

Abstract
CO2 hydrogenation over Rh catalysts comprises multiple reaction pathways, presenting a wide range of possible intermediates and end products, with selectivity toward either CO or methane being of particular interest. We investigate in detail the reaction mechanism of CO2 hydrogenation to the single-carbon (C1) products on the Rh(111) facet by performing periodic density functional theory (DFT) calculations and kinetic Monte Carlo (kMC) simulations, which account for the adsorbate interactions through a cluster expansion approach. We observe that Rh readily facilitates the dissociation of hydrogen, thus contributing to the subsequent hydrogenation processes. The reverse water–gas shift (RWGS) reaction occurs via three different reaction pathways, with CO hydrogenation to the COH intermediate being a key step for CO2 methanation. The effects of temperature, pressure, and the composition ratio of the gas reactant feed are considered. Temperature plays a pivotal role in determining the surface coverage and adsorbate composition, with competitive adsorption between CO and H species influencing the product distribution. The observed adlayer configurations indicate that the adsorbed CO species are separated by adsorbed H atoms, with a high ratio of H to CO coverage on the Rh(111) surface being essential to promote CO2 methanation.
Keywords: CO2 hydrogenation, density functional theory, kinetic Monte Carlo, reaction pathways, product selectivity, rhodium catalyst, temperature effect
Introduction
Energy-efficient catalytic CO2 conversion using renewable energy has attracted considerable attention as a potentially feasible means to mitigate CO2 emissions and produce commodity fuels and chemicals.1−3 It is thermodynamically feasible to hydrogenate CO2 to produce hydrocarbons (olefins, liquid hydrocarbons, and aromatics) and oxygenates (alcohols and dimethyl ether).4,5 One of the most important products is methane (CH4), which can be injected into existing natural gas infrastructure for distribution and storage, and for long-term chemical storage of electricity produced from renewable sources.6,7 In addition, methane can also be used as a feedstock material for the production of chemicals and fuels, including alkenes, gasoline, and aromatic compounds.8−10
Methane can be obtained via the well-known Sabatier reaction,11 a highly exothermic process that nonetheless requires very active catalysts to alleviate the high kinetic barriers arising from the eight-electron reduction of CO2 involved in the reaction process.12,13 Transition metals including Ni,14−17 Rh,18−20 Ru,21−24 and Pd25−27 have been used as catalysts for CO2 methanation, with Ni-based catalysts in particular exhibiting excellent CO2 hydrogenation activity at elevated temperatures, with high CH4 selectivity, although conversion rates are much lower at lower temperatures.11 In contrast, Rh-based catalysts show almost 100% CH4 selectivity and extremely high production rates even at lower temperatures.28,29 Supported Rh catalysts are normally used in experimental studies30 and the low-index Rh(111) facet is usually selected to explore the role of the Rh in catalytic reactions,31−36 since the Rh(111) surface is the most stable facet and therefore accounts for the largest surface area fraction in synthesized Rh particles.37−39 However, the mechanistic routes for CO2 hydrogenation are multiple and complex, with the precise nature of the intermediates remaining poorly understood.5,12,40
There are two categories of reaction mechanism proposed for CO2 methanation: dissociative, whereby C–O bond cleavage takes place before hydrogenation; and associative, in which hydrogenation takes place before C–O bond cleavage. In the former case, CO2 undergoes dissociative adsorption, resulting in the formation of CO* and O* species co-adsorbed on the surface, followed by CO* dissociation to O* and C* species, which subsequently undergo the hydrogenation to methane.41,42 In the latter case, CO2* and CO* can be directly hydrogenated to HXCOY* and HXCOYH* species, with subsequent C–O bond cleavage yielding CHx* intermediates for further hydrogenation to methane.43−48
Several experimental and computational studies have demonstrated the feasibility of dissociation of chemisorbed CO2 into CO species over Rh catalysts.19,28,49−52 Somorjai and co-workers found that CO2 appeared to dissociate to CO upon adsorption on Rh(111) and (100) surfaces, as indicated by the identical ordering and desorption characteristics of these two molecules.51 In addition, by combining scanning tunneling microscopy (STM), X-ray photoelectron spectroscopy (XPS) at near-ambient pressure (NAP), and computational techniques, Park and co-workers observed the cleavage of the O–CO bond on Rh(111) surfaces at room temperature.53 However, it has been found that the subsequent CO* dissociation on Rh catalysts is much less significant than for Ni and Ru catalysts.54−57 Yates et al. have shown that the Rh(111) facet is inactive for CO* dissociation below 870 K at low pressures, by means of isotopic exchange measurements.58 In addition, Solymosi et al. concluded that the adsorbed CO* could undergo dissociation to a limited extent on a supported Rh catalyst above 473 K at high pressures, which was attributed to the influence of the support.54,59 Under hydrogenating conditions, the adsorbed CO* can either desorb to the gas phase, with the remaining O* species being hydrogenated to water to complete the cycle for the reverse water–gas shift (RWGS) mechanism,60 or interact with co-adsorbed H* to form intermediate complexes.61 Jacquemin et al.62 concluded that adsorbed CO2 can undergo dissociation on a Rh/γ-Al2O3 catalyst, with subsequent reaction of CO* with H2, as revealed by in situ DRIFTS experiments. Karelovic and Ruiz studied the reaction mechanisms for CO2 hydrogenation over the supported Rh catalysts at low temperatures and proposed that CO is an important intermediate, with the CO* dissociation barrier being comparable to that of the overall reaction.18,19 Recently, several theoretical studies of the reaction mechanism for Rh-catalyzed CO2 methanation have been published; Kwon and co-workers63 applied DFT techniques to study the reaction pathways for CO2 hydrogenation on Rh(111), demonstrating that Rh can facilitate the direct dissociation of CO2 and that the lowest-energy reaction pathway for CO* hydrogenation to methane was via the formation and dissociation of HCO*, with HCOH* formation and dissociation as a plausible alternative. Similarly, DFT calculations were used to investigate the rate-determining step for CO2 methanation on the Rh(100) surface, which showed that hydrogen can assist the dissociation of CO*, via hydrogenation to CHO* and its subsequent dissociation to CH* and O*.61 In addition, ab initio molecular dynamics was applied to study CO activation on Rh surfaces and concluded that CO* more readily undergoes hydrogenation than dissociation.64 Furthermore, it was found that the strong Rh–CO interaction can impede CO hydrogenation, thus slowing down the overall process.33 The exact role of the intermediate species generated during the reaction process has, however, not been conclusively identified; they may be spectators (having only a minor influence on the mechanistic path), or key reaction intermediates (playing an important role in the reaction mechanism). As a result, further fundamental studies of the reaction mechanism for CO2 methanation over the Rh-based catalysts are necessary.
