

Abstract
The symbiotic interaction of plants with arbuscular mycorrhizal (AM) fungi is ancient and widespread. Plants provide AM fungi with carbon in exchange for nutrients and water, making this interaction a prime target for crop improvement. However, plant–fungal interactions are restricted to a small subset of root cells, precluding the application of most conventional functional genomic techniques to study the molecular bases of these interactions. Here we used single-nucleus and spatial RNA sequencing to explore both Medicago truncatula and Rhizophagus irregularis transcriptomes in AM symbiosis at cellular and spatial resolution. Integrated, spatially registered single-cell maps revealed infected and uninfected plant root cell types. We observed that cortex cells exhibit distinct transcriptome profiles during different stages of colonization by AM fungi, indicating dynamic interplay between both organisms during establishment of the cellular interface enabling successful symbiosis. Our study provides insight into a symbiotic relationship of major agricultural and environmental importance and demonstrates a paradigm combining single-cell and spatial transcriptomics for the analysis of complex organismal interactions.
Subject terms: Arbuscular mycorrhiza, Transcriptomics, RNA sequencing
Using a combination of single-cell and spatial transcriptomic analysis, Serrano et al. uncover transcriptional dynamics during the establishment of arbuscular mycorrhizal symbiosis.
Main
Arbuscular mycorrhizal (AM) fungi occur in all major terrestrial ecosystems1. They are fundamental to agricultural production as they provide plants with nutrients, particularly non-renewable phosphorus, as well as resistance to abiotic stress2 and pathogens3. Plants reward these services by transferring carbohydrates and lipids to AM fungi, which enables extension of extraradical mycelium in the soil4.
Intense coordination between species is required for symbiotic recruitment, development and maintenance within the root. Preceding contact, signalling between plant roots and germinating fungal spores initiates hyphal branching towards the root at which a hyphopodium will form to initiate the symbiosis5. Upon contact, a plant-derived pre-penetration apparatus guides the fungus across and through the epidermis, with hyphae travelling both inter- and intra-cellularly to reach the inner-most cortical cells6,7. Hyphae then differentiate to form branched structures termed arbuscules within cortical cells8. The host plant restructures the cortex cell and builds a peri-arbuscular membrane (PAM)4, creating an apoplastic space across which metabolite exchange occurs between species. As symbiosis develops, the carbon supply from the root allows for expansion of an intraradical and extraradical mycelium, through which soil minerals are transported into the plant5.
Decades of research resulted in much progress regarding functional characterization of genes involved in the AM symbiosis9–17, yet many remain to be characterized. Multiple characteristics of AM symbiosis complicate traditional transcriptomic approaches. As obligate biotrophs, AM fungi cannot complete their life cycles or be cultured independently in asymbiotic conditions. Recent advances in asymbiotic sporulation of mycorrhizal fungi using bacterial fatty acids as stimuli are encouraging18,19, but it remains challenging to develop axenic AM inoculums. Due to asynchronous colonization, many developmental stages exist simultaneously within the cortex. This limits the ability of whole-root transcriptomics to differentiate between transcriptional profiles of each stage20. Arbuscules are extremely transient structures, lasting only a few days before senescence21,22, confounding efforts to distinguish discrete phases of interaction. Furthermore, arbuscule collapse and vesicle or spore formation indicates that the plant and fungus are assimilating exchanged nutrients, which creates the need to analyse root cells that appear to be non-colonized23. Several groups have elegantly addressed these challenges using laser-capture microdissection (LCM) to obtain transcriptomes of cortex cells visually confirmed to be directly adjacent to fungal appressoria (early stage) and colonized cortex cells (CCCs; late stage)24,25. One disadvantage of LCM is that it limits investigation to cell types already known to be involved, which creates the need for an unbiased approach to analyse all root cell types.
The rapid adoption of single-cell RNA sequencing (scRNA-seq) or single-nuclei RNA sequencing (snRNA-seq), with potential to identify novel cell types, model developmental trajectories and analyse transcriptional activity of individual cells26, has revolutionized plant biology. In both scRNA-seq and snRNA-seq, investigation of transcriptomes from all cell types is possible, rather than requiring manual selection of individual cells, as with LCM. Moreover, snRNA-seq’s rapid protocols are robust to diverse organisms and tissue types. In addition, certain cell types are preferentially released as protoplasts during enzymatic digestion for scRNA-seq, but nuclei are extracted uniformly across cell types, leading to a more representative population of cell types in snRNA-seq datasets27,28. For example, arbusculated cells have highly ramified cell membranes, which may be difficult to recover after enzymatic digestion due to their increased surface area. In both scRNA-seq and snRNA-seq, the spatial context of gene expression is lost upon dissociation of cells from the tissue. Spatial transcriptomics allows for sequencing of cell transcriptomes within the tissue context, adding a novel dimension to the data29.
In this Resource, we applied single-nuclei and spatial transcriptomics to the interaction between the model legume Medicago truncatula and the AM fungus Rhizophagus irregularis to create a two-dimensional integrated map of plant and fungal transcriptomes during symbiosis. We provide an unbiased spatial and single-nuclei transcriptomics dataset that profiled a multi-kingdom interaction. The spatially resolved transcriptome provides insight into coordinated gene expression occurring between the two partners across all major M. truncatula root cell types. This transcriptomic map represents a novel resource for AM fungi research and demonstrates the value of novel multi-omics approaches in answering biological questions.
Results
Nuclear RNA profiling identifies M. truncatula cell types
To gain a comprehensive transcriptional profile of the plant/AM fungal interaction, we performed snRNA-seq and spatial RNA-seq on M. truncatula roots, which were mock-inoculated or inoculated with the AM fungus R. irregularis. We isolated and purified nuclei from M. truncatula roots by fluorescence-activated nuclei sorting (FANS, Fig. 1a) before loading the suspension onto a microfluidic chip for snRNA-seq profiling30.
Fig. 1. snRNA profiling in M. truncatula roots colonized by R. irregularis.

a, An overview of the approach. M. truncatula root tissue is flash frozen for nuclei extraction and subsequent snRNA-seq using the 10x Genomics Chromium platform. Intact single nuclei are emulsified with gel beads containing barcoded oligonucleotides within a microfluidic chamber, resulting in a barcoded cDNA library after reverse transcription. b, The UMAP coordinates of 16,890 M. truncatula nuclei from three AM-colonized root harvest timepoints clustered by similarity in transcriptional profiles. The identities of 16 unique clusters are represented by different colours.
Quality filtering and unsupervised clustering resulted in a dataset of 16,890 nuclei grouped in 16 distinct cell clusters (Methods). We assigned cluster identities using cell type-specific gene expression profiles derived from A. thaliana root single-cell datasets31–35 as well as a rhizobia-colonized M. truncatula single-nuclei dataset36 (Supplementary Table 1 and Extended Data Fig. 1). Nine clusters (11,298 cells) exhibited characteristics of cortex cell identity, with one additional cluster (174 cells) composed of cortex cells colonized by R. irregularis (Fig. 1b). We also identified all other major M. truncatula root cell types and generated marker gene sets for each (Supplementary Table 1).
Extended Data Fig. 1. Select marker gene expression for single-nuclei clusters.
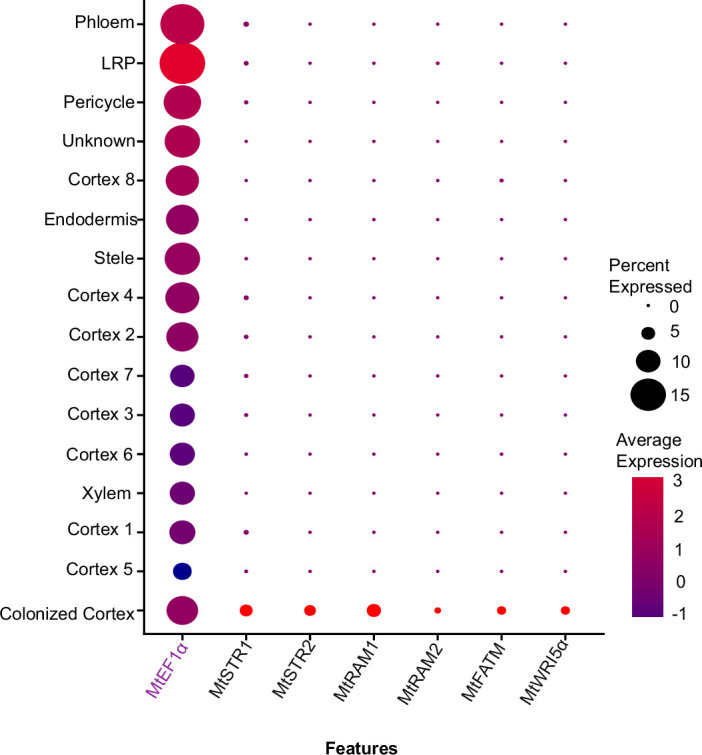
Dot-plot of expression profiles for select M. truncatula marker genes for unique root cell types across labeled cluster identities utilizing hierarchical clustering within the integrated single-nuclei datasets.
Simultaneous spatial capture of plant and fungal transcripts
To investigate gene expression from both symbiotic partners, we performed spatial transcriptomic profiling on inoculated and mock-inoculated M. truncatula roots at 28 dpi (Fig. 2a,b and Supplementary Table 2). An example capture area from an inoculated plant displays numerous root cross-sections fixed and stained on the glass surface (Fig. 2c, i). On average, inoculated capture areas resulted in 20,333 and 5,084 transcripts mapping to the M. truncatula37 and R. irregularis38 genomes, respectively, while mock-inoculated capture areas resulted in an average of 21,987 (M. truncatula) and 23 (R. irregularis) transcripts. Spatial unique molecular identifier (UMI; Fig. 2c, ii) and feature (Fig. 2c, iii) distributions indicate a uniform capture of transcripts across the expected areas of each cryosection, with few hotspots of fungal colonization associated with increased transcript counts from R. irregularis (Fig. 2c, iv) and increased expression of phosphate transporter 4 (MtPT4), a marker gene for arbusculated cells39 (Fig. 1d, v). The spatial technology relies on polyadenylated transcript capture via oligo(dT) primer sequences, which enabled unbiased capture of R. irregularis and M. truncatula transcripts simultaneously. Analysing the distribution of all M. truncatula transcripts within each inoculated capture area (Fig. 3a, i) and comparing it with that of all R. irregularis transcripts (Fig. 3a, ii) showed distinct patterns of messenger RNA capture and expression between the two species, indicating the successful spatially resolved capture of transcriptomes during symbiosis.
Fig. 2. Spatial transcriptomics enables simultaneous capture of M. truncatula and R. irregularis transcripts.

a, M. truncatula root tissue is flash frozen to create 16 µm thick cryosections, each containing numerous root cross-sections. Cryosections are fixed to capture areas, each of which is equipped with ~5,000 spatially barcoded voxels at a resolution of 55 µm. b, Side-by-side images of brightfield tissue image and underlying spatial capture voxels, with a close-up view of a single root cross-section within the capture area (one representative capture area out of nine mycorrhizal capture areas was analysed) highlighting voxel size in relation to the tissue. c, Capture area containing cross-sections from M. truncatula roots infected with R. irregularis at 28 dpi (one representative capture area out of nine mycorrhizal capture areas analysed). Image of root cross-sections within capture area (i). UMI count (ii) and feature count (iii) overlaid onto spots underlying tissue. Expression pattern of all R. irregularis transcripts captured (scale, log2 of UMI counts) (iv). Expression pattern of the arbuscule marker gene post-imputation (v), MtPT4, exhibiting overlap in spots with the highest expression of fungal transcripts (scale, log2 of UMI counts). Visualization done in Loupe Browser.
Fig. 3. Transcriptomic profiling reveals coordinated gene expression between the symbiotic partners.

