

Abstract
The methodology for the repair of critical-sized or non-union bone lesions has unpredictable efficacy due in part to our incomplete knowledge of bone repair and the biocompatibility of bone substitutes. Although human mesenchymal stem cells (hMSCs) differentiate into osteoblasts, which promote bone growth, their ability to repair bone has been unpredictable. We hypothesized that given the multi-stage process of osteogenesis, hMSC-mediated repair might be maximal at a specific time-point of healing. Utilizing a mouse model of calvarial healing, we demonstrate that the osteo-repair capacity of hMSCs can be substantially augmented by treatment with an inhibitor of peroxisome-proliferator-activated-receptor-γ, but efficacy is confined to the rapid osteogenic phase. Upon entry into the bone-remodeling phase, hMSC retention signals are lost, resulting in truncation of healing. To solve this limitation, we prepared a scaffold consisting of hMSC-derived extracellular matrix (ECM) containing the necessary biomolecules for extended site-specific hMSC retention. When inhibitor-treated hMSCs were co-administered with ECM, they remained at the injury well into the remodeling phase of healing, which resulted in reproducible and complete repair of critical-sized defects in 3 weeks. These data suggest that hMSC-derived ECM and inhibitor-treated hMSCs could be employed at optimal times to substantially and reproducibly improve bone repair.
One-sentence summary
Stem cell-generated bone-repair matrices.
Introduction
Non-union defects of bone are a major challenge in orthopedics. Of the 13 million yearly fractures that occur in the United States, about 10% fail to repair (1, 2). In many cases, synthetic implants can temporarily stabilize such injuries, but inadequate cellular responses can delay healing, and poor bone quality can cause rapid loss of fixation (3, 4). Failed implants are notoriously difficult to revise and bones that fail to heal exact a high cost on the medical system in general and on the health of the patient in particular (5). Thus, rapid and reliable healing can prevent costly hardware complications and allow more rapid return to function in patients.
To bridge large defects, autologous bone grafting can be performed where bone is explanted from a different site, often the iliac crest, and implanted at the site of injury (6–8). This procedure is effective, but the available graft material is limited, and involves additional surgery, which has been shown to cause chronic donor-site pain in many patients (9). Bone substitutes taking the form of synthetic material or decellularized bone (10) are abundantly available, but they also have their disadvantages, including poor osteoconductivity, poor host-cell adhesion properties, and immune rejection (7, 11–13). These substitutes provide a well-defined region of containment, but are frequently insufficient to support angiogenesis, proliferation, nutrition, and adherence. They have also had variable success in achieving complete osseointegration with host bone (14).
There is therefore a clear need for self-sustaining biocompatible implants that reflect the osteogenic niche and interact with the surrounding tissue to rapidly achieve homeostasis with the recipient. The most feasible form for this technology is a cell-scaffold composite, which is not a new concept, but achieving reproducibility, biocompatibility, and clinical relevance has hampered translation (15–22). We approached this problem initially by investigating hMSCs from bone marrow and found the peroxisome proliferator-activated receptor γ (PPARγ) inhibitor, GW9662, to be effective in osteogenically enhancing hMSCs (23). These observations were rationalized in terms of an inhibitory crosstalk existing between the adipogenic PPARγ axis and the osteogenic canonical Wnt axis. Upon inhibition of PPARγ, Wnt signaling prevails, resulting in osteogenic gene expression (23–27). In our previous studies, direct application of GW9662-treated hMSCs (GW-hMSCs) to critical-sized calvarial defects resulted in 2–3–fold improvement over untreated controls. However, only about 60% healing of the lesion was observed after 50 days of repeated cell administration. Given that bone heals in a stage-wise manner, with inflammatory, regenerative, and remodeling phases (28–32), we hypothesized that the beneficial effects of hMSCs might be limited to a specific stage of healing.
Utilizing a murine calvarial model of bone healing, we demonstrate here that the osteo-repair capacity of hMSCs is confined to the rapid osteogenic phase that occurs after resolution of inflammation. Upon entry into the later remodeling phase, retention signals were lost, resulting in disruption of hMSC-mediated healing. These observations are of importance because they indicate that hMSCs are efficacious during a distinct temporal window, and thus may explain why bone healing outcomes are variable. In an attempt to extend the retention time of hMSCs at the lesion site, we prepared a scaffold that consisted of hMSC-derived extracellular matrix (ECM), but was distinct from steady-state bone tissue. When GW-hMSCs were co-administered with the ECM, they were retained at the injury well into the remodeling phase, which resulted in reproducible and often complete healing of critical-sized defects. These data suggest that composites of hMSC-derived ECM and GW-hMSC could be used to improve repair strategies for critical-sized bone defects.
Results
Charaterization of hMSCs used in the study.
The hMSCs used in the study were subjected to standard in vitro assays of differentiation and flow cytometry to confirm identity (Supplementary Methods). In accordance with currently accepted hMSC definitions, they exhibited the appropriate immunophenotype and differentiated into osteoblasts, adipocytes, and chondrocytes in vitro (Fig. 1A, B; fig. S1) (33). We then tested the effects of GW9662 on osteogenic differentiation of the hMSCs. In agreement with our previous studies (23), we found that alkaline phosphatase (ALP) activity was increased upon exposure to media containing osteogenic supplementation (Fig. 1C).
Figure 1:
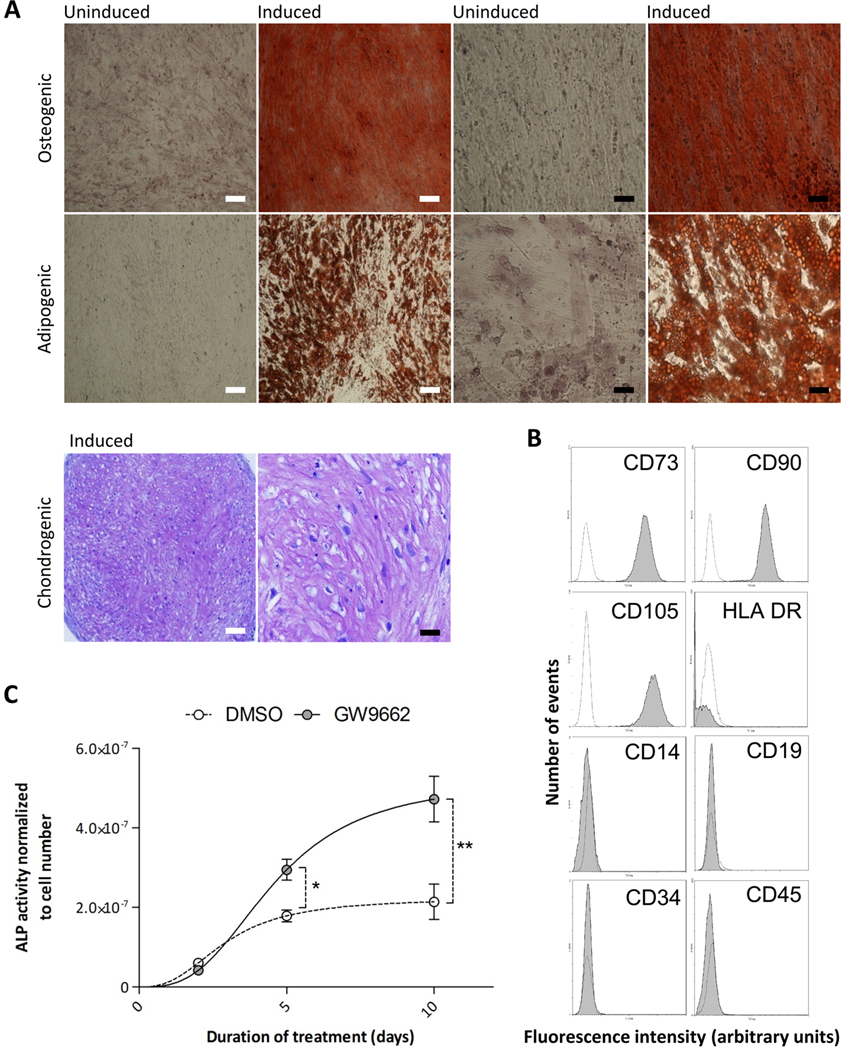
Differentiation of human MSCs. Representative results from one preparation. (A) Phase contrast micrographs of osteogenic cultures stained for calcium with Alizarin Red S, adipogenic cultures stained for lipid vacuoles with Oil Red O, and chondrocytes stained with toludine blue (purple). High and low magnification images are presented scale bars: white= 100μm, black= 25μm (B) immunophenotype of hMScs used in the study (white histograms represent isotype controls) (C) hMSCs were cultured in osteogenic media in the presence or absence of 10 μM GW9662 and alkaline phosphatase (ALP) activity was measured by colorimetric assay over 10 days. Data are means +/− standard deviations (n = 6). *P< 0.05, **P< 0.01 (t-test).
