

Abstract
The elderly population is highly susceptible to developing respiratory diseases, including tuberculosis, a devastating disease caused by the airborne pathogen Mycobacterium tuberculosis (M.tb) that kills one person every 18 seconds. Once M.tb reaches the alveolar space, it contacts alveolar lining fluid (ALF), which dictates host-cell interactions. We previously determined that age-associated dysfunction of soluble innate components in human ALF leads to accelerated M.tb growth within human alveolar macrophages. Here we determined the impact of human ALF on M.tb infection of alveolar epithelial type cells (ATs), another critical lung cellular determinant of infection. We observed that elderly ALF (E-ALF)-exposed M.tb had significantly increased intracellular growth with rapid replication in ATs compared to adult ALF (A-ALF)-exposed bacteria, as well as a dampened inflammatory response. A potential mechanism underlying this accelerated growth in ATs was our observation of increased bacterial translocation into the cytosol, a compartment that favors bacterial replication. These findings in the context of our previous studies highlight how the oxidative and dysfunctional status of the elderly lung mucosa determines susceptibility to M.tb infection, including dampening immune responses and favoring bacterial replication within alveolar resident cell populations, including ATs, the most abundant resident cell type within the alveoli.
INTRODUCTION
The risk of tuberculosis (TB) susceptibility and mortality is significantly increased in individuals aged 65 years and older1–4. TB is caused by airborne Mycobacterium tuberculosis (M.tb), transmitted primarily by inhalation, where it is deposited into the distal portion of the airways and alveoli. In this environment, M.tb encounters the lung mucosa, or alveolar lining fluid (ALF), which contains soluble innate factors such as surfactant proteins A and D (SP-A/SP-D), hydrolytic enzymes, complement, lipids, and others, which activate subsequent innate and adaptive immune responses5–7.
As we age, changes to soluble components of the innate immune system in ALF are anticipated to contribute to the increased susceptibility of the elderly population to TB8,9. Published studies from our group found that ALF from elderly humans and old mice have increased levels of proinflammatory and pro-oxidative mediators8,10, impacting M.tb infection outcomes in vitro and in vivo11. Human macrophages infected with M.tb exposed to elderly human ALF (E-ALF) had reduced control of infection and altered intracellular trafficking with fewer phagosome–lysosome fusion events. These observations were reversed when E-ALF was replenished with functional SP-A/SP-D, supporting the importance of the functionality of ALF innate components in M.tb control11. Similar outcomes were also observed in vivo, where M.tb that had been exposed to E-ALF grew faster within the lungs of young mice and caused increased immunopathology11.
Most studies focus on the role of ALF in altering M.tb-phagocyte interactions5,12,13; however, it is critical to understand the impact of ALF on M.tb infection of non-professional phagocytes, in particular, alveolar epithelial type cells (ATs)14,15. ATs are the most prevalent cell population that covers the internal surface area of the alveolar environment16. Given that ATs are non-professional phagocytes, they are proposed to provide a suitable niche that enables M.tb to replicate and even elude an innate immune response17. Nonetheless, ATs also participate in immune responses involved in controlling M.tb infection by producing pro-inflammatory cytokines (e.g., TNF (Tumor necrosis factor), IL-8 (Interleukin), and GM-CSF (Granulocite macrophage colony stimulating factor)), thereby potentiating cellular crosstalk and activation of alveolar macrophages leading to an increase in their antimycobacterial activity18. An additional host defense mechanism of ATs is the secretion of innate immune molecules, for example, SP-A, SP-D, complement component 3, antimicrobial peptides, antibodies, and hydrolases, among others into the ALF that exhibits an essential role in facilitating cell recruitment, microbial killing19 and even driving the differential outcome of M.tb infection in ATs15. We recently found that M.tb exposed to ALF from healthy adults varies in growth rates within ATs, which was dependent on ALF protein oxidation levels and function15. Here, we aimed to determine the impact of the elderly lung mucosa on M.tb infection of ATs. We provide evidence that M.tb exposure to E-ALF led to increased M.tb replication with consequent growth in ATs, dampened AT immune responses, and evidence for unaltered inflammasome, autophagy, and cell death processes. However, M.tb exposure to E-ALF altered trafficking and promoted endosomal damage within ATs, with subsequent M.tb translocation to the AT cytosolic compartment. These findings suggest that E-ALF promotes M.tb growth within ATs potentially by exploiting the AT cytosol as a favorable replicative niche for M.tb.
RESULTS
M.tb exposure to elderly human ALF drives increased bacterial intracellular growth in ATs in vitro
Our prior studies have shown that M.tb exposure to ALF from elderly individuals (E-ALF) accelerates the growth of M.tb within human alveolar macrophages and monocyte-derived macrophages11. Herein, we observed that E-ALF-exposed M.tb also showed a significantly increased intracellular growth in ATs when compared to A-ALF-exposed M.tb (Fig. 1). This increased bacterial growth of E-ALF-exposed M.tb was not due to differences in the inoculum used and/or uptake by ATs (Figs 1A and 1B). Relative light units (RLU) were a more sensitive method for detecting differences in growth, starting at early time points [24 hours post-infection (hpi)]. Colony-forming unit (CFU) data follow the same trend, supporting the increased growth of E-ALF-exposed M.tb at 72 hpi (Figs 1B and 1C). These intracellular growth differences within ATs were not due to changes in AT cell viability after infection with M.tb exposed to either A-ALF or E-ALF over the infection time (120 hours) (Supplementary Fig. 1). High surface expression of e-Cadherin (CD324) on ATs in all groups studied throughout the infection (Supplementary Fig. 2) indicated that cell-cell interfaces between ATs were stable20, which is consistent with the high cell viability observed. Together these data provide evidence for enhanced E-ALF-exposed M.tb intracellular growth in ATs compared to bacteria exposed to A-ALF.
Fig. 1.
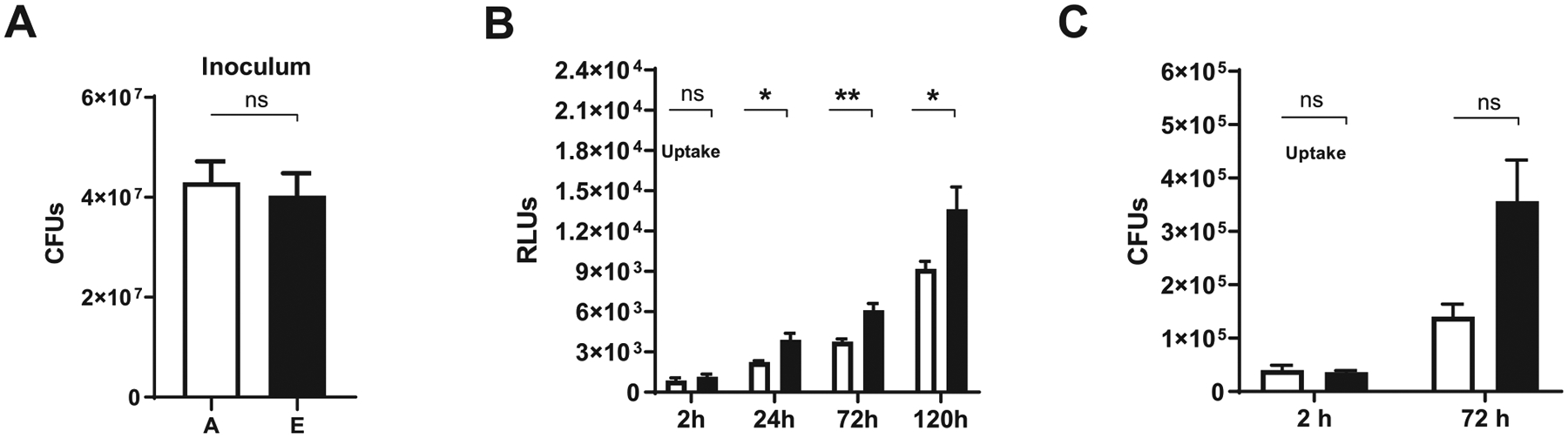
Exposure of M.tb to elderly human ALF is associated with increased intracellular bacterial growth in ATs. ATs were infected with ALF-exposed M.tb (H37Rv-Lux) for 2 hours at MOI of 10:1 followed by 1 hour of gentamicin to kill extracellular M.tb. (A) No differences in bacteria inoculum between conditions. (B) Infected monolayers in 96-well plates were read for increased luminescence (indicative of M.tb H37Rv-Lux intracellular growth in ATs) at the indicated time points, using the GloMax® reading system. Cumulative data of n = 4 (mean ± SEM) in triplicate, using four different A-ALFs and E-ALFs. (C) M.tb intracellular growth in ATs was assessed by colony-forming units (CFUs). Cumulative data of n = 2 (mean ± SEM) in triplicate, using two different A-ALFs and E-ALFs at the indicated time points. Student’s unpaired t test analysis of Adult versus Elderly at each time point, *p < 0.05, **p < 0.01. The “n” values represent the number of biological replicas using an individual ALF sample from different adult or elderly human donors. A = adult ALF-exposed M.tb (white bar); ALF = alveolar lining fluid; ATs = alveolar epithelial type cells; CFUs = colony-forming units; E = elderly ALF-exposed M.tb (black bar); MOI = multiplicity of infection; M.tb = Mycobacterium tuberculosis; ns: no significant differences; RLUs = relative light units; SEM = standard error of the mean.
We next determined if this observed increase in intracellular growth could be explained by changes in the rate of bacterial replication of A-ALF-exposed versus E-ALF-exposed M.tb during AT infection, using the fluorescent replication reporter SSB-GFP (Single-stranded DNA binding - Green Fluorescent Protein), smyc’::mCherry M.tb strain. Results indicate that E-ALF-exposed M.tb already had enhanced replication at 24 hpi (Fig. 2), which supports the increased growth of E-ALF M.tb within ATs over time (Fig. 1B). This increase in replication was maintained until 72 hpi but equivalent to A-ALF-exposed M.tb at this time point (Fig. 2).
Fig. 2.
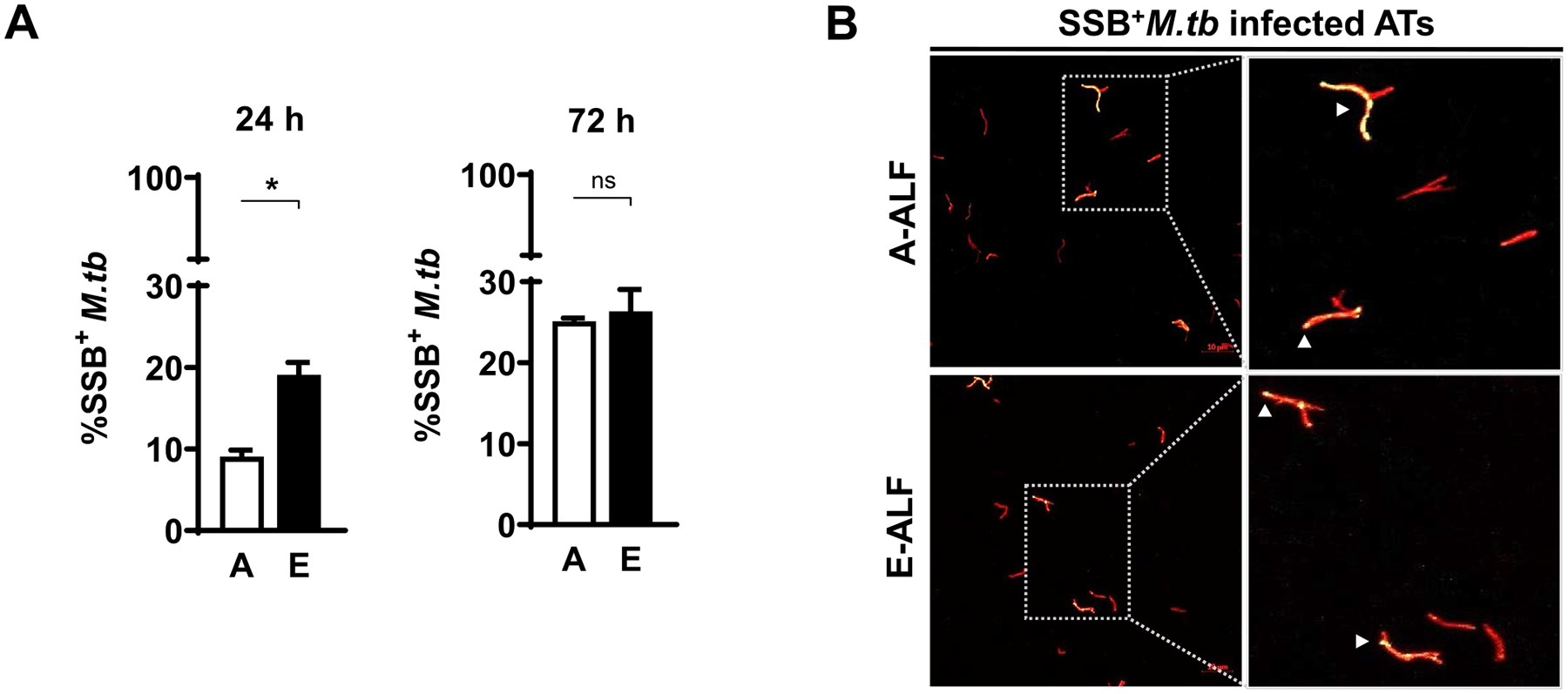
Elderly ALF-exposed M.tb has enhanced early replication during ATs infection. ATs were infected with the reporter SSB-GFP, smyc’:: mCherry M.tb strain at MOI of 10:1 and bacterial replication rate was determined by confocal microscopy at the indicated time points. (A) Percentage of SSB+M.tb exposed to A- and E-ALFs at 24 hpi and 72 hpi, n = 2–4 (mean ± SEM) in replicates, using two different A-ALFs and four different E-ALFs. (B) Representative confocal images of ATs infected with A- and E-ALF-M.tb at 72 hpi. The region indicated by a gray dashed line is shown expanded on the right (top panels, A-ALF; bottom panels, E-ALF). Replicating SSB+M.tb is indicated by white arrowheads, showing merged (yellow) foci. Events were enumerated by counting at least 50 independent events (≥ 50 bacteria). <scale bars represent 10 μm>. Student’s unpaired t test analysis of Adult versus Elderly, *p < 0.05. The “n” values represent the number of biological replicas using an individual ALF sample from different adult or elderly human donors. A = adult ALF-exposed M.tb; ALF = alveolar lining fluid; ATs = alveolar epithelial type cells; E = elderly ALF-exposed M.tb; GFP = green fluorescent protein; hpi = hours post-infection; MOI = multiplicity of infection; M.tb = Mycobacterium tuberculosis; ns: no significant differences; SEM = standard error of the mean; SSB = single-stranded DNA binding protein.