a, The spatial distributions of host and symbiont transcript expression from four unique capture areas containing lateral root cross-sections from M. truncatula plants at 28 dpi (scale bar, 1 mm; scale, log2 of UMI counts). i, Four unique inoculated capture areas exhibiting the spatial distribution of all M. truncatula transcripts within the roots. ii, The same four capture areas exhibiting the spatial distribution of all R. irregularis transcripts within the roots. b, A dot plot of M. truncatula and R. irregularis housekeeping genes along with genes known to be involved in different stages of the symbiosis utilizing hierarchical clustering within the integrated mycorrhizal spatial object. c, A visualization of gene expression imputation from snRNA-seq mycorrhizal-integrated dataset for two lowly expressed genes, MtSKL1 and MtDMI, within a single representative Visium Spatial Gene Expression capture area out of nine mycorrhizal-treated capture areas analysed (scale bar, 1 mm).
Overlapping, symbiosis-responsive transcriptomes
To identify genes associated with AM fungi colonization within our spatial datasets, we performed dimensionality reduction and clustering of all voxel transcriptome profiles from both AM fungi- and mock-inoculated spatial capture areas (Extended Data Fig. 2). Due to the relatively low resolution of the Visium platform (each 55-μm voxel could contain one to five cells), we refrained from assigning cell identities to spatial dataset clusters (referred to as ‘spatial clusters’), as voxels probably represent heterogeneous cell groups. Instead, we identified voxel clusters within the mycorrhizal dataset that could represent sites of AM colonization. To do so, we compiled a list of established AM-responsive marker M. truncatula and R. irregularis genes (Supplementary Table 3) functioning in various stages of AM colonization and analysed their expression across the 13 spatial clusters in reference to a stably expressed housekeeping gene, elongation factor 1 alpha (MtEF1α)40. Spatial clusters 3 and 12 showed high specific expression of the markers from both species and thus were deemed ‘AM responsive’ (Fig. 3b), with spatial cluster 12 showing higher expression of early markers and spatial cluster 3 showing higher expression of late-stage markers. We detected a few AM symbiosis marker genes within the snRNA-seq dataset that were missing or lowly expressed within the spatial datasets. This may be due to lower detection efficiency of unbiased transcriptomic methods as compared with probe-based capture41. To estimate spatial expression of these genes, we integrated nuclear and spatial datasets together and imputed expression values across modalities. Using this approach, we associated the expression of two marker genes, does not make infection 1 (MtDMI1)42 and sickle 1 (MtSKl1)43, with mycorrhizal capture areas, despite their absence from the spatial dataset.
Extended Data Fig. 2. Spatial RNA-seq integrated UMAPs by treatment and dataset.
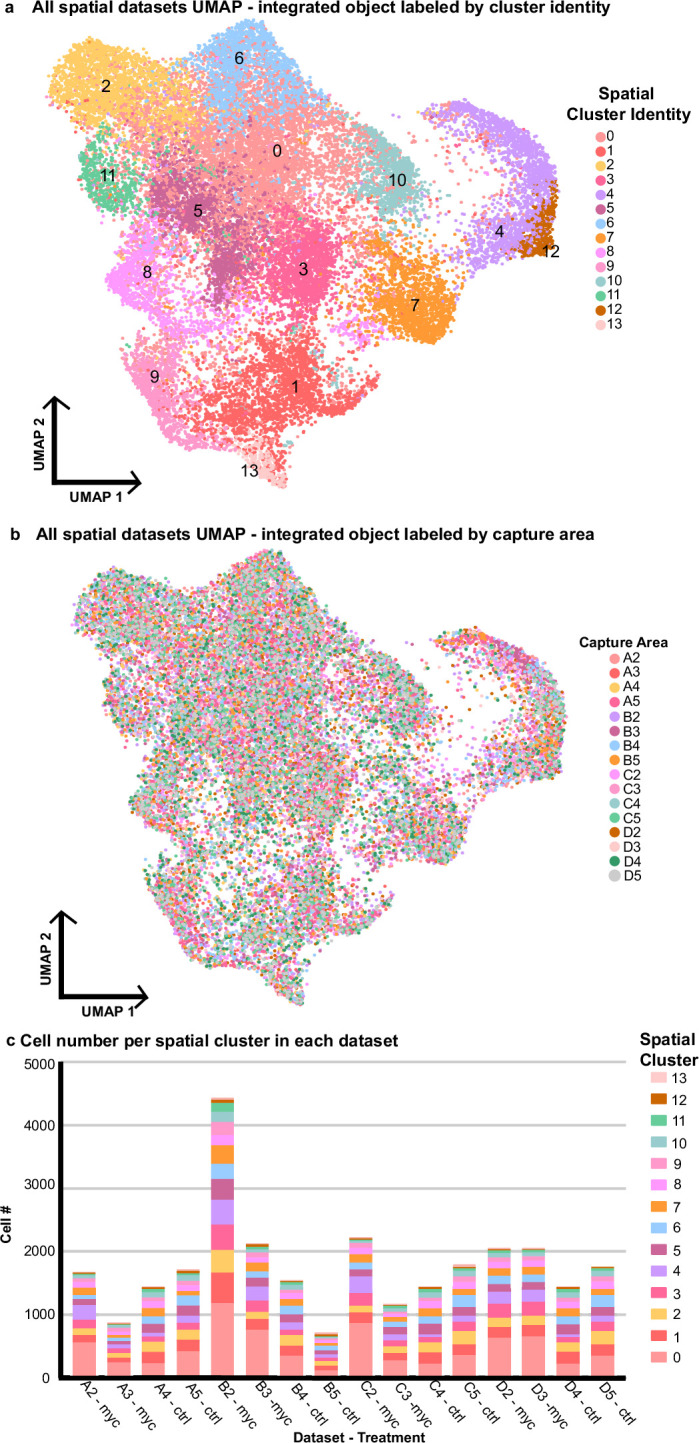
a, UMAP of 28,564 spatial voxels from the integrated Seurat object of all root harvests. The identities of 17 unique clusters are represented by different colors according to cluster identity. b, UMAP of 28,564 spatial voxels from the integrated Seurat object of all root harvests. Different colors correspond to the original dataset. c, Barplot exhibiting the number of cells that can be assigned to a specific cluster for each of the individual capture areas within the integrated spatial dataset.
Colonization stage-specific M. truncatula gene expression
We used stage-specific markers to determine the distribution of cells across arbuscule development within the snRNA-seq datasets. Low phosphate availability stimulates the interaction between plant and AM fungus, resulting in secretion of strigolactones from cortex cells44. ABCG transporter 59 (MtABCG59) is upregulated during phosphate starvation and upon mycorrhizal exposure45. We observed enrichment of MtABCG59 in cells throughout the cortex clusters (Fig. 4a), representing cells that are responding to phosphate starvation. Cluster 14, which specifically expressed 1-deoxy-d-xylulose 5-phosphate synthase (MtDXS2) transcripts, probably represents cells undergoing active AM symbiosis, as MtDXS2 is required for the methyl d-erythritol phosphate pathway-based isoprenoid production to sustain AM colonization (Fig. 4a)46. To determine whether we captured a developmental gradient, we first defined an expression module of AM symbiosis marker genes expressed in the AM symbiosis cluster (cluster 14), and then we computed an AM module score for all cells (Methods). We selected cells in the 98th percentile for this AM module score and re-clustered them into five subclusters (Fig. 4b). Based on the enrichment of marker genes observed, subclusters a, b and e probably represent earlier stages, as these cells are enriched for MtABCG59 transcripts. Clusters c and d may represent later stages of colonization based on the enrichment of MtDXS2 transcripts, and the cells at the edge of cluster d are probably at the most advanced stage of colonization, as they are the only cells in the single-cell datasets that contain high levels of MtPT4 mRNA (Fig. 4b).
Fig. 4. Analysing colonization stage-specific gene expression.

a, The expression of canonical pre- and post-AM marker genes shown in a UMAP plot of single-nuclei dataset, coloured by normalized mRNA counts (scale, log(UMI counts + 1)). b, Module scores based on average expression level of a list of known AM marker genes; cells with module scores in the 98th percentile were selected as ‘CCCs’ and clustering was performed on this subset. Three AM marker genes known to be expressed at different colonization stages are expressed in different subclusters of the colonized cluster. c, Spatial feature plots of four AM marker genes known to be expressed at different colonization stages: MtPDR1, MtCCD1, MtPT4 and MtMYB1. Zoom in red blocks focuses on voxels that switch expression profiles.
We also visualized the spatial dynamics of colonization by tracking the distribution of stage-specific AM symbiosis marker genes. By analysing expression of MtABCG59, carotenoid cleavage dioxygenase 1 (MtCCD1)47, MtPT4 and MYB-like transcription factor 1 (MtMYB1)48, we classified voxels within distinct stages of colonization (Fig. 4c). We repeated this stage-specific analysis with the snRNA-seq data to show the developmental trajectory of AM colonization starting with the phosphate stress response gene pleiotropic drug resistance 1 (MtPDR1)49, which is enriched throughout the cortex cluster. Calcium and Ca2+/calmodulin-dependent protein kinase (MtCCaMK)50 is involved in host–symbiont signalling in early stages and is enriched in the cells closer to the colonized cluster. MtPT4 is enriched in a subset of cells at the furthest edge of the colonized cluster and MtMYB1 is also enriched in these distal cells (Fig. 5c, upper panel).
Fig. 5. Existing and novel transcriptomic studies reveal a robust set of differentially expressed M. truncatula genes during the AM symbiosis.

a, A Venn diagram showing overlap in symbiosis-responsive M. truncatula genes with a log fold change > or <1 between the spatial dataset from this study and the two previously published LCM RNA-seq studies from Gaude et al.15 and Hogekamp et al.14. b, The breakdown of predicted cell types represented in each cluster within the integrated spatial dataset in terms of number of voxels. DEGs between mycorrhizal and control treatments are shown above each bar for each cluster, with the counts of significantly (Sig; see Methods) upregulated M. truncatula genes in red and downregulated gene counts in blue. c, The expression of four canonical pre- and post-AM symbiosis marker genes shown in a UMAP plot of single-nuclei dataset (upper). The expression of four genes enriched in the CCC cluster of this dataset that have yet unknown roles in AMF colonization, shown in a UMAP plot of single-nuclei dataset (lower). The UMAP plots are coloured by normalized mRNA counts (scale, log(UMI counts + 1)) depending on the marker gene, thus reflecting the colonization stage.
Performing differential gene expression analysis, we found 258 genes enriched in the AM symbiosis cluster 14 (log FC >0.25, adjusted P < 0.01, Supplementary Table 4) compared with all other cortex clusters combined. Among these, we recovered known marker genes for AM symbiosis, along with many genes not previously associated with AM signalling as well as genes with no annotated function. These included a gene encoding a monosaccharide transporting ATPase (Medtr8g006790/MtrunA17_Chr8g0335291), which we speculate the plant uses to provide sugars to the fungus, several genes with leucine-rich repeat domains (Medtr6g037750/ MtrunA17_Chr6g0464631 and Medtr3g058840/MtrunA17_Chr3g0102261), which could mediate host–symbiont signalling, and MtABC19, which encodes a xenobiotic-transporting ATPase (Medtr3g093430/ MtrunA17_Chr3g0128391) (Fig. 5c, lower panel). Lastly, we observed high expression of several lipid transfer M. truncatula genes within this cluster (Extended Data Fig. 3).
Extended Data Fig. 3. Lipid transfer gene expression for single-nuclei clusters.

Dot-plot of expression profiles for M. truncatula genes involved in lipid transfer to the fungal symbiont across labeled cluster identities utilizing hierarchical clustering within the integrated single-nuclei datasets.
