

Abstract
Diploid wild oat Avena longiglumis has nutritional and adaptive traits which are valuable for common oat (A. sativa) breeding. The combination of Illumina, Nanopore and Hi-C data allowed us to assemble a high-quality chromosome-level genome of A. longiglumis (ALO), evidenced by contig N50 of 12.68 Mb with 99% BUSCO completeness for the assembly size of 3,960.97 Mb. A total of 40,845 protein-coding genes were annotated. The assembled genome was composed of 87.04% repetitive DNA sequences. Dotplots of the genome assembly (PI657387) with two published ALO genomes were compared to indicate the conservation of gene order and equal expansion of all syntenic blocks among three genome assemblies. Two recent whole-genome duplication events were characterized in genomes of diploid Avena species. These findings provide new knowledge for the genomic features of A. longiglumis, give information about the species diversity, and will accelerate the functional genomics and breeding studies in oat and related cereal crops.
Subject terms: Plant evolution, Phylogenetics, Genome evolution
Background & Summary
Common oat (Avena sativa L.) and its wild relatives (2x, 4x,and 6x) are members of the Aveneae tribe (Poaceae). Clinical studies have shown the beneficial effects of consuming oats that can reduce serum cholesterol and cardiovascular disease, attributed to the soluble β-glucan component1. Oats also exhibit a favourable glycaemic index, with a low value and slow carbohydrate breakdown. Plant oils derived from cereal seeds are vital agricultural commodities used for food, feed, and fuel. Oat endosperm has between 6–18% oil content, which is significantly higher than other cereals [averaging 2.41% in barley (Hordeum vulgare) and 2.18% in wheat (Triticum aestivum)]2,3. The high oil content of oat grain suggests a possible important use for food oils and in animal feeds4. Despite the unique composition, global oat production has steadily declined over the past 50 years to 25 million tons in 2023 (http://www.fao.org/faostat/), suggesting the genetic improvement has lagged behind major cereal crops such as rice, wheat, and maize, making the crop less desirable to grow. There are therefore likely to be substantial opportunities for improvement of oat varieties.
Not least due to the large genome size of A. sativa (10.3 Gb)5, oat genomic research lags behind that of other crops such as rice (Oryza sativa)6, sorghum (Sorghum bicolor)7 or foxtail millet (Setaria italica)8. There is an urgent need for the characterization, exploitation and utilization of wild oat germplasm resources for oat and related crop breeding9,10. A diploid genome of A. longiglumis Durieu (Fig. 1) reveals novelty in target genes and regulatory sequences, such as those for β-glucan synthesis, high linoleic content in grains, drought-adapted phenotypes, and resistance to crown rust disease11. The rapidly developing field of structural variation requires multiple high-quality chromosome-scale assemblies to show the nature of intraspecific variation (individual, variety or populations), polymorphisms within and between diploid species and their related species, and generation of recent structural variations in polyploid species derived from diploid ancestors.
Fig. 1.

The spikelet of Avena longiglumis. Two glumes nearly equal in length (left), the first (middle) and the second (right) florets disarticulated with 2–3 mm awl-shaped callus at the floret base together with 8–12 mm bristles at the lemma tip. Scale bar, 1 cm.
This study utilized a combination of Illumina, Oxford Nanopore Technology (ONT) sequencing, and chromosome conformation capture (Hi-C) data to create a superior chromosome-scale genome assembly of diploid A. longiglumis (ALO; Fig. S1). Its genome assembly had a length of approximately 3,960.97 Mb (Table 1 and S1), which is slightly smaller than the genome size estimated by k-mer analysis (Fig. S2). Through scaffolding contigs into seven super-scaffolds, the 98.84% of reads were anchored. As observed in the Hi-C heatmap, the seven super-scaffolds were mapped to the corresponding seven pseudo-chromosomes (Fig. 2). Among A. longiglumis genome sequences, 87.04% were classified as known repetitive DNA elements (Table 2), showing increased density in broad centromeric regions (Fig. 3 circle b). Compared to the published assembly results of tetraploid A. insularis and hexaploid oat genomes5,9, the diploid A. longiglumis genome in this study exhibits superior sequence continuity, as evidenced by higher contig N50 value of 12.68 Mb and scaffold N50 value of 527.34 Mb, respectively (Table 3), indicating a high assembly quality of the diploid genome, ensuring the reliability of subsequent research.
Table 1.