Time course of hMSC-mediated calvarial healing.
GW9662 treated hMSCs (GW-hMSCs) were prepared by incubating hMSC monolayers with 10 μM GW9662 for 8 days in the presence of β-glycerophosphate and ascorbic acid (Supplementary Methods). Control cultures were incubated in the same way, but with an appropriate volume of vehicle (dimethyl sulphoxide, DMSO). We performed a time-course experiment over 3 weeks where mice were subjected to a calvarial defect and a peri-surgical administration of either GW-hMSCs (n = 4, 1.8×106 cells) or control hMSCs.(n = 4, 1.8×106 cells), followed by weekly local injections of identical cell preparations thereafter. Groups of mice were then euthanized 5, 14, and 21 days after surgery (Fig. 2A, B). After 5 days—which corresponds approximately to the resolution of inflammation (28)—closing of the lesion had not commenced, but the stem cells were clearly detectable at the bone defect and at the skin incision by fluorescent imaging (Fig. 2C). RNA was recovered for analysis and quantitative RT-PCR indicated that approximately 200,000 hMSCs (11% of the original dose) were present at the lesion site (Fig. 2D; fig. S2). At week 2, cell engraftment data were essentially the same as week 1, and the calvarial lesions had started to heal, as determined by densitometric radiography and micro-computed tomography (μCT) scanning (Fig. 2E, F). Administration of GW-hMSCs significantly improved the rate of healing when compared to vehicle treated hMSCs (Fig. 2E, F). After 3 weeks, approximately 60% of the lesion had healed in the group receiving GW-hMSCs compared to approximately 30% in the hMSC-only controls.
Figure 2:

Engraftment and repair of calvarial defects. GFP-positive hMSCs (106) were osteogenically cultured with 10 μM GW9662 or vehicle (DMSO) and administered to calvarial defects. Some mice received 106 additional hMSCs post-surgically after 5 and 14 days. (A) Experimental setup for calvarial lesion model. (A) The phases of intramembranous bone repair in rodents adapted from Hadjiargyrou et al. (28). (B) Timeline, treatment conditions and analyses performed during experiments. (C) After 5 days, hMSCs were detected in the calvaria and incision. Scale bars 10 mm.(D) Relative human GAPDH expression in calvaria normalized to murine Gapdh for control and GW-hMSCs at day 5, 14, and 21. (n = 4). (E) μCT of lesions in cranial (left) and axial (right) reconstructions after 3 weeks. H&E stained sections are presented below (scale bars 1 mm). (F) Radio-opacity was measured by digital radiology followed by densitometry. Healing indices were the ratio of lesioned to contralateral radio-opacity. Complete healing is designated 1 and air is designated 0. Bar plots represent range and mean (n = 4). Mock assays (n = 2) are shown independently with the median. (G) Immunostaining of calvaria (human β2-microglobulin, green) from week 2. Arrows point to hMSCs at the bone surface. The murine cell layer (*) separating the main population of hMSCs (+) from the bone (b) is also indicated (scale bars 50 μm). For (B) and (D), *P < 0.05, **P < 0.01, ***P < 0.005, one-sided ANOVA followed by Tukey post-hoc analysis.
When cell engraftment was assayed at week 3, however, a drastic reduction in cell number was observed for both the control and GW-hMSC groups, with about 0.5% and 1% of the original dose engrafted, respectively (Fig. 2D; fig. S2). Therefore, it appeared that the conditions maintaining viability and/or cell-retention at the lesion site over the prior 2 weeks had disappeared. Although engraftment after 3 weeks was substantially lower than earlier time points, it was apparent that significantly more GW-hMSCs had remained at the lesion compared with vehicle treated controls. Immunocytochemistry of the explanted calvaria from week 2 specimens revealed that there was a split distribution of hMSCs at the lesion site, with populations of cells remaining above the lesion and also adjacent to the bone (Fig. 2G; fig. S3). The two populations were separated by an approximately 10-cell–thick layer of murine fibroblast-like cells (Fig. 2G). Interestingly, calvarial explants that received GW-hMSCs had substantially more cells interacting directly with the bone compared with sporadic bone-surface distribution in the controls (Fig. 2G). These data suggest that GW-hMSCs had a greater affinity for bone tissue and could have been contributing directly to bone repair.
The potential mechanism of hMSC-mediated calvarial healing.
To gain more insight into the superior healing potential of GW-hMSCs and also what might be accounting for their extended engraftment, microarray analyses on the RNA extracted from the calvarial tissue recovered after days 5 and 14 were performed (table S2; table S3). Calvaria that received hMSCs yielded 4–8-fold more RNA than controls that received no cells, and it appeared that most of the RNA was human in origin, thus suggesting that background cross-hybridization from murine transcripts would be low. Microarray data from days 5 and 14 were similar, sharing most of the differentially expressed genes, with a striking prevalence of up-regulated transcripts coding for extracellular matrix (ECM) proteins, particularly collagens; for instance, up-regulation of collagens XI, XII, XV, and XXI by GW-hMSCs (table S2 and S3). Given that this list represented the top collagens up-regulated in cultured GW-hMSCs as well, we postulated that the remaining collagens identified in a separate in vitro (23) might be detectable by a more specific and sensitive quantitative RT-PCR. When the RNA samples were analyzed by quantitative RT-PCR, collagens I, III, V, VI, XI, XII, XIV, XV, and XXI were found to be up-regulated by GW-hMSCs in vivo (Table 1). The increased secretion of particular collagen types at the lesion by GW-hMSCs during the healing process might be partly responsible for their enhanced efficacy and the increased number of GW-hMSCs persisting at week 3. The notion that those collagens were up-regulated during the rapid stage of calvarial bone repair was supported in part by meta-analysis of existing data submitted to the NCBI Gene Expression Omnibus (GEO) (34). These data, accessible through GEO Series accession number GSE20980 (35), indicated that types V, VI, XII, and XV collagen chains were robustly up-regulated at day 5 of calvarial repair when compared to the remodeling phase at day 21 (table S4). Taken together, it appears that hMSC-derived collagens may enhance repair of calvarial lesions by providing an osteogenic retention signal for both exogenous hMSCs and host-derived cells. We aimed to test this notion by co-administering purified hMSC-derived ECM with GW-hMSCs.