Density Functional Theory (DFT), combined with kinetic Monte Carlo (kMC) simulations, are powerful tools for exploring reaction mechanisms under realistic conditions, which can complement operando experimental techniques, and provide a full mechanistic description of CO2 conversion on the catalyst.65−69 In this study, we apply a multiscale approach to investigate the mechanistic pathways of CO2 hydrogenation on the Rh(111) surface, first by calculating activation and reaction energies for all elementary processes from DFT simulations. Secondly, we implement the DFT-calculated energies within the kMC method and are therefore able to identify the most feasible reaction pathways and product selectivity. The simulations incorporate interaction energies for the two-body terms used in the cluster expansion model, along with the rate constants for 52 reversible surface reactions, 8 reversible adsorption processes (involving H2 dissociative adsorption), as well as H atom diffusion process. We investigate the lattice configurations under realistic conditions, as well as the effect of temperature, pressure, and the composition ratio of the gaseous reactant mixtures on the distribution of products over the Rh catalysts.
Computational Methods
Plane-Wave DFT Calculations
Plane-wave DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP) code70,71 in order to explore the reaction mechanisms for CO2 hydrogenation over the Rh(111) surface. Inner electrons were treated as projector-augmented waves (PAW),72 and the valence states were expanded in plane waves with a cutoff energy of 450 eV. Table S1 reports a comparison between the adsorption energies calculated with cutoff energy of 450 and 550 eV, with no significant difference being observed. Hence, a cutoff energy of 450 eV was deemed to be sufficient for the expansion of the valence states in plane waves. The Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional was used throughout the study,73 and a dispersion correction was applied using the D3 scheme,74 in order to account for weak van der Waals interactions. The adsorption energies of the species accounting for vibrational zero-point energies have been calculated with PBE and PBE+D3 methods and compared (Table S1), which confirmed the importance of dispersion correction for molecular adsorption processes. The optimized bulk Rh lattice parameter was determined to be 3.83 Å, in good agreement with the experimental value of 3.79 Å.75
The slab model used for the Rh(111) facet consisted of six layers, separated by 18 Å of vacuum in the z-direction, to avoid spurious interactions between surfaces in adjacent periodic cells. For the purposes of modeling adsorption and reaction processes, a p(3 × 3) supercell was used for the Rh(111) surface. A Monkhorst–Pack k-point sampling scheme was determined commensurately with the slab supercell dimensions,76 with a k-grid of dimensions (3 × 3 × 1) applied. During structural optimization, the top four Rh layers were allowed to relax, while the bottom two were fixed at their optimized bulk lattice positions. A dipole correction was applied for all surface calculations to eliminate any spurious electrostatic interactions arising from the asymmetric relaxation of the surface slab. The six-layer slab model was determined to be suitable from test calculations exploring the relationship between the surface energy and the number of slab layers. Further details can be found in Section S2 in the Supporting Information.
Structural optimizations were regarded as being sufficiently well-converged when all ionic forces were minimized to within 0.01 eV Å–1. The SCF energy convergence threshold for electronic structure optimization was set to 10–5 eV. To explore the elementary processes involved in the reaction mechanism for CO2 hydrogenation, the activation energies for each process were calculated by performing Climbing Image Nudged Elastic Band (CI-NEB)77,78 and Improved Dimer Method (IDM)79,80 calculations, with atomic forces converged to within 0.03 eV Å–1. Vibrational analysis was used to confirm that the obtained transition state represented a true saddle point, indicated by the presence of a single imaginary vibrational frequency, corresponding to the unstable mode.
The activation energy (Ea) and reaction energy (Er) for the elementary steps were obtained from the following:
| 1 |
| 2 |
where EIS, ETS, and EFS are the energies of the optimized initial state, transition state, and final state for each step in the reaction mechanism.
The adsorption energy (Eads) was determined from the following:
| 3 |
where Eads/slab and Eslab are the calculated energies of the optimized surfaces with and without adsorbate, respectively, and Egas is the energy of the optimized gas-phase adsorbate.
The zero-point energy (ZPE) was calculated from the vibrational frequency according to the following:
| 4 |
where h is Planck’s constant and νi is the vibrational frequency.