Genome assembly statistics and gene predictions in the Avena longiglumis genome.
| Features | Number | Size |
|---|---|---|
| Assembly features | ||
| Predicted genome size based on k-mer | 3,965,670,000 bp | |
| Assembly size | 3,960,965,270 bp | |
| Total length of seven pseudo-chromosomes | 3,847,578,604 bp | |
| Scaffold N50 length | 527,343,613 bp | |
| Scaffold N90 length | 6,968,329 bp | |
| Number of scaffolds (>N90) | 9 | |
| Longest scaffold (bp) | 594,546,470 bp | |
| Contig N50 length | 12,682,464 bp | |
| Longest contig | 99,445,397 bp | |
| Repetitive DNAs | ||
| Retrotransposons | 3,198,067,781 bp (80.74%) | |
| DNA transposons | 137,389,012 bp (3.47%) | |
| Total repeats | 3,447,484,807 bp (87.04%) | |
| Gene annotation | ||
| High-confidence (HC) genes | 33,271 | 115,042,134 bp |
| Low-confidence (LC) genes | 7,574 | 18,590,004 bp |
| Total genes | 40,845 | 133,632,138 bp |
| Average length of each gene | 3,272 bp | |
| Non-coding RNAs | 16,439 | 2,222,342 bp |
Fig. 2.
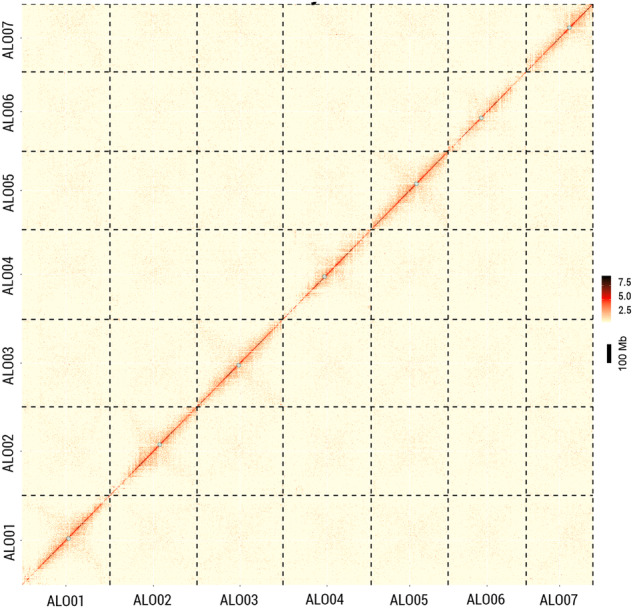
Genome-wide chromatin interaction heatmap (100 kb bins) of diploid A. longiglumis (ALO, PI657387) based on Hi-C data showing chromosome-scale continuity of the assembly. Small shaded circles denoted the centromeric locations.
Table 2.
Repetitive DNA composition of the Avena longiglumis genome.
| Repeat type | Super family | Family | Repeat sequences (bp) | Copy number | Repeat fraction | Genome fraction | |
|---|---|---|---|---|---|---|---|
| Transposable elements | |||||||
| Class I (Retrotransposons) | |||||||
| LTR | Gypsy | 2,045,839,268 | 1,127,011 | 59.34% | 51.65% | ||
| Copia | 1,035,647,971 | 575,382 | 30.04% | 26.15% | |||
| Unknown LTR | 77,372,748 | 61,272 | 2.24% | 1.95% | |||
| Other LTR | 213,586 | 628 | 0.01% | 0.01% | |||
| Total LTR- Retrotransposons | 3,159,073,573 | 1,764,293 | 91.63% | 79.76% | |||
| Non-LTR | LINE | L1 | 38,994,208 | 42,780 | 1.13% | 0.98% | |
| Total Class I retrotransposons | 3,198,067,781 | 1,807,073 | 92.76% | 80.74% | |||
| Class II (DNA transposons)-Subclass 1 | |||||||
| Tc1_Mariner | 21,858,891 | 69,956 | 0.63% | 0.55% | |||
| CACTA | 29,566,991 | 75,699 | 0.86% | 0.75% | |||
| Mutator | 25,388,681 | 82,292 | 0.74% | 0.64% | |||
| PIF_Harbinger | 9,194,639 | 31,402 | 0.27% | 0.23% | |||
| hAT | 7,600,354 | 22,266 | 0.22% | 0.19% | |||
| Class II (DNA transposons)-Subclass II | |||||||
| Helitron | 43,779,456 | 117,248 | 1.27% | 1.11% | |||
| Total Class II DNA transposons | 137,389,012 | 398,863 | 3.99% | 3.47% | |||
| Total transposable elements | 3,335,456,793 | 2,205,936 | 96.75% | 84.21% | |||
| Tandem and simple sequence repeats | 11,144,119 | 162,888 | 0.32% | 0.28% | |||
| Other repeats | 100,883,895 | 369,975 | 2.93% | 2.55% | |||
| Total repetitive DNAs | 3,447,484,807 | 2,728,799 | 100% | 87.04% | |||
Fig. 3.

Genomic features of Avena longiglumis PI657387. (a) Seven chromosomes (scale in 100 Mb) with pink, green and red regions denoting centromere, 5 S (ALO07) and 45 S (ALO01 and ALO07) rDNA positions. (b) Transposable element (TE) density. (c) Long-terminal repeat TE density. (d) Long interspersed nuclear element (LINE) density. (e) Helitron density (cyan). (f) Expanded gene locations. (g) Contracted gene locations. (h) Single copy orthologue gene locations. (i) High-confidence gene locations. (j) Purified selection gene (P-value ≤ 0.05) locations. (k) Gene expression profiling in ALO roots. (l) Gene expression profiling in ALO leaves. (m) Inter-chromosomal synteny. b, d–h & k–l: 100 bp bins; c: 1 Mb bins; i–j: 3 kb bins.