A robust set of symbiosis-responsive M. truncatula genes
Differentially expressed genes (DEGs) between mycorrhizal and control spatial capture areas revealed 2,383 AM-responsive M. truncatula transcripts (Fig. 5a). Of these, 1,464 were upregulated (log2 FC >1.0) and 919 were downregulated (log2 FC <1.0) in the mycorrhizal treatment (Supplementary Table 5). Two LCM-based transcriptomic analyses revealed similar numbers of DEGs in response to mycorrhizal treatment24,25, with 188 genes significantly upregulated across all three datasets, which we refer to as ‘robust’ AM-responsive genes (Fig. 5a and Supplementary Table 5). No genes were found to be significantly downregulated in all three datasets. This set of robust AM-responsive genes includes characterized AM symbiosis marker genes, such as MtMYB1, MtPT4 and MtRAD1 (a positive GRAS transcription regulator of the symbiosis)51. One of the upregulated transcripts cyclin-like 1 (MtCYC1) encodes a putative cyclin-like F-box protein and is a known marker for cell division52, suggesting induction of cortical cell division as a response to AM colonization. However, many of the 188 genes remain to be investigated for their role in AM symbiosis.
The 55-μm resolution of the Visium Spatial Gene Expression platform results in blending of several adjacent cells together into a single profile (that is, it cannot resolve transcripts into single cells). However, we utilized our annotated snRNA-seq and spatial datasets to predict the proportions of each cell type represented in each spatial cluster (Fig. 5b). Differences were observed in the proportion of cell types between spatial clusters. We saw a relatively high proportion of voxels identifying as lateral root primordia, which we hypothesize as resulting from the high amount of mRNA captured from meristematic root tissue on the capture area. We also observed large differences in the amount of DEGs between the individual spatial clusters, indicating the method can discern between cell populations experiencing different degrees of AM colonization.
Functional enrichment depicts a marked symbiotic response
We performed functional enrichment analysis for Gene Ontology terms, including biological process (Extended Data Fig. 4a), molecular function (Extended Data Fig. 4b) and cellular component (Extended Data Fig. 4c) among significantly upregulated M. truncatula genes (I, Supplementary Tables 6 and 7) and the robust gene set (II, Supplementary Tables 8 and 7). As expected, we saw a >20-fold enrichment for the ‘arbuscular mycorrhizal association’ biological process term overall, and >80-fold enrichment for this term within the robust set. We also observed a high enrichment (>100-fold) of the ‘response to symbiotic bacterium’ term within the robust set, indicative of genes common to both fungal and bacterial (rhizobial) symbioses. During rhizobial symbiosis, legumes allow controlled infection by rhizobia, leading to the development of root nodules in which rhizobia directly fix and transfer nitrogen to their hosts. The rhizobial symbiosis coopted numerous components of the ancestral AM symbiosis signalling mechanism53 and this ‘common symbiotic signalling pathway’ is represented within our robust dataset by the differential expression of two co-expressed interacting genes, MtVAPYRIN (MtVPY)54 and MtEXOCYST70 (MtEXO70i)55. These genes function during the intracellular phases of endosymbiosis in both AM and rhizobial symbioses, with MtVPY genetic mutants impaired in arbuscule and infection thread development, respectively56. In the AM symbiosis, MtVPY interacts with MtEXO70i, which is critical to PAM development and arbuscule branching57. Another robust gene, cysteine protease 3 (MtCP3), probably plays a role in arbuscule degeneration, functioning to degrade the PAM48, and also contributes to nodule senescence58, indicating a common functionality. Interestingly, the biological process ‘proline catabolic process to glutamate’ and the molecular function ‘proline dehydrogenase’ showed a >20-fold enrichment (Extended Data Fig. 4a,b). Several studies noted altered proline levels under drought stress in AM-treated plants59,60, and hypothesized that proline confers protection from changes in water availability. As we did not apply a drought stress to plants in our survey, we believe proline metabolism may contribute to a different protective mechanism.
Extended Data Fig. 4. Gene-set enrichment analysis reveals AM-specific gene functionalities within the mycorrhizal spatial datasets and robust gene list.

Gene ontology enrichment scores for the main biological processes (a), molecular functions (b), and cellular components (c) represented within the significantly upregulated genes between the mycorrhizal and control treated i) spatial datasets and ii) robust gene list as compared to the reference genome.
AM fungi convert soil inorganic phosphate (Pi) into inorganic polyphosphate (polyP) and can rapidly accumulate and translocate polyP within hyphae61. AM fungi also depolymerize polyP via fungal endopolyphosphatases and transfer this phosphorus into host plant cells across the PAM62, although the mechanism for this export remains unclear. Some evidence suggests that the majority of this export occurs via the transport of Pi across the apoplastic space and subsequent uptake by the plant via Pi transporters62. However, a growing amount of evidence suggests polyP may be directly exported to the apoplastic space and then hydrolysed by the plant itself61. Nguyen and Saito provided evidence that fungal-derived polyP and plant-derived phosphatases had opposing localizations in mature arbuscules, indicating that plant phosphatase activity could account for assimilation of fungal-derived polyP63. Surprisingly, ‘exopolyphosphatase activity’ exhibited the highest enrichment of all molecular functions, (>30-fold) in our dataset. This adds support to the hypothesis that polyphosphatase activity by the plant plays a larger role in phosphorus export than previously anticipated.
Lastly, we expected that cellular components enriched in our datasets would include those involved in AM symbiosis, such as the PAM and the plant plasma membrane. The robust dataset showed a >100-fold enrichment for the ‘PAM’ category and captured 100% of the M. truncatula genes assigned to that functional category within the genome (Extended Data Fig. 4c). Overall, functional enrichment analysis confirmed a strong symbiotic signature within our mycorrhizal dataset.
Novel symbiosis-responsive R. irregularis gene expression
Bulk64–68 and LCM-based single-cell24,25 RNA-seq studies conducted on M. truncatula in symbiosis with R. irregularis utilized a broad spectrum of cell isolation and transcriptomic techniques. In addition, considerable progress has recently been made68–73 to build our knowledge of the genetic landscape of AM fungi and fungal gene expression occurring within this interaction. However, simultaneous capture of plant and fungal mRNA during symbiosis remains challenging. Our approach built upon existing research by providing the first spatially resolved dataset of simultaneously captured plant and fungal transcriptomes during the AM symbiosis. We detected expression of 12,104 unique fungal transcripts across nine mycorrhizal capture areas (Supplementary Table 9). Fungal gene expression distribution across the capture areas overlaps arbuscules in the tissue (Fig. 6a). Voxels spanning root cross-sections that display a high degree of arbusculation also exhibit high expression of total R. irregularis transcripts. AM fungi provide their hosts with hard-to-access soil nutrients through the actions of transporters across the PAM74 and benefit from the continuous transfer of lipids and sugars from host plant to fungus75. We observed the localized expression of five phosphate transporters (PT1[RIR_1575600/g11592], PT2[RIR_1235500/g7615], PT4[RIR_0355700/g31083], PT5[RIR_3213400/g18438] and PT7[RIR_2900800/g19437]), three ammonium transporters (AMT1[RIR_0149600/g16666], AMT2[RIR_0697800/g1222] and AMT3[RIR_0390200/g18142]), and two sugar transporters (ST2[RIR_2811400/g24501] and ST4[RIR_0496600/g26862]) to spatial cluster 3, the cluster identified as symbiosis-responsive via localization of M. truncatula AM symbiosis markers. We observed lower expression of transporter transcripts relative to that of RiEF1α, although we did observe expression of RiPT1, RiPT4, RiAMT1, RiAMT2 and RiST2. Spatial cluster 3 showed localized expression of M. truncatula transporters, such as MtPT4, MtAMT2 and MtSWEET1b76 (Fig. 3b), indicative of active nutrient transport occurring in both partners in these voxels.
Fig. 6. Spatially resolved R. irregularis transcripts reveal novel AM-specific gene expression patterns.

a, A single root cross-section within capture area (left) and overlapping distributions of all R. irregularis transcript expression (right) visualized in Loupe Browser. i, A representative root cross-section from a mycorrhizal capture area lacking recognizable fungal structures shows low expression of R. irregularis transcripts. ii, A representative root cross-section from a mycorrhizal capture area that contains visible arbuscules (red arrows) shows high expression of R. irregularis transcripts, particularly around arbuscules (scale bar, 250 µm; scale, log(UMI counts + 1)). Approximately 50 root cross-sections across nine mycorrhizal-treated capture areas were analysed. b, A dot plot of RiEF1α and various transporters utilizing hierarchical clustering within integrated mycorrhizal spatial object. c, A dot plot of RiEF1α and various effector proteins utilizing hierarchical clustering within the integrated mycorrhizal spatial object. d, A bar plot depicting top 25 Gene Ontology categories for all expressed R. irregularis transcripts (biological process (i), molecular function (ii) and cellular component (iii)).
The same analysis was performed for eight fungal effectors (Fig. 6c), including secreted protein 7—RIR_3212100/g18424 (RiSP7), an effector protein involved in biotrophic response to AM colonization77, secreted lysin motif—RIR_1359320 (RiSLM), an effector which reduces chitin-triggered immune responses during symbiosis78, SL-induced putative secreted protein 1—RIR_2427800/g2579 (RiSIS1), a secreted protein induced in both AM pre-symbiotic and symbiotic phases69, and nucleus localized effector 1—RIR_2535800/g7021 (RiNLE1), an effector upregulated in arbuscules involved in the suppression of defence responses79, as well as four members of the MycFOLD effector family (RiMycFOLD2—RIR_3103500/g17566, RiMycFOLD9—RIR_2782800/g17548, RiMycFOLD11—RIR_098400/g17317, and RiMycFOLD16—RIR_1383400/g25859)80. Most effectors exhibited specific expression in spatial cluster #3, though relative to RiEF1α, the expression of one of these effectors, RiNLE1, was extremely high (Fig. 6c). In fact, RiNLE1 represented the fifth most highly expressed fungal transcript. Evidence suggests that RiNLE1 translocates to the plant nucleus of arbusculated cells and interacts with the histone H2B protein to suppress defence responses via epigenetic modification of MtH2B (Medtr4g064020/MtrunA17_Chr4g0031671)79. Altogether, the high level of expression and high proportion of cells (60%) that RiNLE1 is expressed in within spatial cluster 3 further supports that this spatial cluster represents arbusculated cells and that other marker genes that specify this cluster are probably involved in AM symbiosis. Supplementary Table 10 describes all plant and fungal transcripts found to be expressed in spatial cluster 3.
Lastly, we identified at least one Gene Ontology classification for 8,559 out of the 12,104 expressed transcripts (Supplementary Table 11). The top biological processes represented include ‘signalling’, ‘transmembrane transport’ and ‘lipid metabolism’, consistent with processes carried out during AM symbiosis (Fig. 6d, i). Similarly, ‘transferase activity’, ‘transporter activity’ and ‘lipid binding’ rank highly among molecular function terms (Fig. 6d, ii). ‘Membrane ‘and ‘nucleus’ were well represented among cellular components, with >1,000 and >800 transcripts classified to each, respectively (Fig. 6d, iii). This dataset of >12,000 symbiosis-responsive spatially resolved R. irregularis genes and corresponding tissue images is the first of its kind and holds immense potential for in-depth characterization.
Discussion
Advances in single-cell transcriptomics transformed molecular genetics by allowing cell type-specific analysis and, more recently, conservation of the tissue context81. Here we combined single-nucleus and spatial RNA-seq to construct the first high-resolution spatially resolved integrated map of a multi-kingdom symbiotic interaction. We successfully adapted the spatial transcriptomics platform for use with plant roots and utilized transcriptome-wide mRNA capture from two species simultaneously. In addition, we identified cell type-specific responses to the AM symbiosis and integrated data from both approaches to discover novel AM-responsive transcripts.
snRNA-seq increases resolution and throughput for the identification of cell-state responses to external treatments and eliminates the hurdle of protoplasting cells82. snRNA-seq provides a potential advantage over scRNA-seq in conceptually allowing profiling of plant and fungal nuclei in the same assay. However, even in batches of nuclei not subjected to gated flow sorting, we failed to recover a substantial number of fungal transcripts and obtained no defined fungal nuclei, possibly because fungal nuclei were not captured or were destroyed by our plant nuclei extraction protocol. AM fungi consist of multinucleate hyphae in a large syncytium but differ from other multinucleated fungi due to the unusually high number of nuclei (up to 35,000) that exist within their cells83. This aspect of their biology may complicate interpretation of snRNA-seq data when compared with spatial or bulk RNA-seq.
