

Abstract
The Cannabis sativa plant has been used for centuries as a recreational drug and more recently in the treatment of patients with neurological or psychiatric disorders. In many instances, treatment goals include relief from posttraumatic disorders, anxiety, or to support treatment of chronic pain. Ligands acting on cannabinoid receptor 1 (CB1R) are also potential targets for the treatment of other health conditions. Using an evidence-based approach, pharmacological investigation of CB1R agonists is timely, with the aim to provide chronically ill patients relief using well-defined and characterized compounds from cannabis. Hexahydrocannabinol (HHC), currently available over the counter in many countries to adults and even children, is of great interests to policy makers, legal administrators, and healthcare regulators, as well as pharmacologists. Herein, we studied the pharmacodynamics of HHC epimers, which activate CB1R. We compared their key CB1R-mediated signaling pathway activities and compared them to the pathways activated by Δ9-tetrahydrocannabinol (Δ9-THC). We provide evidence that activation of CB1R by HHC ligands is only broadly comparable to those mediated by Δ9-THC, and that both HHC epimers have unique properties. Together with the greater chemical stability of HHC compared to Δ9-THC, these molecules have a potential to become a part of modern medicine.
Subject terms: Drug regulation, Public health, Biochemistry, Cell biology, Chemical biology, Drug discovery, Molecular biology, Chemistry
Introduction
The Cannabis sativa plant has been cultivated for recreational and medical use for centuries1. Various psychotropic and therapeutic effects of cannabis have been attributed to the major constituents of the plant, Δ9-tetrahydrocannabinol (Δ9-THC) and cannabidiol (CBD), and these compounds have been extensively studied for medical applications. Cannabis plant extracts include many compounds in addition to Δ9-THC and CBD. Over 400 different compounds have been isolated from the plant, including ligands of cannabinoid receptors, terpenes, alkaloids, and flavonoids2,3. A growing interest in these compounds has resulted in a systematic exploration of the therapeutic potential of other cannabis or cannabinoid-derived compounds. These compounds often have unique chemical, pharmacodynamic, and pharmacokinetic properties, differing from those of Δ9-THC or CBD, which may lead to novel therapeutic uses. Current discussion about the effects and safety of hexahydrocannabinol (HHC) use mandates an examination of its pharmacodynamic properties, including its effect on cannabinoid receptor 1 (CB1R), as there are only limited data concerning its activity, potency, toxicity, and safety4,5.
Δ9-THC-related cannabinoids share the Δ9-THC’s overall structure but differ in the position of a double bond, number and/or orientation of methyl groups, degree of hydrogenation, and length of the side chain. Importantly, these modifications affect their stability and pharmacological characteristics. One “minor” cannabinoid, HHC, is found in trace amounts in the cannabis plant6. This cannabinoid can be easily produced from CBD, initially undergoing acid cyclization to form Δ9-THC, which is subsequently hydrogenated to produce HHC. Due to its straightforward preparation and general availability of CBD, HHC is being manufactured at large scales and widely abused as a novel cannabinoid7.
Synthesis of HHC produces two epimers: (9S)-HHC and (9R)-HHC, which differ in the orientation of the methyl group at atom 9 (Fig. 1A). The (9R)-HHC epimer has superior affinity to cannabinoid receptor 1 over (9S)-HHC8. Early reports comparing HHC and its epimers were compromised by low purity of the compounds9,10. A recent study that used purified HHC epimers showed that the effect of (9R)-HHC on mouse behavior is close to that of Δ9-THC, while (9S)-HHC lacks Δ9-THC-like effects11.
Figure 1.

The structures of the tested cannabinoids and the HHC synthesis scheme. (A) The structures of (9S)-HHC, (9R)-HHC, Δ9-THC, and WIN. (B) The synthesis of HHC from CBD, schematically via transformation of CBD into Δ8-THC and further reduction to obtain HHC epimers.
Cannabinoids activate the cannabinoid receptor family consisting of cannabinoid receptor 1 and cannabinoid receptor 2, peroxisome proliferator-activated receptors (PPAR) α and γ, transient receptor potential cation channel subfamily V member 1 (TRPV1), and orphan receptors GPR55 and GPR1812–16, among others. However, the key target of Δ9-THC in the brain is CB1R, a G protein-coupled receptor that is widely expressed in the brain, especially in the hypothalamus, hippocampus, nucleus accumbens, prefrontal cortex, cerebellum, and the emetic centers in the brainstem17. Neuron-wise, CB1R is located presynaptically, where it may inhibit excitatory or inhibitory synaptic transmission18.
CB1R activation principally results in the activation of Gi/o proteins, decreasing levels of cyclic adenosine triphosphate (cAMP) in the cell and regulating several other signaling pathways. The activity of the receptor is controlled by G protein-coupled receptor kinase 3 (GRK3)-dependent phosphorylation and subsequent binding of β-arrestin; this inhibition of the receptor’s ability to elicit a response is known as desensitization. Further, β-arrestin initiates internalization of the receptor and, at the same time, facilitates activation of signaling pathways such as ERK1/2 or JNK319,20.
Distinct outcomes of GPCR signaling that arise from different compounds activating unique networks of signaling pathways result in signaling bias. In signaling bias, each ligand preferentially activates a suite of particular signaling pathways. Known cannabinoid ligands can preferentially activate subsets of G protein- or β-arrestin-dependent signaling and have different receptor internalization efficacies when the receptors are expressed in heterologous systems21,22.