kMC Simulations
In our study, kMC simulations were performed using the Graph-Theoretical kinetic Monte Carlo (GT-kMC)81,82 approach, as implemented in the Zacros code,83 which has been successfully applied to investigate heterogeneous catalysis reaction on metal surfaces.67−69,81 For example, kMC simulations were applied to show the different reaction orders with respect to O coverage at high and low temperatures for CO oxidation on the Pd(111) surface.67 To account for the impact of lateral interactions, the cluster expansion model has been used to describe the contributions of single- and multibody adsorbates on the surface, allowing the determination of spatial correlations and coverage-dependent activation barriers. The impact of lateral interaction on activation barriers for elementary processes is parametrized in terms of a Brønsted–Evans–Polanyi (BEP) relationship, allowing activation barriers to be adjusted dynamically with surface coverage,82 which was applied in our simulations. For all elementary processes, a proximity factor of ω = 0.5 was applied. The impact of the choice of ω value is discussed in Section S4 in the Supporting Information, and simulations showed that reasonable variations in this parameter have no significant impact on the product distribution. Further information on the cluster expansion model can also be found in Sections S3 and S4 in the Supporting Information. Furthermore, since processes such as adsorption, desorption, and diffusion, typically have rate constants many orders of magnitude higher than those of rate-limiting surface reactions, they will typically be sampled much more frequently in a multiple time-scale-disparate simulation,84 thus hindering kMC time progression. Hence, Zacros version 2.0 implements a temporal acceleration scheme developed by Chatterjee and Voter,85,86 which automatically scales all fast quasi-equilibrated processes. This achieves dynamic detection of time-scale separation and dynamic scaling of the kinetic constants to accelerate the simulation. Hence, our simulations have the advantage of balancing the occurrence frequencies of both fast (like CO2 adsorption, H2 dissociative adsorption, and H atom diffusion) and slow elementary events (like CO2 or CO hydrogenation or dissociation), reducing the computational expense required to achieve useful kMC time progression. The rate constants for the elementary processes were estimated from the Arrhenius expression, including the DFT-calculated activation energies and the pre-exponential factors, resulting from the partition functions based on the DFT predicted vibrational frequencies. The corresponding mathematical expressions can be found in Section S4. All of the reaction steps are treated as reversible processes, including the adsorption and desorption processes. Further details of the general methodology applied can be found in previously published articles.82,87
The reaction network comprises 52 reversible surface reactions, 8 reversible adsorption processes (including H2 dissociative adsorption), as well as an H atom diffusion process over the Rh(111) surface. The lattice model consists of three kinds of active sites, including one top site, three bridge sites, and two hollow sites (with fcc and hcp hollow sites being essentially equivalent) within the Rh(111) surface unit cell, as shown in Figure 1, comprising a total of 15 000 sites on the 50 × 50 kMC lattice. The model comprises 8 gaseous species and 25 surface species, which can occupy one, two, or three neighboring adsorption sites as determined by DFT calculations. The cluster expansion model considers the lateral interactions for the most relevant co-adsorbed species at the nearest neighboring adsorption sites, comprising a total of 57 pairwise interactions, as shown in Table S3–1; the evaluation of additional lateral interactions between CO and the remaining species are provided in Section S3 in the Supporting Information, and validate the cluster expansion model applied. The input gas consisted of a mixture of H2 and CO2 in a ratio of 4:1 over the clean Rh(111) surface at the temperatures of 473.15 and 573.15 K under a pressure of 1 bar, allowing us to consider the effect of temperature on the activity and selectivity. In addition, several kMC simulations were performed using different seeds to minimize errors.
Figure 1.
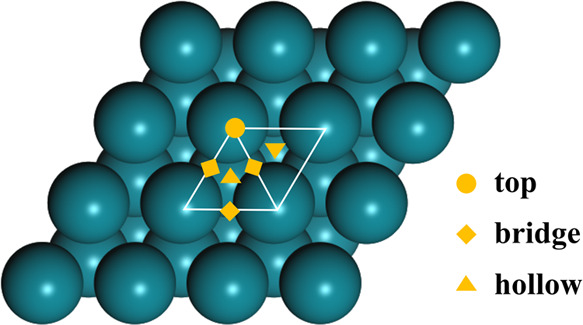
Top view of the Rh(111) surface (teal spheres represent Rh atoms). The defined adsorption sites used in the kMC simulation are indicated by yellow icons. The white parallelogram represents the surface unit cell.
Results and Discussion
DFT Calculations
CO2 Adsorption
The adsorption energies and geometric parameters for different CO2 adsorption modes over Rh(111) are shown in Table 1. Two distinct CO2 adsorption geometries were identified: a linear, physisorbed, CO2 species, and a bent, chemisorbed, CO2 species. CO2 adsorption is slightly exothermic, with ZPE-corrected adsorption energies of −0.23 and −0.33 eV for the physisorbed and chemisorbed CO2, respectively. This value is comparable to the calculated CO2 chemisorption energy (−0.39 eV) over the Rh(111) surface as reported by Kim et al.53 The optimized structure for physisorbed CO2 shows no significant changes compared to the geometry of gaseous CO2. However, for the chemisorbed CO2 species, a significant distortion of CO2 appears, with the O–C–O angle shrinking to 134.5°, and the C–O distance modestly lengthening, implying the weakening of the C=O bonds, corresponding to the activation of CO2 over Rh(111). CO2 chemisorption on the Rh(111) surface thus presents a bent geometry, with one O atom binding to a metal atom, and the C atom binding to the nearest metal atom, which is similar to the results obtained previously.88−93
Table 1. ZPE-Corrected Adsorption Energy (Eads) for Physisorbed and Chemisorbed CO2 over the Rh(111) Surface, with C–Rh Distance (d(C–Rh)), C–O Distance (d(C–O)), and O–C–O Angle (∠(O–C–O)), as well as the Bader Charge Difference (β) for the Physisorbed and Chemisorbed CO2.
| species | Eads (eV) | d(C–Rh) (Å) | d(C–O) (Å) | ∠(O–C–O) (deg) | β (|e|) |
|---|---|---|---|---|---|
| Phys-CO2 | –0.23 | 3.38 | 1.18 | 179.6 | –0.05 |
| Chem-CO2 | –0.33 | 2.06 | 1.22, 1.28 | 134.5 | –0.46 |
To investigate CO2 activation further, a Bader charge analysis was performed, showing that there is a charge transfer of 0.46 e– from the Rh surface to chemisorbed CO2, while the value is only 0.05 e– for physisorbed CO2. In other metallic catalysts, Higham et al. observed an increase in charge transfer of 0.70 e– from the physisorbed to the chemisorbed CO2 on both the Cu(100) and (110) surfaces,93 although in this study the activated CO2 species was metastable, in contrast to the behavior reported in the present work for Rh. In addition, Mulliken charge analysis of the physisorbed and chemisorbed CO2 on the Pd(111), (110), and (100) surfaces was studied by Kowalec et al., which indicates only a limited extent of CO2 reduction for physisorbed CO2 (0.04, 0.10, and 0.11 e– on the Pd (111), (100), and (110) surfaces), and a much greater extent of CO2 reduction for the chemisorbed CO2 on the three Pd surfaces.92 In contrast, the increase of the charge transfer from the physisorbed to the chemisorbed CO2 on metal carbide catalysts is much greater, as Quesne et al. observed that the valence electron count for chemisorbed CO2 increased by one electron on the low-index surfaces of TiC, VC, ZrC and NbC catalysts,94 implying a much greater extent of CO2 reduction for these catalysts. Hence, the Bader charge analysis for CO2 adsorption on Rh(111) can be interpreted as indicating that the chemisorbed CO2 is partially reduced, with the charge accumulation being mainly localized on the C atom. The partial CO2 reduction process can be interpreted in terms of charge transfer from the Rh surface to the C=O π* antibonding orbitals of CO2 species (thus accounting for the modest increase in C–O bond length), which facilitates CO2 activation, and therefore the subsequent reaction of the activated CO2 (i.e., dissociation or hydrogenation). Similarly, by employing the experimental methods of NAP-STM and NAP-XPS, Kim et al.53 have directly observed that the linear geometry of CO2 gas molecules evolves into a chemically active bent structure over the Rh(111) surface, with changes of local charge density at the CO2–Rh(111) interface for the cleavage of C=O bond, thus corroborating our computational results.