Spatial transcriptomics allows for a side-by-side comparison of gene expression and tissue features and enabled the capture of fungal transcripts. Yet limitations exist for this technology as well, notably a super-cellular resolution (large inter-voxel distances that miss many cells and large voxel sizes that blend adjacent cells together). Probe-based capture technologies improve the resolution of spatial transcriptomics, but this limits analysis to a set of pre-defined genes, and may also lead to lower capture efficiency due to fewer primers84. There is a clear need for a high-resolution spatial technology that allows for unbiased mRNA capture from intact tissue.
We found 258 M. truncatula genes that were upregulated in the CCCs versus all other cortex cells of mycorrhizal-treated snRNA-seq datasets. These genes were enriched for Gene Ontology terms related to fungal symbiosis, terpene synthesis pathways and transport proteins. We also found 17 different cytochrome P450-like proteins, whose roles in plant–microbe interactions include hydroxylation of fatty acids and terpene synthesis85, and 19 leucine-rich repeat domain-containing proteins that are typically associated with pathogenesis but have been shown to also be upregulated in AM symbiosis85,86. Several xenobiotic, sugar and amino acid transporters were also upregulated in CCC. The identification of known and novel transcripts within the CCC cluster, along with the 188 genes upregulated among three distinct RNA-seq studies in the same symbiotic system, presents a community resource for characterization of novel AM-associated genes.
R. irregularis forms symbioses with many diverse plant species in natural and agricultural ecosystems87 and serves as a model species for the AM symbiosis. Despite the biological importance of this fungus, functional annotation and characterization of its genes has been slow due to the difficulty in its genetic manipulation88. Recent advances in CRISPR-Cas989 gene editing and continued efforts in creating pure cultures of AM fungi18,19 will hopefully bring forth a new chapter of AM symbiotic research that focuses on the fungal partner. Recent efforts in improving AM fungal genome assemblies90,91 and functional characterization of R. irregularis genes involved in symbiosis69,77–79,92,93 lay the groundwork for this shift. We identified spatial cluster 3 as AM-responsive based on the expression of both M. truncatula and R. irregularis marker genes for the AM symbiosis. The transcripts expressed within this cluster, as well as the thousands of R. irregularis genes expressed within the mycorrhizal capture areas in this study, represent excellent targets for functional characterization studies in both partners and it is our hope that the AM community can use these datasets as a resource to uncover new functionalities.
Methods
Plant growth and inoculation
Seeds of Medicago truncatula Gaertn, cv Jemalong A17 (Noble Foundation) were scarified in concentrated sulfuric acid for 5–10 min and rinsed with distilled water, sterilized in 3.75% sodium hypochlorite solution, then rinsed five times with sterile distilled water and placed on 1∕2 Murashige and Skoog medium, 1% agar plates at 4 °C or room temperature for 48 h. Sand cones were prepared as follows: 8.25-inch cone-tainers (Stuewe and Sons) with 1 cm3 rock wool at base, filled up to 12.7 cm with autoclaved calcined clay (Turface Athletics MVP 50), followed by 2.5 cm of autoclaved horticultural sand (American Soil and Stone) and topped with 2.5 cm fine play sand (SAKRETE). To inoculate seedlings with Rhizophagus irregularis (Błaszk., Wubet, Renker and Buscot) C. Walker and A. Schüßler, 50 ml Agtiv Field Crops liquid mycorrhizal inoculant (PremierTech) spores were captured on a 40-µm filter, rinsed with distilled water and resuspended in 50 ml distilled water. A total of 1 ml of resuspended spores was applied to the horticultural sand layer and an additional 300 µl were applied to the fine play sand layer. Germinated seedlings were transplanted to the top fine sand layer of inoculated and non-inoculated sand cones. Plants were grown in 22–24 °C with 16 h day/8 h night, with 300 μmol m−2 s−1 light intensity and 60% relative humidity. Plants were watered daily and fertilized twice a week with 1/2× Hoagland’s medium modified with 20 μM phosphate to stimulate AM colonization.
Colonization assessment
AM colonization in wild-type roots at 21, 28 or 38 dpi was visualized via staining of AM chitin using 2.5 μg ml−1 wheat germ agglutinin Alexa Fluor 488 (Thermo Fisher Scientific) in 1× phosphate-buffered saline (PBS) solution (pH 7.0). Briefly, roots collected from the fine sand layer were rinsed, fixed in 50% ethanol for 30 min and cleared in 10% KOH at 65 °C for 48 h. Cleared roots were neutralized with 0.1 M HCl and stained with wheat germ agglutinin 488 in 1× PBS at 4 °C for 24 h before imaging. Colonization was quantified using the Trouvelot method94 on a Leica DM6B fluorescence microscope using five biological replicates for each treatment (Extended Data Fig. 5a and Supplementary Table 12).
Extended Data Fig. 5. Root AM colonization analyses.

a, AM colonization quantified by the Trouvelot method at 28 dpi with R. irregularis in roots; Data are presented as mean values +/− SEM (n = 10). F%, frequency of infection; M%, total mycorrhization, A%, total arbuscule abundance; H%, total intraradical hyphae abundance, V%, total vesicle abundance. n represents the number of biologically independent samples. P values were determined using one-way analysis of variance (ANOVA) (* indicates p-value <0.05, ** p<0.10, ***.001). b, Expression of MtPT4 and RiTUB relative to MtEF1-a measured by qRT-PCR; Data are presented as mean values +/− SEM (n = 10). The box plots in a and b indicate the median (line inside the box), the lower and upper quartiles (box), margined by the smallest and largest data within the interval of 1.5x the interquartile range from the box (whiskers); outliers are shown (data points outside of box). n represents the number of biologically independent samples. P values were determined using one-way analysis of variance (ANOVA)(* indicates p-value <0.05, ** p<0.10, ***.001).
Quantitative real-time PCR of target genes
To quantify expression of target genes, 100 mg of roots from the fine sand layer were flash frozen in liquid nitrogen. Total RNA was extracted using the RNeasy Plant Mini Kit (Qiagen) and corresponding DNase. Complementary DNA synthesis was conducted using the SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific) from 500 ng of total RNA, and quantiative polymerase chain reaction (qPCR) was conducted from cDNA diluted 1:5 using the PowerUp SYBR Green Master Mix (Thermo Fisher Scientific). A 200 nM primer concentration and the following protocol were used for qPCR for all targets: 2 min at 50 °C and 2 min at 95 °C, followed by 39 repeats of 15 s at 95 °C, 15 s at 60 °C and 1 min at 72 °C, and ending with 5 s at 95 °C. A melting curve (55–95 °C; at increments of 0.5 °C) was generated to verify the specificity of primer amplification. Five biological replicates and three technical replicates of all targets (MtPT4 and RiTUB) were quantified for gene expression levels relative to the housekeeping gene MtEF1α using the ΔΔCT method (Extended Data Fig. 5b). All primer sequences used for qPCR can be found in Supplementary Table 13. Raw ΔΔCT values used for statistical analysis can be found in Supplementary Table 14.
Nuclei and bulk root tissue RNA profiling
M. truncatula roots from three plants from each treatment were harvested at 21, 28 and 38 days post-inoculation; 150 mg of roots grown in the inoculated fine sand layer was either flash frozen in liquid nitrogen, or nuclei extraction was performed up to the 20 µm filtration step and then flash frozen. RNA was extracted using the RNeasy Plant Mini Kit (Qiagen). Library preparation and sequencing were performed at the QB3 UC Berkeley Genomics Core Sequencing Facility.
Nuclei extraction and sequencing
M. truncatula roots from three plants per condition were harvested at 21, 28 or 38 dpi. A total of 150 mg of roots growing in inoculated fine sand layer was weighed out and placed in the lid of a petri dish and chopped rapidly with a razor blade for 3 min in 600 µl NIBAM (1x NIB, Sigma CELLYTPN1-1KT; 4% BSA; 1 mM DTT; 0.4 U µl−1 Superase RNAse inhibitor, Sigma; 1:100 Protease Inhibitor Cocktail for plant tissues, Sigma). NIBAM root slurry was strained through 40 µm and 20 µm filters (CellTrics). SYBR Green (1:10,000) was used to visualize nuclei during purification on the Influx Flow Cytometer. A total of 20,000 nuclei were sorted into 19 µl of ‘landing buffer’ (PBS with 0.4 U µl−1 Superase RNAse inhibitor) with a final volume of 43 µl. DAPI was applied to 2 µl of nuclei suspension to evaluate the quality of nuclei on a Leica AxioObserver at 20× magnification. The remaining 41 µl was mixed with 10× Genomics Chromium RT Master Mix with no additional water added and loaded onto a Chromium Chip G, and thereafter the standard manufacturer’s protocol was followed (V3.1 Dual Index). Twelve cycles were used for cDNA amplification, and the completed cDNA library was quantified using an Agilent Bioanalyzer. Sequencing was performed at the QB3 UC Berkeley Genomics Core Sequencing Facility on a single NovaSeq SP lane with the sequencing parameters 28 bp (read 1 length), 10 bp (index 1 length), 10 bp (index 2 length) and 90 bp (read 2 length), or at Novogene (Sacramento, CA) using the sequencing parameters 150 bp (read 1 length), 10 bp (index 1), 10 bp (index 2) and 150 bp (read 2 length).
Tissue preparation for spatial transcriptomics
Spatial transcriptomics was performed with the Visium Spatial Gene Expression platform from 10x Genomics. Harvested plant roots were rinsed with deionized H2O and cryopreserved in optimal cutting temperature compound via submerging of optimal cutting temperature-embedded molds into a dewar of isopentane chilled liquid nitrogen for even freezing. Cryomolds of roots were stored at −80 °C until cryosectioning. Cryosectioning was performed on an Epredia CryoStar NX70 Cryostat with a blade temperature of 14 °C and a sample head temperature of −12 °C with a section thickness of 16 °C. Cryosections were placed onto the surface of the chilled Visium Spatial Gene Expression slide and adhered to the slide using heat from the sectioner’s finger placed on the back surface of the capture area. Prepared slides were stored at 80 °C before processing for 10× Visium spatial transcriptome sequencing according to the manufacturer’s instructions with the following modifications: first, cryosections were stained using an incubation of 0.05% toluidine blue O in 1× PBS for 1 min and rinsed three times with 1× PBS. Second, a pre-permeabilization step was added as suggested by Giacomello et al.95. The pre-permeabilization mix for each slide (48 µl exonuclease I, 4.5 µl of bovine serum albumin and 428 µl of 2% PVP 40) was then prepared, and 70 µl was pipetted into each well. Pre-permeabilization occurred for 30 min at 37 °C after which the manufacturer’s protocol for tissue permeabilization was followed. Permeabilization enzyme (70 µl) was added to each capture area and incubation at 37 °C occurred for 12 min on the basis of the results of the manufacturer’s tissue optimization protocol (Extended Data Fig. 6).
Extended Data Fig. 6. Visium tissue optimization.

a, Brightfield images of eight capture areas on Visium Tissue Optimization slide containing 16 - thick M. truncatula lateral root cryo-sections after methanol fixation and staining with Toluidine Blue-O (scale bar = 1mm). Experiment was repeated twice, for a total of 16 capture areas. b, Corresponding cDNA footprint after fluorescently-tagged nucleotides were incorporated into reverse transcription reaction across 6 different permeabilization timepoints (scale bar = 1mm). Timepoint of 12 minutes was chosen for subsequent Visium Gene Expression workflows.
Data processing and analysis
Cellranger and Spaceranger software (10x Genomics) were used to preprocess single-nuclei and spatial transcriptomic sequencing libraries, respectively. A formatted reference genome was generated using Cellranger or Spaceranger’s ‘mkref’ function using the Medicago truncatula MedtrA17_4.0 (ref. 37) whole genome sequence and annotation and the Rhizophagus irregularis Rir_HGAP_ii_V2 (DAOM 181602, DAOM 197198)38 whole genome sequence and annotation using default parameters. Single-nuclei and spatial reads were aligned to the genome references using the ‘count’ function in Cellranger 7.0 and Spaceranger 1.3 software packages (10x Genomics), respectively. Brightfield tissue images were aligned to the spatial capture area fiducial frame and voxels corresponding to overlaying tissue were manually selected for all capture areas in Loupe Browser (10x Genomics). Data analysis for both the single-nuclei and spatial data was performed using the Seurat22 4.3.0 package in R 4.2.1 available at https://www.R-project.org.