Table 3.
Summary of genome assemblies of Avena longiglumis of this study and published tetraploid A. insularis and hexaploid A. sativa. –: unavailable data.
| Species | Avena longiglumis PI 657387 | A.insularis BYU2095 | A.sativa cv. Sang | A. insularis CN108634 | A. sativa ssp. nuda cv. Sanfensan9 | A. sativa_OT3098v.2 |
|---|---|---|---|---|---|---|
| Number of contigs | 2,381 | 6,523 | 1,823,168 | 2,732 | 436 | 1,343 |
| Number of scaffolds | 414 | 15 | 22 | — | — | 84 |
| Assembled sequences | 3,960,965,270 bp | 7,256,293,586 bp | 11,012,379,496 bp | 7,519,018,440 bp | 10,757,433,345 bp | 10,839,200,031 bp |
| Contig N50 length | 12.682 Mb | 5.157 Mb | 21.001 kb | 5.637 Mb | 75.273 Mb | 71.000 Mb |
| Scaffold N50 length | 583.925 Mb | 481.348 Mb | 490.397 Mb | — | — | 374.00 Mb |
| BUSCO | 99.00% | 99.60% | 99.40% | 99.32% | 99.44% | 99.38% |
The BUSCO12 results revealed the retrieval of 99.0% of the complete single-copy genes, of which 16.3% were duplicated, indicating high genome assembly completeness of our A. longiglumis_CN58138 (Table S2). Compared to other diploid assemblies of A. longiglumis_CN58138 (93.0%) and A. eriantha (94.0%) (Extended Data Fig. 2a of ref. 5), our diploid A. longiglumis_PI657387 genome exhibited a higher proportion of complete orthologous genes, comprising 99.0% of the genome assembly (Fig. 4). Compared to tetraploid A. insularis (7.9%) and hexaploid A. sativa (11.2%), the A. longiglumis genome in our study exhibits a higher proportion of single-copy orthologous genes, comprising 82.7% of the genome assembly (Fig. 4). In addition, the fragmented genes in this diploid genome display a similarity (0.2%) to those found in A. sativa.
Fig. 4.

BUSCO scores of the assembled genomes of Avena longiglumis, A. insularis (Kamal et al.22), and A. sativa (Kamal et al.22). Our A. longiglumis genome assembly stored on GenBank https://identifiers.org/ncbi/insdc.gca: GCA_030063025.1 (2023); Genome assemblies of A. insularis and A. sativa from the European Nucleotide Archive (ENA) under accession numbers PRJEB45088 and PRJEB44810, respectively.
A total of 40,845 protein-coding genes were annotated for A. longiglumis using databases of NCBI NR (Non-redundant protein)13, EggNOG (Evolutionary genealogy of genes: non-supervised orthologous groups)14, Pfam (Pfam protein families)15, COG (Clusters of orthologous groups)16, SwissProt (Swiss Institute of Bioinformatics and Protein Information Resource)17, GO (Gene ontology)18, KOG (EuKaryotic orthologous groups)19, KEGG (Kyoto encyclopedia of genes and genomes)20, PlantTFDB (Plant transcription factor)21, and CAZy (Carbohydrate-Active enZYmes)22 (Table S3). Dotplots of our A. longiglumis assembly were compared with two published genomes of A. longiglumis5,9, indicating the conservation of gene order and equal expansion of all syntenic blocks among three ALO genome assemblies (Fig. 5a,b).
Fig. 5.

Pairwise comparisons of dotplots for three Avena longiglumis (ALO) genome assemblies and the diploid Avena species genomes. (a) ALO_PI657387and ALO_CN58138 (Kamal et al.22). (b) ALO_PI657387 and ALO_ CN58139 (Peng et al.9). The dotplots provide insights into the conservation of gene order and the genomic rearrangements among three A. longiglumis genome assemblies. The x- and y-axes represent the genomic coordinates of each species.
Methods
Plant Materials
Young leaf samples were collected from an A. longiglumis plant (ALO, accession PI 657387; US Department of Agriculture at Beltsville, https://www.ars-grin.gov/, originally collected in Morocco) grown in climatic box conditions (16 h light / 8 h dark and day/night temperatures of 25°C/15°C) at the South China National Botanical Garden, Guangzhou, China. Young leaves were collected for DNA isolation and whole-genome sequencing. The leaves and roots were collected for RNA-sequencing (RNA-seq) and transcriptome assembly. The samples were immediately flash-frozen in liquid nitrogen after harvest, and stored at −80 °C for subsequent nucleic acid extraction. The extraction and purification of RNA were carried out utilizing the Qiagen RNeasy Plant Mini Kit (Qiagen, CA, USA), following the instructions of the manufacturer, one of 8 Gb and one of 10 Gb pair-end read data were obtained. A total of 511.4 Gb Oxford Nanopore Technology (ONT) long reads (~128.9 × coverage), 435.6 Gb Hi-C reads (~109.8 × coverage), 268.6 Gb (~67.7 × coverage) paired-end Illumina reads, and 99.0 Gb RNA-seq reads were generated for the genome assembly, genome survey, and transcriptome assembly (Table S1).