H2 Dissociative Adsorption
In addition to CO2 adsorption and activation, H2 dissociative adsorption is a key prerequisite for any CO2 hydrogenation catalyst. Hence, it is necessary to establish the adsorption and dissociation behavior of H2 over the Rh catalyst surface. Previous studies have found that for H2 molecular adsorption, binding at the top site, oriented parallel to the metal surface is most stable, and that subsequent dissociation of H2 is more thermodynamically favorable than desorption on the Rh(100) surface.95 Additionally, in situ DRIFTS studies performed at 300 °C have shown that the dissociative adsorption of H2 readily proceeds on Rh catalysts.96
We found the ZPE-corrected physisorption energy of H2 to be −0.09 eV, with the molecule located 3.00 Å away from the Rh surface. However, the H–H bond is elongated from 0.76 to 0.96 Å when H2 chemisorbs at the top site of the Rh(111) surface, and the H–Rh distance is 1.66 Å, which is in agreement with the previously reported results.97 The calculated structures, ZPE-corrected relative energies, and geometric parameters for H2 adsorption and dissociation are reported in Figure 2. As noted, the adsorption of H2 at the top site results in considerable H–H bond elongation (d(H–H) = 0.96 Å), suggesting that the Rh catalyst readily promotes H2 activation, with full dissociation; the transition from the physisorbed H2 to the chemisorbed H2 at the top site has a negligible activation energy of 0.01 eV. To better understand the electronic structures for H2 dissociative adsorption at the top site, Density of States (DOS) and Crystal Orbital Hamilton Population (COHP) methods were applied to analyze the change of the H–H bond from physisorption to chemisorption, as described in Sections S5 and S6 in the Supporting Information. In addition, H atoms were found to adsorb more exothermically at the hollow sites on the Rh(111) surface, and the activation energy for H diffusion from the top site to the hollow site is only 0.03 eV, suggesting that the elongated H2 molecule at the top site readily dissociates to yield two H* species on the adjacent hollow sites. These results, therefore, clearly show that the Rh(111) surface promotes H2 dissociative adsorption, thus facilitating the subsequent hydrogenation processes.
Figure 2.

ZPE-corrected relative energies and the structures for the processes of H2 adsorption and dissociation. The transition states (T.S.) for H2 dissociative adsorption and H diffusion processes are also shown here. d(H–H) and d(H–Rh) represent the H–H distance and H–Rh distance, respectively. The teal and white spheres are the Rh and H atoms, respectively. The zero-energy state corresponds to physisorbed H2.
Reaction Network for CO2 Hydrogenation
For CO2 hydrogenation, there are multiple, often overlapping, reaction pathways leading to product formation. Hence, all possible elementary processes are considered to obtain a complete reaction network (Figure 3). The favored product results from the most kinetically feasible (i.e., least energy-demanding) pathway proceeding via the most stable intermediates. To provide further insight into the reaction mechanisms, the ZPE-corrected activation energies (Ea) and reaction energies (Er) for all of the possible relevant reaction pathways are summarized in Table 2.
Figure 3.

Reaction network for CO2 hydrogenation into C1 species, with possible products comprising gaseous CO, CH4, CH3OH, HCOOH, and CH2O. The black, blue, and red lines signify the adsorption and desorption processes, hydrogenation and dehydrogenation processes, and reversible decomposition processes, respectively. Species appearing in more than one mechanistic pathway are identified with colored labels for clarity (e.g., adsorbed HCOOH is denoted by text in green). All species are adsorbed on the Rh(111) surface, except for those marked with (g), which indicates gaseous entities.