In the present study, we aimed to synthesize and purify (9S)-HHC and (9R)-HHC in larger amounts than previously accomplished, separate the epimers, and clarify their proportions in each procedure, thus allowing us to thoroughly explore the pharmacodynamics of the compounds at CB1R.
Methods
CB1R ligands
WIN 55,212-2 mesylate (WIN) was obtained from Tocris R&D (USA). Δ9-tetrahydrocannabinol (Δ9-THC) was synthesized as described previously23. Hexahydrocannabinol (HHC) was synthesized as described below.
Synthesis and purification of HHC
Commercially-available CBD isolate (CBDepot, Czech Republic), p-toluenesulphonic acid (P-Lab, Czech Republic), and 5% palladium on activated charcoal (Merck KGaA, Germany) were used for the reaction. Solvents were purchased from a local distributor (Lach-Ner, Czech Republic) and were used without further purification. Solvents were evaporated using a vacuum rotary evaporator. Argon (5N) was used as an inert gas, and hydrogen (3.5N) was used for the reduction. Polar silica 40–63 µm (Merck KGaA, Germany) was used for ∆8-THC purification. The Aldrich® Kugelrohr™ short-path distillation apparatus (Merck KGaA, Germany) was used for HHC vacuum distillation. HHC epimers were separated using COMBIFLASH RF200 UV/VIS (Teledyne ISCO, United States) and RediSep Gold® Silica Gel Disposable Flash Columns (Teledyne ISCO, United States). HPLC/UV spectra were measured using LC/MS Agilent Technologies, 1290 Infinity DAD. The ratios of HHC epimers were determined based on signal characteristics in 1H NMR spectra (δ 3.03 ppm for (9R)-HHC and δ 2.87 for (9S)-HHC).
The scale of the reaction ranged from 10 g of CBD up to 1 kg. CBD was dissolved in DCM to achieve a concentration of 50 g/L. For every gram of CBD, 0.5 g of p-toluenesulphonic was added. The mixture was flushed with argon and stirred for 48 h at room temperature. The reaction mixture was filtered through the silica column using 3 g of silica for every gram of CBD. The silica was washed with DCM until no more product was eluted. The solution of ∆8-THC in DCM was concentrated to 1/10 of its original volume. An equal amount of MeOH was added diluting the solution approximately two times and the solution was evaporated once again to half of its volume. This procedure was repeated until no DCM signal (δ 5.30 ppm) was present on 1H NMR. The resulting mixture of ∆8-THC and MeOH was used for the reduction without further purification.
The corresponding conditions are listed in the Table 1. Palladium on activated charcoal was added to the solution of ∆8-THC in MeOH. The reaction vessel was flushed with argon and then the argon was replaced by hydrogen. The mixture was stirred, and the pressure of hydrogen was maintained at around 1 atm. The mixture was filtered through celite and the celite was washed with MeOH until no more product was eluted. The MeOH was evaporated and the crude HHC was vacuum distilled using Kugelrohr™ (220 °C, 0.4 torr). A mixture of epimers (9R/S)-HHC (HPLC/UV purity 96%) was produced by this procedure. Samples of pure (9R)-HHC and (9S)-HHC were obtained from a 3:2 mixture (entry 2) using FLASH chromatography (hexane: t-BuOMe, 1–2%).
Table 1.
The synthesis of (9R)-HHC and (9S)-HHC at different scales.
| Entry | Synthesis scale (∆8-THC mass), g | Mass of 5% Pd/C per 1 g of ∆8-THC, mg | ∆8-THC concentration, g/L | Duration of the reduction, days | 9R:9S molar ratio |
|---|---|---|---|---|---|
| 1 | 10 | 50 | 200 | 2 | 3:1 |
| 2 | 10 | 20 | 50 | 2 | 3:2 |
| 3 | 250 | 50 | 200 | 5 | 3:1 |
| 4 | 1000 | 50 | 250 | 14 | 3:1 |
NMR characterization of HHC epimers
The NMR spectra were measured with Agilent 400 MR DDR2 (Agilent Technologies Inc., United States) using CDCl3 (Merck KGaA, Germany) as a solvent and referenced on residual CDCl3 signal (1H δ 7.26 ppm). The spectra of corresponding epimers were identical to NMR spectra published by Russo et al.11.
(9S)-HHC
1H NMR (400 MHz, CDCl3) δ 6.25 (d, J = 1.6 Hz, 1H), 6.07 (d, J = 1.6 Hz, 1H), 4.70 (s, 1H), 2.91–2.85 (m, 1H), 2.71–2.64 (m, 1H), 2.47–2.37 (m, 2H), 2.15–2.07 (m, 1H), 1.69–1.61 (m, 3H), 1.56 (p, J = 7.6 Hz, 2H), 1.51–1.44 (m, 1H), 1.36 (s, 3H), 1.35–1.27 (m, 6H), 1.13 (d, J = 7.3 Hz, 3H), 1.09 (s, 3H), 0.88 (t, J = 7.0 Hz, 3H).
(9R)-HHC
1H NMR (400 MHz, CDCl3) δ 6.25 (d, J = 1.7 Hz, 1H), 6.08 (d, J = 1.6 Hz, 1H), 4.69 (s, 1H), 3.06–3.00 (m, 1H), 2.49–2.38 (m, 3H), 1.88–1.81 (m, 2H), 1.68–1.59 (m, 1H), 1.58–1.50 (m, 2H), 1.49–1.38 (m, 1H), 1.37 (s, 3H), 1.35–1.24 (m, 4H), 1.17–1.02 (m, 2H), 1.07 (s, 3H), 0.94 (d, J = 6.6 Hz, 3H), 0.88 (t, J = 7.0 Hz, 3H), 0.83–0.74 (m, 1H).