Illumina sequencing and genome survey
Pair-end genome sequencing with a 350 bp insert size used Illumina TruSeq® Nano DNA library preparation kit (Illumina, San Diego, CA, USA) and libraries were sequenced on an Illumina NovaSeq 6000 platform (Table S1). Fastp v.0.23.223 was utilized to remove contaminants, Illumina adapters, and low-quality reads. The 268.60 Gb clean data were processed via Kmerfreq_AR v.2.0.424. The 17-bp k-mers with Illumina reads counted using Jellyfish v.2.2.625 with default parameters. The genome size of 3.966 Gb, a heterozygosity of 0.48%, and repeat content were estimated using GenomeScope v.2.026 (Fig. S2).
ONT sequencing and genome assembly
The genomic DNA (10 μg) was broken into fragments around 10–50 kb long with the use of a g-TUBE device (Covaris, Inc., MA, USA) and size selection with BluePippin (Sage Science, Inc., MA, USA). To prepare the ONT PromethION (Genome Centre of Grandomics, Wuhan, China) sequencing libraries, DNA end repair was carried out by utilizing the NEBNext End Repair/dA-Tailing Module (New England Biolabs, MA, UK), and the ligation sequencing kit (SQK-LSK109, ONT, UK) (Table S1).
ONT reads were subjected to self-correction using three tools, NextDenovo v.2.4.0 (https://github.com/Nextomics/NextDenovo), wtdbg2.huge v.1.2.827 and SMARTdenovo v.1.0.028. The corrected reads were then passed on to NextDenovo for additional read correction. Subsequently, we evaluated several parameters and detected that utilizing the corrected reads in combination with SMARTdenovo v.1.0.028 and assembler parameters “-c 3” and “-k 11” produced desired outcomes by generating a preliminary assembly. The contigs were polished with ONT raw data thrice using NextPolish v.1.0129 and four times with filtered Illumina reads.
Hi-C sequencing and chromosome-level genome assembly
For Hi-C sequencing, 3-week-old leaves of A. longiglumis seedlings were fixed in 2% formaldehyde solution to obtain nuclear/chromatin samples. DpnII enzyme (Cat. E0543L, NEB, UK) was utilized to digest these fixed tissues. Hi-C libraries were then constructed and sequenced on the Illumina Novaseq 6000 platform to generate 150 bp paired-end reads (Table S1). High-quality reads were extracted and aligned to the reference genome assembly using Bowtie2 v.2.3.230. Juicer v.2.031 was utilized to create a de-duplicated listing of alignments of Hi-C reads to the draft A. longiglumis assembly. HiC-Pro v.2.7.832 was used to determine the ligation site for each unmapped read, after which the 5’ fragments were aligned to the genome assembly.
A single alignment file was generated by merging the results of both mapping steps, and low-quality reads were discarded, which included reads with multiple matches, singletons, and mitochondrial DNA. Valid pairs of interaction were employed in scaffolding the assembled contigs into 7 pseudo-chromosomes utilizing the LACHESIS pipeline33. The quality and completeness of the genome assembly were evaluated by utilizing BUSCO v.5.4.612 (Table S2). In addition, the chromosome matrix was depicted as a heatmap that manifested diagonal patches of robust linkage.
Identification and characterization of repetitive elements
De novo repeat prediction of the ALO assembly was carried out by EDTA v.1.7.0 (Extensive de-novo TE Annotator)34, which was composed of eight software. LTRharvest33,34, LTR_FINDER_parallel v.1.235, LTR_retriever v. 2.9.036 (it was incorporated to identify LTR retrotransposons); Generic Repeat Finder v.1.7.037 and TIR-Learner v.1.7.038 were included to identify TIR transposons; HelitronScanner v.1.039 was identified Helitron transposons; RepeatModeler v.2.0.2a40 was used to identify transposable elements (TEs, such as LINEs); Finally, RepeatMasker v.4.1.141 was used to annotate fragmented TEs based on homology to structurally annotated TEs. In addition, TEsorter v.1.1.442 was used to identify TE-related genes.