Table 1: Mesenchymal stem cell-derived collagen transcripts differentially regulated by GW9662 treatment when implanted into murine calvarial defects.
Data are mean fold-changes compared with DMSO (control)-treated MSCs with ± 1SD (n = 4 animals). Statistical comparisons of week 1 and 2 data performed by one-sided t-test on arcsine-transformed data. PCR fold changes in human GAPDH were derived using the ΔΔCT method and normalized to the mouse Gapdh transcript.
| Collagen transcript | Microarray | PCR | ||||
|---|---|---|---|---|---|---|
| Microarray probe and target collagen subunit | Week 1 fold increase | Week 2 fold increase | Target collagen subunit | Week 1 fold increase | Week 2 fold increase | |
| COL I | 217430_x_at_(a1) 202311_s_at_(a1) |
3* 2.25* |
NM NM |
a1 | 27.8±19.5 | 8.2±3.9 |
| COLIII | 232458_at_(a1) | 2.25* | NM | a1 | 7.1±1.03 | 3.08±1.04 |
| COL V | 52255_s_at_(a3) 218975_at_(a3) |
3.34* 2.17* |
NM NM |
a1 | 13.53±8.74 | 11.69±6.23 |
| COLVI | 212937_s_at_(a1) 212091_s_at_(a1) 212940_at_(a1) 209156_s_at_(a2) 213290_at_(a2) |
3.23* 3.22* 2.99* 2.71* 2.29* |
NM NM NM NM NM |
a2 | 14.94± 10. 5 | 3.24±2.73 |
| COLXI | 37892_at_(a1) 204320_at_(a1) 229271_x_at_(al) |
3.75 3.32 2.23 |
4.16 3.95 3.13 |
a1 | 24.73±3.79 | 11.04±8.42 |
| COLXII | 231766_s_at_(a1) 225664_at_(a1) |
2.47 2.37 |
ndet 2.07 |
a1 | 10.95±6.18 | 4.82±1.82 |
| COLXIV | 203477_at_(a1) | 2.98 | ndet | a1 | 4.7±2.3 | 2.17±1.12 |
| COLXV | 203477_at_(a1) | 2.98 | 3.00 | a1 | 11.8±4.73 | 5.27±1.79 |
| COLXXI | 208096_s_at_(a1) | 4.63 | 4.1 | a1 | 6.99±6.6 | 4.03±1.82 |
Detected in vitro after 8 days in culture [from Krause et al. (23)], but not by in vivo microarrays in this study. NM, not measured; ndet, measured but not detected.
To generate hMSC-derived ECM, cells were cultured as a monolayer and subjected to a two-stage osteogenic protocol initially optimized for ECM-yield (Supplementary Methods). This consisted of a pre-osteogenic, non-mineralizing stage where the cells were exposed to osteogenic media in the absence of dexamethasone, but the presence of GW9662 or vehicle; then a mineralizing stage, where the hMSCs received osteogenic media with dexamethasone, but the GW9662 was absent. The monolayers were then recovered and the cells and contaminating proteins removed, while maintaining the collagen-rich components of the ECM. After processing, the material had a fibrous appearance, with regularly oriented fibrils (Fig. 3A). Immunoblotting confirmed that the process had depleted the presence of intracellular proteins, such as GAPDH and β-actin, but enriched the collagen-containing extracellular component (Fig. 3B). Complete proteolytic digestion of the ECM resulted in an inorganic residue that could be identified as brushite, a hydrated form of calcium phosphate (CaHPO4·2H2O) (Fig. 3C). When the purified ECM was co-cultured with GFP-labeled hMSCs, we observed that the cells oriented themselves on the surface of the material in a manner similar to the osteoblast layer on a periosteal surface (Fig. 3D). The resemblance to osteoblasts was also supported by the presence of high ALP activity (Fig. 3D). Although the cultures were viable for 15 days, initial cell loss was observed until equilibrium was achieved, suggesting that binding sites favoring long-term survival are present, but can be saturated by addition of excess numbers of cells (Fig. 3E).
Figure 3:

Matrix extraction and partial characterization. (A) Decellularized, insoluble matrix recovered from osteogenic monolayer cultures of hMSCs. Unprocessed and processed ECM was imaged by electron microscopy. Electron micrograph images show fibril orientation. Scale bars 5 μm. (B) Immunoblotting for cellular proteins GAPDH and β-actin, and matrix protein collagen type I. (C) X-ray diffraction analysis of the inorganic component of the matrix. (D) Processed matrix seeded with GFP-expressing hMSCs shows cells inhabiting the external surface. hMSCs were analyzed for alkaline phosphatase (ALP) activity. Scale bars 100 μm. (E) Stabilization of cell number over 5 days in matrix constructs initially seeded with 106 GFP-positive hMSCs. Data are means +/− standard deviations.
Effect of GW9662 on ECM generation by hMSCs.
We then compared ECM preparations generated by GW-hMSCs to those generated by untreated hMSCs and found that 7 of hMSC preparations tested (n=10) were highly responsive to 10 μM GW9662 treatment, as shown by a dose-dependent increase in ALP activity (Fig. 4A) and formation of mineralized nodules that are an indicator of bone matrix deposition (Fig. 4B). The remaining cultures had very high inherent osteogenic activity and thus were marginally improved by GW92662. For all hMSC preparations tested, GW9662 treatment caused a greater volume of ECM to be formed (Fig. 4C), with elevated calcium levels, indicative of mineralizing osteogenesis (Fig. 4D). Therefore, the data show that GW9662 had a positive effect on ECM yield, but it was unclear whether composition had been affected. Conventional proteomic analysis proved impossible owing to the physical characteristics of the ECM and its lack of solubility even under common denaturing conditions. We therefore employed amino acid analysis, hypothesizing that significant changes in the composition of ECM preparations would be detectable by shifts in the various proportions of amino acids present. In terms of collagen content, these changes would be expected to manifest themselves as variations in glycine, proline, and hydroxyproline levels. Amino acid analysis revealed that ECMs generated from GW9662-treated (GW-ECM) and control hMSCs (DMSO-ECM) were remarkably similar in terms of their amino acid composition, with the exception of hydroxyproline, which was slightly reduced (by 14–28%) in GW-ECM compared to controls (table S5). These reductions occurred without an associated change in non-hydroxylated proline levels. There was also a slight but reproducible increase in lysine content. Altogether, amino acid analysis suggested that the effect of GW9662 on hMSC-derived ECM was predominantly on yield rather than composition. We next examined whether GW-ECM differed from DMSO-ECM in terms of cell binding and differentiation. GW-ECM and DMSO-ECM constructs (4 mm3) were prepared and incubated with GW-hMSCs or control-hMSCs for up to 5 days. Although both GW-ECM and DMSO-ECM exhibited similar cell-binding characteristics, a combination of GW-hMSCs with DMSO-ECM provided the best cell retention and ALP activity after 5 days of co-culture (Fig. 4E, F). Overall, these data demonstrated that although yield of ECM was about 2-fold higher from GW-hMSCs, the ECM generated from control hMSCs exhibited superior cell binding and osteogenic support.