Table 2. Calculated ZPE-Corrected Activation Energies (Ea) and Reaction Energies (Er) for the Elementary Steps.
| elementary steps | ZPE-corrected Ea (eV) | ZPE-corrected Er (eV) | |
|---|---|---|---|
| R1 | CO2 + * ↔ CO + O | 0.45 | –0.98 |
| R2 | CO + * ↔ C + O | 2.74 | 0.93 |
| R3 | CO + H ↔ COH | 1.27 | 0.71 |
| R4 | CO + H ↔ HCO | 1.35 | 1.21 |
| R5 | COH + H ↔ HCOH | 1.31 | 1.07 |
| R6 | COH ↔ C + OH | 1.68 | 0.55 |
| R7 | HCO + H ↔ CH2O | 0.70 | 0.51 |
| R8 | HCO + H ↔ HCOH | 0.69 | 0.56 |
| R9 | HCO + * ↔ CH + O | 1.17 | –0.49 |
| R10 | CH2O + H ↔ CH2OH | 0.75 | 0.27 |
| R11 | CH2O + H ↔ CH3O | 0.74 | 0.37 |
| R12 | CH2O + * ↔ CH2 + O | 0.95 | –0.64 |
| R13 | HCOH + * ↔ CH + OH | 0.47 | –0.72 |
| R14 | HCOH + H ↔ CH2OH | 0.59 | 0.21 |
| R15 | CH2OH ↔ CH2 + OH | 0.75 | –0.58 |
| R16 | CH3O + * ↔ CH3 + O | 1.12 | –0.64 |
| R17 | C + H ↔ CH | 0.67 | –0.20 |
| R18 | CH + H ↔ CH2 | 0.64 | 0.36 |
| R19 | CH2 + H ↔ CH3 | 0.63 | 0.38 |
| R20 | CH3 + H ↔ CH4 | 0.45 | 0.23 |
| R21 | CO2 + H ↔ COOH | 0.72 | –0.03 |
| R22 | COOH + * ↔ CO + OH | 0.36 | –0.62 |
| R23 | COOH + H ↔ HCOOH | 1.34 | 0.51 |
| R24 | HCOOH + * ↔ HCO + OH | 0.42 | 0.09 |
| R25 | HCOOH + H ↔ H2COOH | 0.36 | 0.13 |
| R26 | H2COOH + * ↔ CH2O + OH | 0.44 | 0.00 |
| R27 | CO2 + H ↔ HCOO | 0.69 | –0.13 |
| R28 | HCOO + * ↔ HCO + O | 1.27 | 0.37 |
| R29 | HCOO + H ↔ HCOOH | 0.87 | 0.61 |
| R30 | HCOO + H ↔ H2COO | 2.24 | 1.56 |
| R31 | H2COO + H ↔ H2COOH | 0.53 | –0.35 |
| R32 | CH2OH + H ↔ CH3OH | 0.73 | 0.15 |
| R33 | CH3O + H ↔ CH3OH | 0.78 | 0.05 |
| R34 | CH3OH ↔ CH3+OH | 1.67 | –0.35 |
| R35 | O + H ↔ OH | 0.80 | 0.33 |
| R36 | OH + H ↔ H2O | 0.78 | 0.01 |
| R37 | OH + OH ↔ H2O + O | 0.33 | –0.32 |
| R38 | CO2 + OH ↔ COOH + O | 0.10 | –0.36 |
| R39 | CO2 + H2O ↔ HCOO + OH | 1.49 | –0.14 |
| R40 | CO2 + H2O ↔ COOH + OH | 0.43 | –0.04 |
| R41 | CO2 + OH ↔ HCOO + O | 1.51 | –0.46 |
| R42 | COOH + H ↔ COHOH | 0.70 | 0.51 |
| R43 | COHOH ↔ COH + OH | 0.73 | –0.43 |
| R44 | CO + H2O ↔ COH + OH | 1.01 | 0.69 |
| R45 | CO + CO ↔ CO2 + C | 2.92 | 1.90 |
| R46 | CO + COOH ↔ CO2 + COH | 0.74 | 0.73 |
| R47 | CO + HCOO ↔ CO2 + COH | 0.94 | 0.83 |
| R48 | CO + OH ↔ O + HCO | 1.87 | 0.88 |
| R49 | CO + H2O ↔ OH + HCO | 1.63 | 1.20 |
| R50 | HCOO + HCO ↔ HCOOH + CO | 0.35 | –0.61 |
| R51 | HCOO + HCO ↔ H2COO + CO | 0.95 | 0.35 |
| R52 | HCOOH + HCO ↔ H2COOH + CO | 0.42 | –0.62 |
| A1 | CH4 ↔ CH4(g) + * | 0.20 | 0.20 |
| A2 | CH3OH ↔ CH3OH(g) + * | 0.67 | 0.67 |
| A3 | CO ↔ CO(g) + * | 2.28 | 2.28 |
| A4 | CH2O ↔ CH2O(g) + * | 1.19 | 1.19 |
| A5 | HCOOH ↔ HCOOH(g) + * | 0.79 | 0.79 |
| A6 | H2O ↔ H2O(g) + * | 0.53 | 0.53 |
| A7 | CO2(g) + * ↔ CO2 | 0.00 | –0.33 |
| A8 | H2(g) + * ↔ H + H | 0.01 | –0.58 |
| D1 | H(top)+ * ↔ H(hollow) + * | 0.03 | –0.36 |
Reverse Water–Gas Shift (RWGS)
As shown in Figure 4, three different pathways for the RWGS reaction are considered: the redox, formate, and carboxyl mechanisms. We will address each of these mechanisms in turn, starting from adsorbed CO2.
Figure 4.
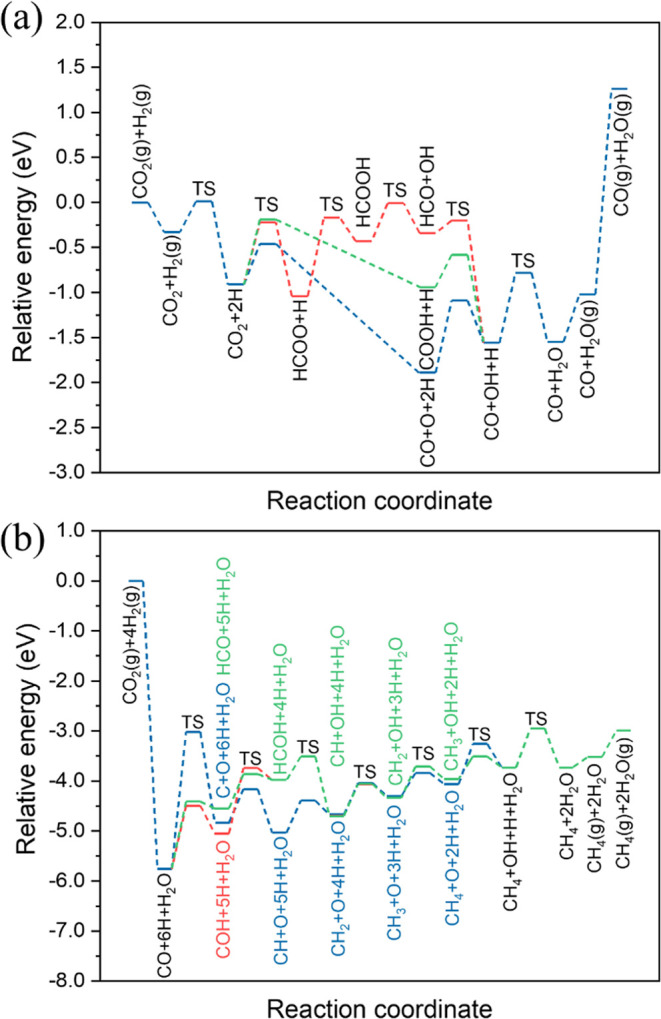
(a) Energy profiles for the RWGS reaction, including redox mechanism (blue line), formate mechanism (red line), and carboxyl mechanism (green line). (b) Energy profiles for CO2 methanation, including the pathways through the carbon (blue line), COH (red line), and HCO (green line). Transition states (TS) are labeled accordingly.