Gene prediction and functional annotation
Gene structure prediction relied on three distinct approaches that were applied, including ab initio prediction, homology-based prediction, and RNA-seq-assisted prediction43. The de novo-based gene prediction was carried out using Augustus v.3.4.044 with default parameters, to predict A. longiglumis-assembled genes. Furthermore, the homology-based prediction was performed by GeMoMa v.1.6.145 with default parameters, utilizing filtered proteins from genomes of six species (Arabidopsis thaliana46, Brachypodium distachyon47, Hordeum vulgare48, Sorghum bicolor7, Triticum aestivum49 and Zea mays50). The RNA-seq-based gene prediction was executed using TransDecoder v.5.5.051. High-confidence (HC) genes refer to both homology-based prediction supported by ≥ two species (1,083) and by RNA-seq-assisted prediction if the FPKM (Fragments Per Kilobase of exon model per Million mapped fragments) value > 0 (32,188). The predicted gene structures from each of these three approaches were integrated into consensus gene models using EVidenceModeler v.1.1.152. The resulting gene models were then filtered to obtain a precise gene set, whereby genes with transposable element sequences were removed using TransposonPSI v.1.0.0 (http://transposonpsi.sourceforge.net/).
Functional annotation was performed for the predicted protein-coding genes via comparing with public databases including NCBI NR13, EggNOG14, Pfam15, COG16, SwissProt17, GO18, KOG19, KEGG20, PlantTFDB21, and CAZy22 (Table S3). Protein sequences were aligned to NCBI NR13, SwissProt17 and KOG19 by BLASTP v.2.10.153 (E-value ≤ 1e-15). EggNOG14, Pfam15, and COG16 annotations were performed with eggNOG v.5.014. GO18 ID for each gene were determined using Blast2GO v.1.4454. Genes were mapped to KEGG database20 (Fig. S3). Additionally, transcription factor annotation was carried out using PlantTFDB v.5.021, while gene annotation used CAZy22 (Table S3).
Non-coding RNA annotation
The prediction of the non-coding RNA gene set (ncRNA) was carried out across the genome. Initially, the data was aligned with the noncoding database of Rfam library v.11.055, for the annotation of genes encoding various non-coding RNAs including small nuclei RNAs (snRNAs), ribosomal RNAs (rRNAs), and microRNAs (miRNAs). The transfer RNA (tRNA) sequences were subsequently identified using tRNAscan-SE v.2.056 (Table 1).
Pairwise comparisons of genome assemblies
To create the dotplots of A. longiglumis, the reference sequence of CN581385 and CN581399 were aligned with the de novo assembly of PI 657387 using Minigraph v. 2.2557, respectively, with the ‘-ax asm5’ option, resulting in a PAF alignment file. The PAF file was uploaded to D-Genies v.1.5.058 to create the dotplot using their default setting. Dotplots of the assembly (accession PI657387) were compared with two published genomes of A. longiglumis, indicating the conservation of gene order and equal expansion of all syntenic blocks among three genome assemblies (Fig. 5a,b).
Data Records
Sequencing reads for Avena longiglumis are available on the NCBI Sequence Read Archive (SRA) https://identifiers.org/ncbi/insdc.sra: SRR1927951859 for genome survey data; SRR19279519-SRR19279520 and SRR19279522-SRR1927953159 for ONT data; SRR19279511-SRR19279517, SRR19279521, and SRR19279532-SRR1927953359 for Hi-C data; and SRR24234795-SRR24234797 and SRR24234802-SRR2423480460 for RNA sequencing data. Genome assembly for A. longiglumis is available on the GenBank https://identifiers.org/ncbi/insdc.gca: GCA_030063025.161.
Technical Validation
The chromosome-level genome assembly was 3,960.97 Mb with a scaffold N50 of 527.34 Mb. The interaction contact pattern was organized around the principal diagonal in the Hi-C heatmap (Fig. 2), directly supporting the accuracy of the chromosome assembly. The completeness of the final assembled genome was assessed using BUSCO v.5.4.612 by searching embryophyta_odb10 databases. The results revealed the retrieval of 99.0% of the complete single-copy genes, of which 16.3% were duplicated. Only 0.2% of BUSCO genes were fragmented, and 0.8% were missing from the A. longiglumis genome (Fig. 4).
Supplementary information
Acknowledgements
This work was supported by the grants from National Natural Science Foundation of China (32070359, 32370402), Guangdong Flagship Project of Basic and Applied Basic Research (2023B0303050001), Chinese Academy of Sciences (CAS) President’s International Fellowship Initiative (2024PVA0028), UK Research and Innovation (UKRI) via the Engineering and Physical Sciences Research Council (EPSRC; EP/Y00597X/1-project RP13W471907), Overseas Distinguished Scholar Project of South China Botanical Garden, Chinese Academy of Sciences (Y861041001), UK Biotechnology and Biological Sciences Research Council (BB/R022828/1), and Innovation Training Programs for Undergraduates, Chinese Academy of Sciences (KCJH-80107-2023-148).
Author contributions
Q.L. and J.S.H.H. conceived and designed the study. G.X. and T.Y.T. collected the samples. Z.W.W., Y.X.W. and T.S. assembled the genome. Q.L., Z.W.W., Y.X.W. and T.Y.T. performed gene annotation and supported the software. Q.L., T.S. and J.S.H.H. wrote the manuscript. All authors contributed and approved the final manuscript.