Filtering and normalization
For both single-nuclei and spatial datasets, normalization and scaling were performed using the SCTransform R function in Seurat before clustering. Metrics used for filtering of the data during quality control steps can be found in Supplementary Table 2.
Principal components analysis and K-means clustering
Principal components analysis was performed on both snRNA-seq and spatial RNA-seq datasets using the RunPCA function in Seurat with the ‘SCT’ assay specified. The FindNeighbors function was applied to construct a shared nearest neighbour graph for the data using the first 30 principal components. Clustering was performed using the FindClusters function, which utilizes the shared nearest neighbour graph from the previous step. Finally, the RunUMAP function was utilized to construct the uniform manifold approximation and projection (UMAP) dimensionality reduction and visualize the dataset in two dimensions. Of the seven snRNA-seq datasets generated (Extended Data Fig. 7 and Supplementary Table 2), we selected five for further characterization, resulting in a final dataset of 16,890 nuclei with an average of 1,120 mRNA molecules per cell after quality filtering. The remaining two datasets were not included due to poor apparent colonization by R. irregularis. All spatial datasets were analysed.
Extended Data Fig. 7. Single-nuclei RNA-seq integrated UMAPs by treatment and dataset.
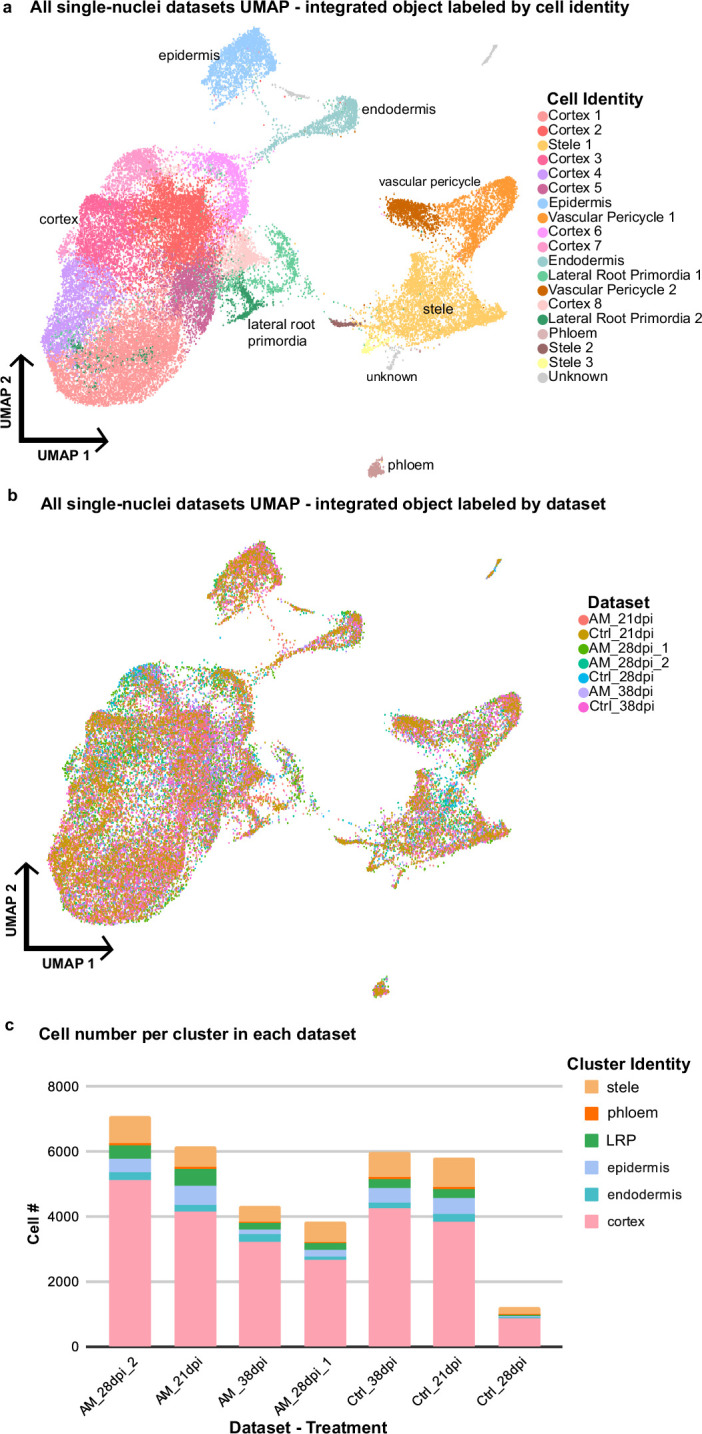
a, UMAP of 38,096 M. truncatula nuclei from the integrated Seurat object of all root harvests. Identities of 19 unique clusters are represented by different colors according to cell type. Clusters without a known identity are marked as ‘unknown’. b, UMAP of 38,096 M. truncatula nuclei from the integrated Seurat object of all root harvests. Different colors correspond to the original dataset. c, Barplot exhibiting the number of cells that can be assigned to a specific cell type identity for each of the individual datasets within the integrated single nuclei dataset.
Integration of replicate datasets
Replicate capture areas or samples from each treatment (mycorrhizal or mock-inoculated) were integrated into a two sets of Seurat objects (snRNA-seq, spatial) using the data integration pipeline in Seurat96. First, we applied the PrepSCTIntegration function using all transcripts for integration. We then identified a set of integration anchors with the FindIntegrationAnchors function. Finally, we applied the IntegrateData function. Principal component analysis and dimensionality reduction were performed on the integrated objects in the same manner as the individual objects with the following adjustments: (1) number of principal components, 30; (2) metric, cosine; and (3) resolution was set to 0.5 for clustering. UMAP plots for all datasets were created using the DimPlot() function and are displayed in Extended Data Fig. 7 (snRNA-seq) and Extended Data Fig. 2 (spatial).
Cluster identification for snRNA-seq
We determined that clusters 0, 1, 2, 4, 6, 8, 9, 14 and 16 are cortical cells based on enrichment of isoflavone synthase 1 (MtIFS1, Medtr4g088195) and isoflavone synthase 3 (MtIFS3, Medtr4g088160)97, as well as ATP-binding cassette transporter 59 (MtABCG59, Medtr3g107870)45, which encodes a strigolactone transporter that is expressed in cortical cells under phosphate-depleted conditions. Cluster 14 represents cortical cells that are colonized by AM fungi, based on a range of known marker genes, including ATP-binding cassette transporter (MtNOPE1, Medtr3g093270)98, two isoforms of ABCB for mycorrhization and nodulation (MtAMN2, Medtr4g081190 and MtAMN3, Medtr8g022270)99, 1-deoxy-d-xylulose 5-phosphate synthase (MtDXS2, Medtr8g068265)46, reduced arbuscular mycorrhiza 1 (MtRAM1, Medtr7g027190)48 and vapyrin (MtVPY, Medtr1g089180)100. We specifically focused on more mature roots, excluding meristematic or developing cells for most cell types as arbuscules do not form in these cell types. As a result, we did not observe evidence for developmental variation (trajectories) that are typically captured in single-cell studies of embryonic or meristematic tissues. Marker genes for quiescent centre and lateral root primordia, however, were used to identify cluster 12 as meristematic cells, including four homologues of plethora (MtPLT1-4, Medtr2g098180, Medtr4g065370, Medtr5g031880 and Medtr7g080460)101 and yucca (MtYUC8, Medtr7g099330). MtYUC8102 and MtPLT genes tend to be associated with nodule formation as well, but other genes that are upregulated by nodulation, such as nodule inception (MtNIN, Medtr5g099060)103, were either absent from our dataset or expressed at low levels and not specific to any cluster. Respiratory burst oxidase homologues (MtRboHF, Medtr7g060540)104, which is specifically expressed in root hairs, defined a small cluster adjacent to the LRP cluster as root hairs. The presence of scarecrow (MtSCR, Medtr7g074650)105 indicated that cluster 7 represents endodermal cells. Clusters 3, 5 and 11 were predicted to be vascular tissue, with several stele-specific A. thaliana homologues such as three homologues of transcription factor MYB domain protein (MtMYB071, Medtr5g014990, MtMYB113, Medtr2g096380, and MtMYB112, Medtr4g063100), as well as functionally characterized M. truncatula marker genes enriched in these clusters: three phosphate transporter homologues: (MtPHO1.1-1.3, Medt1g041695, Medtr1g075640, Medtr8g069955)48,106 and sugar transport protein 13 (MtSTP13, Medtr1g104780)25. Based on homologues from Arabidopsis marker genes, such as peroxidase 13 (MtPrx13, Medtr1g101830)36 and LOB-domain protein (MtLBD18, Medtr8g036085)36, cluster 17 represents xylem cells. FE/altered phloem development (MtFe, Medtr6g444980) and Arabidopsis phloem early DOF1 homologues (PEAR1, Medtr3g077750 and Medtr4g461080) were enriched in cluster 13, suggesting that these are phloem cells107. We defined cluster 15 as representing companion cells, as it is enriched for Arabidopsis phloem marker homologue super numeric nodules (MtSUNN, Medtr4g070970)108 and homologues of Arabidopsis companion cell markers Arabidopsis sucrose proton symporter 2 (AtSUC2, Medtr1g096910) and Arabidopsis FT interacting protein1 (AtFTIP1, Medtr0291s0010)109. Clusters 5 and 11 were enriched for MtPHO1.1-1.3 (Medtr1g041695, Medtr1g075640 and Medtr8g069955)106, suggesting that these are central cylinder/pericycle cells. Marker genes for all snRNA-seq clusters can be found in Supplementary Table 13 and a dot plot showing expression of markers for each cluster can be found in Extended Data Fig. 1.
Differential gene expression
Differentially expressed genes (upregulated and downregulated) between the mycorrhizal and control integrated datasets for all clusters were identified using the Likelihood Ratio Test from the DESeq2 (ref. 110) package with an adjusted P value of <0.05 and a log2 fold change threshold of −1.0 or 1.0.
Module score analysis
To determine which cells represented CCCs, we generated a list of genes (Supplementary Table 3) that are known to be involved in colonization, and used these as input to assign a module score to each cell using the Seurat AddModuleScore function. Cells with a score above the threshold of 98th percentile were selected as ‘colonized’ cells and subsetted to a new object for further subclustering analysis.
Gene expression imputation
Using the annotated snRNA-seq integrated object as a reference and the spatial integrated object as a query, we performed a data transfer using the UMI counts from the RNA assay within the single-nuclei object as the reference data and stored the new data under a new assay called ‘imputation’. We then were able to predict gene expression within the spatial dataset using the expression values from the snRNA-seq dataset by specifying the assay to ‘imputation’ during the analysis.
Voxel cell type proportion prediction in spatial RNA-seq
Using the annotated snRNA-seq integrated object as a reference and the spatial integrated object as a query, we performed a label transfer using the cell type annotations as the reference data. The query dataset is then projected onto the PCA of the reference dataset and the labels are predicted.
Comparison with previous datasets
We compared our dataset with two prior studies (Gaude et al. 2012 and Hogekamp et al. 2013)24,25 that improved our understanding of gene expression changes during the mycorrhizal symbiosis between M. truncatula and R. irregularis. We wanted to include these datasets in our analysis to identify a core set of DEGs between the three different RNA-seq techniques and be able to compare and contrast the various methods in spatial transcriptomics. One major hurdle to this comparison resulted from the use of the Affymetrix Medicago GeneChip array by these two studies leading to a difference in feature identifications (IDs). We constructed an ID converter (Supplementary Table 14) to convert between the Affymetrix GeneChip, MedtrA17_4.0 and the M. truncatula A17 r5.0 gene IDs for a certain locus in bulk fashion using data available at refs. 111,112. Genes identified as common between datasets can be found in Supplementary Table 5.
