinetic symptoms in parkinsonism. However, there are discrepancies in the changes in the overall neuronal activity of the motor thalamus and the motor cortex. While some studies showed decreased firing frequency of neurons in the thalamus in a parkinsonian state, others reported different or even opposing results (Nakamura et al., 2021). Similarly, discrepancies also exist in changes in the overall firing rate of pyramidal neurons of the motor cortex in parkinsonism (Goldberg et al., 2002; Pasquereau and Turner, 2011). In contrast, changes in the firing pattern of thalamic and cortical neurons, particularly an abnormally synchronized bursting pattern of firing, are more prominent and consistent among studies using nonhuman primate and rodent models of parkinsonism (Goldberg et al., 2002; Pasquereau and Turner, 2011; Li et al., 2012). Together, these observations raise the question – what are the cellular and synaptic mechanisms that underlie the altered output of cortical neurons in parkinsonism. Building on lessons from the basal ganglia field, we designed a series of experiments to investigate how the loss of dopamine affects cellular and synaptic properties of cortical neurons that may contribute to the emergence of the altered output of the motor cortex in a parkinsonian state.
We employed the mice with 6-hydroxydopamine lesions of the medial forebrain bundle as a model of parkinsonism (Chen et al., 2023). This model recapitulates key features of neural network activity and behavior phenotypes of parkinsonism, as seen in people with Parkinson’s disease, making it suitable for mechanistic studies of cellular and synaptic adaptation after the loss of dopamine. Combining viral-mediated optogenetics and ex vivo electrophysiology, we found that following the loss of dopamine, there is a significant reduction of thalamic excitation to pyramidal tract (PT) neurons, but not the intratelencephalic (IT) neurons, in the layer 5 of the primary motor cortex (M1). Further anatomical studies also found that PT, but not IT, neurons exhibit a cortical layer-specific loss of dendritic spines in dopamine-depleted mice, supporting a cell subtype-specific decrease of thalamic excitation to M1 pyramidal neurons. Moreover, the connection strength of the corticocortical projections was not affected by dopamine depletion. Altogether, these results suggest that loss of dopamine triggers circuit-specific changes in M1. A recent report from the MPTP-treated monkeys also showed the same conclusion (Villalba et al., 2021). In parkinsonian monkeys, Villalba et al. (2021) demonstrated a robust decrease of thalamic axon terminals in the deep layers of M1 and a loss of layer-specific pattern of synaptic innervation of postsynaptic structure in the M1 by the thalamic inputs. Together with our earlier study (Chen et al., 2021), compelling evidence from both rodent and primate models of parkinsonism suggests that loss of midbrain dopamine neurons triggers a series of cell subtype- and inputs-selective changes in the M1 circuits, which may underlie the abnormal cortical output in the state of parkinsonism.
What are the mechanisms that underlie the layer-, cell subtype-, and synapse-specific changes in the M1 microcircuits after loss of dopamine? To address this question, we compared the basic synaptic properties of thalamic inputs to PT and IT neurons (i.e., thalamo-PT and thalamo-IT, respectively), as well as sensory cortical input to PT neurons (i.e., SC-PT). We found a significantly higher contribution of N-methyl-D-aspartate receptors (NMDAR)-mediated transmission at thalamo-PT synapses, relative to those at the thalamo-IT and SC-PT synapses. Additional ex vivo studies suggest that the exceptionally higher NMDAR levels at the thalamo-PT synapses make these connections more vulnerable relative to other synapses within the M1 local circuits. Similar NMDARs-dependent mechanisms that mediate circuit impairment have been reported in other neurological disorders. Thus, it seems plausible to propose that abnormally synchronized burst firing of thalamic neurons after the loss of dopamine (Nakamura et al., 2021) triggers excessive glutamate release in the cerebral cortex, which in turn chronically overstimulates the NMDARs of cortical neurons, leading to a reduced thalamocortical connectivity in parkinsonian state (Figure 1).
Figure 1.
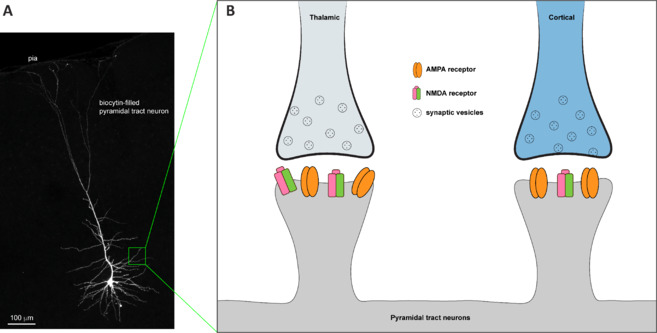
Thalamic inputs to pyramidal tract neurons in M1 express high levels of NMDA receptors relative to cortical inputs.
(A) A representative pyramidal tract neuron in the M1 that was labeled with biocytin during whole-cell patch clamp recording. (B) A diagram showing higher levels of NMDA receptor expression at the thalamic synapse to PT neurons, relative to cortical inputs to PT neurons. Refer to Chen et al. (2023) for details. Created with Adobe Illustrator. AMPA: α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; M1: primary motor cortex; NMDA: N-methyl-D-aspartate; PT: pyramidal tract.