Cell culture and transfection
Human Embryonic Kidney 293 (HEK293) cells (ATCC, USA, CRL-1573) were cultured in high glucose Dulbecco’s Modified Eagle’s Medium (DMEM) (Sigma) supplemented with 10% fetal bovine serum (Gibco) at 37 °C, 5% CO2 in the air, and 95% humidity. The cells were plated in 96-well plates (Greiner BioOne, UK) at 50,000 cells per well and transfected with 150 ng of DNA per well using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The transfected cells were tested 24 h after transfection.
Bioluminescence resonance energy transfer assay
Bioluminescence resonance energy transfer (BRET) assay was used to measure CB1R-induced G protein dissociation and β-arrestin interaction with CB1R, as described previously24,25. To evaluate G protein dissociation, we transfected the cells with Gαi1-Rluc8 or GαoA-Rluc8, Gβ2-Flag, Gγ2-EYFP, and SNAP-CB1R plasmids in a mass ratio of 1:1:1:2. To measure β-arrestin2 interaction with CB1R, we transfected the cells with β-arrestin2-Rluc and CB1R-EYFP plasmids in a mass ratio of 1:2. To study GRK3-CB1R interaction, the cells were transiently transfected with GRK3-Rluc8 and CB1R-EYFP plasmids (1:2 ratio). Before the measurements, the transfected cells were washed with phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 1.8 mM KH2PO4) and incubated in Tyrode’s solution (137 mM NaCl, 0.9 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 11.9 mM NaHCO3, 3.6 mM NaH2PO4, 5.5 mM D-glucose, 25 mM HEPES, pH 7.4) at 37 °C for at least 30 min. Next, we added coelenterazine h (NanoLight) at a final concentration of 5 µM to the cells, followed by the addition of increasing concentrations of compounds (9S)-HHC, (9R)-HHC, Δ9-THC, WIN, or their vehicles. BRET donor and acceptor emission was measured 12 min after the addition of the compounds using Mithras LB940 plate reader (Berthold Biotechnologies, Germany). The BRET ratio was obtained by dividing the acceptor emission (540 ± 20 nm) by the donor emission (480 ± 10 nm). After subtracting the BRET ratio of the vehicle addition from the BRET ratio of the compounds, we obtained deltaBRET (ΔBRET). Data analysis was performed using GraphPad Prism 9.3.1 for Windows (GraphPad Software, USA). The concentration–response curves were fitted using a non-linear regression function.
CB1R internalization assay
Cell surface receptor internalization was studied using the Homogenous Time-Resolved FRET (HTRF) technology as described previously26. First, HEK293 cells were seeded on a 96-well plate (Merck, Germany) and transiently transfected with SNAP-tagged CB1R plasmid together with empty vector pRK6 (1:2 DNA mass ratio) using Lipofectamine™ 2000 (Thermo Fisher Scientific) according to the manufacturer's protocol. Twenty-four hours post-transfection, the cell culture medium was removed, and the cells were labeled with 100 nM SNAPLumi4-Tb (PerkinElmer—CisBio, France), diluted in Tag-lite labeling medium (PerkinElmer—CisBio, France) and incubated for 1 h at 37 °C, 5% CO2. Subsequently, labeled cells were washed four times with Tag-Lite buffer solution. The receptor internalization experiment was performed by adding Tag-lite buffer containing 24 μM fluorescein (Merck, Germany) and corresponding CB1R agonist or vehicle (dimethyl sulfoxide, or in the case of Δ9-THC, ethanol). HTRF signal was recorded over 60 min at 37 °C using the Mithras LB 940 microplate reader (Berthold Technologies, Germany) equipped with the HTRF module and relevant filters. The donor fluorophore (terbium cryptate) was excited at 340 ± 26 nm and emission was measured at 520 ± 10 nm. The acceptor (fluorescein) emission was measured at 620 ± 10 nm. The HTRF ratio was calculated as the donor emission divided by the acceptor emission multiplied by 10,000.
Extracellular signal-regulated kinases 1/2 phosphorylation assay
Phosphorylation levels of endogenous extracellular signal-regulated kinases 1/2 (ERK1/2) were detected using the Phospho-ERK1/2 (Thr202/Tyr204) kit (Cisbio Bioassays, France). The transfected cells plated in 96-well plates (Greiner BioOne, UK) were serum-starved for 16 h prior to the experiment in serum-free DMEM media. Afterwards, the cells were stimulated for the indicated times by CB1R ligand diluted in serum-free DMEM and then lysed in 50 μl of supplemented lysis buffer. After homogenization, 16 μl of the cell lysate was transferred from the 96-well plate to a 384-well black plate (Greiner BioOne, UK) and incubated with 4 μl of detection buffer containing anti-ERK1/2-Eu3+cryptate and anti-Phospho-ERK1/2-d2 for at least 4 h in dark. The fluorescence emissions at 665 nm and 620 nm were read on HTRF® compatible Mithras LB 940 microplate reader (Berthold Technologies, Germany). Data are presented as the ratio of 665 nm emission and 620 nm emission multiplied by 10,000.