Figure 4:

ALP activity predicts the effect of GW9662 on ECM yield. (A) ALP activity exhibited by hMSCs in response to escalating GW9662 dose. (B) ECM nodules detectable by phase microscopy in vehicle- and GW9662-treated hMSCs. Scale bar, 10 μm. (C) Yield of recoverable matrix per 154 mm2 tissue culture dish. (D) Calcium mineral levels in recoverable matrix. (E) Cell binding characteristics of ECM generated by control hMSCs (DMSO-ECM) or GW-hMSCs (GW-ECM) when loaded with 1×105 control hMSCs or GW-hMSCs. (F) ALP activity of control- or GWhMSCs on GW-ECM or DMSO-ECM. Data are means +/− standard deviations and *P < 0.05, **P < 0.01, ***P < 0.005. For panels C and D, t-test, N=6. For panels A, E and F, one-sided ANOVA followed by Tukey post-hoc analysis, for A, N=6, for C and D, N=3.
The effect of hMSC derived ECM on calvarial healing.
To test the effects of the hMSC-derived ECM in vivo, calvarial lesions were generated in nude mice as described previously. During surgery, one group of animals (n = 4) received GW-hMSCs only, one group received ECM only (n = 4), and the final group received both GW-hMSCs and DMSO-ECM (n = 4). Subsequent treatments were administered 1 and 2 weeks thereafter by injection. At week 3, mice were euthanized for analysis. Groups that received matrix only or GW-hMSCs only exhibited marginal and 60% healing, respectively (Fig. 5A). In contrast, mice that received GW-hMSCs and ECM had healed 80–100%, with some animals generating levels of bone in excess of the contralateral side (Fig. 5A, B). When cell engraftment was assayed by RT-PCR, lesions that received only cells had generally low engraftment, reflecting about 2% of the original final dose. In one instance, engraftment was substantially higher than mean levels, but this was not accompanied by increased bone healing. Overall, cell engraftment was substantially higher at week 3 in animals that received the cell-matrix composite (Fig. 5C), reaching levels equivalent to engraftment measured at day 5 in experiment 1. In vivo, GW-hMSCs alone became elongated and clustered sparsely in groups adjacent to the bone tissue (Fig. 5D, left). In contrast, when co-administered with ECM, GW-hMSCs were matrix-localized, engrafting in much larger clusters that did not appear to be associated with the remodeling bone surface (Fig. 5D, right). Since the ECM provides additional cell binding sites that are not directly adjacent to or in the repairing bone, the mechanism of action appears to be through trophic support rather than direct tissue contribution. Indeed, the paracrine action of hMSCs on endogenous repair responses has been proposed as a mechanism of efficacy in numerous biological systems (36), including bone. (37) To examine this notion further, we generated a list of transcripts up-regulated in GW-hMSCs after 1 week of injury engraftment that encode secreted proteins (table S6). Using the Database for Annotation, Visualization and Integrated Discovery (38, 39), a comparison was then performed against various tissue expression lists to determine whether the up-regulated transcripts of GW-hMSCs could support osteogenesis. The results of the search confirmed that transcripts up-regulated in GW-hMSCs that encoded secreted proteins corresponded closest to bone and vascular expression profiles with strong statistical significance (table S7).
Figure 5:

Efficacy of GW-hMSC–ECM composites in healing calvarial defects in mice. Groups received GW-hMSCs only (n=4), ECM only (n=4), and one received both (composite) (n=4) at the time of surgery. Cells were administered 1 and 2 weeks thereafter. After 3 weeks, calvaria were analyzed. (A) μCT of lesions in explanted crania (left) and axial reconstructions (right) with H&E-stained sections directly below. Scale bar 1 mm.(B) Healing of the bone lesions as a function of radio-opacity of the bone (0= no helaing, 1= fully healed). The mock group refers to no treatment. (C) Human GAPDH expression at week 3 normalized to mouse Gapdh in calvaria of GW9662-treated and composite-treated animals (n=4 per treatment group). The arbitrary value 1.0 on the y-axis represents engraftment measured at week 1 in the original experiments (Fig. 2B). (D to E) Human β2-microglobulin (green) staining in calvarial specimens in the absence of matrix (D) and in presence of composite (E). Statistics for (b) and (c), *P < 0.05, **P < 0.01, ***P < 0.005, one-sided ANOVA followed by Tukey post-hoc analysis.
We explored the possibility that integrins at the cell surface might be directly regulated by GW9662, in synergy with the differentially expressed collagens. However, immunophenotyping demonstrated that integrins commonly associated with osteoblasts (40, 41), such as β1 (CD29), α1 (CD49a), α2 (CD49b), α5 (CD49e), and αυ (CD51), were presented at equally high levels on the membranes of GW-hMSCs and untreated hMSCs (table S8). The potential expression of CD36 (glycoprotein 3b) on the surface of hMSCs was also of interest because it is a known ligand of type V collagen, originally discovered as part of the platelet aggregation pathway (42). However, we could not detect expression on the membranes of hMSCs irrespective of GW9662 treatment. Taken together, these results do not support GW9662-mediated receptor-matrix synergy for the most likely candidate collagen receptors.
Discussion
Bone healing is a multi-stage process involving the temporal regulation of a number of distinct phases that have been well-characterized in rodent models (28–32). For example, after fracture of long bones, there is rapid reorganization of the tissue architecture with initiation of inflammation. Shortly after, angiogenesis is initiated, feeding the lesion with nutrients and necessary cytokines. Thereafter, simultaneous processes of intramembranous and endochondral ossification (IO and EO, respectively) occur. IO occurs at the periosteum, where mesenchymal progenitors differentiate directly to osteoblasts. This process involves the prompt deposition of a bony “hard callus” adjacent to cortical bone, but the process is generally attenuated after two weeks, resulting in a relatively low volume of new tissue and limited range in terms of gap closure (29). EO is initiated by rapid expansion of mesenchymal cells adjacent to the fracture that differentiate into chondrocytes, forming a cartilaginous “soft callus” rich in type II collagen, and over a few weeks the callus is calcified to bone. When cultured MSCs are induced to generate a mineralized matrix, they do so by a process that resembles IO in that the cells differentiate directly to mineralizing osteoblast-like cells in the absence of an intermediate chondrocytic phase (23, 43, 44). We therefore selected a model of rodent calvarial healing that occurs exclusively by IO to examine potential stage-wise effects of hMSC-mediated osteo-repair. This model recapitulates the inflammatory, rapid osteogenic, and remodeling phases without a chondrogenic stage characteristic of EO (45).
Over the 21-day time course we found that retention of hMSCs at the site of injury was confined to the first two weeks of repair and rapidly dwindled between 2 and 3 weeks, resulting in up to 60% closure of the lesions (Fig. 2). Because injections of fresh hMSCs were administered at each week following surgery, the reduction in engraftment was attributed to the microenvironment rather than the inherent longevity of the cells. Given that the rapid repair phase of IO occurs for about two weeks post-injury (28, 29), it seemed that engraftment was coincident with this phase and was attenuated during remodeling which is initiated after about 2 weeks of repair in rodents (28). One explanation for these results arises from reports of strong mitotic cues persisting for 2 weeks post-lesion that cause a rapid expansion of local osteoprogenitor cells (46). However, it is unlikely that this process entirely explains the observed engraftment kinetics, because the presence of hMSC-derived matrix substantially improves engraftment 1 week after the predicted end of the mitotic burst.