Code availability
Parameters of software tools involved in the methods are described below:1) Fastp: version 0.23.2, default parameters;2) Kmerfreq_AR: version 2.0.4, parameters: (k-mer size of 17);3) Jellyfish: version 2.2.6, parameters: (count -m 17 -s 10 G -t 10 -C);4) GenomeScope: version 2.0, parameters: (k-mer size of 17, read length of 100, maximum k-mer coverage of 1000);5) NextDenovo: version 2.4.0, parameters: (read_cutoff = 3k, seed_cutoff = 27k, blocksize = 5 g);6) wtdbg 2.huge: version 1.2.8, parameters: (wtdbg-1.2.8 -k 0 -p 21 -S 2, wtdbg-cns -c 0 -k 13, kbm-1.2.8 -k 0 -p 19 -S 2 -O 0, wtdbg-cns -k 11 -c 3);7) SMARTdenovo: version 1.0.0, parameters: (-c 3 and -k 11);8) NextPolish: version 1.01, default parameters;9) Bowtie2: version 2.3.2, parameters: (-end-to-end,–very-sensitive –L 30);10) Juicer: version 2.0, default parameters;11) HiC-Pro: version 2.7.8, default parameters;12) LACHESIS: latest version, parameters: (CLUSTER MIN RE SITES = 100; CLUSTER MAX LINK DENSITY = 2.5; CLUSTER NONINFORMATIVE RATIO = 1.4; ORDER MIN N RES IN TRUNK = 60; ORDER MIN N RES IN SHREDS = 60);13) BUSCO: version 5.4.6, parameters: (embryophyta_odb10);14) EDTA: version 1.7.0, parameters: (sudo docker run -it -v $PWD:/in -w /in oushujun/edta:1.9.5);15) LTRharvest: lastest version, parameters: (-minlenltr 100 -maxlenltr 7000 -mintsd 4 -maxtsd 6 -motif TGCA -motifmis 1 -similar 85 -vic 10 -seed 20 -seqids yes);16) LTR_FINDER_parallel: version 1.2, default parameters;17) LTR_retriever: version 2.9.0, default parameters;18) Generic Repeat Finder: version 1.7.0, default parameters;19) TIR-Learner: version 1.7.0, default parameters;20) HelitronScanner: version 1. 0, default parameters;21) RepeatModeler: version 2.0.2a, default parameters;22) RepeatMasker: version 4.1.1, parameters: (-pa 30 -lib –no_is -poly -html -gff -dir masker);23) TEsorter: version 1.1.4, default parameters;24) Augustus: version 3.4.0, default parameters;25) GeMoMa: version 1.6.1, default parameters;26) TransDecoder: version 5.5.0, parameters: (-G universal, -m 100);27) EVidenceModeler: version 1.1.1, default parameters;28) TransposonPSI: version 1.0.0, default parameters;29) BLASTP: version 2.10.1, parameters: (-outfmt 6, -evalue 1e-15);30) eggNOG: version 5.0, default parameters;31) Blast2GO: version 1.44, default parameters;32) PlantTFDB: version 5.0, default parameters;33) CAFE: version 4.2.1, default parameters;34) Rfam library: version 11.0, default parameters;35) tRNAscan-SE: version 2.0, default parameters;36) Minigraph2: version 2.25 (r1173), parameters: (-ax asm5);37) D-GENIES: version 1.5.0, default parameters;
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Qing Liu, Email: liuqing@scib.ac.cn.
John Seymour Heslop-Harrison, Email: phh4@le.ac.uk.
Supplementary information
The online version contains supplementary material available at 10.1038/s41597-024-03248-6.
References
- 1.Grundy MML, Fardet A, Tosh SM, Rich GT, Wilde PJ. Processing of oat: the impact on oat’s cholesterol lowering effect. Food Funct. 2018;9:1328–1343. doi: 10.1039/C7FO02006F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu KS. Comparison of lipid content and fatty acid composition and their distribution within seeds of 5 small grain species. J. Food Sci. 2011;76:C334–C342. doi: 10.1111/j.1750-3841.2010.02038.x. [DOI] [PubMed] [Google Scholar]
- 3.White DA, Fisk ID, Gray DA. Characterisation of oat (Avena sativa L.) oil bodies and intrinsically associated E-vitamers. J. Cereal Sci. 2006;43:244–249. doi: 10.1016/j.jcs.2005.10.002. [DOI] [Google Scholar]
- 4.Yang Z, et al. Oat: current state and challenges in plant-based food applications. Trends Food Sci. Technol. 2023;134:56–71. doi: 10.1016/j.tifs.2023.02.017. [DOI] [Google Scholar]
- 5.Kamal N, et al. The mosaic oat genome gives insights into a uniquely healthy cereal crop. Nature. 2022;606:113–119. doi: 10.1038/s41586-022-04732-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ouyang S, et al. The TIGR rice genome annotation resource: improvements and new features. Nucleic Acids Res. 2007;35:D883–D887. doi: 10.1093/nar/gkl976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCormick RF, et al. The Sorghum bicolor reference genome: improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J. 2018;93:338–354. doi: 10.1111/tpj.13781. [DOI] [PubMed] [Google Scholar]
- 8.Yang ZR, et al. A mini foxtail millet with an Arabidopsis-like life cycle as a C4 model system. Nat. Plants. 2020;6:1167–1178. doi: 10.1038/s41477-020-0747-7. [DOI] [PubMed] [Google Scholar]
- 9.Peng YY, et al. Reference genome assemblies reveal the origin and evolution of allohexaploid oat. Nat. Genet. 2022;54:1248–1258. doi: 10.1038/s41588-022-01127-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Q, et al. Genome-wide expansion and reorganization during grass evolution: from 30 Mb chromosomes in rice and Brachypodium to 550 Mb in Avena. BMC Plant Biol. 2023;23:627. doi: 10.1186/s12870-023-04644-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saini, P. et al. Disease Resistance in Crop Plants: Molecular, Genetic and Genomic Perspectives (ed. Wani, S. H.) Ch. 9 (Springer Nature, 2019).