For the redox mechanism, CO2 undergoes direct dissociation to yield co-adsorbed CO and O, with this process being exothermic and having only a moderate activation barrier (Ea = 0.45 eV and Er = −0.98 eV). The surface O atom resulting from CO2 dissociation can react with the co-adsorbed H to produce OH, with this process having an activation energy of 0.80 eV and endothermic reaction energy of 0.33 eV. Subsequently, water can be produced by two pathways in all three mechanisms. The first is the direct hydrogenation of OH (Ea = 0.78 eV and Er = +0.01 eV) to form H2O. The second is the reaction of two co-adsorbed OH species (Ea = 0.33 eV and Er = −0.32 eV) to form H2O and adsorbed O, affording a lower activation barrier; hence, it is possible that OH disproportionation is the predominant mechanism by which water is formed.
For the formate mechanism, CO2 must first undergo hydrogenation, which can proceed via direct CO2 hydrogenation by co-adsorbed H, with this process being slightly exothermic and having a moderate activation barrier (Ea = 0.69 eV and Er = −0.13 eV), although the activation barrier is greater than for direct CO2 dissociation as discussed in the previous paragraph. Alternatively, CO2 hydrogenation could proceed via hydrogenation by OH or H2O; however, the calculations reveal that both of these processes have much higher activation barriers (Ea = 1.51 eV and Er = −0.46 eV for R41; Ea = 1.49 eV and Er = −0.14 eV for R39 in Table 2). Hence, it is likely that most formate on the Rh(111) surface originates from direct CO2 hydrogenation. HCOO can then subsequently undergo further hydrogenation or dissociation; the calculations suggest that HCOO hydrogenation to HCOOH (Ea = 0.87 eV and Er = +0.61 eV) is more kinetically accessible compared to both direct dissociation of HCOO into HCO and O (Ea = 1.27 eV and Er = +0.37 eV), and H2COO formation via HCOO hydrogenation (Ea = 2.24 eV and Er = +1.56 eV); although subsequent H2COO hydrogenation to H2COOH (Ea = 0.53 eV and Er = −0.35 eV) has a lower activation energy, the H2COO species is likely to be kinetically inaccessible due to the high activation barrier for its formation. The mechanism can then proceed via the dissociation of HCOOH to HCO and OH (Ea = 0.42 eV and Er = +0.09 eV), with the subsequent dissociation of HCO to produce CO having an even lower barrier and being highly exothermic (Ea = 0.14 eV and Er = −1.21 eV). Notably, both of these processes have lower activation barriers than the HCOOH desorption energy (0.79 eV), suggesting that formic acid is likely to undergo further reactive processes, rather than being desorbed to the gas phase. Formic acid may also undergo hydrogenation to yield the H2COOH intermediate, although subsequent processes will probably result in the formation of formaldehyde, and subsequently methoxy, as will be discussed later. Hence, formic acid is likely to be a key intermediate in forming CO by the formate mechanism.
Finally, the carboxyl mechanism involves first the formation of COOH from hydrogenation of CO2. Direct hydrogenation by co-adsorbed H, like the corresponding process for formate formation, is slightly exothermic and has a moderate activation barrier (Ea = 0.72 eV and Er = −0.03 eV). However, hydrogenation of CO2 to COOH via OH (Ea = 0.10 eV and Er = −0.36 eV) or H2O (Ea = 0.43 eV and Er = −0.04 eV) affords considerably lower activation barriers, potentially making the carboxyl RWGS pathway competitive with the redox mechanism already discussed. The mechanism proceeds via the dissociation of COOH to yield CO and OH, which has a modest activation barrier and is appreciably exothermic (Ea = 0.36 eV and Er = −0.62 eV). By contrast, COOH hydrogenation to HCOOH is highly activated and endothermic (Ea = 1.34 eV and Er = +0.51 eV), thus rendering the HCOOH dissociation mechanism to yield CO described above inaccessible from COOH.
In summary, the calculations suggest that the redox mechanism is the most likely RWGS pathway, in agreement with experimental results,51,53 although the calculations also suggest that CO2 hydrogenation to COOH via OH or H2O may render the carboxyl pathway a viable alternative. For all of the possible RWGS mechanisms described, it should be noted that the resulting CO is strongly bound to the Rh(111) surface, with an adsorption energy of −2.28 eV. The DFT-calculated adsorption energy at a temperature of 0 K and a coverage of 1/9 ML is more exothermic than the experimental value of −1.47 eV, which was determined at a temperature of 500 K and a coverage of 1/4 ML.98 This reflects the universal impact that surface coverage and temperature have on adsorption energies. The impact of surface coverage on the adsorption of CO on the Rh surface was corroborated by experimental investigations of the heat of CO adsorption on a reduced 3% Rh/Al2O3 varying with coverage, which varied from 195 kJ/mol at low coverage (θ = 0) to 103 kJ/mol at high coverage (θ = 1) for the linear CO species.99 Furthermore, the general tendency of the PBE functional to overestimate heats of adsorption on metal surfaces is well known,100−102 with adsorbates typically overbound by about 0.6 eV per adsorbate.103 Hence, the calculated adsorption energy is qualitatively consistent with the strong binding of CO on Rh(111), and quantitatively consistent with similar theoretical studies, as well as being commensurate with the experimentally determined values within the confines and limitations of the models and methods applied.51,104−106 The impact of CO binding energy on the reaction mechanisms and kMC product distribution will be discussed in more detail below and in Section S7 in the Supporting Information. Hence, the highly exothermic adsorption of CO on Rh(111) will therefore largely preclude its evolution to the gas phase, with CO undergoing either further dissociation or hydrogenation, to yield methanol or methane, which will be discussed in the following section.
Pathways for Methane Formation
For methane formation, we propose three mechanistic pathways starting from the intermediate CO produced from the RWGS reaction. They proceed through carbon, COH, and HCO, respectively (Figure 4b). The carbon hydrogenation pathway involves surface C species formed via CO dissociation (Ea = 2.74 eV and Er = +0.93 eV) and COH dissociation (Ea = 1.68 eV and Er = +0.55 eV); both of these processes have very high activation barriers, suggesting that surface atomic C species are unlikely to be formed or play a significant role in the overall methanation mechanism. Similarly, the process involving CH formation via HCO dissociation (Ea = 1.17 eV and Er = −0.49 eV) is also highly activated. The subsequent hydrogenation processes for surface C species, however, all have moderate activation barriers and are either modestly exo- or endothermic, proceeding via hydrogenation to CH (Ea = 0.67 eV and Er = −0.20 eV), CH2 (Ea = 0.64 eV and Er = +0.36 eV), CH3 (Ea = 0.63 eV and Er = +0.38 eV), and finally CH4 (Ea = 0.45 eV and Er = +0.23 eV). We note that while the calculations suggest that the surface C and CH species are unlikely to be formed via CO or HCO dissociation, the CH, CH2, and CH3 species may well be formed as a result of other processes, and their subsequent conversion to methane is likely to be accessible under typical conditions, as shown by the series of processes detailed above.