The hypothesis that an extended hMSC retention signal might be beneficial arose from the observations that, irrespective of the relatively low level of engraftment at week 3, it remained 2-fold higher with GW-hMSCs, that GW-hMSCs secrete higher levels of ECM collagens when compared to untreated controls (23), and that GW-hMSCs exhibit greater efficacy in bone repair. These observations therefore raised the possibility that ECM collagens themselves might be contributing to a retention signal and may explain the enhanced efficacy observed. This hypothesis was confirmed when purified ECM from hMSCs was co-administered with GW-hMSCs and retention was extended throughout week 3, resulting in substantially improved lesion closure that was complete in most cases (Fig. 5). Based on the near-exclusive distribution of GW-hMSCs on the matrix—rather than at the surface of the remodeling bone—it is reasonable to assume that at later stages of healing, the GW-hMSCs adopt a bystander or trophic role rather than direct contribution to tissue repair when administered with matrix. This hypothesis is strongly supported by the observation that transcripts up-regulated by engrafted GW-hMSCs encoding secreted proteins closely match expression profiles of bone, vasculature and cartilage (table S6, 7).
We speculated that GW-hMSCs generate ECM of a distinct composition that favors retention, but biochemical data strongly indicated that GW9662 affected matrix yield rather than altered composition (Fig. 4C, D; table S4). Nevertheless, this possibility could not be completely ignored because practical constraints limited compositional characterization to amino acid analysis and qualitative immunoblotting. Transcriptomic assays of calvarial samples containing untreated and GW-hMSCs identified a short-list of up-regulated collagen genes that could represent retention signal candidates including types I, III, V, VI, XI, XII, XIV, XV, and XXI (Table 1; tables S2-S4). With the exception of types I and III collagen, which are abundant in bone, types V (47–50), VI (51–54), XI (55, 56), XII (57–59), XIV(57), and XV(60) have been reported to play a role in skeletal development and/or accelerated osteoblast activity. Furthermore, meta-analysis of a transcriptome array dataset present on the NCBI Gene Expression Omnibus identified collagen types V, VI, XII, and XV to be up-regulated in early rather than late healing of a calvarial defect in rats (table S4). Collagen type XXI was also found to be highly up-regulated in GW-hMSCs and, because expression is highest in highly vascularized tissues such as heart, stomach, and placenta (61, 62), it may play a role in supporting angiogenesis. Indeed, both GW-hMSCs and untreated hMSCs appear to enhance angiogenesis in healing murine calvaria (23). Nevertheless, the observation that a combination of DMSO-ECM and GW-hMSCs resulted in the best combined cell-retention and osteogenesis suggested that factors in addition to collagen composition may also contribute to the observed ECM efficacy (Fig. 4).
This study suggests that the ECM generated by osteogenic hMSCs provides a biologically complex extracellular microenvironment that mimics repairing rather than homeostatic bone tissue, making it distinct from common clinically-utilized bone-repair scaffolds such as demineralized bone matrix, hydroxyapatite, and collagen foam. This matrix is sufficient to extend the retention of osteoblasts and their progenitors at the lesion site by at least 1 week, resulting in substantially improved osteogenesis when administered with an appropriate cellular component (MSCs). We propose that hMSC-derived ECM could be employed for surface “bio-conditioning” of commonly used synthetic orthopedic materials to improve osteogenesis and bio-integration. Because the ECM matrix provides a retention signal to hMSCs, a favorable biologic niche for the cells to reside, and promotes osteogenesis, it can be thought of as a self-sustaining graft material when used in conjunction with hMSCs. This notion is also supported by the observation that MSC-derived ECM has been reported to directly enhance the osteogenic functionality of hMSCs. (63)
Translational applications of this technology include the manufacture of cryo-preserved, live GW-hMSC/ECM composites for bone bridging applications. Furthermore, bio-conditioning of inert materials commonly used in orthopedics with hMSC-derived ECM has great potential. This is especially noteworthy given the reported biocompatibility issues associated with many synthetic or cadaveric materials currently employed in orthopedics (7, 10–13). Bio-conditioned products may be utilized in a variety of orthopedic settings, in particular, vertebral arthrodeisis (spinal fusion) procedures. Although vertebral arthrodeisis is one of the most common orthopedic procedures, it has a failure rate of up to 40% (14). The use of bone morphogenic proteins can substantially improve the probability of success, but their use is associated with potentially fatal safety concerns such as ectopic bone growth, paralysis and inflammation (64–68). Live bone allograft is the gold standard substrate for spinal fusion, the procedure is associated with donor site morbidity and a limited volume of available material (6–8). Given the well-known superiority of live bone auto-grafting for spinal fusion, it is attractive to speculate that a material that resembles live, anabolic bone tissue may exhibit equivalent efficacy to allograft when utilized in combination with GW-hMSCs or even whole autologus bone marrow. Furthermore, scaffolds bioconditioned by hMSCs are not limited by volume and do not necessarily require an autologous cell-source, given their limited immunogenicity in allogeneic recipients (36, 69).
Materials and Methods
Culture of human mesenchymal stem cells
hMSCs were acquired from the Tulane University/Texas A&M Health Science Center adult stem cell distribution facility in accordance with institutionally approved protocols. Cells were cultured according to standard protocols (70). In brief, cells were cultured in complete culture medium (CCM) consisting of alpha minimal essential medium (α-MEM, GIBCO, Invitrogen) containing 20% (v/v) FBS (Hyclone and Altanta Biologicals), 2 mM L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin (GIBCO, Invitrogen). Media was changed every 48 h. For each passage, cells were seeded at 100 cm-2. Cells were then recovered by trypsinization (GIBCO, Invitrogen) followed by cryopreservation in α-MEM containing 50% (v/v) FBS and 5% (v/v) DMSO (Hybrimax, Sigma-Aldrich) in the vapor of liquid nitrogen. Passage 1 cells were used in lentiviral labeling experiments and passage 3 or 4 cells were used in all other experiments. Phase contrast and fluorescence microscopy of live cultures was performed using an inverted microscope (Nikon Eclipse, TE200) fitted with a Nikon DXM1200F digital camera.
Immunophenotyping
MSCs were recovered by brief trypsinization and incubated with fluorophore-tagged antibodies or respective isotype controls (Becton Dickinson Pharmingen or Beckman Coulter) for 30 min in PBS containing 2% (v/v) FBS. A minimum of 20,000 cells was analyzed on a Cytomics FC500 flow cytometer (Beckman Coulter) and data were processed using the manufacturer’s software (CXP). A list of clones is provided in Supplemental Methods.
Differentiation of human multipotent stromal cells in vitro
Differentiation assays for MSCs into mineralizing osteoblasts, adipocytes, and chondroblasts were performed using standard in vitro protocols detailed in Supplemental Methods (23, 70, 71).