Results
Synthesis of (9S)-HHC and (9R)-HHC
We produced the HHC epimers by employing the ∆8-THC reduction reaction, described previously by Russo and colleagues (Fig. 1B)11. They found that using ∆8-THC for the reaction provides predominantly (9R)-HHC in a 3:1 ratio. They also claimed that using ∆9-THC as a precursor leads to an excess of (9S)-HHC in a 2:1 ratio11. Because we used ∆8-THC in the reduction reaction, we were able to confirm that, when a high concentration of ∆8-THC and a relatively high amount of palladium on carbon is used, the reaction indeed predominantly produces (9R)-HHC at a ratio of approximately 3:1 (Table 1, entry 1). However, when only a small amount of palladium on carbon and a low concentration was used, (9R)-HHC was produced in a lower ratio of 3:2 (entry 2). This reaction was also carried out in separate preparations at larger scales of 250 g (entry 3) and 1 kg (entry 4). For the scale-up reaction, a high concentration of ∆8-THC and high amount of palladium on carbon was used, yielding predominantly (9R)-HHC in a 3:1 ratio. Achieving complete hydrogenation at large scales took significantly longer than at a small scale. Yields of HHC were not calculated as the hydrogenation reaction is quantitative and yields are mostly dependent on the scale of the reaction due to losses during distillation.
G protein activation induced by CB1R stimulation
To test whether HHC induces signaling via CB1R, we first measured the G protein activation in the transfected cells. We tested the effect of the studied cannabinoids (9S)-HHC, (9R)-HHC, Δ9-THC, and WIN on G protein activation by employing a BRET-based assay that monitors the dissociation of Gα and Gβγ subunits of the Gi/o protein upon its activation by CB1R. We tested Gi1 and GoA activation mediated by CB1R stimulation with increasing concentrations of (9S)-HHC, (9R)-HHC, Δ9-THC, and WIN. In all cases, agonist engagement of the receptor was followed by a prompt decrease in the BRET ratio, reflecting activation of the G proteins (Fig. 2).
Figure 2.

CB1R-driven G protein activation induced by the tested cannabinoids. HEK293 cells were transiently transfected with Gαi1-Rluc8 or GαoA-Rluc8, Gβ2-Flag, Gγ2-VENUS, and SNAP-CB1R. The cells were stimulated with the indicated concentrations of (9S)-HHC, (9R)-HHC, Δ9-THC, WIN, or their vehicles. BRET donor and acceptor emission was measured 12 min after receptor stimulation. (A) Concentration–response relationship of Gαi1 subunit dissociation from the G protein complex after CB1R stimulation. (B) Concentration–response relationship of GαoA subunit dissociation from the G protein complex after CB1R stimulation. The data are presented as means ± SEM from three independent experiments. The data analysis is disclosed in Supplementary Tables 1 and 2.
In the Gi1 and GoA activation assays, (9S)-HHC had a potency and efficacy lower than (9R)-HHC (Fig. 2 and Supplementary Tables 1 and 2). The potency and efficacy of (9R)-HHC were similar to those of Δ9-THC. Overall, the results demonstrate that the effect of (9R)-HHC epimer on the Gi and Go signaling pathways is similar to that of Δ9-THC, while (9S)-HHC induces lower levels of the G protein activation.
GRK3 and β-arrestin2 interactions with the activated CB1R
We next studied the recruitment of GRK3 and β-arrestin2 to CB1R, as stimulated by the tested cannabinoids. The employed BRET-based interaction assays monitor the association of GRK3 and CB1R following receptor phosphorylation and the interaction of β-arrestin2 with the phosphorylated CB1R. Agonist activation of the receptor increased the BRET ratio, reflecting increased GRK3-CB1R and β-arrestin2-CB1R interactions (Fig. 3).
Figure 3.

GRK3-CB1R and β-arrestin2-CB1R interactions elicited by the tested cannabinoids. HEK293 cells were transiently transfected with CB1R-EYFP and β-arrestin2-Rluc or GRK3-Rluc8 (1:2 ratio). The cells were stimulated with increasing concentrations of compounds (9S)-HHC, (9R)-HHC, Δ9-THC, WIN, or their vehicles. BRET donor and acceptor emission was measured 12 min after the addition of the compounds. (A) Concentration–response relationship of GRK3-CB1R association mediated by CB1R stimulation. (B) Concentration–response relationship of β-arrestin2 recruitment to CB1R after CB1R stimulation. The data are presented as means ± SEM from three independent experiments. The data analysis is disclosed in Supplementary Tables 3 and 4.
WIN application in these assays elicited the strongest responses and also showed the highest potency and efficacy (Fig. 3 and Supplementary Tables 3 and 4). On the other hand, the interactions induced by Δ9-THC were negligible. The potency of (9R)-HHC was higher than that of (9S)-HHC, but the curve fitting demonstrated that these epimers have similar efficacies. Overall, the results indicate that the (9R)-HHC epimer stimulates GRK3-CB1R and β-arrestin2-CB1R interactions more effectively than Δ9-THC or the (9S)-HHC epimer.
Internalization of activated CB1R
β-arrestin interaction with the desensitized receptor initiates receptor internalization and activates specific signaling cascades. We used the HTRF-based approach to monitor the kinetics of receptor internalization upon activation by the tested cannabinoids. In this approach, internalization results in increased emission of the terbium cryptate fluorophore that is covalently attached to the receptor.
Application of the cannabinoids initiated prompt and massive internalization of CB1R, but the extent of internalization varied. WIN had the highest effect on CB1R internalization rate (Fig. 4). The HHC epimers and Δ9-THC had comparable effects on CB1R internalization, which were about half of the WIN effect.
Figure 4.