For the COH hydrogenation pathway, COH resulting from CO hydrogenation can undergo subsequent hydrogenation processes before C–O bond cleavage taking place to enable methane formation. While the direct hydrogenation of CO to COH is highly activated and moderately endothermic (Ea = 1.27 eV and Er = +0.71 eV), it is more feasible that COH species are formed via the COOH hydrogenation to COHOH (Ea = 0.70 eV and Er = +0.51 eV), which subsequently dissociates to COH (Ea = 0.73 eV and Er = −0.43 eV). Other alternative pathways to COH formation, such as the interactions of CO with H2O (Ea = 1.01 eV and Er = +0.69 eV), COOH (Ea = 0.74 eV and Er = +0.73 eV), or HCOO (Ea = 0.94 eV and Er = +0.83 eV), have higher activation barriers. The subsequent hydrogenation of COH to HCOH, however, has a higher activation barrier and is highly endothermic (Ea = 1.31 eV and Er = +1.07 eV). The resulting HCOH intermediate could then either dissociate to CH (Ea = 0.47 eV and Er = −0.72 eV) or undergo further hydrogenation to CH2OH (Ea = 0.59 eV and Er = +0.21 eV), with both of these processes having much lower activation barriers. In the event of HCOH dissociation, CH can be sequentially hydrogenated to the final product CH4, as has already been discussed within the context of the surface carbon mechanism for methane formation. If CH2OH is formed, the intermediate can then undergo dissociation to CH2 (Ea = 0.75 eV and Er = −0.58 eV), again followed by the further hydrogenation to the ultimate product CH4; hydrogenation of CH2OH to methanol will be discussed separately later in this work.
For the HCO pathway, HCO formation, as discussed previously, is likely to occur more readily via HCOOH decomposition (Ea = 0.42 eV and Er = +0.09 eV), compared to the much less accessible direct CO hydrogenation (Ea = 1.35 eV and Er = +1.21 eV). We also consider the interactions of CO with OH or H2O to yield HCO (CO+OH, Ea = 1.87 eV and Er = +0.88 eV; CO+H2O, Ea = 1.63 eV and Er = +1.20 eV), which are highly activated and endothermic. HCO can then undergo hydrogenation to formaldehyde (CH2O, Ea = 0.70 eV and Er = +0.51 eV), which can then undergo hydrogenation to methoxy (CH3O, Ea = 0.74 eV and Er = +0.37 eV), and CH2OH (Ea = 0.75 eV and Er = +0.27 eV). Methoxy can then undergo dissociation to yield CH3 (Ea = 1.12 eV and Er = −0.64 eV), and finally hydrogenation to CH4. For CH2OH species, the subsequent processes have been discussed for the COH hydrogenation mechanism. If CH2O undergoes dissociation (Ea = 0.95 eV and Er = −0.64 eV), the resulting CH2 can then undergo further hydrogenation to the ultimate product CH4, in a manner analogous to that already discussed for the carbon pathway. While the calculated activation barriers for HCO hydrogenation and CH3O dissociation are higher than those for surface C hydrogenation, the relative ease of formation of the HCO intermediates means that its subsequent hydrogenation is likely to be of greater importance for the overall methanation mechanism. For example, HCOH resulting from HCO hydrogenation (Ea = 0.69 eV and Er = +0.56 eV), is moderately activated and endothermic; and subsequent progress from HCOH to methane formation has been discussed within the context of the COH hydrogenation mechanism. In addition, HCO can react with other important co-adsorbed intermediates. HCO interacts with HCOO to yield HCOOH (Ea = 0.35 eV and Er = −0.61 eV) with a lower activation energy, compared with the formation of H2COO (Ea = 0.95 eV and Er = +0.35 eV). The further process involving HCOOH reacting with HCO to H2COOH (Ea = 0.42 eV and Er = −0.62 eV), has a moderate activation energy, which is also the case for HCOOH direct hydrogenation to H2COOH (Ea = 0.36 eV and Er = +0.13 eV). H2COOH subsequently dissociates thermoneutrally to yield CH2O (Ea = 0.44 eV and Er = 0.00 eV) with a lower activation energy, and further processes from CH2O to methane formation have been shown above.
In summary, the DFT results suggest that Rh(111) can facilitate the H2 dissociative adsorption to surface atomic H species, which can react with activated CO2 and CO for further hydrogenation. The RWGS reaction appears to occur predominantly via the redox mechanism (i.e., the dissociation of the adsorbed CO2 to the adsorbed CO). However, the most favorable pathway for methane formation appears to be via the HCOO and HCOOH intermediates, with CO2 hydrogenation to HCOO having an energy barrier of 0.69 eV and reaction energy of −0.13 eV, which is then followed by further hydrogenation to HCOOH (Ea = 0.87 eV and Er = +0.61 eV). HCOOH dissociation to yield HCO is slightly endothermic and has a modest activation barrier (Ea = 0.42 eV and Er = +0.09 eV). The adsorbed HCO can subsequently undergo hydrogenation to HCOH (Ea = 0.69 eV and Er = +0.56 eV), followed by its dissociation to CH (Ea = 0.47 eV and Er = −0.72 eV), which can be hydrogenated to the ultimate product methane. It should be noted that the intermediate HCO can either hydrogenate to HCOH or dissociate to CO. Given the numerous competing pathways, the extent to which other mechanisms contribute to methane formation remains unclear. Kinetic Monte Carlo techniques, however, can elucidate many subtleties in complex reaction mechanisms that are not obvious after initial analysis of the DFT results. Hence, we will return to this topic and discuss the competition between these mechanistic pathways as revealed by the kinetic Monte Carlo simulations in the corresponding section later.