Gene Ontology and functional enrichment analysis
For M. truncatula, we conducted Gene Ontology and functional enrichment analyses utilizing the PANTHER Classification system (www.pantherdb.org)113,114. For R. irregularis, we conducted Gene Ontology analysis with the Blast2Go software115.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Legends for Supplementary Tables 1–15.
Supplementary Table 1 Marker genes for identifying cell types for snRNA-seq dataset. Supplementary Table 2 sNuc-RNA-seq and spatial RNA-seq data overview. Supplementary Table 3 List of AM marker genes used as an AM-specific expression module. Supplementary Table 4 Nuclei CCC markers. Supplementary Table 5 Multi-dataset DEGs comparison. Supplementary Table 6 Gene Ontology analysis results for M. truncatula DEGs found within the spatial datasets. Supplementary Table 7 Gene Ontology enrichment analysis for M. truncatula DEGs. Supplementary Table 8 Gene Ontology for robust M. truncatula genes. Supplementary Table 9 All expressed R. irregularis genes. Supplementary Table 10 Raw and normalized values for M. truncatula and R. irregularis genes expressed in spatial cluster 3. Supplementary Table 11 R. irrregularis Gene Ontology terms. Supplementary Table 12 Raw data for colonization scoring of AM fungal structures in colonization analysis. Supplementary Table 13 Primer sets used for reverse transcription qPCR in this study. Supplementary Table 14 Raw data for reverse transcription qPCR of AM symbiosis marker genes in colonization analysis. Supplementary Table 15 M. truncatula marker genes for all single-nuclei clusters. Supplementary Table 16 Matched IDs for the Mt4.0v1 genome, MtrunA17r5.0-ANR genome and the Medicago GeneChip Affymetrix IDs. Supplementary Table 17 R objects for single and multiple integrated datasets.
Acknowledgements
We acknowledge C. Gee, L. Washington, V. Vera, J. Dalton, M. Harrison, T. Tivey, B. Guillotin and R. Hunter for their advice, expertise and technical help. We also thank the QB3 UC Berkeley Genomics Core Sequencing Facility (QB3 Genomics, UC Berkeley, Berkeley, CA, RRID:SCR_022170) for their help in cryosectioning and sequencing. This study was performed at the DOE Joint BioEnergy Institute (http://www.jbei.org) and the DOE Joint Genome Institute (https://ror.org/04xm1d337) and was supported by the US Department of Energy, Office of Science, Biological and Environmental Research Program, through contract DE-AC02-05CH11231 with Lawrence Berkeley National Laboratory. This study was supported by a Laboratory Directed Research and Development award to B.C. at Lawrence Berkeley National Laboratory, and by an Early Career Research Program award to B.C. K.S. and H.V.S. were funded by The Novo Nordisk Foundation grant no. NNF19SA0059362 (InRoot).
Extended data
Author contributions
K.S., M.B., H.V.S. and B.C. planned experiments; K.S., M.B., T.D. and D.G. performed experiments; R.O., R.M. and A.V. provided consultation; K.S., M.B. and B.C. analysed the data; K.S., M.B., H.V.S. and B.C. wrote the manuscript.
Peer review
Peer review information
Nature Plants thanks Andrea Genre, Manuel Becana, Pierre-Marc Delaux and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
Raw feature and UMI counts for all datasets are displayed in Supplementary Table 2. Data availability on NCBI GEO (Gene Expression Omnibus) is available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE240107 (ref. 116). Genome assemblies for MedtrA17_4.0 (ref. 37) and the Rhizophagus irregularis Rir_HGAP_ii_V2 (DAOM 181602, DAOM 197198)38 were accessed at https://www.ebi.ac.uk/ena/browser/view/GCA_000219495.2 and https://www.ebi.ac.uk/ena/browser/view/GCA_002897155.1 (refs. 117,118), respectively.
Code availability
All scripts used for data analysis are available on GitHub at https://github.com/kserrano109/Medicago_Rhizophagus_RNAseq119 and archived at Zenodo. Information on all Seurat objects used within the data analysis for all datasets can be found in Supplementary Table 15.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Karen Serrano, Margaret Bezrutczyk.
Extended data
is available for this paper at 10.1038/s41477-024-01666-3.
Supplementary information
The online version contains supplementary material available at 10.1038/s41477-024-01666-3.
References
- 1.Chen M, Arato M, Borghi L, Nouri E, Reinhardt D. Beneficial services of arbuscular mycorrhizal fungi—from ecology to application. Front. Plant Sci. 2018;9:1270. doi: 10.3389/fpls.2018.01270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Begum N, et al. Role of arbuscular mycorrhizal fungi in plant growth regulation: implications in abiotic stress tolerance. Front. Plant Sci. 2019;10:1068. doi: 10.3389/fpls.2019.01068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jacott CN, Murray JD, Ridout CJ. Trade-offs in arbuscular mycorrhizal symbiosis: disease resistance, growth responses and perspectives for crop breeding. Agronomy. 2017;7:75. doi: 10.3390/agronomy7040075. [DOI] [Google Scholar]
- 4.Keymer A, et al. Lipid transfer from plants to arbuscular mycorrhiza fungi. eLife. 2017;6:e29107. doi: 10.7554/eLife.29107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacLean AM, Bravo A, Harrison MJ. Plant signaling and metabolic pathways enabling arbuscular mycorrhizal symbiosis. Plant Cell. 2017;29:2319–2335. doi: 10.1105/tpc.17.00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gutjahr C, Parniske M. Cell and developmental biology of arbuscular mycorrhiza symbiosis. Annu. Rev. Cell Dev. Biol. 2013;29:593–617. doi: 10.1146/annurev-cellbio-101512-122413. [DOI] [PubMed] [Google Scholar]
- 7.Lanfranco L, Bonfante P, Genre A. The mutualistic interaction between plants and arbuscular mycorrhizal fungi. Microbiol. Spectr. 2016;4:6. doi: 10.1128/microbiolspec.FUNK-0012-2016. [DOI] [PubMed] [Google Scholar]
- 8.Harrison MJ. Molecular and cellular aspects of the arbuscular mycorrhizal symbiosis. Annu. Rev. Plant Biol. 1999;50:361–389. doi: 10.1146/annurev.arplant.50.1.361. [DOI] [PubMed] [Google Scholar]
- 9.Wang E, et al. A H+-ATPase that energizes nutrient uptake during mycorrhizal symbioses in rice and Medicago truncatula. Plant Cell. 2014;26:1818–1830. doi: 10.1105/tpc.113.120527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang P, et al. Medicago SPX1 and SPX3 regulate phosphate homeostasis, mycorrhizal colonization, and arbuscule degradation. Plant Cell. 2021;33:3470–3486. doi: 10.1093/plcell/koab206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindsay PL, Williams BN, MacLean A, Harrison MJ. A phosphate-dependent requirement for transcription factors IPD3 and IPD3L during arbuscular mycorrhizal symbiosis in Medicago truncatula. Mol. Plant Microbe Interact. 2019;32:1277–1290. doi: 10.1094/MPMI-01-19-0006-R. [DOI] [PubMed] [Google Scholar]
- 12.Hartmann RM, et al. Insights into the complex role of GRAS transcription factors in the arbuscular mycorrhiza symbiosis. Sci. Rep. 2019;9:3360. doi: 10.1038/s41598-019-40214-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uhe M, Hogekamp C, Hartmann RM, Hohnjec N, Küster H. The mycorrhiza-dependent defensin MtDefMd1 of Medicago truncatula acts during the late restructuring stages of arbuscule-containing cells. PLoS One. 2018;13:e0191841. doi: 10.1371/journal.pone.0191841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gibelin‐Viala C, et al. The Medicago truncatula LysM receptor‐like kinase LYK9 plays a dual role in immunity and the arbuscular mycorrhizal symbiosis. N. Phytol. 2019;223:1516–1529. doi: 10.1111/nph.15891. [DOI] [PubMed] [Google Scholar]
- 15.Breuillin-Sessoms F, et al. Suppression of arbuscule degeneration in Medicago truncatula phosphate transporter4 mutants is dependent on the ammonium transporter 2 family protein AMT2;3. Plant Cell. 2015;27:1352–1366. doi: 10.1105/tpc.114.131144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang Y, et al. Medicago AP2-domain transcription factor WRI5a is a master regulator of lipid biosynthesis and transfer during mycorrhizal symbiosis. Mol. Plant. 2018;11:1344–1359. doi: 10.1016/j.molp.2018.09.006. [DOI] [PubMed] [Google Scholar]
- 17.Irving TB, et al. KIN3 impacts arbuscular mycorrhizal symbiosis and promotes fungal colonisation in Medicago truncatula. Plant J. 2022;110:513–528. doi: 10.1111/tpj.15685. [DOI] [PubMed] [Google Scholar]
- 18.Sugiura Y, et al. Myristate can be used as a carbon and energy source for the asymbiotic growth of arbuscular mycorrhizal fungi. Proc. Natl Acad. Sci. 2020;117:25779–25788. doi: 10.1073/pnas.2006948117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kameoka H, et al. Stimulation of asymbiotic sporulation in arbuscular mycorrhizal fungi by fatty acids. Nat. Microbiol. 2019;4:1654–1660. doi: 10.1038/s41564-019-0485-7. [DOI] [PubMed] [Google Scholar]
- 20.Limpens, E. in The Model Legume Medicago Truncatula (ed. de Bruijn, F.) 501–512 (John Wiley & Sons, 2020).