Calvarial lesions and cell administration
Protocols were approved by the Texas A&M Health Science Institutional Animal Care and Use Committee. Two month-old female Nu/J mice were acquired from Jackson Laboratories. Under isoflurane anesthesia, a 4 mm diameter circular lesion in the frontal calvarial bone was generated 1–2 mm from the sagittal and coronal sutures using a 3 mm osteotomy burr. In each case, the depth and extent of the lesion was checked by gentle probing around the circumference of the cut so as to ensure the hole was the entire depth of the calvarial bone. After irrigation with PBS, cells were administered. Once the mixture clotted, the scalp was sutured closed and mice were allowed to recover from anesthesia with additional warming and oxygen for 2–3 min. For 2 days after surgery, saline and analgesia was administered subcutaneously. For experiments involving the hMSC-derived matrix, the matrix was placed in the lesion and cells were overlaid.
Green fluorescent protein (GFP)-positive hMSCs were plated at 100 cm−2, then cultured in CCM until 80% confluence. To osteogenically enhance hMSCs, cells were treated for 8 days with osteogenic medium (CCM with 50 μg/ml ascorbic acid and 5 mM β-glycerol phosphate [Sigma-Aldrich]) containing 10 μM GW9662 in DMSO. Control cells received DMSO only. Cells were recovered on the day of surgery by trypsinization and suspended in Hank’s buffered salt solution (Invitrogen) containing 2% (v/v) FBS on ice until administration. At the point of administration, 1.8×106 cells were pelleted by centrifugation, suspended in 45 μL cold human plasma (Sigma Aldrich), added to 45 μL cold 2x thromboplastin C (Fisher Scientific), and immediately pipetted onto the lesion. Subsequent doses (fig S2) were prepared in the same manner and they were injected under the scalp while the mice were anesthetized.
Cranial bone extractions and processing
In accordance with IACUC procedures, mice were humanely euthanized and cranial bones were cut away using a rotary tool fitted with a 10 mm diameter diamond-cutting wheel. The top of the skull was gently removed, washed in PBS, and cleared of excess connective tissue. For RNA extractions, the tissue was flash-frozen in liquid nitrogen and for histology, calvaria were fixed in 10% (v/v) neutral buffered formalin. (VWR International). Details on RNA extraction, decalcification, and sectioning are in Supplementary Methods.
Standard and real time quantitative RT-PCR
Total RNA (1 μg) was used to make cDNA (Superscript III cDNA kit, Invitrogen). For conventional PCR, 0.5 μg of cDNA was amplified in a 25-μL reaction using PCR SuperMix (Invitrogen) on a standard thermocycler (Biorad C1000). Amplified DNA was visualized by agarose gel electrophoresis. For quantitative RT-PCR, 0.5 μg of cDNA was amplified in a 25-μL reaction containing SYBR-green PCR master mix (Fast SYBR Green, Applied Biosystems) on a C1000 thermocycler fitted with a real time module (CFX96, Biorad). Human GAPDH and mouse Gapdh expression was calculated using the 2-deltaCT method. Murine GAPDH expression levels was used as a constant to normalize human cell engraftment measurements. Collagen expression data were calculated using the 2-Δ, ΔCT method using human GAPDH as a reference. PCR conditions and primers are given in table S1.
Immunoblotting
Immunoblotting was performed in the usual manner using Novex reagents (Invitrogen). Antibodies were: mouse anti-GAPDH (MAB374, Chemicon-Millipore), mouse anti-β-actin (AC-15, Sigma-Aldrich), goat anti-type I collagen (8788, Santa Cruz Biotechnology), goat anti-mouse IgG (Biomeda), and rabbit anti-goat IgG (Biomeda).
Immunocytochemistry
Sections were deparaffinized and blocked for 16 h in PBS containing 2% (v/v) donkey serum and 0.1% (v/v) Triton X100 (Sigma-Aldrich). Human MSCS were stained with a rabbit anti-human β2-microglobulin antibody (ab15976, Abcam) at 1:100 dilution in PBS blocking buffer over 48 h at 4oC. Controls were performed with non-specific rabbit IgG (Sigma). Bound antibody was detected by donkey anti-rabbit IgG conjugated to AlexaFluor 488 (Invitrogen). Sections were mounted with 4’,6-diamidino-2-phenylindole-containing mounting media (Vector Laboratories) and visualized with fluoresce microscopy (Nikon Eclipse 80i fitted with a Retiga 2000 camera) running digital imaging software (Elements, Nikon). Owing to the high incidence of autofluorescence in bone tissue, true green fluorescence was detected using a 475–490/540–565 nm dual-band filter.
Statistics
Statistical tests and data-plotting were performed using GraphPad Prism version 5.00 for Windows. For multiple tests of means within datasets, data were statistically analyzed by one way analysis of variance (ANOVA). Data involving ratios were statistically tested on arcsine transformed data and post-analysed by Tukey’s method. Single means were compared using T-test. Data were regarded significant if p values were <0.05.
Supplementary Material
Methods
Figure S1: Representative immunophenotype analysis of hMSCs used in the study.
Figure S2: Cell engraftment assays by conventional RT-PCR and gel electrophoresis.
Figure S3: Immunohistochemistry staining of calvarial defects with staining controls
Table S1: Primers used in this study
Table S2: Upregulated genes determined by microarray analysis after 1 week of engraftment.
Table S3: Up-regulated genes determined by microarray analysis after 2 weeks of engraftment.
Table S4: Meta-analysis of calvarial healing time course.
Table S5: Amino acid analysis of ECM.
Table S6: Potential trophic mediators expressed by GW-hMSCs are found in bone, cartilage and vascular tissue.
Table S7: Comparison of the secreted protein list against various tissue expression databases using the DAVID analysis system.
Table S8: Expression of integrins commonly associated with osteoblasts.
Acknowledgments:
We thank K. Coble and J. Ylostalo for help with the microarray analysis and the Veterinary Integrative Biosciences Histology Laboratory at Texas A & M University, College Station, for support with histology.
Funding:
The Institute for Regenerative Medicine Program Funds and Scott & White RGP grant #90172 (C.A.G.).
Footnotes
Competing interests: The authors declare no conflicts of interest. Accession numbers: Microarray data GEO Series accession number GSE35503 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE35503).