- 12.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 13.Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts, and proteins. Nucleic Acids Res. 2005;33:D501–D504. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huerta-Cepas J, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;44:D309–D314. doi: 10.1093/nar/gky1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finn RD, et al. The Pfam protein family’s database. Nucleic Acids Res. 2014;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kristensen DM, et al. A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics. 2010;26:1481–1487. doi: 10.1093/bioinformatics/btq229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashburner M, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2001;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tatusov RL, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanehisa M, et al. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51:D587–D592. doi: 10.1093/nar/gkac963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin J, et al. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2016;45:D1040–D1045. doi: 10.1093/nar/gkw982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels. 2013;6:41. doi: 10.1186/1754-6834-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo R, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020;11:1–10. doi: 10.1038/s41467-020-14998-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruan J, Li H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods. 2020;17:155–158. doi: 10.1038/s41592-019-0669-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu H, Wu S, Li A, Ruan J. SMARTdenovo: a de novo assembler using long noisy reads. GigaByte. 2021;15:1–9. doi: 10.46471/gigabyte.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu J, Fan JP, Sun ZY, Liu SL. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 2020;36:2253–2255. doi: 10.1093/bioinformatics/btz891. [DOI] [PubMed] [Google Scholar]
- 30.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Durand NC, et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016;3:95–98. doi: 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Servant N, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burton JN, et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013;31:1119–1125. doi: 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ou SJ, Jiang N. LTR_FINDER_parallel: parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mob. DNA. 2019;10:48. doi: 10.1186/s13100-019-0193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellinghaus D, Kurtz S, Willhoeft U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics. 2008;9:18. doi: 10.1186/1471-2105-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ou SJ, Jiang N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018;176:1410–1422. doi: 10.1104/pp.17.01310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi JM, Liang C. Generic repeat finder: a high-sensitivity tool for genome-wide de novo repeat detection. Plant Physiol. 2019;180:1803–1815. doi: 10.1104/pp.19.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su W, Gu X, Peterson T. TIR-Learner, a new ensemble method for TIR transposable element annotation, provides evidence for abundant new transposable elements in the maize genome. Mol. Plant. 2016;12:447–460. doi: 10.1016/j.molp.2019.02.008. [DOI] [PubMed] [Google Scholar]
- 39.Xiong W, He LM, Lai JS, Dooner HK, Du CG. HelitronScanner uncovers a large overlooked cache of Helitron transposons in many plant genomes. Proc. Natl. Acad. Sci. USA. 2014;111:10263–10268. doi: 10.1073/pnas.1410068111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flynn JM, et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA. 2020;117:9451–9457. doi: 10.1073/pnas.1921046117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tarailo-Graovac M, Chen NS. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics. 2009;4:1–14. doi: 10.1002/0471250953.bi0410s25. [DOI] [PubMed] [Google Scholar]
- 42.Zhang RG, et al. TEsorter: an accurate and fast method to classify LTR-retrotransposons in plant genomes. Hortic. Res. 2022;9:uhac017. doi: 10.1093/hr/uhac017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yandell M, Ence D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012;13:329–342. doi: 10.1038/nrg3174. [DOI] [PubMed] [Google Scholar]
- 44.Stanke M, et al. Augustus: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34:W435–W439. doi: 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keilwagen J, Hartung F, Grau J. GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol. Biol. 2019;1962:161–177. doi: 10.1007/978-1-4939-9173-0_9. [DOI] [PubMed] [Google Scholar]