E-ALF versus A-ALF exposure decreases M.tb bacilli trafficking to late endosomes and endolysosomes within ATs
Differential trafficking of M.tb within ATs may allow for bacterial evasion of killing or serve as a host-killing mechanism15, depending on intracellular location. Using GFP expressing M. tb, we quantified co-localization events of A-ALF versus E-ALF-exposed M.tb with AT intracellular compartment markers by confocal microscopy. E-ALF-exposed M.tb showed decreased co-localization events for the late endosomal marker Rab7 (together with Rab5+ or alone) as early as 6 hpi, reaching statistical significance at the later time point studied (72 hpi, Fig. 3). Thus, the data indicated that E-ALF-exposed M.tb trafficked away from Rab5+ and/or Rab7+ vacuoles. This trend was also observed for LAMP-1+ lysosomal vacuoles, where up to 50% reduction for E-ALF-exposed versus A-ALF-exposed M.tb was observed during the time period studied (Fig. 4). For additional intracellular vacuoles studied [autophagosome (LC3+), multivesicular body (ABCA1+), and lamellar body (ABCA3+)], there were no significant differences observed for the location of E-ALF-exposed versus A-ALF-exposed M.tb (Fig. 4, Supplementary Fig. 3). Overall, our data indicate that exposure of M.tb to E-ALF alters intracellular trafficking resulting in an increased number of bacteria not associated with late endosome and endolysosome compartments.
Fig. 3.
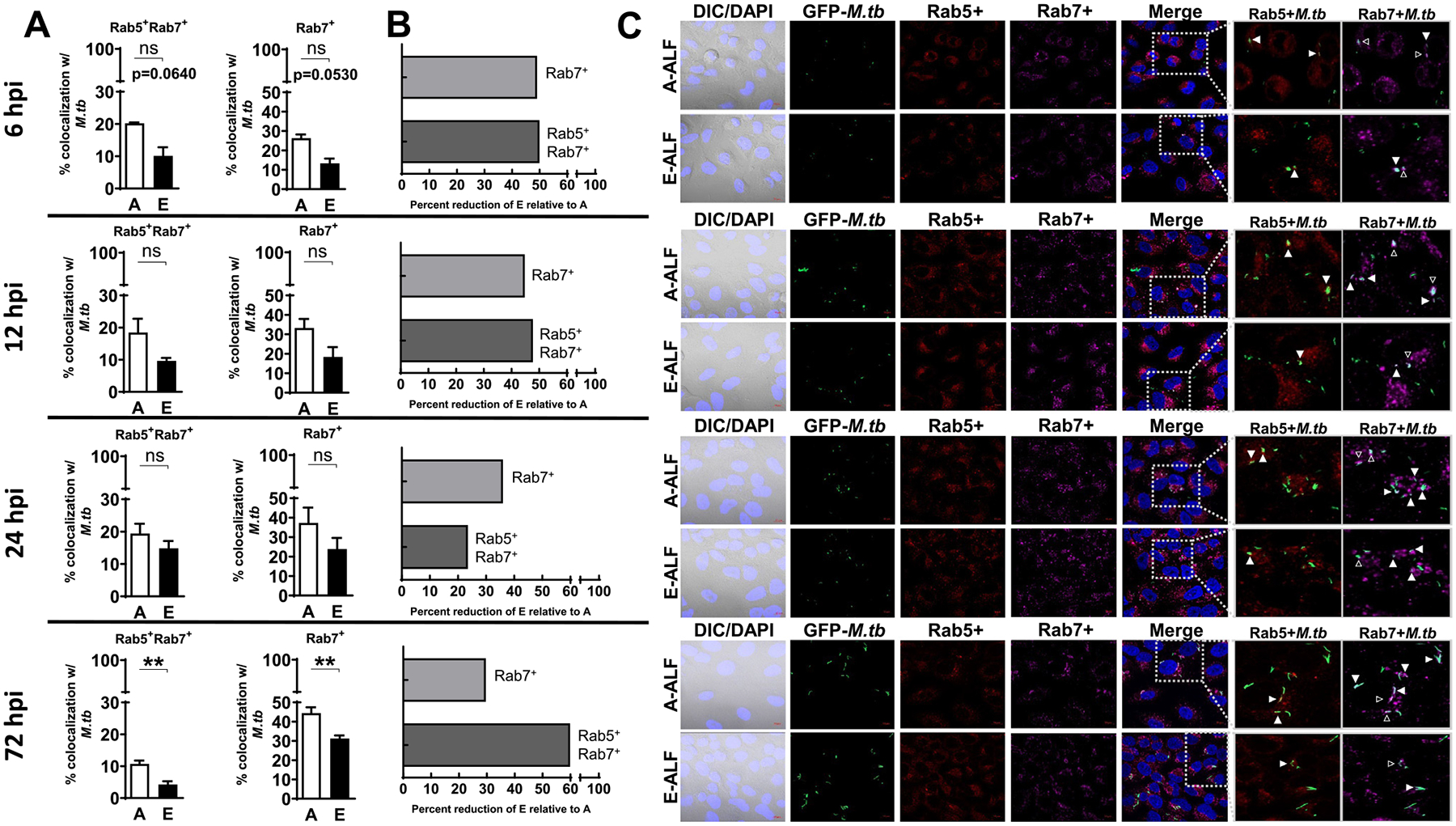
E-ALF-exposed M.tb has decreased trafficking to late endosomes within ATs. ATs were infected with either A-ALF or E-ALF-exposed GFP-M.tb for 2 hours at MOI of 100:1 followed by 1 hour of gentamicin to kill extracellular M.tb. Monolayers were stained with different intracellular markers at the indicated time points (6 hpi, 12 hpi, 24 hpi, and 72 hpi). (A) Co-localization events indicative of compartment fusion of M.tb with Rab5+Rab7+ (represents M.tb movement from early to late endosomes) or Rab7+ (represents M.tb already in late endosomes), n = 4 (mean ± SEM) in duplicate, using pooling of four different A-ALFs or E-ALFs. (B) Percent reduction in co-localization of E-ALF-exposed M.tb relative to A-ALF-exposed M.tb at the indicated time points. (C) Representative confocal images of ATs infected with A-ALF-exposed and E-ALF-exposed M.tb stained with intracellular markers Rab5 and Rab7 at the indicated time points. Events were enumerated by counting at least 200 independent events (200–300 bacteria). The region indicated by the gray dashed line is shown expanded on the right; and co-localization events are indicated by white arrowheads. Open arrowheads indicate double co-localization events (Rab5+Rab7+). <scale bars represent 10 μm>. Student’s unpaired t test analysis of Adult versus Elderly, *p < 0.05, **p < 0.01. The “n” values represent the number of biological replicas using pooling of ALF samples from different adult or elderly human donors. A = adult ALF-exposed M.tb; ALF = alveolar lining fluid; ATs = alveolar epithelial type cells; DAPI = 4’,6-diamidino-2-phenylindole (ATs nuclear DNA); DIC = differential interference contrast; E = elderly ALF-exposed M.tb; GFP = green fluorescent protein; hpi = hours post-infection; MOI = multiplicity of infection; M.tb = Mycobacterium tuberculosis; ns = no significant differences; Rab = Ras-associated binding protein; SEM = standard error of the mean.
Fig. 4.