At the circuitry level, chemogenetic disruption of basal ganglia output could rescue the altered thalamo-PT transmission in dopamine-depleted mice, but had a litter effect on normal dopamine-intact animals. Since the restoration of cortical synaptic changes was induced in dopamine depleted state, it suggests that decreased dopaminergic modulation of cortical circuits does not play a major role in the impaired thalamo-PT synaptic transmission in mice under parkinsonian state. This conclusion raises the possibility that the reported thalamo-PT synaptic changes are triggered by the pathological output of the basal ganglia. However, this hypothesis requires further testing. It is worth noting that both levodopa and deep brain stimulation can effectively suppress the bursting and synchronized neuronal activity at the basal ganglia output. It is likely that these symptomatic treatments may partially restore cortical function, which in turn contributes to the beneficial effects on motor function. Together, this study indicates that an in-depth understanding of cortical circuit adaptations after a complete loss of dopamine has a translational significance.
Further studies are needed to interrogate circuit dysfunction in Parkinson’s disease. Given that Parkinson’s disease is a network disorder, circuit studies are not necessarily limited to the basal ganglia. Instead, they should be extended to downstream targets of the basal ganglia, including both the cerebral cortex and the brain stem. Our work highlights that cellular and synaptic changes resulting from the loss of dopamine are not limited to the basal ganglia circuits. The cerebral cortex is also involved and affected (Chen et al., 2021, 2023), likely through its close interaction with the basal ganglia nuclei. However, the results discussed above do not exclude a potential involvement of local dopaminergic neuromodulation of cortical circuit structure, function, and plasticity in both health and parkinsonism (Guo et al., 2015; Cousineau et al., 2020). A better understanding of cortical circuit changes at multiscale levels will provide novel insight into the molecular and cellular bases of the aberrant network activities throughout the cortico-basal ganglia-thalamocortical circuits (e.g., burst firing, beta oscillation, and phase-amplitude coupling). Moreover, Braak staging theory states that Lewy pathology attacks cerebral cortices at the end stage of the disease, thus during a long period of Parkinson’s progression M1 circuit dysfunction is more likely driven by abnormal subcortical feedbacks (e.g., those from the basal ganglia), resulting from a robust loss of dopamine and other neuromodulators. A better understanding of cortical circuit adaptations is absolutely needed for Parkinson’s pathophysiology. It also will generate new knowledge required to optimize the existing symptomatic treatments (e.g., deep brain electric stimulation), or identify novel druggable targets for future individualized strategies of Parkinson’s disease treatment, such as noninvasive neuromodulation approaches.
This work was supported in part by the research grant from the National Institute of Neurological Disorders and Stroke, No. R01NS12137 (to HYC) and was funded in whole or in part by Aligning Science Across Parkinson’s (No. ASAP-0205720 through the Michael J. Fox Foundation for Parkinson’s Research (MJFF).
Footnotes
C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
References
- Chen L, Daniels S, Kim Y, Chu HY. Cell type-specific decrease of the intrinsic excitability of motor cortical pyramidal neurons in parkinsonism. J Neurosci. 2021;41:5553–5565. doi: 10.1523/JNEUROSCI.2694-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Daniels S, Dvorak R, Chu HY. Reduced thalamic excitation to motor cortical pyramidal tract neurons in parkinsonism. Sci Adv. 2023;9:eadg3038. doi: 10.1126/sciadv.adg3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HY. Synaptic and cellular plasticity in Parkinson’s disease. Acta Pharmacol Sin. 2020;41:447–452. doi: 10.1038/s41401-020-0371-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousineau J, Lescouzères L, Taupignon A, Delgado-Zabalza L, Valjent E, Baufreton J, Bon-Jégo ML. Dopamine D2-like receptors modulate intrinsic properties and synaptic transmission of parvalbumin interneurons in the mouse primary motor cortex. Eneuro. 2020;7 doi: 10.1523/ENEURO.0081-20.2020. ENEURO.0081-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Boraud T, Maraton S, Haber SN, Vaadia E, Bergman H. Enhanced synchrony among primary motor cortex neurons in the 1-methyl-4-Phenyl-1,2,3,6-tetrahydropyridine primate model of Parkinson’s disease. J Neurosci. 2002;22:4639–4653. doi: 10.1523/JNEUROSCI.22-11-04639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Xiong H, Kim JI, Wu YW, Lalchandani RR, Cui Y, Shu Y, Xu T, Ding JB. Dynamic rewiring of neural circuits in the motor cortex in mouse models of Parkinson’s disease. Nat Neurosci. 2015;18:1299–1309. doi: 10.1038/nn.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Ke Y, Chan Danny CW, Qian ZM, Yung Ken KL, Ko H, Arbuthnott Gordon W, Yung WH. Therapeutic deep brain stimulation in parkinsonian rats directly influences motor cortex. Neuron. 2012;76:1030–1041. doi: 10.1016/j.neuron.2012.09.032. [DOI] [PubMed] [Google Scholar]
- McGregor MM, Nelson AB. Circuit mechanisms of Parkinson’s disease. Neuron. 2019;101:1042–1056. doi: 10.1016/j.neuron.2019.03.004. [DOI] [PubMed] [Google Scholar]
- Nakamura KC, Sharott A, Tanaka T, Magill PJ. Input zone-selective dysrhythmia in motor thalamus after dopamine depletion. J Neurosci. 2021;41:10382–10404. doi: 10.1523/JNEUROSCI.1753-21.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquereau B, Turner RS. Primary motor cortex of the parkinsonian monkey: differential effects on the spontaneous activity of pyramidal tract-type neurons. Cereb Cortex. 2011;21:1362–1378. doi: 10.1093/cercor/bhq217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba RM, Behnke JA, Pare JF, Smith Y. Comparative ultrastructural analysis of thalamocortical innervation of the primary motor cortex and supplementary motor area in control and MPTP-treated parkinsonian monkeys. Cereb Cortex. 2021;31:3408–3425. doi: 10.1093/cercor/bhab020. [DOI] [PMC free article] [PubMed] [Google Scholar]