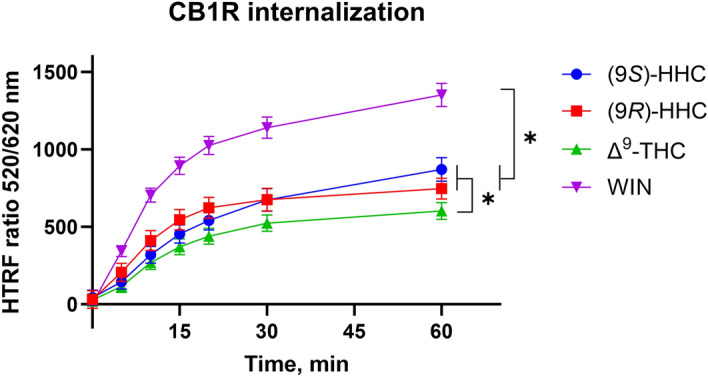
Internalization of CB1R induced by the tested cannabinoids. Internalization was elicited by the application of 10 µM of (9S)-HHC, (9R)-HHC, Δ9-THC, and WIN. HEK293 cells were transiently transfected with the plasmids coding SNAP-CB1R or mock plasmid pRK6 (1:2 DNA mass ratio). Data represent net receptor internalization by each drug treatment (i.e. receptor internalization by the indicated drug minus receptor internalization by vehicle). The data are presented as means ± SEM of three independent experiments performed in 3 technical replicates. The statistical analysis is disclosed in Supplementary Table 5. *, p < 0.05 by ANOVA.
ERK1/2 phosphorylation induced by CB1R stimulation
G proteins and β-arrestin both contribute to the activity of the ERK1/2 signaling cascade. We determined the extent of ERK1/2 activity driven by the tested cannabinoids by measuring its phosphorylation in the HTRF-based sandwich ELISA. In this assay, ERK1/2 phosphorylation is detected as an increase in the HTRF ratio.
WIN activation of CB1R led to a rapid but transient increase in ERK1/2 phosphorylation that peaked at 5 min after agonist stimulation and then progressively diminished (Fig. 5). Application of (9R)-HHC, (9S)-HHC, and Δ9-THC induced lower levels of ERK1/2 phosphorylation, peaking at 10 min.
Figure 5.
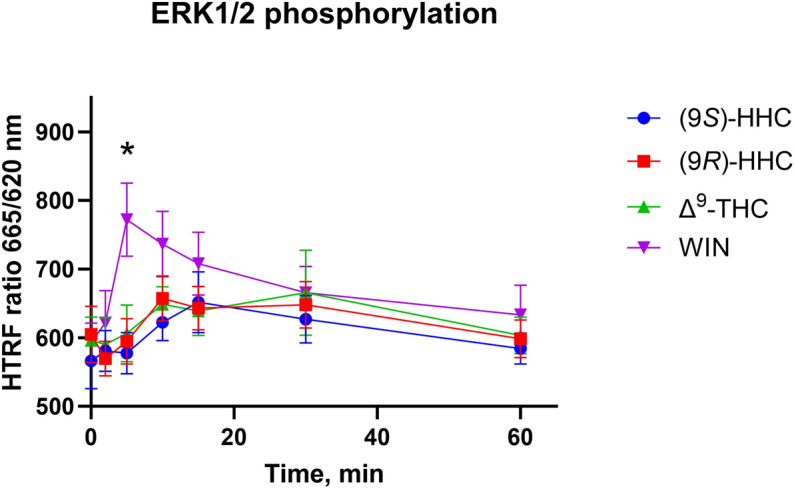
ERK1/2 phosphorylation elicited by the cannabinoids. HEK293 cells were transiently transfected with CB1R and empty vector (1:2 ratio). 24 h after transfection, cells were stimulated by 10 µM of (9S)-HHC, (9R)-HHC, Δ9-THC or WIN, and kinetics of ERK1/2 phosphorylation were measured at the indicated times. The data are presented as means ± SEM of three independent experiments performed in 3 technical replicates. The statistical analysis is disclosed in Supplementary Table 6. *, p < 0.05 (9S)-HHC vs. WIN by ANOVA.
Discussion
CB1R is the principal receptor of the central nervous system endocannabinoid system (ECS)27. CB1R is expressed in all brain regions, including those important for processing anxiety, fear, stress, and cognitive functions. CB1R is abundant in the basal ganglia, hippocampus, cerebellum, prefrontal cortex, and amygdala28. The neuronal ECS, with its central receptor, CB1R, is important for synaptic plasticity, strength, and maintenance. In addition to neurons, CB1R is also expressed in the central nervous system in astrocytes, microglia, and oligodendrocytes, where it modulates synaptic transmission, glucose metabolism, and immunomodulator production18,29. Furthermore, CB1R is also present in the peripheral nervous system, as well as in skeletal muscle, bone, skin, eyes, adipose tissue, and the reproductive system30. Subcellularly, CB1R is typically, but not exclusively, located presynaptically in many glutamatergic, GABAergic, cholinergic, serotonergic, and noradrenergic neurons. Endocannabinoids are synthesized on demand on the postsynaptic side and suppress neurotransmitter release via activation of presynaptic CB1R31–33. CB1R is primarily directed to cell surface; however, an important discrete pool of CB1Rs is in the outer mitochondrial membrane34.
ECS is involved in appetite stimulation, energy balance regulation, learning and memory, pain processing, neurogenesis and neuroprotection, immune responses, and many other physiological regulations including neurohumoral system homeostasis. CB1R also plays an important role in pathological conditions including schizophrenia, multiple sclerosis, anxiety, depression, epilepsy, Parkinson's disease, Huntington's disease, Alzheimer's disease, addiction, stroke, inflammation, glaucoma, cancer, as well as musculoskeletal and liver disorders16,35.