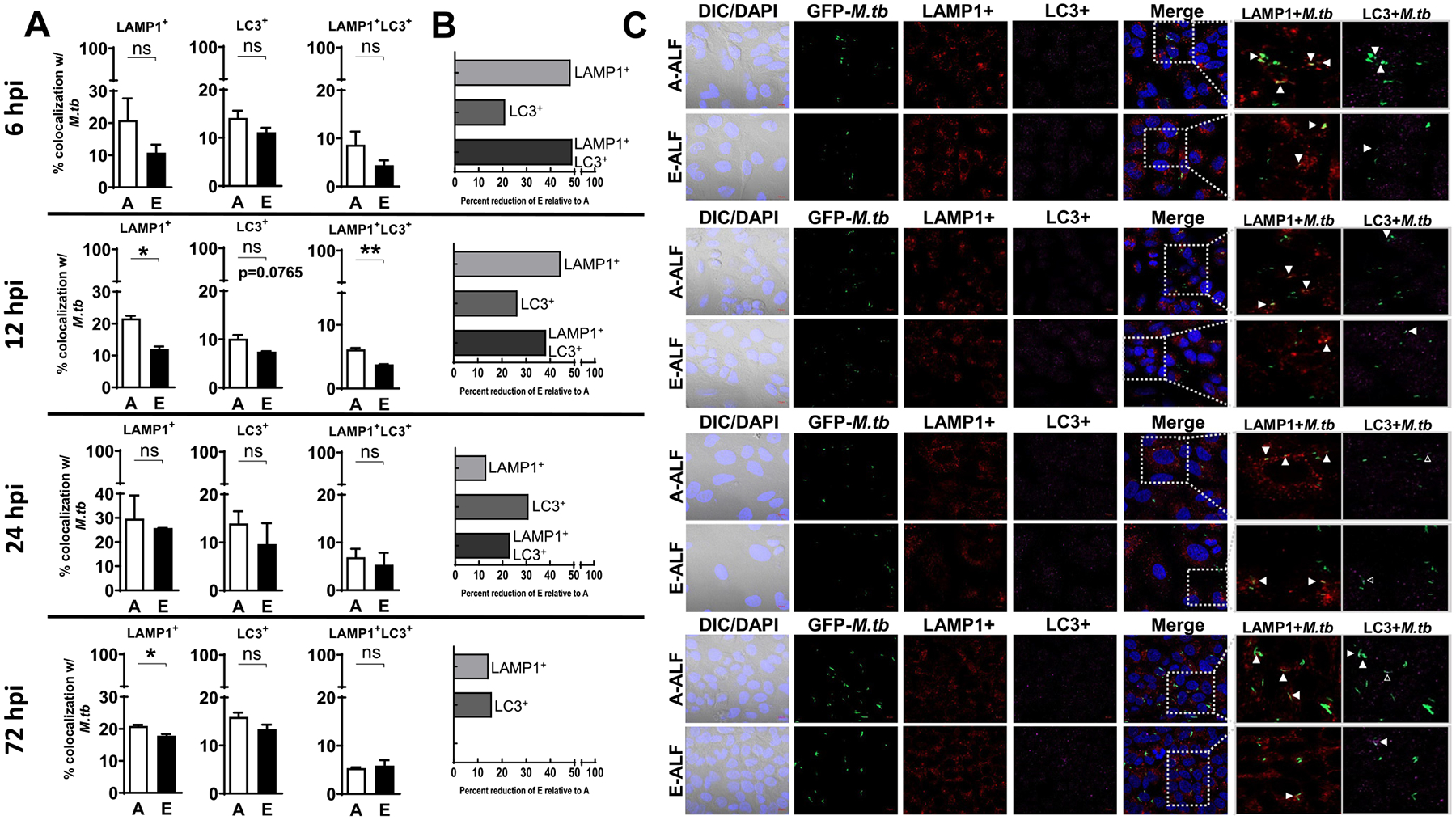
E-ALF-exposed M.tb has decreased endolysosomal trafficking within ATs. ATs were infected with either A-ALF or E-ALF-exposed GFP-M. tb for 2 hours at MOI of 100:1 followed by 1 hour of gentamicin to kill extracellular M.tb. Monolayers were stained with different intracellular markers at the indicated time points (6 hpi, 12 hpi, 24 hpi, and 72 hpi). (A) Co-localization events indicative of compartment fusion of M.tb with LAMP-1+ (represents M.tb in lysosomes) or LC3+ (represents M.tb in autophagosomes), n = 4 (mean ± SEM) in duplicate, using pooling of four different A-ALFs or E-ALFs. (B) Percent reduction in co-localization of E-ALF-exposed M.tb relative to A-ALF-exposed M.tb at the indicated time points. (C) Representative confocal images of ATs infected with A-ALF and E-ALF-M.tb stained with intracellular markers LAMP-1 and LC3 at the indicated time points. Events were enumerated by counting at least 200 independent events (200–300 bacteria). The region indicated by the gray dashed line is shown expanded on the right; and co-localization events are indicated by white arrowheads. Open arrowheads indicate double co-localization events (LAMP-1+LC3+). <scale bars represent 10 μm>. Student’s unpaired t test analysis of Adult versus Elderly, *p < 0.05, **p < 0.01. The “n” values represent the number of biological replicas using pooling of ALF samples from different adult or elderly human donors. A = adult ALF-exposed M.tb; ALF = alveolar lining fluid; ATs = alveolar epithelial type cells; DAPI = 4’,6-diamidino-2-phenylindole (ATs nuclear DNA); DIC = differential interference contrast; DNA = deoxyribonucleic acid; E = elderly ALF-exposed M.tb; GFP = green fluorescent protein; hpi = hours post-infection; LC3 = microtuble-associated protein light chain 3; LAMP = Lysosomal-associated membrane protein; MOI = multiplicity of infection; M.tb = Mycobacterium tuberculosis; ns = no significant differences; SEM = standard error of the mean.
E-ALF-exposed M.tb increases endosomal membrane damage leading to bacterial escape into the ATs cytosol
While it is well accepted that M.tb traffics through the endosomal pathway in phagocytic cells, less is known about M.tb intracellular localization within non-phagocytic cells such as ATs. There is some evidence to suggest that bacteria, including M. tb, can escape from phagosomes into the host-cell cytosol favoring survival within phagocytes21,22. Thus, we explored if the decreased intracellular trafficking of E-ALF-exposed M.tb to late endosomes and endolysosomes provided a survival advantage. Our transmission electron microscopy (TEM) results indicate that, at 72 hpi, in contrast to A-ALF-exposed M.tb, E-ALF-exposed M.tb was primarily located in the cytosol (56.3% vs. 16.9%) (Fig. 5). The designation of cytosolic bacteria was determined by a lack of vacuolar (endosomal) membranes surrounding bacteria (Fig. 5B). To complement our TEM data, we further showed that E-ALF-exposed M.tb significantly increased co-localization with the endolysosomal damage marker Galectin 3 (Fig. 6A)23,24. This suggests that when M.tb is exposed to E-ALF it is able to escape endolysosomes through membrane damage and subsequently translocate into the cytosol. The same trend was also observed for Galectin 8 alone or in combination with Galectin 3, albeit not significant (Fig. 6B, Supplementary Fig. 4). In addition to galectins, damaged phagosomal membranes can also be marked by ubiquitinated proteins (FK2 +)25,26. Despite the fact that this marker did not show differences in co-localization between E-ALF-exposed versus A-ALF-exposed M.tb (Fig. 6A), its dual staining with Gal-3 showed increased co-localization for the E-ALF-exposed M.tb at 120 hpi (Supplementary Fig. 4). Together, the TEM and galectins confocal microscopy results indicate that the majority of E-ALF-exposed M.tb were increasingly present in the cytosol, while A-ALF-exposed M.tb remained in endosomal compartments and that membrane damage is the potential mechanism for the finding with E-ALF-exposed M.tb.
Fig. 5.
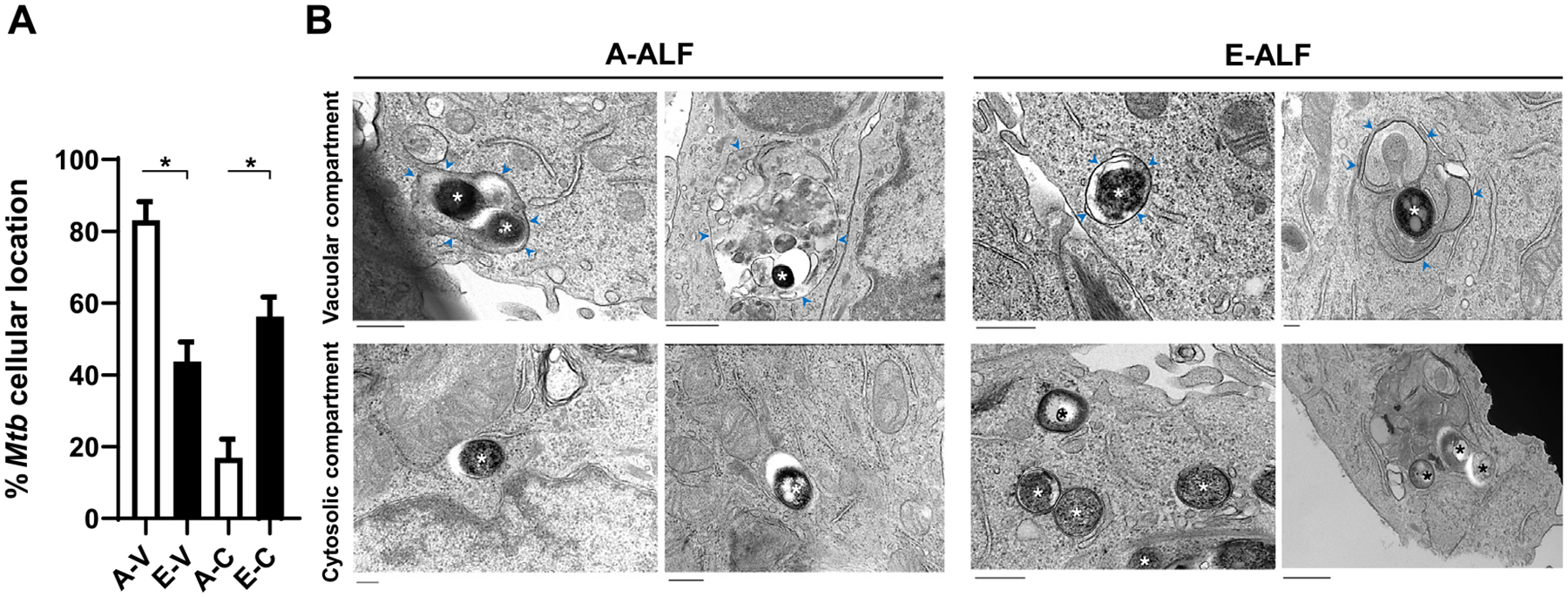
ALF-exposed M.tb drives differences in intracellular localization within ATs. ATs were infected with either A-ALF or E-ALF-exposed M.tb for 2 hours at MOI of 100:1 followed by 1 hour of gentamicin to kill extracellular M.tb. Relative proportion of intracellular bacteria located within membrane-bound vesicles or free in the cytosol by TEM. (A) Coded samples were scored by blinded analysis to quantify A-ALF-exposed and E-ALF-exposed M.tb located in vacuolar (endosomal/lysosomal) or cytosolic compartments at 72 hpi, n = 2 (mean ± SEM). (B) TEM micrographs of ALF-exposed M.tb. A-ALF-exposed and E-ALF-exposed M.tb were scored as “cytosolic, C” if they were not enclosed within a membrane or scored as “vacuolar, V” if they were surrounded by a vacuolar membrane. Vacuole membranes are indicated by blue arrowheads. Bacteria are indicated by asterisks. Values were determined by counting at least 100 independent events (bacteria). Student’s unpaired t test analyses of A-V versus E-V and A-C versus E-C; *p < 0.05. A-V: adult ALF-exposed M.tb in vacuolar compartment (Size bar 400 nm and 800 nm, respectively), A-C: adult ALF-exposed M.tb in cytosolic compartment (Size bar 400 nm), E-V: elderly ALF-exposed M.tb in vacuolar compartment (Size bar 600 nm and 200 nm, respectively), E-C: elderly ALF-exposed M.tb in cytosolic compartment (Size bar 400 nm and 600 nm, respectively). The “n” values represent the number of biological replicas using an individual ALF sample from different adult or elderly human donors. A = adult ALF-exposed M.tb; ALF = alveolar lining fluid; ATs = alveolar epithelial type cells; E = elderly ALF-exposed M.tb; hpi = hours post-infection; MOI = multiplicity of infection; M.tb = Mycobacterium tuberculosis; SEM = standard error of the mean; TEM = transmission electron microscopy.
Fig. 6.