References
- 1.Marsh D, Concepts of fracture union, delayed union, and nonunion. Clin Orthop Relat Res, S22–30 (1998). [DOI] [PubMed]
- 2.Rosemont IL, “United States Bone and Joint Decade: The burden of musculoskeletal diseases and musculoskeletal injuries. ” (American Academy of Orthopedic Surgeons;, 2008). [Google Scholar]
- 3.Ingham E, Fisher J, Biological reactions to wear debris in total joint replacement. Proc Inst Mech Eng H 214, 21–37 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Schwarz EM, O’Keefe RJ, Looney RJ, Bone implant interface, osteolysis and potential therapies. J Musculoskelet Neuronal Interact 4, 390–392 (2004). [PubMed] [Google Scholar]
- 5.Hak DJ, McElvany M, Removal of broken hardware. J Am Acad Orthop Surg 16, 113–120 (2008). [DOI] [PubMed] [Google Scholar]
- 6.de Boer HH, The history of bone grafts. Clin Orthop Relat Res, 292–298 (1988). [PubMed]
- 7.Rihn JA, Kirkpatrick K, Albert TJ, Graft options in posterolateral and posterior interbody lumbar fusion. Spine (Phila Pa 1976) 35, 1629–1639 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Rubery PT, Enhancing allograft bone healing through gene therapy. Spine (Phila Pa 1976) 35, 1640–1647 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Banwart JC, Asher MA, Hassanein RS, Iliac crest bone graft harvest donor site morbidity. A statistical evaluation. Spine (Phila Pa 1976) 20, 1055–1060 (1995). [DOI] [PubMed] [Google Scholar]
- 10.Kao ST, Scott DD, A review of bone substitutes. Oral Maxillofac Surg Clin North Am 19, 513–521, vi (2007). [DOI] [PubMed] [Google Scholar]
- 11.Aro HT, Aho AJ, Clinical use of bone allografts. Ann Med 25, 403–412 (1993). [DOI] [PubMed] [Google Scholar]
- 12.Aho AJ, Ekfors T, Dean PB, Aro HT, Ahonen A, Nikkanen V, Incorporation and clinical results of large allografts of the extremities and pelvis. Clin Orthop Relat Res, 200–213 (1994). [PubMed]
- 13.Shegarfi H, Reikeras O, Review article: bone transplantation and immune response. J Orthop Surg (Hong Kong) 17, 206–211 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Boden SD, Overview of the biology of lumbar spine fusion and principles for selecting a bone graft substitute. Spine (Phila Pa 1976) 27, S26–31 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Cuomo AV, Virk M, Petrigliano F, Morgan EF, Lieberman JR, Mesenchymal stem cell concentration and bone repair: potential pitfalls from bench to bedside. J Bone Joint Surg Am 91, 1073–1083 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Mauney JR, Volloch V, Kaplan DL, Role of adult mesenchymal stem cells in bone tissue engineering applications: current status and future prospects. Tissue Eng 11, 787–802 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Gregory CA, in Stem Cell Repair and Regeneration., Levicar H. N.N.A., Gordon MY, Dimarakis I, Ed. (Imperial College Press, London, 2008), vol. 3, pp. 21–44. [Google Scholar]
- 18.Phinney DG, Kopen G, Isaacson RL, Prockop DJ, Plastic adherent stromal cells from the bone marrow of commonly used strains of inbred mice: variations in yield, growth, and differentiation. J Cell Biochem 72, 570–585 (1999). [PubMed] [Google Scholar]
- 19.Phinney DG, Kopen G, Righter W, Webster S, Tremain N, Prockop DJ, Donor variation in the growth properties and osteogenic potential of human marrow stromal cells. J Cell Biochem 75, 424–436 (1999). [PubMed] [Google Scholar]
- 20.Phinney DG, Prockop DJ, Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair--current views. Stem Cells 25, 2896–2902 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Phinney DG, Biochemical heterogeneity of mesenchymal stem cell populations: clues to their therapeutic efficacy. Cell Cycle 6, 2884–2889 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Derubeis AR, Cancedda R, Bone marrow stromal cells (BMSCs) in bone engineering: limitations and recent advances. Ann Biomed Eng 32, 160–165 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Krause U, Harris S, Green A, Ylostalo J, Zeitouni S, Lee N, Gregory CA, Pharmaceutical modulation of canonical Wnt signaling in multipotent stromal cells for improved osteoinductive therapy. Proc Natl Acad Sci U S A 107, 4147–4152 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bennett CN, Ross SE, Longo KA, Bajnok L, Hemati N, Johnson KW, Harrison SD, MacDougald OA, Regulation of Wnt signaling during adipogenesis. J Biol Chem 277, 30998–31004 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Farmer SR, Regulation of PPARgamma activity during adipogenesis. Int J Obes (Lond) 29 Suppl 1, S13–16 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Krause U, Gregory CA, Potential of Modulating Wnt Signaling Pathway Toward the Development of Bone Anabolic Agent. Curr Mol Pharmacol, (2011). [DOI] [PubMed]
- 27.Moldes M, Zuo Y, Morrison RF, Silva D, Park BH, Liu J, Farmer SR, Peroxisome-proliferator-activated receptor gamma suppresses Wnt/beta-catenin signalling during adipogenesis. Biochem J 376, 607–613 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hadjiargyrou M, Lombardo F, Zhao S, Ahrens W, Joo J, Ahn H, Jurman M, White DW, Rubin CT, Transcriptional profiling of bone regeneration. Insight into the molecular complexity of wound repair. J Biol Chem 277, 30177–30182 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Einhorn TA, The cell and molecular biology of fracture healing. Clin Orthop Relat Res, S7–21 (1998). [DOI] [PubMed]
- 30.Marsell R, Einhorn TA, The biology of fracture healing. Injury 42, 551–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schindeler A, McDonald MM, Bokko P, Little DG, Bone remodeling during fracture repair: The cellular picture. Semin Cell Dev Biol 19, 459–466 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Shapiro F, Bone development and its relation to fracture repair. The role of mesenchymal osteoblasts and surface osteoblasts. Eur Cell Mater 15, 53–76 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E, Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Edgar R, Domrachev M, Lash AE, Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30, 207–210 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.G. E. O. National Center for Biotechnology Information.
- 36.Caplan AI, Why are MSCs therapeutic? New data: new insight. J Pathol 217, 318–324 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tasso R, Gaetani M, Molino E, Cattaneo A, Monticone M, Bachi A, Cancedda R, The role of bFGF on the ability of MSC to activate endogenous regenerative mechanisms in an ectopic bone formation model. Biomaterials, (2011). [DOI] [PubMed]
- 38.Huang da W, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Huang da W, Sherman BT, Lempicki RA, Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gronthos S, Stewart K, Graves SE, Hay S, Simmons PJ, Integrin expression and function on human osteoblast-like cells. J Bone Miner Res 12, 1189–1197 (1997). [DOI] [PubMed] [Google Scholar]
- 41.Ode A, Duda GN, Glaeser JD, Matziolis G, Frauenschuh S, Perka C, Wilson CJ, Kasper G, Toward biomimetic materials in bone regeneration: functional behavior of mesenchymal stem cells on a broad spectrum of extracellular matrix components. J Biomed Mater Res A 95, 1114–1124 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Kehrel B, Kronenberg A, Rauterberg J, Niesing-Bresch D, Niehues U, Kardoeus J, Schwippert B, Tschope D, van de Loo J, Clemetson KJ, Platelets deficient in glycoprotein IIIb aggregate normally to collagens type I and III but not to collagen type V. Blood 82, 3364–3370 (1993). [PubMed] [Google Scholar]