The Cannabis sativa plant produces a vast repertoire of chemically and biologically interesting and diverse compounds. Over 400 compounds, about a quarter of which unique, have been detected in the plant. This remarkable mixture includes phytocannabinoids, terpenes and other compound classes2,3. Recent efforts to use a scientific approach to marijuana for medical purposes, namely in Canada, Israel, the USA, and the Czech Republic, have led to an approach in which two main substances, Δ9-THC and CBD, were evaluated by controlled trials in a broad cohort of patients with favorable outcomes. However, it is known that Cannabis sativa chemistry is not limited to only these two compounds, and many more structures must be taken into an account.
One historically-overlooked CB1R ligand, HHC, share a similar chemical structure with Δ9-THC and CBD. Herein we show that they activate CB1R in a unique way, most likely by favoring differential active conformational states of CB1R than those favored by Δ9-THC. Recent studies in mice have shown that HHC compounds are psychoactive, namely in the cannabinoid tetrad tests. Many results from behavioral analyses highlight generally overlapping, but not entirely parallel impacts, on the performance in the tests. The pharmacodynamic analyses presented here, together with subsequent pharmacokinetic studies may help us to understand these differences.
Various examples of ligands that exert divergent effects on CB1R signaling pathways have been described. Certain cannabinoids favor G protein-mediated signaling over the β-arrestin pathway, as in the case of novel compounds PNR-4-20 and PNR-4-02 that selectively activate the Gαi pathway, while eliciting significantly less β-arrestin2 recruitment36. On the other hand, the allosteric modulator ORG27569 induces CB1R conformation state that selectively activates the ERK1/2 cascade via β-arrestin137. Distinct ligands induce and stabilize different conformations of a given GPCR. Consequently, these conformations could preferentially activate a particular signaling cascade over others, a phenomenon called “biased signaling”. Activation of a pathway resulting in desired therapeutic efficacy, together with a decrease of signaling pathways leading to undesired effects, typically psychoactivity or tolerance, may have profound consequences in drug discovery of molecules with potential medicinal uses, including those acting via CB1R. CB1R-mediated signaling is complex, and its outcome depends on the cellular environment, associated protein network, and ligand that activates the receptor in a particular way, or modulates its signaling in a unique way for each HHC enantiomer.
In neurons and other naïve cells, CB1R-interacting proteins also bias the signaling of the receptor, for example, SH3-containing GRB2-like protein 3-interacting protein 1 (SGIP1)24,25,38,39, Cannabinoid Receptor Interacting Protein 1a and 1b (CRIP1a/b)40,41, and G Protein-Coupled Receptor Associated Protein 1 (GASP1)42,43. The situation may become yet more complex with heterodimers of CB1R44,45. For example, CB1R was reported to form dimers with dopamine receptor 2. Activation of these heterodimeric receptors may activate the Gαs pathway leading to the increase of cAMP, thus generating the opposite effect as when CB1R is signaling alone44.
The urgent need for better pharmaceutical management in patients prompts investigations for novel therapeutic agents. The ECS is involved in a plethora of nervous system physiology and pathophysiology. However, implementing medical applications achieved by manipulating the ECS has been challenging, mainly due to the pleiotropic functions of the ECS. These include psychoactive and other undesired side effects of drugs acting on CB1R. Pharmacological approaches based on tinkering with the pleiotropic nature of CB1R signaling are one way to avoid undesired side effects. The biased CB1R-mediated signaling of the two HHC epimers, compared with each other and that of Δ9-THC (Fig. 6), together with the greater stability of HHC, represents an emerging prospective treatment via the ECS with possibly limited side effects.
Figure 6.

Pharmacological profiles of (9S)-HHC, (9R)-HHC, and Δ9-THC. (A) Calculated maximum response values or the time-course peaks were plotted on the axes of the radar plot. For clathrin-mediated internalization (CME), the 60 min time points were used; for ERK1/2 phosphorylation, the 10 min time points were used. (B) Calculated maximum response values of the tested cannabinoids were represented as fractions of WIN and normalized to (9R)-HHC. The normalized values for β-arrestin interaction were plotted on the x-axis, and the means of the normalized values for Gi1/GoA activation were plotted on the y-axis. The values represent only the maximum responses elicited by the ligands.
Supplementary Information
Acknowledgements
We thank Irina Cheveleva for cell culture maintenance and DNA preparation and Aaron Rulseh for text editing.
Abbreviations
- HHC
Hexahydrocannabinol
- (9S)-HHC
(9S)-6,6,9-Trimethyl-3-pentyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromen-1-ol
- (9R)-HHC
(9R)-6,6,9-Trimethyl-3-pentyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromen-1-ol
- MeOH
Methanol
- Δ8-THC
Δ8-Tetrahydrocannabinol
- Δ9-THC
Δ9-Tetrahydrocannabinol
- BRET
Bioluminescence resonance energy transfer
- cAMP
Cyclic adenosine triphosphate
- CB1R
Cannabinoid receptor 1
- CRIP1a/b
Cannabinoid receptor interacting protein 1a and 1b
- DNA
Deoxyribonucleic acid
- DCM
Dichloromethane
- DMEM
Dulbecco’s modified Eagle’s medium
- ECS
Endocannabinoid system
- GASP1
G protein-coupled receptor associated protein 1
- GRK3
G protein-coupled receptor kinase 3
- HEK293
Human embryonic kidney 293
- HTRF
Homogenous time-resolved FRET
- HPLC
High-performance liquid chromatography
- NMR
Nuclear magnetic resonance
- PPAR
Peroxisome proliferator-activated receptor
- SGIP1
SH3-containing GRB2-like protein 3-interacting protein 1
- TRPV1
Transient receptor potential cation channel subfamily V member 1
- EYFP
Enhanced yellow fluorescent protein
Author contributions
M.K. and J.B. developed and supervised the project, conceived the experiments plan and drafted the manuscript; P.P. carried out the synthesis and purification and the spectroscopic isolation, and characterization of HHC epimers; O.D., M.G. performed the pharmacodynamic studies; K.M. contributed to writing, editing and formal validation of the text and data interpretation. All authors reviewed the manuscript.