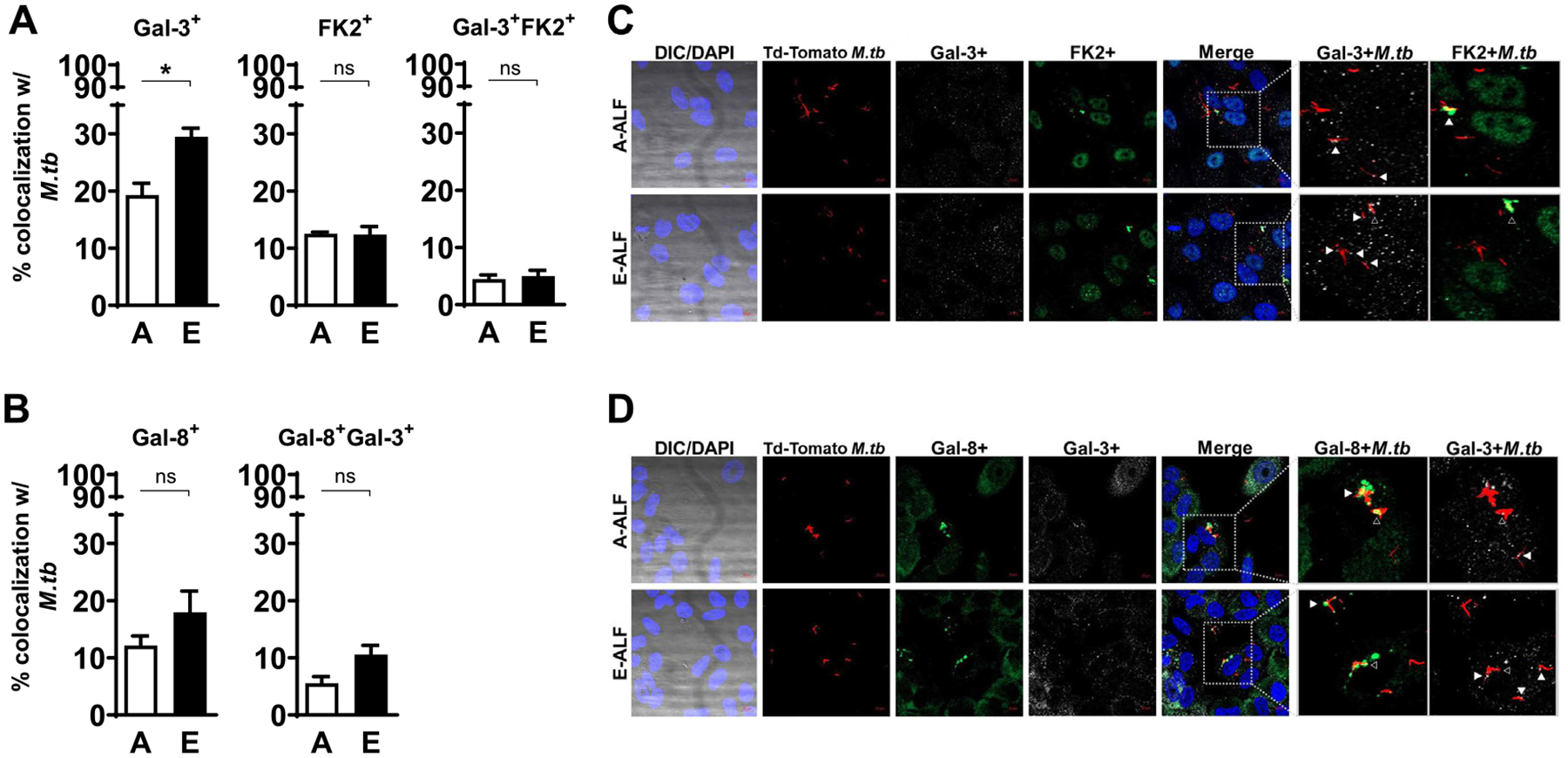
E-ALF exposure increases M.tb endosomal membrane damage within ATs. ATs were infected with either A-ALF or E-ALF-exposed Td-Tomato M.tb for 2 hours at MOI of 100:1 followed by 1 hour of gentamicin to kill extracellular M.tb. Monolayers were stained with different intracellular markers at 72 hpi. (A-B) Co-localization events indicative of compartment fusion of Mtb-Galectin-3+, Mtb-Galectin-8+, and M.tb-FK2+ (accumulation of ubiquitinated proteins), representing damaged endosomal membranes; n = 3–4 (mean ± SEM) in duplicate, using pooling of three different A-ALFs donors or four different E-ALFs. (C-D) Representative confocal images of ATs infected with A-ALF-exposed and E-ALF-exposed M.tb stained with intracellular markers Gal-3 and FK2 (C) or Gal-8 and Gal-3 (D) at 72 hours post-infection. Events were enumerated by counting 100–300 independent events (bacteria). The region indicated by the gray dashed line is shown expanded on the right; and co-localization events are indicated by white arrowheads. Open arrowheads indicate double co-localization events (Gal-3+FK2+ or Gal-8+Gal-3+). <scale bars represent 10 μm>. Student’s unpaired t test analysis of Adult versus Elderly, *p < 0.05. The “n” values represent the number of biological replicas using pooling of ALF samples from different adult or elderly human donors. A = adult ALF-exposed M.tb; ALF = alveolar lining fluid; ATs = alveolar epithelial type cells; DAPI = 4’,6-diamidino-2-phenylindole (ATs nuclear DNA); DIC = differential interference contrast; E = elderly ALF-exposed M.tb; FK2 = ubiquitinylated proteins (clone FK2); Gal = Galectin; hpi = hours post-infection; MOI = multiplicity of infection; M.tb = Mycobacterium tuberculosis; ns = no significant differences; SEM = standard error of the mean.
Effect of A-ALF and E-ALF M.tb on the production of immune mediators
Considering that M.tb exposure to E-ALF drives increased intracellular bacterial growth in ATs, we next tested whether E-ALF-exposed M.tb increased levels of AT pro-inflammatory cytokines and chemokines, reflecting increased AT activation. We quantified the production of immune mediators responsible for immune cell infiltration toward the site of infection and/or for promoting immune cell proliferation and maturation in the AT cultures17. Our results indicate that E-ALF-exposed M.tb drives decreased production of AT immune mediators, including some growth factors, chemokines, inflammasome mediators, interferons, and Th1 analytes, relative to A-ALF-exposed group (Fig. 7). Both A-ALF and E-ALF-exposed M.tb infection induced mainly pro-inflammatory cytokines by ATs when compared to uninfected ATs, where levels significantly differed at the latest time points (Supplementary Fig. 5). A significant decreased production of growth factors and chemokines was observed from AT cultures infected with E-ALF-exposed M.tb compared to A-ALF-exposed M.tb (Supplementary Figs 5A and 5B). This was particularly the case for the following analytes, growth factors: G-CSF, GM-CSF, FGF-2, FGF-1, TGF-α, IL-17, and chemokines: VEGF, MDC, MIP-1a, MIP-1b, IP-10 at later time points post-infection (Supplementary Fig 5A and 5B). There was also decreased production of IFNγ (Supplementary Fig. 5D) and TH1 proinflammatory mediators, including TNF, IL-12(p40), IL-12(p70), IL-2, IL-15 (Supplementary Fig. 5E), at 120 hours after infection. No significant differences were reported in most inflammasome and TH2/Th17 mediator production during infection between A-ALF or E-ALF-exposed M.tb. (Supplementary Figs 5C and 5F). With the exception of decreased levels of IL-1β at 120 hpi for E-ALF-exposed M.tb that could suggest decreased inflammasome activation (Supplementary Fig. 5C). Indeed M.tb infection reduced inflammasome activation in ATs at the latest time points, but there were no differences between ALF groups (Supplementary Fig. 6). Overall, we observed significant differences in production of several AT immune mediators during infection between A-ALF and E-ALF-exposed M.tb at later time points.
Fig. 7.
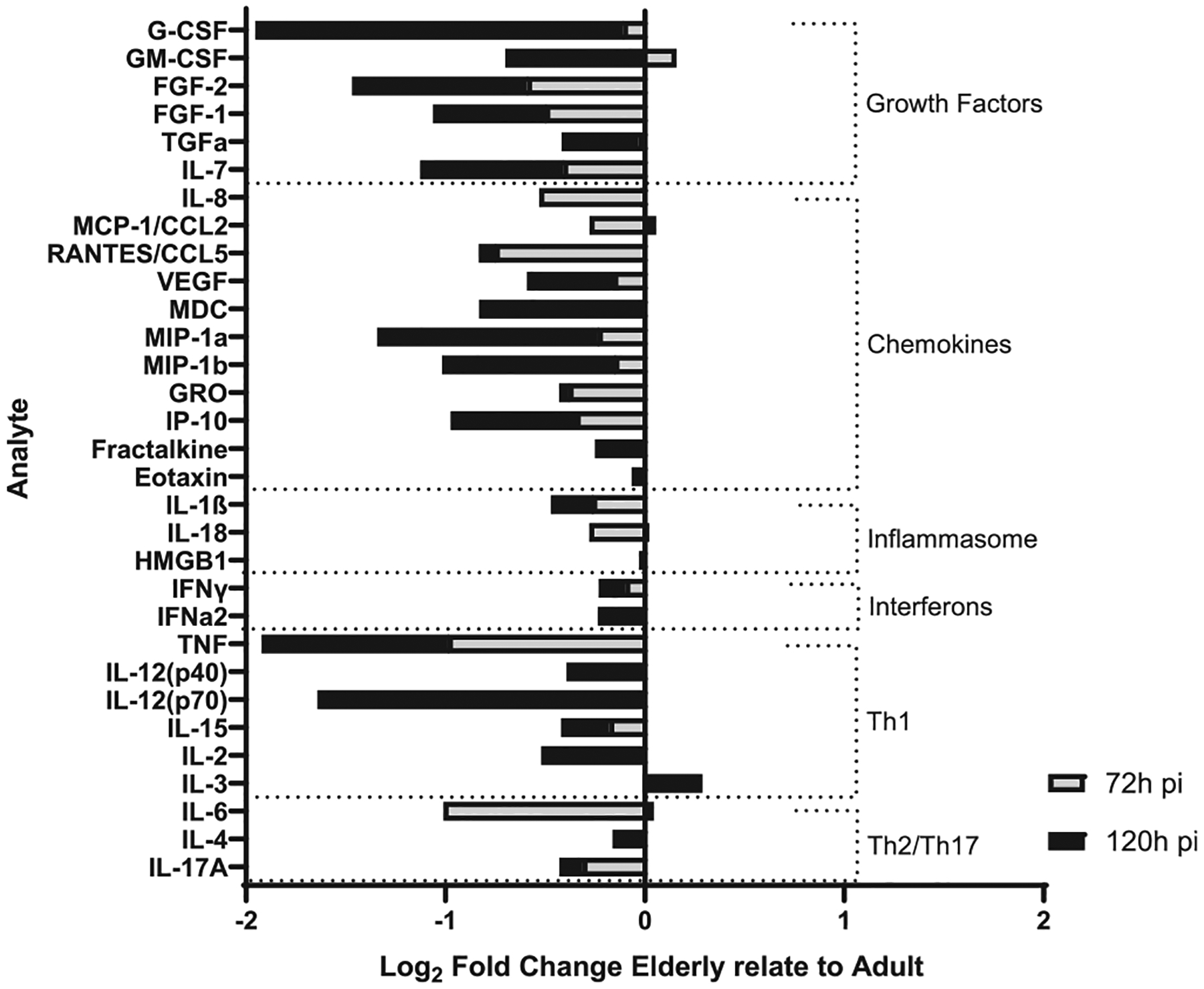
E-ALF-exposed M.tb drives a decrease in production of ATs immune mediators. ATs were infected with either A-ALF or E-ALF-exposed M.tb for 2 hours at MOI of 100:1 followed by 1 hour of gentamicin to kill extracellular M.tb. AT supernatants from infected ATs with either A-ALF-exposed or E-ALF-exposed M.tb were assessed for growth factors, cytokines, and chemokines production and measured at 72 hpi and 120 hpi by multiplexed biomarker assay following the manufacturer’s instructions. The graph shows the log2 fold changes of supernatants from E-ALF-exposed M.tb infected ATs relative to A-ALFs; and categorized into the following groups: growth factors, chemokines, inflammasome, interferons, Th1 and Th2/Th17 mediators. Values correspond to log2 (median total [pg/ml] E-ALF / median total [pg/ml] A-ALF) for n = 4, using pooling of four different A-ALF or E-ALF donors from three separate experiments. A = adult ALF-exposed M.tb; ALF = alveolar lining fluid; ATs = alveolar epithelial type cells; E = elderly ALF-exposed M.tb; hpi = hours post-infection; MOI = multiplicity of infection; M. tb = Mycobacterium tuberculosis; Th = T helper cell mediators.
DISCUSSION
The elderly population (65 years or older) with inherent compromised immunity is at higher risk of developing active TB disease2. Our published studies have proven that the interactions of human ALF components, at their physiological concentrations within the lung, with M.tb determine the outcome of infection in vitro and in vivo11. We also established that E-ALF from the elderly population has a significant degree of oxidative stress driving the dysfunctionality of some of these components (e.g., SP-D) favoring M.tb infection in vitro (phagocytes) and in vivo (mouse model)8,10,11. However, it remained unclear how E-ALF-exposed M.tb impacts AT infection, the major resident cell in the alveoli. Here we determined that E-ALF-exposed or A-ALF-exposed M.tb similarly enters ATs. However, E-ALF-exposed M.tb replicated faster resulting in more growth inside ATs, which was associated with altered endosomal trafficking marked by endosomal membrane damage and bacterial translocation into the cytosolic compartment, suggesting that this location is a favorable niche for M.tb to establish an initial infection, averting local alveolar host immune responses (Fig. 8). This finding supports the importance of altered ALF functions in old age, as a result of a different composition and functional status8,11. Finally, our results indicate that E-ALF-exposed M.tb does not significantly alter AT cell viability, enabling the continued intracellular growth of E-ALF-exposed M.tb within ATs over time.
Fig. 8.