- 21.Harrison MJ. Signaling in the arbuscular mycorrhizal symbiosis. Annu Rev. Microbiol. 2005;59:19–42. doi: 10.1146/annurev.micro.58.030603.123749. [DOI] [PubMed] [Google Scholar]
- 22.Kobae Y, Fujiwara T. Earliest colonization events of Rhizophagus irregularis in rice roots occur preferentially in previously uncolonized cells. Plant Cell Physiol. 2014;55:1497–1510. doi: 10.1093/pcp/pcu081. [DOI] [PubMed] [Google Scholar]
- 23.Montero H, Choi J, Paszkowski U. Arbuscular mycorrhizal phenotyping: the dos and don’ts. N. Phytol. 2019;221:1182–1186. doi: 10.1111/nph.15489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hogekamp C, Küster H. A roadmap of cell-type specific gene expression during sequential stages of the arbuscular mycorrhiza symbiosis. BMC Genomics. 2013;14:306. doi: 10.1186/1471-2164-14-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaude N, Bortfeld S, Duensing N, Lohse M, Krajinski F. Arbuscule-containing and non-colonized cortical cells of mycorrhizal roots undergo extensive and specific reprogramming during arbuscular mycorrhizal development. Plant J. 2012;69:510–528. doi: 10.1111/j.1365-313X.2011.04810.x. [DOI] [PubMed] [Google Scholar]
- 26.Shaw R, Tian X, Xu J. Single-cell transcriptome analysis in plants: advances and challenges. Mol. Plant. 2021;14:115–126. doi: 10.1016/j.molp.2020.10.012. [DOI] [PubMed] [Google Scholar]
- 27.Giacomello S. A new era for plant science: spatial single-cell transcriptomics. Curr. Opin. Plant Biol. 2021;60:102041. doi: 10.1016/j.pbi.2021.102041. [DOI] [PubMed] [Google Scholar]
- 28.Guillotin B, et al. A pan-grass transcriptome reveals patterns of cellular divergence in crops. Nature. 2023;617:785–791. doi: 10.1038/s41586-023-06053-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larsson L, Frisén J, Lundeberg J. Spatially resolved transcriptomics adds a new dimension to genomics. Nat. Methods. 2021;18:15–18. doi: 10.1038/s41592-020-01038-7. [DOI] [PubMed] [Google Scholar]
- 30.Thibivilliers S, Anderson D, Libault M. Isolation of plant root nuclei for single cell RNA sequencing. Curr. Protoc. Plant Biol. 2020;5:e20120. doi: 10.1002/cppb.20120. [DOI] [PubMed] [Google Scholar]
- 31.Shahan R, et al. A single-cell Arabidopsis root atlas reveals developmental trajectories in wild-type and cell identity mutants. Dev. Cell. 2022;57:543–560.e9. doi: 10.1016/j.devcel.2022.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ryu KH, Huang L, Kang HM, Schiefelbein J. Single-cell RNA sequencing resolves molecular relationships among individual plant cells. Plant Physiol. 2019;179:1444–1456. doi: 10.1104/pp.18.01482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Denyer T, et al. Spatiotemporal developmental trajectories in the arabidopsis root revealed using high-throughput single-cell RNA sequencing. Dev. Cell. 2019;48:840–852.e5. doi: 10.1016/j.devcel.2019.02.022. [DOI] [PubMed] [Google Scholar]
- 34.Shulse CN, et al. High-throughput single-cell transcriptome profiling of plant cell types. Cell Rep. 2019;27:2241–2247.e4. doi: 10.1016/j.celrep.2019.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang T-Q, Xu Z-G, Shang G-D, Wang J-W. A single-cell RNA sequencing profiles the developmental landscape of Arabidopsis root. Mol. Plant. 2019;12:648–660. doi: 10.1016/j.molp.2019.04.004. [DOI] [PubMed] [Google Scholar]
- 36.Cervantes-Pérez SA, et al. Cell-specific pathways recruited for symbiotic nodulation in the Medicago truncatula legume. Mol. Plant. 2022;15:1868–1888. doi: 10.1016/j.molp.2022.10.021. [DOI] [PubMed] [Google Scholar]
- 37.Tang H, et al. An improved genome release (version Mt4.0) for the model legume Medicago truncatula. BMC Genomics. 2014;15:312. doi: 10.1186/1471-2164-15-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maeda T, et al. Evidence of non-tandemly repeated rDNAs and their intragenomic heterogeneity in Rhizophagus irregularis. Commun. Biol. 2018;1:87. doi: 10.1038/s42003-018-0094-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrison MJ, Dewbre GR, Liu J. A phosphate transporter from Medicago truncatula involved in the acquisition of phosphate released by arbuscular mycorrhizal fungi. Plant Cell. 2002;14:2413–2429. doi: 10.1105/tpc.004861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bravo A, York T, Pumplin N, Mueller LA, Harrison MJ. Genes conserved for arbuscular mycorrhizal symbiosis identified through phylogenomics. Nat. Plants. 2016;2:15208. doi: 10.1038/nplants.2015.208. [DOI] [PubMed] [Google Scholar]
- 41.Moses L, Pachter L. Museum of spatial transcriptomics. Nat. Methods. 2022;19:534–546. doi: 10.1038/s41592-022-01409-2. [DOI] [PubMed] [Google Scholar]
- 42.Ané J-M, et al. Medicago truncatula DMI1 required for bacterial and fungal symbioses in legumes. Science. 2004;303:1364–1367. doi: 10.1126/science.1092986. [DOI] [PubMed] [Google Scholar]
- 43.Varma Penmetsa R, et al. The Medicago truncatula ortholog of Arabidopsis EIN2, sickle, is a negative regulator of symbiotic and pathogenic microbial associations. Plant J. 2008;55:580–595. doi: 10.1111/j.1365-313X.2008.03531.x. [DOI] [PubMed] [Google Scholar]
- 44.Besserer A, et al. Strigolactones stimulate arbuscular mycorrhizal fungi by activating mitochondria. PLoS Biol. 2006;4:e226. doi: 10.1371/journal.pbio.0040226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banasiak J, Borghi L, Stec N, Martinoia E, Jasiński M. The full-size ABCG transporter of Medicago truncatula is involved in strigolactone secretion, affecting arbuscular mycorrhiza. Front. Plant Sci. 2020;11:18. doi: 10.3389/fpls.2020.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Floss DS, et al. Knock-down of the MEP pathway isogene 1-deoxy-d-xylulose 5-phosphate synthase 2 inhibits formation of arbuscular mycorrhiza-induced apocarotenoids, and abolishes normal expression of mycorrhiza-specific plant marker genes. Plant J. Cell Mol. Biol. 2008;56:86–100. doi: 10.1111/j.1365-313X.2008.03575.x. [DOI] [PubMed] [Google Scholar]
- 47.Floss DS, Schliemann W, Schmidt J, Strack D, Walter MH. RNA interference-mediated repression of MtCCD1 in mycorrhizal roots of Medicago truncatula causes accumulation of C27 apocarotenoids, shedding light on the functional role of CCD1. Plant Physiol. 2008;148:1267–1282. doi: 10.1104/pp.108.125062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Floss DS, et al. A transcriptional program for arbuscule degeneration during AM symbiosis is regulated by MYB1. Curr. Biol. CB. 2017;27:1206–1212. doi: 10.1016/j.cub.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 49.Kretzschmar T, et al. A petunia ABC protein controls strigolactone-dependent symbiotic signalling and branching. Nature. 2012;483:341–344. doi: 10.1038/nature10873. [DOI] [PubMed] [Google Scholar]
- 50.Singh S, Parniske M. Activation of calcium- and calmodulin-dependent protein kinase (CCaMK), the central regulator of plant root endosymbiosis. Curr. Opin. Plant Biol. 2012;15:444–453. doi: 10.1016/j.pbi.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 51.Rey T, et al. The Medicago truncatula GRAS protein RAD1 supports arbuscular mycorrhiza symbiosis and Phytophthora palmivora susceptibility. J. Exp. Bot. 2017;68:5871–5881. doi: 10.1093/jxb/erx398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Russo G, et al. Ectopic activation of cortical cell division during the accommodation of arbuscular mycorrhizal fungi. N. Phytol. 2019;221:1036–1048. doi: 10.1111/nph.15398. [DOI] [PubMed] [Google Scholar]
- 53.Chen C, Zhu H. Are common symbiosis genes required for endophytic rice–rhizobial interactions? Plant Signal. Behav. 2013;8:e25453. doi: 10.4161/psb.25453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pumplin N, et al. Medicago truncatula Vapyrin is a novel protein required for arbuscular mycorrhizal symbiosis. Plant J. 2010;61:482–494. doi: 10.1111/j.1365-313X.2009.04072.x. [DOI] [PubMed] [Google Scholar]
- 55.Zhang X, Pumplin N, Ivanov S, Harrison MJ. EXO70I is required for development of a sub-domain of the periarbuscular membrane during arbuscular mycorrhizal symbiosis. Curr. Biol. 2015;25:2189–2195. doi: 10.1016/j.cub.2015.06.075. [DOI] [PubMed] [Google Scholar]
- 56.Lindsay PL, Ivanov S, Pumplin N, Zhang X, Harrison MJ. Distinct ankyrin repeat subdomains control VAPYRIN locations and intracellular accommodation functions during arbuscular mycorrhizal symbiosis. Nat. Commun. 2022;13:5228. doi: 10.1038/s41467-022-32124-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heck C, et al. Symbiotic fungi control plant root cortex development through the novel GRAS transcription factor MIG1. Curr. Biol. 2016;26:2770–2778. doi: 10.1016/j.cub.2016.07.059. [DOI] [PubMed] [Google Scholar]
- 58.Van Wyk SG, Du Plessis M, Cullis CA, Kunert KJ, Vorster BJ. Cysteine protease and cystatin expression and activity during soybean nodule development and senescence. BMC Plant Biol. 2014;14:294. doi: 10.1186/s12870-014-0294-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zou Y-N, Wu Q-S, Huang Y-M, Ni Q-D, He X-H. Mycorrhizal-mediated lower proline accumulation in Poncirus trifoliata under water deficit derives from the integration of inhibition of proline synthesis with increase of proline degradation. PLoS One. 2013;8:e80568. doi: 10.1371/journal.pone.0080568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu H-H, Zou Y-N, Rahman MM, Ni Q-D, Wu Q-S. Mycorrhizas alter sucrose and proline metabolism in trifoliate orange exposed to drought stress. Sci. Rep. 2017;7:42389. doi: 10.1038/srep42389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ohtomo R, Saito M. Polyphosphate dynamics in mycorrhizal roots during colonization of an arbuscular mycorrhizal fungus. N. Phytol. 2005;167:571–578. doi: 10.1111/j.1469-8137.2005.01425.x. [DOI] [PubMed] [Google Scholar]
- 62.Ezawa T, Saito K. How do arbuscular mycorrhizal fungi handle phosphate? New insight into fine‐tuning of phosphate metabolism. N. Phytol. 2018;220:1116–1121. doi: 10.1111/nph.15187. [DOI] [PubMed] [Google Scholar]
- 63.Nguyen CT, Saito K. Role of cell wall polyphosphates in phosphorus transfer at the arbuscular interface in mycorrhizas. Front. Plant Sci. 2021;12:725939. doi: 10.3389/fpls.2021.725939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Camps C, et al. Combined genetic and transcriptomic analysis reveals three major signalling pathways activated by Myc‐LCOs in Medicago truncatula. N. Phytol. 2015;208:224–240. doi: 10.1111/nph.13427. [DOI] [PubMed] [Google Scholar]
- 65.Bonneau L, Huguet S, Wipf D, Pauly N, Truong H. Combined phosphate and nitrogen limitation generates a nutrient stress transcriptome favorable for arbuscular mycorrhizal symbiosis in Medicago truncatula. N. Phytol. 2013;199:188–202. doi: 10.1111/nph.12234. [DOI] [PubMed] [Google Scholar]
- 66.Hohnjec N, Vieweg MF, Pühler A, Becker A, Küster H. Overlaps in the transcriptional profiles of Medicago truncatula roots inoculated with two different glomus fungi provide insights into the genetic program activated during arbuscular mycorrhiza. Plant Physiol. 2005;137:1283–1301. doi: 10.1104/pp.104.056572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cope KR, et al. Physiological and transcriptomic response of Medicago truncatula to colonization by high- or low-benefit arbuscular mycorrhizal fungi. Mycorrhiza. 2022;32:281–303. doi: 10.1007/s00572-022-01077-2. [DOI] [PubMed] [Google Scholar]
- 68.Apelt F, et al. Shoot and root single cell sequencing reveals tissue- and daytime-specific transcriptome profiles. Plant Physiol. 2022;188:861–878. doi: 10.1093/plphys/kiab537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tsuzuki S, Handa Y, Takeda N, Kawaguchi M. Strigolactone-induced putative secreted protein 1 is required for the establishment of symbiosis by the arbuscular mycorrhizal fungus Rhizophagus irregularis. Mol. Plant-Microbe Interact.®. 2016;29:277–286. doi: 10.1094/MPMI-10-15-0234-R. [DOI] [PubMed] [Google Scholar]
- 70.Kamel L, et al. The comparison of expressed candidate secreted proteins from two arbuscular mycorrhizal fungi unravels common and specific molecular tools to invade different host plants. Front. Plant Sci. 2017;8:124. doi: 10.3389/fpls.2017.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dallaire A, et al. Transcriptional activity and epigenetic regulation of transposable elements in the symbiotic fungus Rhizophagus irregularis. Genome Res. 2021;31:2290–2302. doi: 10.1101/gr.275752.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zeng T, et al. Host‐ and stage‐dependent secretome of the arbuscular mycorrhizal fungus Rhizophagus irregularis. Plant J. 2018;94:411–425. doi: 10.1111/tpj.13908. [DOI] [PubMed] [Google Scholar]