Funding
This work was supported by the Czech Science Foundation, grant number 21-02371S, Ministry of Interior of the Czech Republic, grant number VK01010212, the institutional funding (RVO68378050), and the United States National Institutes of Health, grant number AT011162.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding authors on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Oleh Durydivka, Petr Palivec, Jaroslav Blahos and Martin Kuchar.
Contributor Information
Oleh Durydivka, Email: oleh.durydivka@img.cas.cz.
Martin Kuchar, Email: kuchara@vscht.cz.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-58845-7.
References
- 1.Mechoulam R. Cannabinoids as Therapeutic Agents. CRC Press; 1986. pp. 1–19. [Google Scholar]
- 2.ElSohly MA, Slade D. Chemical constituents of marijuana: The complex mixture of natural cannabinoids. Life Sci. 2005;78:539–548. doi: 10.1016/j.lfs.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Radwan MM, Chandra S, Gul S, ElSohly MA. Cannabinoids, phenolics, terpenes and alkaloids of cannabis. Molecules. 2021 doi: 10.3390/molecules26092774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casati S, et al. Hexahydrocannabinol on the light cannabis market: The latest "new" entry. Cannabis Cannabinoid. 2022 doi: 10.1089/can.2022.0253. [DOI] [PubMed] [Google Scholar]
- 5.Ujvary, I. et al. Hexahydrocannabinol (HHC) and related substances. 1–106 (2023).
- 6.Qureshi MN, Kanwal F, Afridi M, Akram M. Estimation of biologically active cannabinoids in Cannabis indica by gas chromatography-mass spectrometry (GC-MS) World Appl. Sci. J. 2012;19:918–923. doi: 10.5829/idosi.wasj.2012.19.07.1922. [DOI] [Google Scholar]
- 7.Gaoni Y, Mechoulam R. Isomerization of cannabidiol to tetrahydrocannabinols. Tetrahedron. 1966;22:1481. doi: 10.1016/S0040-4020(01)99446-3. [DOI] [Google Scholar]
- 8.Reggio PH, Greer KV, Cox SM. The importance of the orientation of the C9 substituent to cannabinoid activity. J. Med. Chem. 1989;32:1630–1635. doi: 10.1021/jm00127a038. [DOI] [PubMed] [Google Scholar]
- 9.Mechoulam R, et al. Stereochemical requirements for cannabinoid activity. J. Med. Chem. 1980;23:1068–1072. doi: 10.1021/jm00184a002. [DOI] [PubMed] [Google Scholar]
- 10.Adams R, et al. Structure of cannabidiol. VIII. Position of the double bonds in cannabidiol. Marihuana activity of tetrahydro-cannabinols. J. Am. Chem. Soc. 1940;62:2566–2567. doi: 10.1021/ja01866a510. [DOI] [Google Scholar]
- 11.Russo F, et al. Synthesis and pharmacological activity of the epimers of hexahydrocannabinol (HHC) Sci. Rep. 2023;13:11061. doi: 10.1038/s41598-023-38188-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cristino L, et al. Immunohistochemical localization of anabolic and catabolic enzymes for anandamide and other putative endovanilloids in the hippocampus and cerebellar cortex of the mouse brain. Neuroscience. 2008;151:955–968. doi: 10.1016/j.neuroscience.2007.11.047. [DOI] [PubMed] [Google Scholar]
- 13.Gray RA, Whalley BJ. The proposed mechanisms of action of CBD in epilepsy. Epilept. Disord. 2020;22:10–15. doi: 10.1684/epd.2020.1135. [DOI] [PubMed] [Google Scholar]
- 14.Penumarti A, Abdel-Rahman AA. The novel endocannabinoid receptor GPR18 is expressed in the rostral ventrolateral medulla and exerts tonic restraining influence on blood pressure. J. Pharmacol. Exp. Ther. 2014;349:29–38. doi: 10.1124/jpet.113.209213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villapol S. Roles of peroxisome proliferator-activated receptor gamma on brain and peripheral inflammation. Cell Mol. Neurobiol. 2018;38:121–132. doi: 10.1007/s10571-017-0554-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou SL, Kumar U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int. J. Mol. Sci. 2018 doi: 10.3390/ijms19030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mackie K. Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb. Exp. Pharmacol. 2005 doi: 10.1007/3-540-26573-2_10. [DOI] [PubMed] [Google Scholar]
- 18.Castillo PE, Younts TJ, Chavez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shenoy SK, Lefkowitz RJ. beta-arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 2011;32:521–533. doi: 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rueda D, Galve-Roperh I, Haro A, Guzman M. The CB1 cannabinoid receptor is coupled to the activation of c-Jun N-terminal kinase. Mol. Pharmacol. 2000;58:814–820. doi: 10.1124/mol.58.4.814. [DOI] [PubMed] [Google Scholar]