- 46.The Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 47.International Brachypodium Initiative Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463:763–768. doi: 10.1038/nature08747. [DOI] [PubMed] [Google Scholar]
- 48.Mascher M, et al. A chromosome conformation capture ordered sequence of the barley genome. Nature. 2017;544:427–433. doi: 10.1038/nature22043. [DOI] [PubMed] [Google Scholar]
- 49.International Wheat Genome Sequencing Consortium (IWGSC) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science. 2018;361:eaar7191. doi: 10.1126/science.aar7191. [DOI] [PubMed] [Google Scholar]
- 50.Schnable PS, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 51.Haas BJ, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haas BJ, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9:1–22. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lavigne R, Seto D, Mahadevan P, Ackermann HW, Kropinski AM. Unifying classical and molecular taxonomic classification: analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008;159:406–414. doi: 10.1016/j.resmic.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 54.Conesa A, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 55.Griffiths-Jones S, et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–D124. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chan PP, Lin BY, Mak AJ, Lowe TM. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021;49:9077–9096. doi: 10.1093/nar/gkab688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H, Feng X, Chu C. The design and construction of reference pangenome graphs with minigraph. Genome Biol. 2020;21:1–19. doi: 10.1186/s13059-020-02168-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cabanettes F, Klopp C. D-GENIES: dot plot large genomes in an interactive, efficient and simple way. Peer J. 2018;6:e4958. doi: 10.7717/peerj.4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.2022. NCBI Sequence Read Archive. SRP375311
- 60.2023. NCBI RNA Sequencing Data. SRP433645
- 61.2023. NCBI Assembly. GCA_030063025.1
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- 2022. NCBI Sequence Read Archive. SRP375311
- 2023. NCBI RNA Sequencing Data. SRP433645
- 2023. NCBI Assembly. GCA_030063025.1
Supplementary Materials
Data Availability Statement
Parameters of software tools involved in the methods are described below:1) Fastp: version 0.23.2, default parameters;2) Kmerfreq_AR: version 2.0.4, parameters: (k-mer size of 17);3) Jellyfish: version 2.2.6, parameters: (count -m 17 -s 10 G -t 10 -C);4) GenomeScope: version 2.0, parameters: (k-mer size of 17, read length of 100, maximum k-mer coverage of 1000);5) NextDenovo: version 2.4.0, parameters: (read_cutoff = 3k, seed_cutoff = 27k, blocksize = 5 g);6) wtdbg 2.huge: version 1.2.8, parameters: (wtdbg-1.2.8 -k 0 -p 21 -S 2, wtdbg-cns -c 0 -k 13, kbm-1.2.8 -k 0 -p 19 -S 2 -O 0, wtdbg-cns -k 11 -c 3);7) SMARTdenovo: version 1.0.0, parameters: (-c 3 and -k 11);8) NextPolish: version 1.01, default parameters;9) Bowtie2: version 2.3.2, parameters: (-end-to-end,–very-sensitive –L 30);10) Juicer: version 2.0, default parameters;11) HiC-Pro: version 2.7.8, default parameters;12) LACHESIS: latest version, parameters: (CLUSTER MIN RE SITES = 100; CLUSTER MAX LINK DENSITY = 2.5; CLUSTER NONINFORMATIVE RATIO = 1.4; ORDER MIN N RES IN TRUNK = 60; ORDER MIN N RES IN SHREDS = 60);13) BUSCO: version 5.4.6, parameters: (embryophyta_odb10);14) EDTA: version 1.7.0, parameters: (sudo docker run -it -v $PWD:/in -w /in oushujun/edta:1.9.5);15) LTRharvest: lastest version, parameters: (-minlenltr 100 -maxlenltr 7000 -mintsd 4 -maxtsd 6 -motif TGCA -motifmis 1 -similar 85 -vic 10 -seed 20 -seqids yes);16) LTR_FINDER_parallel: version 1.2, default parameters;17) LTR_retriever: version 2.9.0, default parameters;18) Generic Repeat Finder: version 1.7.0, default parameters;19) TIR-Learner: version 1.7.0, default parameters;20) HelitronScanner: version 1. 0, default parameters;21) RepeatModeler: version 2.0.2a, default parameters;22) RepeatMasker: version 4.1.1, parameters: (-pa 30 -lib –no_is -poly -html -gff -dir masker);23) TEsorter: version 1.1.4, default parameters;24) Augustus: version 3.4.0, default parameters;25) GeMoMa: version 1.6.1, default parameters;26) TransDecoder: version 5.5.0, parameters: (-G universal, -m 100);27) EVidenceModeler: version 1.1.1, default parameters;28) TransposonPSI: version 1.0.0, default parameters;29) BLASTP: version 2.10.1, parameters: (-outfmt 6, -evalue 1e-15);30) eggNOG: version 5.0, default parameters;31) Blast2GO: version 1.44, default parameters;32) PlantTFDB: version 5.0, default parameters;33) CAFE: version 4.2.1, default parameters;34) Rfam library: version 11.0, default parameters;35) tRNAscan-SE: version 2.0, default parameters;36) Minigraph2: version 2.25 (r1173), parameters: (-ax asm5);37) D-GENIES: version 1.5.0, default parameters;