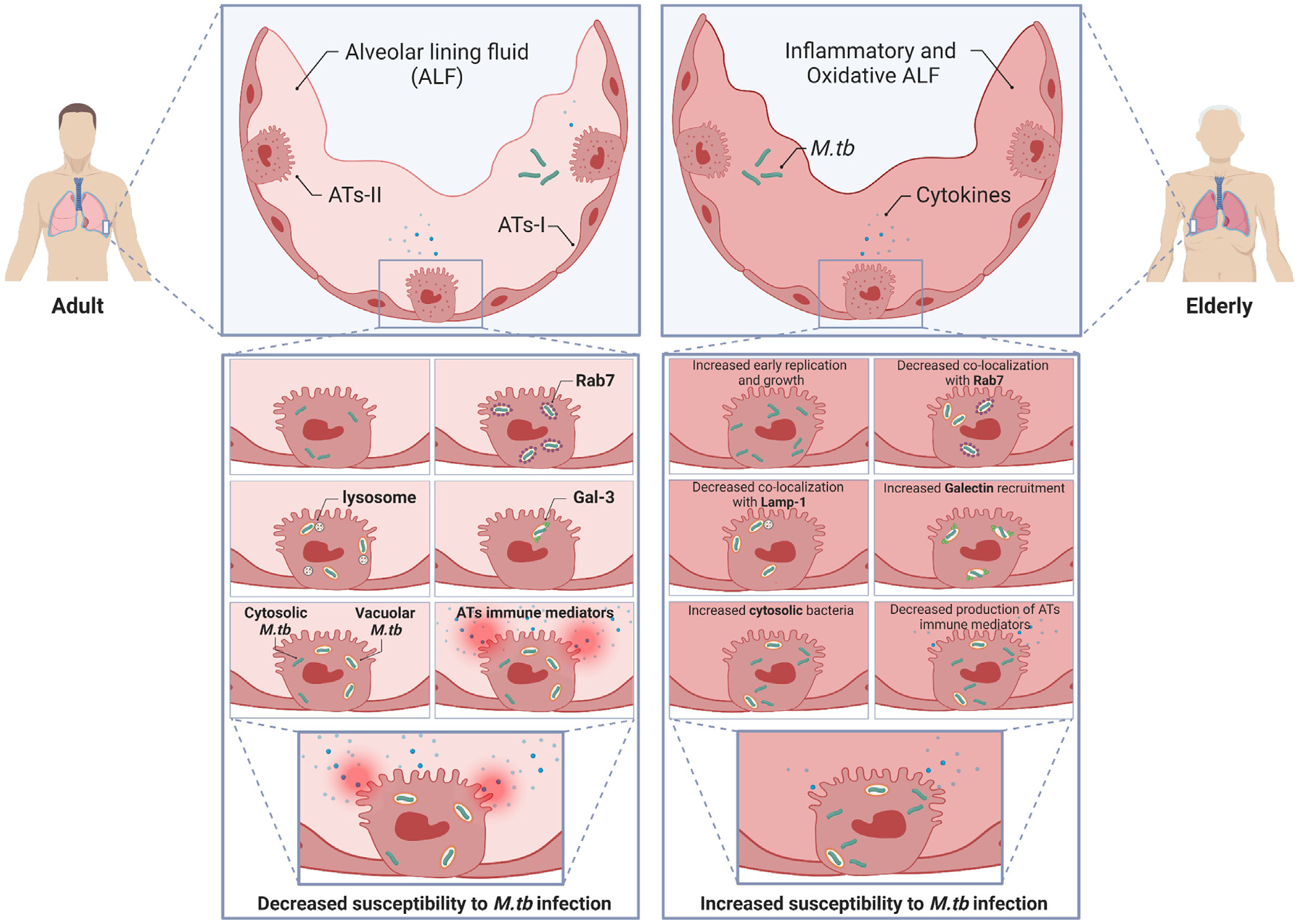
Schematic overview of the main findings in this study. M.tb exposure to E-ALF from elderly individuals enhances early replication and consequent intracellular growth in ATs. Moreover, AT infection with E-ALF-exposed M.tb alters bacterial endosomal trafficking and increases galectin recruitment suggesting increased endosomal membrane damage driving increased bacterial translocation into the cytosol. E-ALF-exposed M.tb also shows altered production of some inflammatory mediators (cytokines and chemokines) without impacting host cell viability. Overall, A-ALF-exposed M.tb is mainly located in endosomal/lysosomal (vacuoles), whereas; E-ALF-exposed M.tb is located in both vacuolar and cytosolic compartments. In short, exposure to E-ALF promotes M.tb growth within ATs and diminishes AT immune responses, preventing host cell activation and death. E-ALF-exposed M.tb exploits the AT cytosol as a niche for bacterial survival. This illustration was created with BioRender (https://biorender.com/). A = adult ALF-exposed M.tb; ALF = alveolar lining fluid; ATs = alveolar epithelial type cells; E = elderly ALF-exposed M.tb; M.tb = Mycobacterium tuberculosis.
The M.tb phagosome in macrophages shares features with early endosomes due to blockage of EEA1 and Rab7 recruitment27,28, whereas M.tb was found to traffic to late endosomes (Rab7+) in epithelial cells29. Our results with A-ALF-exposed M. tb are consistent with previous studies, in which M.tb resides mainly in Rab7+ late endosomes in ATs29,30. However, exposure to E-ALF decreased M.tb endosome association with Rab7+ late endosomes in ATs. This result was independent of the infection dose used (data not shown). Further, M.tb exposure to E-ALF drove a significant decrease in association with LAMP-1. As expected, M.tb exposure to A-ALF did not alter bacterial trafficking to other compartments of the vesicular network such as autophagosomes, multivesicular bodies, and lamellar bodies15. This was also the case for E-ALF-exposed M.tb. When assessing the overall intracellular distribution, our results indicate that E-ALF-exposed M.tb bacilli are located in both vacuolar and cytosolic compartments, whereas A-ALF-exposed M.tb bacilli are located mainly in vacuoles where ATs could potentially limit their replication and consequent growth. Greater escape of E-ALF-exposed M.tb into the cytosol is further supported by an accumulation of galectins (3 and 8), which are considered to be important indicators of endolysosomal membrane damage23–25.
There is evidence that M.tb can translocate from the phagolysosome into the cytosol facilitated by the ESAT-6 secretion complex-1 (ESX-1 type VII secretion system) in macrophages31. While more studies are needed to elucidate whether the M.tb ESX-1 type VII secretion system mediates M. tb translocation into the cytosol in non-professional phagocytes, M.tb genes encoding ESAT-6 proteins are upregulated during M. tb infection of ATs32. Given that E-ALF exposure promoted M.tb replication and growth within ATs and greater translocation into the cytosol, we speculate the M.tb ESX-1 type VII secretion system plays a role. Overall, it will be important to further determine the M.tb metabolic status within ATs at the time of its translocation into the cytosol to elucidate the bacterial mechanism(s) involved in this process.
ATs can activate infiltrating myeloid cells and lymphocytes by acting as antigen-presenting cells through surface expression of major histocompatibility complexes I and II (MHCI/II)33,34. Incidentally, we observed that AT infection with E-ALF-exposed or A-ALF-exposed M.tb had similar MHC surface marker expression (HLA-ABC and HLA-DR/DP/DQ) in ATs (Supplementary Fig. 7). Interestingly, E-ALF-exposed M.tb decreased production of several AT immunological mediators, some significantly reduced (e.g., G-CSF, GM-CSF, FGF-2, and IL-12, among others), some critical to the initiation of local inflammatory responses (TNF, IL-1β), host-cell recruitment (G-CSF, GM-CSF, FGF-2, MDC, GRO), activation of host alveolar innate cells (IFNγ, IL-7, MIP-1a, MIP-1b) and induction of cell-mediated immunity (IL-2, IL-12, IP-10, IL-15) after M.tb infection35–40. Thus, this decrease in AT immune responses by E-ALF-exposed M.tb indicates that altered activation of alveolar tissue responses (e.g., cell activation, infiltration, and differentiation) in the initial stages of the infection could reduce effective local immunity and thus favor M.tb infection and spread41.
ATs are distinguished by their significant role in participating in the host response to M.tb, including but not limited to potentiating cellular crosstalk (regulating alveolar macrophage activation) and secreting innate immune molecules which promote anti-M.tb activity17,42. In addition, there is increasing evidence that M.tb can invade ATs during infection and disease18,30,43–47. For example, reports show alveolar epithelial cells positive for M.tb DNA in infected human and mouse lung tissue43,44,48. Other reports show the presence of M.tb in the alveolar epithelium in infected mouse lung sections by electron microscopy18 as well as using lung-on-chip models46,47. Herein we include additional evidence by the visualization of M.tb within the alveolar epithelium in the mouse lung at day 15 post-infection46,47 (Fig. 9), and in ex vivo human lung tissue from a patient with TB (Fig. 10). Finally, TEM studies using in vitro M.tb systems with A549 cell lines49–51 demonstrate M.tb within these epithelial cells.
Fig. 9.
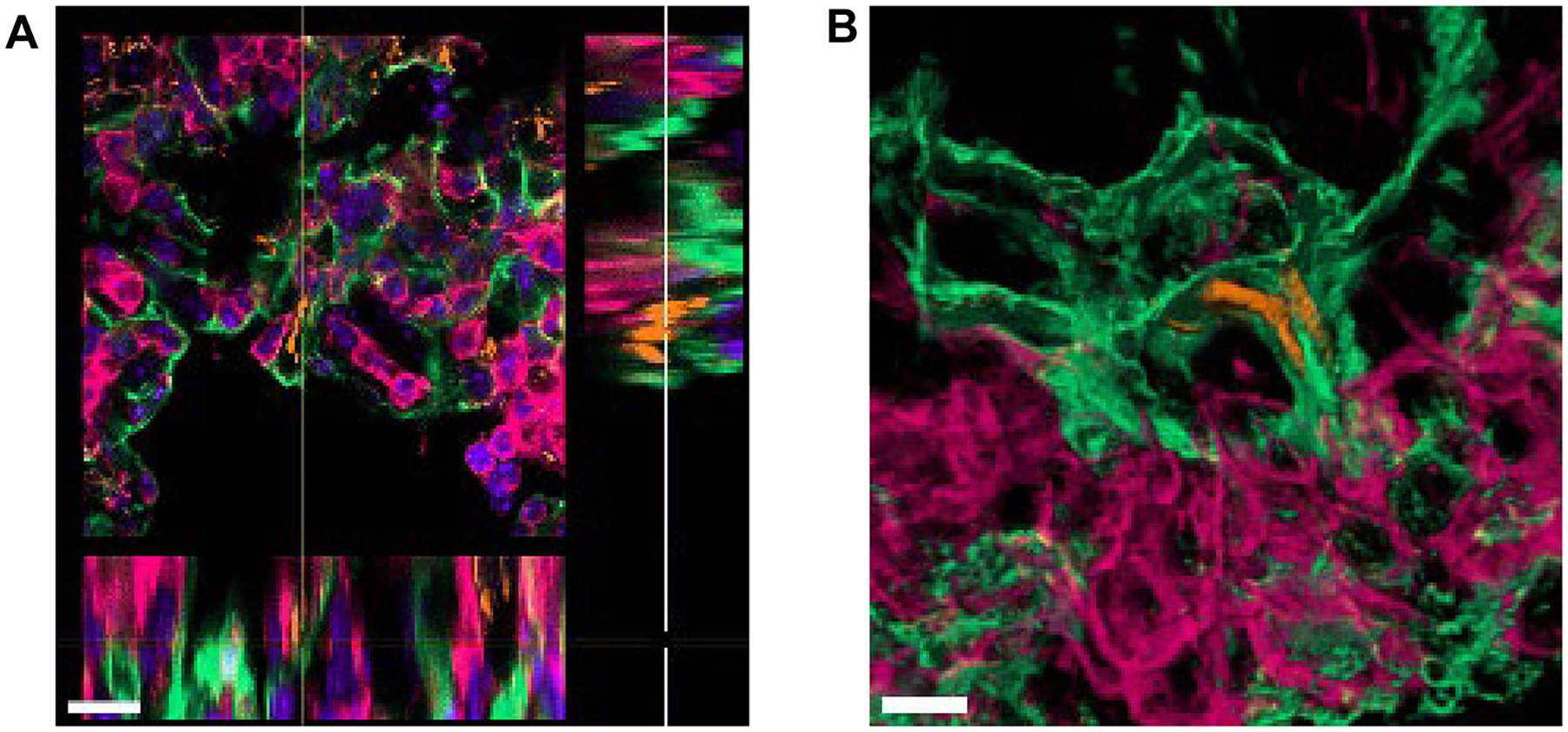
M.tb-infected ATs in early infection in the mouse model. (A) Extended ortho-section and (B) 3D view of M.tb bacilli intracellular in AT cells in the lungs of C57BL/6 mice infected with M.tb Erdman constitutively expressing Td-Tomato (orange) at 15 days post-infection. Podoplanin, PDPN (positive type I ATs, green); CD45 (leukocyte marker, pink), and DAPI (DNA/ nuclei marker, blue in panel A only). <scalebars (A) 15 μm, (B) 5 μm>. 3D = three-dimensional; ATs = alveolar epithelial type cells; DAPI = 4’,6-diamidino-2-phenylindole; DNA = deoxyribonucleic acid; M.tb = Mycobacterium tuberculosis; PDPN = podoplanin.