- 21.Leo LM, Abood ME. CB1 cannabinoid receptor signaling and biased signaling. Molecules. 2021 doi: 10.3390/molecules26175413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ibsen MS, Connor M, Glass M. Cannabinoid CB1 and CB2 receptor signaling and bias. Cannabis Cannabinoid. 2017;2:48–60. doi: 10.1089/can.2016.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marzullo P, et al. Cannabidiol as the substrate in acid-catalyzed intramolecular cyclization. J. Nat. Prod. 2020;83:2894–2901. doi: 10.1021/acs.jnatprod.0c00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hajkova A, et al. SGIP1 alters internalization and modulates signaling of activated cannabinoid receptor 1 in a biased manner. Neuropharmacology. 2016;107:201–214. doi: 10.1016/j.neuropharm.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Gazdarica M, et al. SGIP1 modulates kinetics and interactions of the cannabinoid receptor 1 and G protein-coupled receptor kinase 3 signalosome. J. Neurochem. 2021 doi: 10.1111/jnc.15569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levoye A, et al. A broad G protein-coupled receptor internalization assay that combines SNAP-tag labeling, diffusion-enhanced resonance energy transfer, and a highly emissive terbium cryptate. Front. Endocrinol. 2015 doi: 10.3389/fendo.2015.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herkenham M, et al. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J. Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herkenham M, et al. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. U. S. A. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jimenez-Blasco D, et al. Glucose metabolism links astroglial mitochondria to cannabinoid effects. Nature. 2020;583:603–608. doi: 10.1038/s41586-020-2470-y. [DOI] [PubMed] [Google Scholar]
- 30.Maccarrone M, et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015;36:277–296. doi: 10.1016/j.tips.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur. J. Neurosci. 1999;11:4213–4225. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- 32.Haring M, Marsicano G, Lutz B, Monory K. Identification of the cannabinoid receptor type 1 in serotonergic cells of raphe nuclei in mice. Neuroscience. 2007;146:1212–1219. doi: 10.1016/j.neuroscience.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 33.Kirilly E, Hunyady L, Bagdy G. Opposing local effects of endocannabinoids on the activity of noradrenergic neurons and release of noradrenaline: Relevance for their role in depression and in the actions of CB1 receptor antagonists. J. Neural Transm. 2013;120:177–186. doi: 10.1007/s00702-012-0900-1. [DOI] [PubMed] [Google Scholar]
- 34.Hebert-Chatelain E, Marsicano G. A new link between cannabinoids and memory—The mitochondria. M. S-Med. Sci. 2017;33:579–581. doi: 10.1051/medsci/20173306007. [DOI] [PubMed] [Google Scholar]
- 35.Joshi N, Onaivi ES. Endocannabinoid system components: Overview and tissue distribution. Adv. Exp. Med. Biol. 2019;1162:1–12. doi: 10.1007/978-3-030-21737-2_1. [DOI] [PubMed] [Google Scholar]
- 36.Ford BM, et al. Characterization of structurally novel G protein biased CB1 agonists: Implications for drug development. Pharmacol. Res. 2017;125:161–177. doi: 10.1016/j.phrs.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahn KH, Mahmoud MM, Kendall DA. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. J. Biol. Chem. 2012;287:12070–12082. doi: 10.1074/jbc.M111.316463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dvorakova M, et al. SGIP1 is involved in regulation of emotionality, mood, and nociception and modulates in vivo signalling of cannabinoid CB1 receptors. Br. J. Pharmacol. 2021;178:1588–1604. doi: 10.1111/bph.15383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Durydivka O, Mackie K, Blahos J. SGIP1 in axons prevents internalization of desensitized CB1R and modifies its function. Front. Neurosci. 2023 doi: 10.3389/fnins.2023.1213094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niehaus JL, et al. CB1 cannabinoid receptor activity is modulated by the cannabinoid receptor interacting protein CRIP 1a. Mol. Pharmacol. 2007;72:1557–1566. doi: 10.1124/mol.107.039263. [DOI] [PubMed] [Google Scholar]
- 41.Blume LC, et al. Cannabinoid receptor interacting protein 1a competition with beta-arrestin for CB1 receptor binding sites. Mol. Pharmacol. 2017;91:75–86. doi: 10.1124/mol.116.104638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martini L, et al. Ligand-induced down-regulation of the cannabinoid 1 receptor is mediated by the G-protein-coupled receptor-associated sorting protein GASP1. Faseb J. 2007;21:802–811. doi: 10.1096/fj.06-7132com. [DOI] [PubMed] [Google Scholar]
- 43.Martini L, Thompson D, Kharazia V, Whistler JL. Differential regulation of behavioral tolerance to WIN55, 212–2 by GASP1. Neuropsychopharmacology. 2010;35:1363–1373. doi: 10.1038/npp.2010.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jarrahian A, Watts VJ, Barker EL. D2 dopamine receptors modulate Galpha-subunit coupling of the CB1 cannabinoid receptor. J. Pharmacol. Exp. Ther. 2004;308:880–886. doi: 10.1124/jpet.103.057620. [DOI] [PubMed] [Google Scholar]
- 45.Hudson BD, Hebert TE, Kelly ME. Ligand- and heterodimer-directed signaling of the CB(1) cannabinoid receptor. Mol. Pharmacol. 2010;77:1–9. doi: 10.1124/mol.109.060251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding authors on reasonable request.