- 43.Gregory CA, Prockop DJ, Spees JL, Non-hematopoietic bone marrow stem cells: molecular control of expansion and differentiation. Exp Cell Res 306, 330–335 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Doi M, Nagano A, Nakamura Y, Genome-wide screening by cDNA microarray of genes associated with matrix mineralization by human mesenchymal stem cells in vitro. Biochem Biophys Res Commun 290, 381–390 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Dishowitz MI, Terkhorn SP, Bostic SA, Hankenson KD, Notch signaling components are upregulated during both endochondral and intramembranous bone regeneration. J Orthop Res, (2011). [DOI] [PMC free article] [PubMed]
- 46.Iwaki A, Jingushi S, Oda Y, Izumi T, Shida JI, Tsuneyoshi M, Sugioka Y, Localization and quantification of proliferating cells during rat fracture repair: detection of proliferating cell nuclear antigen by immunohistochemistry. J Bone Miner Res 12, 96–102 (1997). [DOI] [PubMed] [Google Scholar]
- 47.Roulet M, Ruggiero F, Karsenty G, LeGuellec D, A comprehensive study of the spatial and temporal expression of the col5a1 gene in mouse embryos: a clue for understanding collagen V function in developing connective tissues. Cell Tissue Res 327, 323–332 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Yamaguchi K, Matsuo N, Sumiyoshi H, Fujimoto N, Iyama KI, Yanagisawa S, Yoshioka H, Pro-alpha3(V) collagen chain is expressed in bone and its basic N-terminal peptide adheres to osteosarcoma cells. Matrix Biol 24, 283–294 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Imamura Y, Scott IC, Greenspan DS, The pro-alpha3(V) collagen chain. Complete primary structure, expression domains in adult and developing tissues, and comparison to the structures and expression domains of the other types V and XI procollagen chains. J Biol Chem 275, 8749–8759 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Yamazaki M, Majeska RJ, Yoshioka H, Moriya H, Einhorn TA, Spatial and temporal expression of fibril-forming minor collagen genes (types V and XI) during fracture healing. J Orthop Res 15, 757–764 (1997). [DOI] [PubMed] [Google Scholar]
- 51.Alexopoulos LG, Youn I, Bonaldo P, Guilak F, Developmental and osteoarthritic changes in Col6a1-knockout mice: biomechanics of type VI collagen in the cartilage pericellular matrix. Arthritis Rheum 60, 771–779 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marvulli D, Volpin D, Bressan GM, Spatial and temporal changes of type VI collagen expression during mouse development. Dev Dyn 206, 447–454 (1996). [DOI] [PubMed] [Google Scholar]
- 53.Keene DR, Sakai LY, Burgeson RE, Human bone contains type III collagen, type VI collagen, and fibrillin: type III collagen is present on specific fibers that may mediate attachment of tendons, ligaments, and periosteum to calcified bone cortex. J Histochem Cytochem 39, 59–69 (1991). [DOI] [PubMed] [Google Scholar]
- 54.Becker J, Schuppan D, Benzian H, Bals T, Hahn EG, Cantaluppi C, Reichart P, Immunohistochemical distribution of collagens types IV, V, and VI and of pro-collagens types I and III in human alveolar bone and dentine. J Histochem Cytochem 34, 1417–1429 (1986). [DOI] [PubMed] [Google Scholar]
- 55.Niyibizi C, Eyre DR, Identification of the cartilage alpha 1(XI) chain in type V collagen from bovine bone. FEBS Lett 242, 314–318 (1989). [DOI] [PubMed] [Google Scholar]
- 56.Morris NP, Oxford JT, Davies GB, Smoody BF, Keene DR, Developmentally regulated alternative splicing of the alpha1(XI) collagen chain: spatial and temporal segregation of isoforms in the cartilage of fetal rat long bones. J Histochem Cytochem 48, 725–741 (2000). [DOI] [PubMed] [Google Scholar]
- 57.Walchli C, Koch M, Chiquet M, Odermatt BF, Trueb B, Tissue-specific expression of the fibril-associated collagens XII and XIV. J Cell Sci 107 (Pt 2), 669–681 (1994). [DOI] [PubMed] [Google Scholar]
- 58.Sugrue SP, Gordon MK, Seyer J, Dublet B, van der Rest M, Olsen BR, Immunoidentification of type XII collagen in embryonic tissues. J Cell Biol 109, 939–945 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gregory KE, Keene DR, Tufa SF, Lunstrum GP, Morris NP, Developmental distribution of collagen type XII in cartilage: association with articular cartilage and the growth plate. J Bone Miner Res 16, 2005–2016 (2001). [DOI] [PubMed] [Google Scholar]
- 60.Lisignoli G, Codeluppi K, Todoerti K, Manferdini C, Piacentini A, Zini N, Grassi F, Cattini L, Piva R, Rizzoli V, Facchini A, Giuliani N, Neri A, Gene array profile identifies collagen type XV as a novel human osteoblast-secreted matrix protein. J Cell Physiol 220, 401–409 (2009). [DOI] [PubMed] [Google Scholar]
- 61.Chou MY, Li HC, Genomic organization and characterization of the human type XXI collagen (COL21A1) gene. Genomics 79, 395–401 (2002). [DOI] [PubMed] [Google Scholar]
- 62.Fitzgerald J, Bateman JF, A new FACIT of the collagen family: COL21A1. FEBS Lett 505, 275–280 (2001). [DOI] [PubMed] [Google Scholar]
- 63.Sun Y, Li W, Lu Z, Chen R, Ling J, Ran Q, Jilka RL, Chen XD, Rescuing replication and osteogenesis of aged mesenchymal stem cells by exposure to a young extracellular matrix. FASEB J 25, 1474–1485 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lewandrowski KU, Nanson C, Calderon R, Vertebral osteolysis after posterior interbody lumbar fusion with recombinant human bone morphogenetic protein 2: a report of five cases. Spine J 7, 609–614 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Perri B, Cooper M, Lauryssen C, Anand N, Adverse swelling associated with use of rh-BMP-2 in anterior cervical discectomy and fusion: a case study. Spine J 7, 235–239 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Robin BN, Chaput CD, Zeitouni S, Rahm MD, Zerris VA, Sampson HW, Cytokine-mediated inflammatory reaction following posterior cervical decompression and fusion associated with recombinant human bone morphogenetic protein-2: a case study. Spine (Phila Pa 1976) 35, E1350–1354 (2010). [DOI] [PubMed] [Google Scholar]
- 67.Wong DA, Kumar A, Jatana S, Ghiselli G, Wong K, Neurologic impairment from ectopic bone in the lumbar canal: a potential complication of off-label PLIF/TLIF use of bone morphogenetic protein-2 (BMP-2). Spine J 8, 1011–1018 (2008). [DOI] [PubMed] [Google Scholar]
- 68.Smucker JD, Rhee JM, Singh K, Yoon ST, Heller JG, Increased swelling complications associated with off-label usage of rhBMP-2 in the anterior cervical spine. Spine (Phila Pa 1976) 31, 2813–2819 (2006). [DOI] [PubMed] [Google Scholar]
- 69.Uccelli A, Moretta L, Pistoia V, Immunoregulatory function of mesenchymal stem cells. Eur J Immunol 36, 2566–2573 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Gregory CA, Prockop DJ, in Culture of Human Stem Cells, Freshney RIS, N G. and Auerbach JM, Ed. (Wiley-Liss, Hoboken NJ, 2007), pp. 208. [Google Scholar]
- 71.Gregory CA, Gunn WG, Peister A, Prockop DJ, An Alizarin red-based assay of mineralization by adherent cells in culture: comparison with cetylpyridinium chloride extraction. Anal Biochem 329, 77–84 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methods
Figure S1: Representative immunophenotype analysis of hMSCs used in the study.
Figure S2: Cell engraftment assays by conventional RT-PCR and gel electrophoresis.
Figure S3: Immunohistochemistry staining of calvarial defects with staining controls
Table S1: Primers used in this study
Table S2: Upregulated genes determined by microarray analysis after 1 week of engraftment.
Table S3: Up-regulated genes determined by microarray analysis after 2 weeks of engraftment.
Table S4: Meta-analysis of calvarial healing time course.
Table S5: Amino acid analysis of ECM.
Table S6: Potential trophic mediators expressed by GW-hMSCs are found in bone, cartilage and vascular tissue.
Table S7: Comparison of the secreted protein list against various tissue expression databases using the DAVID analysis system.
Table S8: Expression of integrins commonly associated with osteoblasts.