Fig. 10.
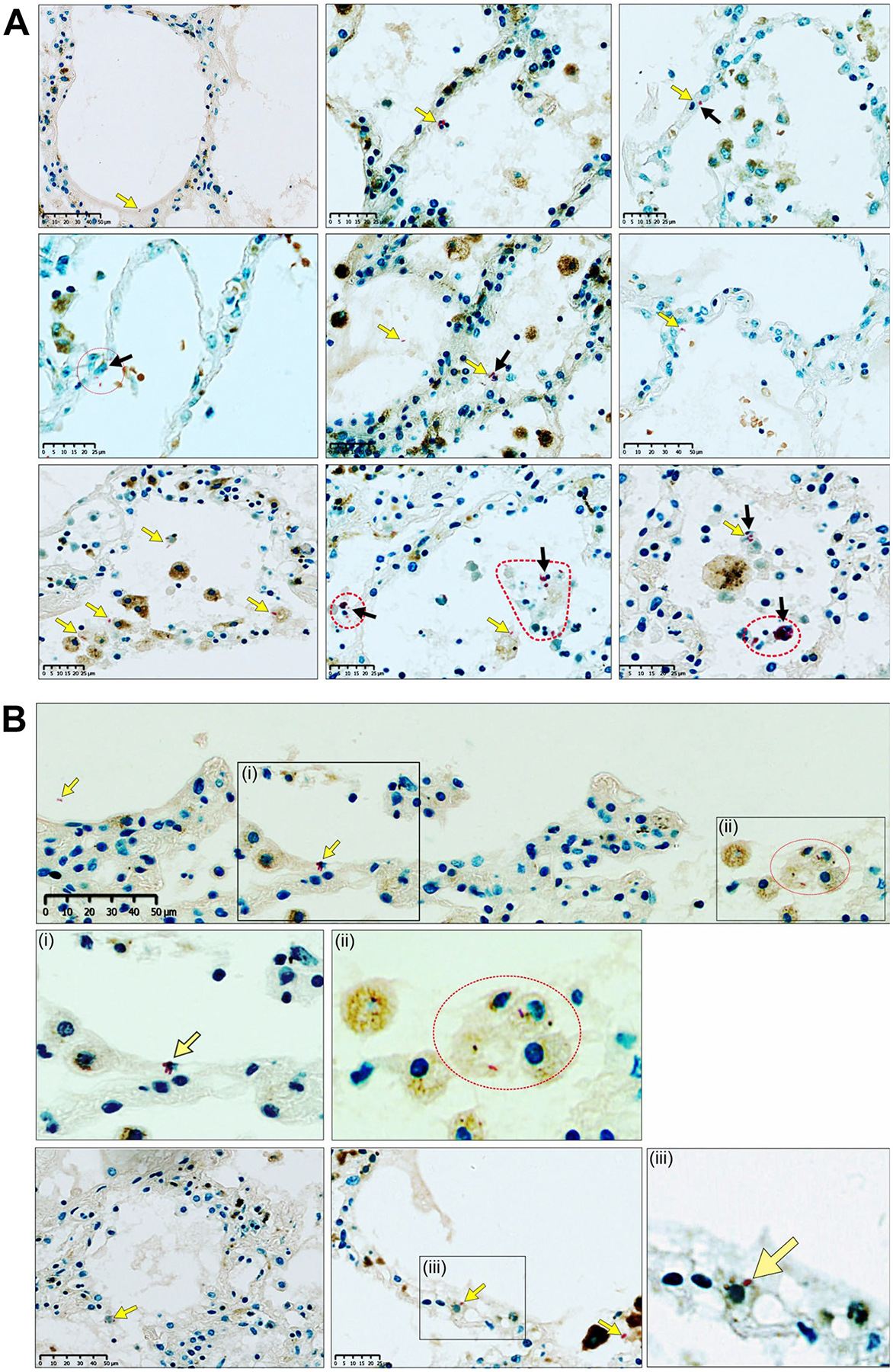
Visualization of M.tb within the alveoli and alveolar epithelium in the human lung. (A) Combined Ziehl-Neelsen(ZN)/CD68 and (B) ZN/Heme Oxygenase-1 (HO-1) staining of multiple sections of the lung parenchyma from a patient with microbiologically confirmed TB: Low-power images and high-power magnifications demonstrate acid-fast bacilli (AFBs) (yellow arrows) in extracellular and intracellular locations, including AFBs within alveolar epithelial cells (black arrows). Dotted red line shapes indicate M.tb-infected cells with multiple AFBs. The ZN method stains the AFBs of M.tb red in color, including cross-sections thereof that appear as red dot-like structures. CD68 is a macrophage immunomarker. A dilute HO-1 immunohistochemical stain permitted an improved visualization of cellular outlines. In (B), (i), (ii), and (iii) are magnifications. AFB = acid-fast bacilli; HO-1 = Heme-oxygenase 1; M.tb = Mycobacterium tuberculosis; TB = tuberculosis; ZN = Ziehl-Neelsen.
Altogether, M.tb exposure to the inflammatory and oxidative E-ALF environment8,11 alters intracellular trafficking of M.tb promoting its translocation into the cytosol, which enhances M.tb intracellular replication and consequent growth in ATs. Our study highlights the impact of the status and composition of elderly lung mucosa on M.tb infection of ATs, critical non-professional phagocytes that impact TB as well as other respiratory infectious diseases. These findings support our previous publications, which suggest that as we age, the oxidative status of the lung environment increases, and this could be related to the increased susceptibility to respiratory infections8,10,11,52. Thus, reducing the oxidative status in the aging lung might be an effective host-directed therapeutic platform for TB and other respiratory infections, whereby direct delivery of antioxidants into the lungs could restore the lung oxidative balance, re-establish the levels and function of soluble innate components present in ALF, and subsequently increase the resistance of the elderly individual to respiratory infections. Our recent study showed that the transfer of young mitochondria in CD4+ T cells of old mice reduces T cell oxidative stress, increases their immune function, and results in old mice being able to better control both M.tb and influenza A infections52.
MATERIALS AND METHODS
Ethics statement and human subjects
Human subject studies were carried out in strict accordance with the US Code of Federal and Local Regulations (The Ohio State University Institutional Review Board numbers 2012H0135 and 2008H0119, and Texas Biomedical Research Institute/UT-Health San Antonio/South Texas Veterans Health Care System Institutional Review Board number HSC20170673H). Participants, adults (aged 18–45 years, n = 8) and elderly (aged 60 years and older, n = 9), were enrolled from both sexes without discrimination of race or ethnicity after informed written consent to obtain bronchoalveolar lavage fluid. Demographic and clinical characteristics of the human donors are shown in Supplementary Table 1. All participants with comorbidities were excluded and not enrolled (see Supplementary Material for details). A pneumonectomy specimen was chosen from a study cohort that was approved by the University of KwaZulu-Natal Biomedical Research Ethics Committee (class approval study number: BCA 535/16). Patients undergoing lung resection for TB (study identifier: BE019/13) were recruited from King Dinizulu Hospital Complex, a tertiary center for TB patients in Durban, South Africa. Written informed consent was obtained from all study participants.
AT culture
For all experimental procedures, we utilized the human AT type II-like cell line A549 (ATCC® CCL-185™), a lung carcinoma cell line that exhibits many AT type II cell traits and is an established model of ATs. Cell cultures were prepared as we previously described15 with minor changes. Briefly, the A549 cell line was cultured at 37°C with 5% CO2 in DMEM/F12 (Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12) supplemented with 10% FBS (Atlas Biologicals, Fort Collins, CO) and 1% PenStrep (Sigma, St. Louis, MO). Cells were maintained in an antibiotic-free growth medium one week before M.tb infection.
Human ALF isolation
ALF was obtained, concentrated, and normalized to its physiological concentrations within the human lung (at 1 mg/mL of phospholipid) as we previously described in detail5,10–13,15,53,54.
M.tb cultures
M.tb H37Rv-Lux (27294) (kindly provided by Drs. Azad and Schlesinger, Texas Biomedical Research Institute) was grown as described55,56. SSB-GFP, smyc’::mCherry M.tb Erdman, and Td-Tomato M.tb Erdman (kindly provided by Dr. Russell, Cornell University) were grown as described57. GFP-M.tb Erdman (kindly provided by Dr. Horwitz, University of California, Los Angeles) was grown as previously described5. Single bacterial suspensions were prepared as we described11,15. For infection in mouse models, WT M.tb strain Erdman with constitutive Td-Tomato expression46 was cultured at 37°C in Middlebrook 7H9 base (Difco) supplemented with 0.5% albumin, 0.2% glucose, 0.085% NaCl, 0.5% glycerol, and 0.02% tyloxapol for liquid culture in 100 mL roller bottles until exponential phase.
Exposure of M.tb bacteria to human ALF
Preparation of adult and elderly ALF-exposed M.tb was performed using single bacterial suspensions (1×108 bacteria/exposure) as we described in detail5,12,13,15,53. Freshly exposed M.tb were gently washed (to remove traces of ALF) and prepared immediately for bacteria inoculum and subsequent infection. ALF-exposed M.tb inoculums were serially diluted in 7H9 broth and used for AT infection or plated on 7H11 agar to determine M.tb viability and specifically to confirm no differences in viable bacterial counts among E-ALF versus A-ALF-exposed M.tb.
M.tb infection of ATs: luciferase-based intracellular growth by relative light units and colony-forming units
M.tb infection of ATs was performed as previously described15. Briefly, single-cell suspensions of ALF-exposed M.tb in DMEM/F12/FBS media (bacteria inoculum) were added to the ATs culture at various multiplicity of infections (MOIs), and cells were incubated for 2 hours with the first 30 minutes on a platform shaker. Alternatively, cold synchronization (synchronized phagocytosis) was performed to facilitate bacterial internalization58. After infection, unbound bacteria were removed by washing, and gentamicin (50 μg/mL)-supplemented medium was added for 1 hour to kill extracellular bacteria. Then, infected ATs were washed and incubated with 10 μg/mL gentamicin-supplemented medium for the indicated times to assess intracellular growth. For luciferase-based M.tb growth assays56, ATs were infected with M.tb H37Rv-Lux at MOI 10:1, and bacterial bio-luminescence was measured every 24 hours for up to 120 hours with a GloMax® Multi Detection System (Promega, Madison, WI). For determination of intracellular M.tb by the CFUs method, infected ATs at MOI 10:1 were lysed and enumerated as described previously11,59. For CFUs, intracellular trafficking, and electron microscopy experiments, ATs were infected with GFP-M.tb Erdman strain, unless otherwise specified in the figure legends. For the M.tb replication rate determination, ATs were infected with SSB-GFP, smyc’::mCherry M.tb Erdman strain.
AT cell viability assay
At indicated times post-infection, AT cytotoxicity was determined by CellTiter-Glo® luminescent cell viability assay (Promega Cat. #G7570) following the manufacturer’s instructions. The assay consists of determining the number of viable cells in culture based on the quantification of ATP present in the media (the amount of ATP [Adenosine triphosphate] is directly proportional to the number of viable cells present in the culture media). The luminescent signal generated by the thermostable luciferase was measured every 24 hours post-infection (hpi) for up to 120 hpi with a GloMax Multi Detection System (Promega, Madison, WI).
Immunocytochemistry and confocal microscopy (in vitro studies)
ATs monolayers on glass coverslips were infected for 2 hours with A-ALF-exposed or E-ALF-exposed M.tb at MOI 100:1 and processed as described15. Briefly, at 6 hpi, 12 hpi, 24 hpi, and 72 hpi, ATs were washed and fixed with cold 4% paraformaldehyde for 15 minutes at room temperature and permeabilized with 0.1% Triton-X100 in phosphate-buffered saline (PBS) for 10 minutes at room temperature. To evaluate the M.tb intracellular trafficking, cellular compartments of infected and uninfected cells were stained and evaluated by confocal microscopy as described15. For specific antibody information, refer to Supplementary Material. The nucleus was stained with 50 ng/ml 4’,6-diamidino-2-phenylindole (DAPI) (Invitrogen Cat. # D1306) for 10 minutes at room temperature. After multiple washes to remove the excess DAPI solution, coverslips were mounted on slides using ProLong Gold Antifade Reagent (Invitrogen Cat. #P36934).