- 73.Beaudet D, et al. Ultra-low input transcriptomics reveal the spore functional content and phylogenetic affiliations of poorly studied arbuscular mycorrhizal fungi. DNA Res. 2018;25:217–227. doi: 10.1093/dnares/dsx051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Banasiak J, Jamruszka T, Murray JD, Jasiński M. A roadmap of plant membrane transporters in arbuscular mycorrhizal and legume–rhizobium symbioses. Plant Physiol. 2021;187:2071–2091. doi: 10.1093/plphys/kiab280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang W, et al. Nutrient exchange and regulation in arbuscular mycorrhizal symbiosis. Mol. Plant. 2017;10:1147–1158. doi: 10.1016/j.molp.2017.07.012. [DOI] [PubMed] [Google Scholar]
- 76.An J, et al. A Medicago truncatula SWEET transporter implicated in arbuscule maintenance during arbuscular mycorrhizal symbiosis. N. Phytol. 2019;224:396–408. doi: 10.1111/nph.15975. [DOI] [PubMed] [Google Scholar]
- 77.Kloppholz S, Kuhn H, Requena N. A secreted fungal effector of glomus intraradices promotes symbiotic biotrophy. Curr. Biol. 2011;21:1204–1209. doi: 10.1016/j.cub.2011.06.044. [DOI] [PubMed] [Google Scholar]
- 78.Zeng T, et al. A lysin motif effector subverts chitin‐triggered immunity to facilitate arbuscular mycorrhizal symbiosis. N. Phytol. 2020;225:448–460. doi: 10.1111/nph.16245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang P, et al. A nuclear‐targeted effector of Rhizophagus irregularis interferes with histone 2B mono‐ubiquitination to promote arbuscular mycorrhization. N. Phytol. 2021;230:1142–1155. doi: 10.1111/nph.17236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Teulet A, et al. A pathogen effector FOLD diversified in symbiotic fungi. N. Phytol. 2023;239:1127–1139. doi: 10.1111/nph.18996. [DOI] [PubMed] [Google Scholar]
- 81.Marx V. Method of the year: spatially resolved transcriptomics. Nat. Methods. 2021;18:9–14. doi: 10.1038/s41592-020-01033-y. [DOI] [PubMed] [Google Scholar]
- 82.Oh J-M, et al. Comparison of cell type distribution between single-cell and single-nucleus RNA sequencing: enrichment of adherent cell types in single-nucleus RNA sequencing. Exp. Mol. Med. 2022;54:2128–2134. doi: 10.1038/s12276-022-00892-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kokkoris V, Stefani F, Dalpé Y, Dettman J, Corradi N. Nuclear dynamics in the arbuscular mycorrhizal fungi. Trends Plant Sci. 2020;25:765–778. doi: 10.1016/j.tplants.2020.05.002. [DOI] [PubMed] [Google Scholar]
- 84.Asp M, Bergenstråhle J, Lundeberg J. Spatially resolved transcriptomes—next generation tools for tissue exploration. BioEssays. 2020;42:1900221. doi: 10.1002/bies.201900221. [DOI] [PubMed] [Google Scholar]
- 85.Minerdi D, Savoi S, Sabbatini P. Role of cytochrome P450 enzyme in plant microorganisms’ communication: a focus on grapevine. Int. J. Mol. Sci. 2023;24:4695. doi: 10.3390/ijms24054695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ji L, Yang X, Qi F. Distinct responses to pathogenic and symbionic microorganisms: the role of plant immunity. Int. J. Mol. Sci. 2022;23:10427. doi: 10.3390/ijms231810427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Walker C, Schüßler A, Vincent B, Cranenbrouck S, Declerck S. Anchoring the species Rhizophagus intraradices (formerly Glomus intraradices). Fungal Syst. Evol. 2021;8:179–201. doi: 10.3114/fuse.2021.08.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Manley BF, et al. A highly contiguous genome assembly reveals sources of genomic novelty in the symbiotic fungus Rhizophagus irregularis. G3. 2023;13:jkad077. doi: 10.1093/g3journal/jkad077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Muñoz IV, Sarrocco S, Malfatti L, Baroncelli R, Vannacci G. CRISPR–Cas for fungal genome editing: a new tool for the management of plant diseases. Front. Plant Sci. 2019;10:135. doi: 10.3389/fpls.2019.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sperschneider J, et al. Arbuscular mycorrhizal fungi heterokaryons have two nuclear populations with distinct roles in host–plant interactions. Nat. Microbiol. 2023;8:2142–2153. doi: 10.1038/s41564-023-01495-8. [DOI] [PubMed] [Google Scholar]
- 91.Yildirir G, et al. Long reads and Hi‐C sequencing illuminate the two‐compartment genome of the model arbuscular mycorrhizal symbiont Rhizophagus irregularis. N. Phytol. 2022;233:1097–1107. doi: 10.1111/nph.17842. [DOI] [PubMed] [Google Scholar]
- 92.Voß S, Betz R, Heidt S, Corradi N, Requena N. RiCRN1, a crinkler effector from the arbuscular mycorrhizal fungus Rhizophagus irregularis, functions in arbuscule development. Front. Microbiol. 2018;9:2068. doi: 10.3389/fmicb.2018.02068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang S, et al. A transcriptional activator from Rhizophagus irregularis regulates phosphate uptake and homeostasis in AM symbiosis during phosphorous starvation. Front. Microbiol. 2023;13:1114089. doi: 10.3389/fmicb.2022.1114089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Trouvelot, A. Mesure du taux de mycorhization va d’un systeme radiculaire. Recherche de methodes d’estimation ayant une signification fonctionnelle. Physiological And genetical aspects of mycorrhizae. In Proc. 1st European Symposium on Mycorrhizae, 217–221 (Institut national de la recherche agronomique, 1986).
- 95.Giacomello S, Lundeberg J. Preparation of plant tissue to enable spatial transcriptomics profiling using barcoded microarrays. Nat. Protoc. 2018;13:2425–2446. doi: 10.1038/s41596-018-0046-1. [DOI] [PubMed] [Google Scholar]
- 96.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Biała W, Banasiak J, Jarzyniak K, Pawela A, Jasiński M. Medicago truncatula ABCG10 is a transporter of 4-coumarate and liquiritigenin in the medicarpin biosynthetic pathway. J. Exp. Bot. 2017;68:3231–3241. doi: 10.1093/jxb/erx059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nadal M, et al. An N-acetylglucosamine transporter required for arbuscular mycorrhizal symbioses in rice and maize. Nat. Plants. 2017;3:17073. doi: 10.1038/nplants.2017.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Roy S, et al. Three common symbiotic ABC subfamily B transporters in Medicago truncatula are regulated by a NIN-independent branch of the symbiosis signaling pathway. Mol. Plant Microbe Interact. 2021;34:939–951. doi: 10.1094/MPMI-02-21-0036-R. [DOI] [PubMed] [Google Scholar]
- 100.Murray JD, et al. Vapyrin, a gene essential for intracellular progression of arbuscular mycorrhizal symbiosis, is also essential for infection by rhizobia in the nodule symbiosis of Medicago truncatula. Plant J. Cell Mol. Biol. 2011;65:244–252. doi: 10.1111/j.1365-313X.2010.04415.x. [DOI] [PubMed] [Google Scholar]
- 101.Franssen HJ, et al. Root developmental programs shape the Medicago truncatula nodule meristem. Development. 2015;142:2941–2950. doi: 10.1242/dev.120774. [DOI] [PubMed] [Google Scholar]
- 102.Cao X, et al. The roles of auxin biosynthesis YUCCA gene family in plants. Int. J. Mol. Sci. 2019;20:6343. doi: 10.3390/ijms20246343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Marsh JF, et al. Medicago truncatula NIN is essential for rhizobial-independent nodule organogenesis induced by autoactive calcium/calmodulin-dependent protein kinase. Plant Physiol. 2007;144:324–335. doi: 10.1104/pp.106.093021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chapman J, Muhlemann J, Gayomba S, Muday G. RBOH-dependent ROS synthesis and ROS scavenging by plant specialized metabolites to modulate plant development and stress responses. Chem. Res. Toxicol. 2019;32:370–396. doi: 10.1021/acs.chemrestox.9b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dong W, et al. An SHR–SCR module specifies legume cortical cell fate to enable nodulation. Nature. 2021;589:586–590. doi: 10.1038/s41586-020-3016-z. [DOI] [PubMed] [Google Scholar]
- 106.Nguyen NNT, et al. PHO1 family members transport phosphate from infected nodule cells to bacteroids in Medicago truncatula. Plant Physiol. 2021;185:196–209. doi: 10.1093/plphys/kiaa016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Miyashima S, et al. Mobile PEAR transcription factors integrate positional cues to prime cambial growth. Nature. 2019;565:490–494. doi: 10.1038/s41586-018-0839-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Karlo M, et al. The CLE53–SUNN genetic pathway negatively regulates arbuscular mycorrhiza root colonization in Medicago truncatula. J. Exp. Bot. 2020;71:4972–4984. doi: 10.1093/jxb/eraa193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kim J-Y, et al. Distinct identities of leaf phloem cells revealed by single cell transcriptomics. Plant Cell. 2021;33:511–530. doi: 10.1093/plcell/koaa060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Medicago truncatula A17 r5.0 genome portal. INRAhttps://medicago.toulouse.inra.fr/MtrunA17r5.0-ANR (2018).
- 112.Pecrix Y, et al. Whole-genome landscape of Medicago truncatula symbiotic genes. Nat. Plants. 2018;4:1017–1025. doi: 10.1038/s41477-018-0286-7. [DOI] [PubMed] [Google Scholar]
- 113.Thomas PD, et al. PANTHER: making genome‐scale phylogenetics accessible to all. Protein Sci. 2022;31:8–22. doi: 10.1002/pro.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Conesa A, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 115.BLAST2GO. BioBamhttps://www.biobam.com/blast2go/ (2023).
- 116.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Assembly: GCA_000219495.2. ENAhttps://www.ebi.ac.uk/ena/browser/view/GCA_000219495.2 (2024).
- 118.Assembly: GCA_002897155.1. ENAhttps://www.ebi.ac.uk/ena/browser/view/GCA_002897155.1 (2024).
- 119.Medicago_Rhizophagus_RNAseq. GitHubhttps://github.com/kserrano109/Medicago_Rhizophagus_RNAseq(2023).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Legends for Supplementary Tables 1–15.
Supplementary Table 1 Marker genes for identifying cell types for snRNA-seq dataset. Supplementary Table 2 sNuc-RNA-seq and spatial RNA-seq data overview. Supplementary Table 3 List of AM marker genes used as an AM-specific expression module. Supplementary Table 4 Nuclei CCC markers. Supplementary Table 5 Multi-dataset DEGs comparison. Supplementary Table 6 Gene Ontology analysis results for M. truncatula DEGs found within the spatial datasets. Supplementary Table 7 Gene Ontology enrichment analysis for M. truncatula DEGs. Supplementary Table 8 Gene Ontology for robust M. truncatula genes. Supplementary Table 9 All expressed R. irregularis genes. Supplementary Table 10 Raw and normalized values for M. truncatula and R. irregularis genes expressed in spatial cluster 3. Supplementary Table 11 R. irrregularis Gene Ontology terms. Supplementary Table 12 Raw data for colonization scoring of AM fungal structures in colonization analysis. Supplementary Table 13 Primer sets used for reverse transcription qPCR in this study. Supplementary Table 14 Raw data for reverse transcription qPCR of AM symbiosis marker genes in colonization analysis. Supplementary Table 15 M. truncatula marker genes for all single-nuclei clusters. Supplementary Table 16 Matched IDs for the Mt4.0v1 genome, MtrunA17r5.0-ANR genome and the Medicago GeneChip Affymetrix IDs. Supplementary Table 17 R objects for single and multiple integrated datasets.
Data Availability Statement
Raw feature and UMI counts for all datasets are displayed in Supplementary Table 2. Data availability on NCBI GEO (Gene Expression Omnibus) is available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE240107 (ref. 116). Genome assemblies for MedtrA17_4.0 (ref. 37) and the Rhizophagus irregularis Rir_HGAP_ii_V2 (DAOM 181602, DAOM 197198)38 were accessed at https://www.ebi.ac.uk/ena/browser/view/GCA_000219495.2 and https://www.ebi.ac.uk/ena/browser/view/GCA_002897155.1 (refs. 117,118), respectively.
All scripts used for data analysis are available on GitHub at https://github.com/kserrano109/Medicago_Rhizophagus_RNAseq119 and archived at Zenodo. Information on all Seurat objects used within the data analysis for all datasets can be found in Supplementary Table 15.