Cells were visualized by laser scanning confocal microscopy using a ZEISS LSM 800 microscope set at appropriate parameters and a final magnification of 600X. For the M.tb replication rate experiment, SSB-GFP, smyc’::mCherry M.tb Erdman strain was quantified by blinded analysis performed by two independent researchers, counting at least 50 events (≥ 50 bacteria) per condition in duplicate. For intracellular trafficking, co-localization events of the different cellular compartments containing GFP-M.tb Erdman or Td-Tomato M.tb Erdman were quantified by blinded analysis performed by two independent researchers by counting at least 100 events (100–300 bacteria) per condition in duplicate. All microscopy data were analyzed with Zeiss ZEN Software.
Transmission electron microscopy (TEM)
ATs monolayers were infected for 2 hours with A-ALF-exposed M.tb or E-ALF-exposed M.tb at MOI 100:1. Infected ATs, at 72 hpi were fixed in 2.5% glutaraldehyde and 2% formaldehyde (in 0.1 M Na Cacodylate pH 7.3) and analyzed by TEM as previously described60,61 with minor changes. Briefly, after the primary fixative (overnight), bacteria were rinsed with 0.1M phosphate buffer and post-fixed with 1% Zetterqvist’s buffered Osmium Tetroxide for 30 minutes. Then, a stepwise prolonged dehydration procedure with a graded series of alcohols, with all steps being once for 10 minutes, except the 100% alcohol steps, which were 10 minutes twice, and finally dehydrated with propylene oxide twice for 10 minutes each. For resin infiltration and embedding, cell blocks went from propylene oxide to 3:1 propylene oxide:Epon for 4 hours of incubation followed by two more incubations in 1:1 propylene oxide:Epon and 2:1 propylene oxide:Epon on a rotator. Cell blocks were transferred to neat Epon for 4 hours and cured at 60°C for 48 hours. Ultra-thin sections were cut and post-stained on grid with 1% uranyl acetate in 50% MeOH (Methanol) for 1 hour at room temperature in the dark followed by staining with lead citrate for 3 minutes at room temperature.
Samples were shipped to Dr. Daniel L. Clemens, an expert in the study of intracellular compartmentalization of M.tb by TEM62–64 for analyses in a blinded manner. The relative proportion of bacilli located within membrane-bound vesicles or located in the cytosol compartment (lacking membrane bilayers around the bacilli) was quantified. As individual host cells often had M.tb inside different compartments, data were scored on a bacterium level rather than a host-cell level. For each condition, at least 100 events (≥ 100 bacteria) were imaged and scored in a blinded fashion using the JEOL 100CX transmission electron microscope (Brain Research Institute Electron Microscopy Core Facility; Brain Research Institute, UCLA).
AT immune mediators
Protein levels in supernatants from infected (A-ALF-exposed M. tb and E-ALF-exposed M.tb) or non-infected ATs at 24 hpi were determined using a multiplex panel human magnetic bead Luminex® Assay for IL-6, CCL5/RANTES, GM-CSF, TNF, IL-1β, IL-18/IL-1F4, and IL-12/IL-23 (R&D, Human 10-Plex Cat. #LXSAHM-10, Lot #L134898); and using human ELISA kits for IL-8/CXCL8 (dilution 1:5; R&D Cat. # DY208–05, Lot #P105639) and CCL2/MCP-1 (dilution 1:15; R&D Cat. # DY279–05, Lot #333900) following the manufacturer’s instructions. Human Luminex analysis was performed using Luminex200 (SN LX10009028406) with the xPONENT 4.3 Software version by the Texas Biomed Molecular Core, following these parameters: DD gate 8000–16,500, 50 μl of sample volume, 50–100 events per bead/region, and Low PMT (LMX100/200: Default).
Protein levels in supernatants from infected (A-ALF-exposed M.tb and E-ALF-exposed M.tb) or non-infected ATs at 72 hpi and 120 hpi were determined using Luminex multiplexed biomarker assays for G-CSF, GM-CSF, FGF-2, TGFα, FGF-1, IL-7, IL-8, MCP-1, RANTES, VEGF, MIP-1α, MIP-1b, GRO, IP-10, Fractalkine, Eotaxin, IL-1β, IL-18, HMGB1, TNFα, IL-12 (p40), IL-12 (p70), IL-15, IL-2, IL-3, IFNγ, IFNα2, IL-6, IL-4, and IL-17A (EMD Millipore’s Milliplex® Cat. #HCYTA-60K-11 and #HCYP4MAG-64K-02) following the manufacturer’s instructions and using the Luminex Flex-MAP 3D instrument, Milliplex Analyst software version 5.1 by the University of Texas Health Center at San Antonio’s Bioanalytics and Single-Cell Core. The analysis software was configured to collect a total of 1000 beads with an average of 50 events per bead in 100 μL of sample/well. The median fluorescence intensity (MFI) was used to acquire raw data. The lowest standard which was at least three times above the background was used to define the lower limit of quantitation. The MFI and the acquired standard curve were used to compute the concentrations of each analyte.
AT inflammasome activity
AT inflammasome activity was determined by the Caspase-Glo® 1 Inflammasome assay (Promega Cat. # G9951) following the manufacturer’s instructions. The assay consists of selectively measuring the activity of caspase-1, among other caspases. The luminescent signal generated by the thermostable luciferase was measured at 24 hpi and 72 hpi with a GloMax Multi Detection System (Promega, Madison, WI).
Mouse infection
Female C57BL/6 mice aged 8 weeks were purchased from a commercial supplier (Charles River) and housed in a specific pathogen-free facility. Animal protocols were reviewed and approved by EPFL’s Chief Veterinarian, by the Service de la Consommation et des Affaires Vétérinaires of the Canton of Vaud, and by the Swiss Office Vétérinaire Fédéral under license VD3472. Littermates of the same sex were randomly assigned to experimental groups.
Mice were infected by the aerosol route with between 50–100 CFU of exponential phase M.tb using a custom-built aerosol machine, as previously described47. The infectious dose was verified by measurements of the total pulmonary bacterial burden via CFU assays on 7H11 agar plates containing the MGITTM PANTATM antibiotic mix (BD).
Immunofluorescence and imaging (lung tissue sections)
At 2 weeks post-infection, mice were euthanized by CO2 over-dose, and the lungs were removed aseptically. For imaging of thick infected tissue sections, lung lobes were immersed in 20 mL 4% neutral-buffered paraformaldehyde (PFA) solution (Biosystems) and incubated for 2 hours at room temperature followed by 24 hours at 4°C for fixation. The tissue section was then equilibrated in 30% sucrose for a subsequent 24–48 hours and cryofrozen for sectioning. 150 μm-thick sections of fixed frozen infected animal tissues were cut using a cryostat (Leica CM3050S). After a blocking step of 2 hours in 1% BSA with 1% Triton X-100 in PBS, sections were incubated for 72 hours with primary antibodies directly labeled with fluorophores [CD45 (30-F11)]; anti-mouse; AF647 conjugated; rat monoclonal (Biolegend) and Podoplanin (eBio 8.1.1); anti-mouse; AF488 conjugated; Syrian hamster monoclonal (Thermo Fisher Scientific) diluted in 1% BSA with 0.1% Triton X-100 in PBS. After counterstaining with DAPI, sections were mounted on Superfrost+ slides using a frame (BioRad, Frame-Seal™ Slide Chambers, #SLF1201) and FluoromountG (Bioconcept, 0100–01). Fixed, stained sections were imaged using a Leica SP8 confocal microscope with a 40x glycerol immersion objective (NA = 1.25, Leica). Z-stacks were subsequently deconvolved using the Huygens Deconvolution Software (Scientific Volume Imaging). Imaris 9.9 (Bitplane) was used to render 3D images and extended ortho-sections.
Ziehl-Neelsen and IHC visualization of M.tb within the alveoli, macrophages, and alveolar epithelium in the human lung
Combined Ziehl-Neelsen (ZN)/CD68 and ZN/heme-oxygenase 1 (HO-1) stains were performed on multiple representative human lung tissue samples from the selected case. Histopathological appraisal confirmed the presence of intracellular acid-fast bacilli (AFBs) within ATs and macrophages. In addition, extracellular AFBs were identified within alveolar spaces, including immediately adjacent to the alveolar wall. Human pulmonary samples of interest were aseptically removed and fixed in 10% neutral-buffered formalin (10% NBF). These samples were routinely processed in a vacuum filtration tissue processor using a xylene-free protocol. Tissue sections were embedded and blocked in paraffin wax. Sections were cut at 4 μm, baked at 56°C for 15 minutes, dewaxed through two changes of xylene, and rehydrated through descending grades of alcohol to water. Routine H&E (Hematoxylin and Eosin) staining was performed whereby slides were placed in hematoxylin for 5 minutes, washed in tap water for 2 minutes, blued in lithium carbonate for 1 minute, rinsed in tap water for 2 minutes, and counterstained with eosin for 5 minutes before a final rinse in tap water for 2 minutes. Slides were dehydrated in ascending grades of alcohol, cleared in xylene, and mounted with Distyrene, Plasticizer, and Xylene. For the combined IHC (Immunohistochemistry) and ZN staining, human lung tissues were cut into 2 mm thick sections and were mounted on charged slides, and heated at 56°C for 15 minutes. Mounted sections were dewaxed in xylene followed by rinse in 100% ethanol and one change of SVR (95%). Slides were then washed under running water for 2 minutes followed by antigen retrieval via Heat Induced Epitope Retrieval in Tris-sodium chloride (pH 6.0) for 30 minutes. Slides were then cooled for 15 minutes and rinsed under running water for 2 minutes. Endogenous peroxide activity was blocked using 3% hydrogen peroxide for 10 minutes at room temperature (RT). Slides were then washed in PBST and blocked with protein block (Novolink) for 5 minutes at RT. Sections were incubated with primary antibodies for CD68 (M0814-CD68-KP1, DAKO, 1:3000) or HO-1 (ab13248, Abcam; 1:100) followed by washing and incubation with the polymer (Novolink) for 30 minutes at RT. Slides were then washed and stained with DAB for 5 minutes and washed under running water for 5 minutes. Slides were then incubated with heated carbol fuchsin for 10 minutes and washed in running tap water. 3% acid alcohol was applied to the slide to decolorize for 30 seconds or until sections appeared clear. Slides were then washed in running tap water for 2 minutes followed by counterstaining with methylene blue. Slides were then rinsed under running water, dehydrated, and mounted in Distyrene, Plasticiser, and Xylene.
Slide scanning
Tissue specimens were digitized using a Hamamatsu NDP slide scanner (Hamamatsu NanoZoomer RS2, Model C10730–12) and its viewing platform (NDP.View2). The red, green, and color balance was kept at 100% whereas gamma correction was maintained between 0.7 and 2. Brightness and contrast settings are very modest between slides and depend on staining quality. For a detailed list of the reagents used in this process, see Supplementary Material.
Statistical analysis
GraphPad Prism 8 Software was used to prepare the graphs and determine the statistical significance between experimental groups. Two-tailed Student’s t test (unpaired), One-way analysis of variance, or two-way analysis of variance with Tukey post hoc correction for multiple testing was applied where applicable and as described in the figure legends. In this study, the “n” values represent the number of biological replicas using an individual (or pooled) ALF sample from different adult or elderly human donors as described in figure legends. Statistical differences between groups were reported as significant (*) when the p value was less than or equal to 0.05.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. David Russell (Cornell University), Marcus Horwitz (UCLA), and Abul Azad (Texas Biomed) for kindly providing us with the SSB-GFP smyc’::mCherry M.tb Erdman, GFP-M.tb Erdman, and Lux-M.tb H37Rv strains, respectively. We thank Dr. Morwan Osman (University of Cambridge) for advice on galectin staining. We acknowledge Dr. Reagan Meredith (Texas Biomed) for biostatistics consultation and the BSL-3 Operations Program at Texas Biomed for their services and support.
FUNDING
This study was supported by the National Institute on Aging (NIA), National Institutes of Health (NIH) (Grant number P01 AG-051428 to JT, JBT, BIR and LSS, and F99 AG-079802 to AMO-F), the NIH/National Institute of Allergy and Infectious Diseases (NIAID) R33 AI-138280 to AJCS; the HFSP Long-Term Fellowship (LT000231/2016-L) and support from the Holcim Stiftung zur Förderung der Wissenschaftlichen and to VVT. JBT was also partially supported by the Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation. This study was also partially supported by the Office of the Director, NIH, under Award Number S10 OD-028653. AMO-F was partially supported by the Douglass Graduate Fellowship at Texas Biomed. This research has been facilitated by the infrastructure and resources provided by the Texas Biomedical Research Institute Interdisciplinary NexGen TB Research Advancement Center (IN-TRAC), an NIH/NIAID-funded program (P30 AI-168439). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
DECLARATION OF COMPETING INTEREST
The authors have no competing interests to declare.
APPENDIX A. SUPPLEMENTARY DATA
Supplementary data to this article can be found online at https://doi.org/10.1016/j.mucimm.2024.01.001.
DATA AVAILABILITY
All data needed to evaluate the conclusions of the paper are present in this manuscript or the Supplementary Material.
REFERENCES
- 1.Guerra-Laso JM et al. Macrophages from elders are more permissive to intracellular multiplication of Mycobacterium tuberculosis. Age (Dordr) 35, 1235–1250 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schaaf HS, Collins A, Bekker A & Davies PD Tuberculosis at extremes of age. Respirology 15, 747–763 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Olmo-Fontánez AM & Turner J. Tuberculosis in an aging world. Pathogens 11, 1101 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martini M, Barberis I, Gazzaniga V & Icardi G The fight to end tuberculosis: a global challenge in strong partnership. J. Prev. Med. Hyg 61(Suppl. 1), E1–E2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arcos J et al. Human lung hydrolases delineate Mycobacterium tuberculosis-macrophage interactions and the capacity to control infection. J. Immunol 187, 372–381 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferguson JS, Voelker DR, McCormack FX & Schlesinger LS Surfactant protein D binds to Mycobacterium tuberculosis bacilli and lipoarabinomannan via carbohydrate-lectin interactions resulting in reduced phagocytosis of the bacteria by macrophages. J. Immunol 163, 312–321 (1999). [PubMed] [Google Scholar]
- 7.Gaynor CD, McCormack FX, Voelker DR, McGowan SE & Schlesinger LS Pulmonary surfactant protein A mediates enhanced phagocytosis of Mycobacterium tuberculosis by a direct interaction with human macrophages. J. Immunol 155, 5343–5351 (1995). [PubMed] [Google Scholar]
- 8.Moliva JI et al. Molecular composition of the alveolar lining fluid in the aging lung. Age (Dordr) 36, 9633 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piergallini TJ & Turner J Tuberculosis in the elderly: why inflammation matters. Exp. Gerontol 105, 32–39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Vilanova A et al. The aging human lung mucosa: a proteomics study. J. Gerontol. A. Biol. Sci. Med. Sci 77, 1969–1974 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moliva JI et al. The lung mucosa environment in the elderly increases host susceptibility to mycobacterium tuberculosis infection. J. Infect. Dis 220, 514–523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arcos J et al. Mycobacterium tuberculosis cell wall released fragments by the action of the human lung mucosa modulate macrophages to control infection in an IL-10-dependent manner. Mucosal Immunol 10, 1248–1258 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scordo JM et al. Mycobacterium tuberculosis cell wall fragments released upon bacterial contact with the human lung mucosa alter the neutrophil response to infection. Front. Immunol 8, 307 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bermudez LE & Goodman J Mycobacterium tuberculosis invades and replicates within Type II alveolar cells. Infect. Immun 64, 1400–1406 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scordo JM et al. The human lung mucosa drives differential Mycobacterium tuberculosis infection outcome in the alveolar epithelium. Mucosal Immunol 12, 795–804 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ward HE & Nicholas TE Alveolar type I and type II cells. Aust. N. Z. J. Med 14 (Suppl. 3), 731–734 (1984). [PubMed] [Google Scholar]
- 17.Scordo JM, Knoell DL & Torrelles JB Alveolar epithelial cells in Mycobacterium tuberculosis infection: active players or innocent bystanders? J. Innate Immun 8, 3–14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato K et al. Type II alveolar cells play roles in macrophage-mediated host innate resistance to pulmonary mycobacterial infections by producing proinflammatory cytokines. J. Infect. Dis 185, 1139–1147 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Torrelles JB & Schlesinger LS Integrating lung physiology, immunology, and tuberculosis. Trends Microbiol 25, 688–697 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carterson AJ et al. A549 lung epithelial cells grown as three-dimensional aggregates: alternative tissue culture model for Pseudomonas aeruginosa pathogenesis. Infect. Immun 73, 1129–1140 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jamwal SV et al. Mycobacterial escape from macrophage phagosomes to the cytoplasm represents an alternate adaptation mechanism. Sci. Rep 6, 23089 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Wel N et al. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129, 1287–1298 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Jia J et al. Galectin-3 coordinates a cellular system for lysosomal repair and removal. Dev. Cell 52, 69–87.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osman MM et al. The C terminus of the mycobacterium ESX-1 secretion system substrate ESAT-6 is required for phagosomal membrane damage and virulence. Proc. Natl Acad. Sci. U. S. A 119:e2122161119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong KW & Jacobs WR Jr Critical role for NLRP3 in necrotic death triggered by Mycobacterium tuberculosis. Cell. Microbiol 13, 1371–1384 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dupont N et al. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe 6, 137–149 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Fratti RA, Backer JM, Gruenberg J, Corvera S & Deretic V Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J. Cell Biol 154, 631–644 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Via LE et al. Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J. Biol. Chem 272, 13326–13331 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Fine KL et al. Involvement of the autophagy pathway in trafficking of Mycobacterium tuberculosis bacilli through cultured human type II epithelial cells. Cell. Microbiol 14, 1402–1414 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Harriff MJ et al. Human lung epithelial cells contain Mycobacterium tuberculosis in a late endosomal vacuole and are efficiently recognized by CD8(+) T cells. PLoS One 9, e97515 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Houben D et al. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell. Microbiol 14, 1287–1298 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Ryndak MB, Singh KK, Peng Z & Laal S Transcriptional profile of Mycobacterium tuberculosis replicating in type II alveolar epithelial cells. PLoS One 10, e0123745 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chuquimia OD et al. The role of alveolar epithelial cells in initiating and shaping pulmonary immune responses: communication between innate and adaptive immune systems. PLoS One 7, e32125 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corbière V et al. Phenotypic characteristics of human type II alveolar epithelial cells suitable for antigen presentation to T lymphocytes. Respir. Res 12, 15 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Domingo-Gonzalez R, Prince O, Cooper A & Khader SA Cytokines and chemokines in Mycobacterium tuberculosis infection. Microbiol. Spectr 4. 10.1128/microbiolspec.TBTB2-0018-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nambiar JK, Ryan AA, Kong CU, Britton WJ & Triccas JA Modulation of pulmonary DC function by vaccine-encoded GM-CSF enhances protective immunity against Mycobacterium tuberculosis infection. Eur. J. Immunol 40, 153–161 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Mvubu NE, Pillay B, McKinnon LR & Pillay M Mycobacterium tuberculosis strains induce strain-specific cytokine and chemokine response in pulmonary epithelial cells. Cytokine 104, 53–64 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Lin Y, Zhang M & Barnes PF Chemokine production by a human alveolar epithelial cell line in response to Mycobacterium tuberculosis. Infect. Immun 66, 1121–1126 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richter JR et al. Macrophage-derived chemokine (CCL22) is a novel mediator of lung inflammation following hemorrhage and resuscitation. Shock 42, 525–531 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saukkonen JJ et al. Beta-chemokines are induced by Mycobacterium tuberculosis and inhibit its growth. Infect. Immun 70, 1684–1693 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y, Wang Y & Liu X The role of airway epithelial cells in response to mycobacteria infection. Clin. Dev. Immunol 2012:791392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olmo-Fontánez AM & Torrelles JB Alveolar epithelial cells. In Advances in Host-Directed Therapies Against Tuberculosis (Karakousis PC, Hafner R & Gennaro ML, eds) 247–255 (Springer International Publishing, Cham, 2021). [Google Scholar]
- 43.Hernández-Pando R et al. Persistence of DNA from Mycobacterium tuberculosis in superficially normal lung tissue during latent infection. Lancet 356, 2133–2138 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Arriaga AK, Orozco EH, Aguilar LD, Rook GA & Hernández PR Immunological and pathological comparative analysis between experimental latent tuberculous infection and progressive pulmonary tuberculosis. Clin. Exp. Immunol 128, 229–237 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maertzdorf J et al. Mycobacterium tuberculosis invasion of the human lung: first contact. Front. Immunol 9, 1346 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thacker VV et al. A lung-on-chip model of early Mycobacterium tuberculosis infection reveals an essential role for alveolar epithelial cells in controlling bacterial growth. Elife 9, e59961 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mishra R et al. Mechanopathology of biofilm-like Mycobacterium tuberculosis cords. Cell 186, 5135–5150.e28 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wells G et al. Micro-computed tomography analysis of the human tuberculous lung reveals remarkable heterogeneity in three-dimensional granuloma morphology. Am. J. Respir. Crit. Care Med 204, 583–595 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hingley-Wilson SM, Sambandamurthy VK & Jacobs WR Jr Survival perspectives from the world’s most successful pathogen. Mycobacterium tuberculosis. Nat. Immunol 4, 949–955 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Guo XG, Ji TX, Xia Y & Ma YY Autophagy protects type II alveolar epithelial cells from Mycobacterium tuberculosis infection. Biochem. Biophys. Res. Commun 432, 308–313 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Fine-Coulson K, Giguère S, Quinn FD & Reaves BJ Infection of A549 human type II epithelial cells with Mycobacterium tuberculosis induces changes in mitochondrial morphology, distribution and mass that are dependent on the early secreted antigen, ESAT-6. Microbes Infect 17, 689–697 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Headley CA et al. Extracellular delivery of functional mitochondria rescues the dysfunction of CD4(+) T cells in aging. Adv. Sci. (Weinh) 2023. 10.1002/advs.202303664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arcos J et al. Lung mucosa lining fluid modifies Mycobacterium tuberculosis to reprogram human neutrophil killing mechanisms. J. Infect. Dis 212, 948–958 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moliva JI et al. Exposure to human alveolar lining fluid enhances Mycobacterium bovis BCG vaccine efficacy against Mycobacterium tuberculosis infection in a CD8(+) T-cell-dependent manner. Mucosal Immunol 11, 968–978 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arnett E et al. PPARgamma is critical for Mycobacterium tuberculosis induction of Mcl-1 and limitation of human macrophage apoptosis. PLOS Pathog 14, e1007100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salunke SB et al. Design and synthesis of novel anti-tuberculosis agents from the celecoxib pharmacophore. Bioorg. Med. Chem 23, 1935–1943 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Sukumar N, Tan S, Aldridge BB & Russell DG Exploitation of Mycobacterium tuberculosis reporter strains to probe the impact of vaccination at sites of infection. PLOS Pathog 10, e1004394 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leopold Wager CM et al. Activation of transcription factor CREB in human macrophages by Mycobacterium tuberculosis promotes bacterial survival, reduces NF-kB nuclear transit and limits phagolysosome fusion by reduced necroptotic signaling. PLOS Pathog 19, e1011297 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Olakanmi O, Britigan BE & Schlesinger LS Gallium disrupts iron metabolism of mycobacteria residing within human macrophages. Infect. Immun 68, 5619–5627 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kushida H Propylene oxide as a dehydrating agent for embedding with epoxy resins. J. Electron Microsc 10, 203–204 (1961). [Google Scholar]
- 61.Glauert AM Fixation, Dehydration and Embedding of Biological Specimens 1st edn. (Elsevier Science, Amsterdam, 1975). [Google Scholar]
- 62.Clemens DL Characterization of the Mycobacterium tuberculosis phagosome. Trends Microbiol 4, 113–118 (1996). [DOI] [PubMed] [Google Scholar]
- 63.Clemens DL, Lee BY & Horwitz MA Deviant expression of Rab5 on phagosomes containing the intracellular pathogens Mycobacterium tuberculosis and Legionella pneumophila is associated with altered phagosomal fate. Infect. Immun 68, 2671–2684 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clemens DL, Lee BY & Horwitz MA Mycobacterium tuberculosis and Legionella pneumophila phagosomes exhibit arrested maturation despite acquisition of Rab7. Infect. Immun 68, 5154–5166 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions of the paper are present in this manuscript or the Supplementary Material.