Methanol Formation, Desorption, and Decomposition
As discussed in the preceding section, many of the intermediates relevant to the various mechanistic pathways for CO2 methanation are common to methanol formation. Methanol formation via the hydrogenation of CH2OH is only modestly endothermic and has a moderate activation energy (CH3OH, Ea = 0.73 eV and Er = +0.15 eV). Similarly, methanol can also result from CH3O hydrogenation with a comparable reaction energy and activation barrier (Ea = 0.78 eV and Er = +0.05 eV). Hence, it is likely that elementary processes leading to the formation of methanol on the Rh(111) surface are feasible. As such, it is of interest to consider next the fate of any methanol molecules that may be formed.
Clearly, there are two possibilities: either methanol can desorb to the gas phase, or undergo some dissociation process. While methanol desorption is endothermic by 0.67 eV, the dissociation of methanol to CH2OH (i.e., the reverse of the process for CH2OH hydrogenation discussed in the preceding paragraph), is exothermic by −0.15 eV and has a lower activation energy, 0.58 eV. Conversely, methanol dissociation to CH3 and OH is highly activated (Ea = 1.67 eV and Er = −0.35 eV). Hence, it is highly likely that methanol decomposition to CH2OH will out-compete the desorption of methanol to the gas phase. As discussed in the previous section pertaining to the HCO and COH mechanisms for methane formation, the dissociation of CH2OH to CH2 and OH is exothermic, by −0.58 eV, although this process has a slightly higher activation barrier of 0.75 eV compared to reforming methanol (Ea = 0.73 eV). Hence, it appears likely that over longer time scales, CH2OH dissociation to CH2 will predominate, since methanol desorption is more activated than methanol dissociation to CH2OH, and the CH2OH dissociation process is more exothermic (by −0.58 eV) to yield CH2, which undergoes further hydrogenation to the final product CH4. Furthermore, the alternative dissociation of CH2OH to HCOH has a lower activation energy of 0.38 eV, which is exothermic by −0.21 eV. HCOH can then dissociate to CH with an activation energy of 0.47 eV, and the subsequent activation energies for CH successive hydrogenation are low (no greater than 0.64 eV for CH successive hydrogenation to CH4, which is lower than the effective activation barrier of 1.20 eV for reforming CH2OH from CH), and the corresponding reaction energies are all endothermic, suggesting that, as noted above, CH can undergo successive hydrogenation to CH4. Unlike methanol, methane can easily desorb to the gas phase, since the desorption energy (0.20 eV) is lower than the activation barriers for the reverse of the methane formation processes already discussed. Hence, the computational results presented above offer a potential explanation for why methane is observed to be a significant CO2 hydrogenation product instead of methanol over the Rh surface, based on the reaction mechanisms explored herein.
kMC simulations
It is clear from the discussion above that the product distribution is controlled by a complex balance between activation, reaction, and desorption energies. To explore further the distribution of products and reaction mechanism, we performed kMC simulations including all elementary steps under the experimental operating conditions. The kMC simulations involve a gas mixture consisting of H2 and CO2 in a 4:1 ratio over the clean Rh(111) surface at two different temperatures: first at 473.15 K and a pressure of 1 bar, corresponding to typical experimental conditions,52,107 and second at 573.15 K, to investigate the impact of temperature on product selectivity.
Gas Products and Reaction Mechanism
The results show that under these conditions, gaseous H2O, CH4, and HCOOH are evolved as products, with H2O predominating (Figure 5a,b). The evolution of significant quantities of H2O without a corresponding amount of C-containing products can be rationalized by the strong binding of CO to the Rh(111) surface, and thus the high CO desorption energy, which is supported by the high coverage of CO on the adlayer configurations in Figure S8. Hence, under these conditions, the RWGS reaction predominates.
Figure 5.

Time evolution of the gas selectivity from the CO2 hydrogenation to C1 products under the temperatures of (a) 473.15 K and (b) 573.15 K, with a pressure of 1 bar (PH2 = 0.8 bar, PCO2 = 0.2 bar). Time evolution of the selectivity for carbon-based gas under the temperatures of (c) 473.15 K and (d) 573.15 K, with a pressure of 1 bar (PH2 = 0.8 bar, PCO2 = 0.2 bar).
The selectivity for evolved carbon-containing gases is shown in Figure 5c,d. At 473.15 K, the selectivity to methane is initially 100% and reaches the steady-state value of 75.9% around 5900 s. While the selectivity to methane begins to decrease from a kMC time of 1300 s, gaseous HCOOH emerges concurrently, with no gaseous CO being evolved at any point. To explore the gas product distribution in more detail, we plotted the occurrence frequency of all of the elementary steps between the time intervals 0–1300 s (Figure 6) and 1300–2600 s (Figure S9). Figure 6 shows that all three pathways identified are potentially feasible means to produce adsorbed CO, including direct dissociation of CO2, and the carboxyl and formate mechanisms (via formic acid), in agreement with the rationalization of the DFT simulations discussed previously. The resulting adsorbed CO predominantly undergoes hydrogenation, first to COH and subsequently to HCOH, rather than desorbing to the gas phase; while the DFT calculations show that the CO hydrogenation process to yield COH has a high activation barrier (+1.27 eV) and is moderately endothermic (+0.71 eV), the barrier is lower than that for HCO formation (+1.35 eV), which is considerably more endothermic (+1.21 eV). Furthermore, the activation barrier for COH formation is lower than both the activation barriers for reverting back to CO2 (+1.43 eV) and CO desorption (+2.28 eV). Hence, the process statistics and persistent CO surface coverage reported from the kMC simulation are consistent with the DFT calculations, despite the high activation barrier for COH formation. Furthermore, the process statistics show that formic acid produced via the formate route tends to dissociate to HCO and OH, with most HCO in turn dissociating to yield CO, and some forming HCOH or formaldehyde. Hence, the CO2 direct dissociation mechanism, and formate and carboxyl mechanisms, all ultimately converge at the formation of adsorbed CO and HCOH species. The process statistics also show that most of the HCOH formed dissociates to yield CH, which can be sequentially hydrogenated to the product methane. Hence, adsorbed CO is the central intermediate for CO2 methanation, leading to further hydrogenation to COH and HCOH, being pivotal steps in methane production.
Figure 6.
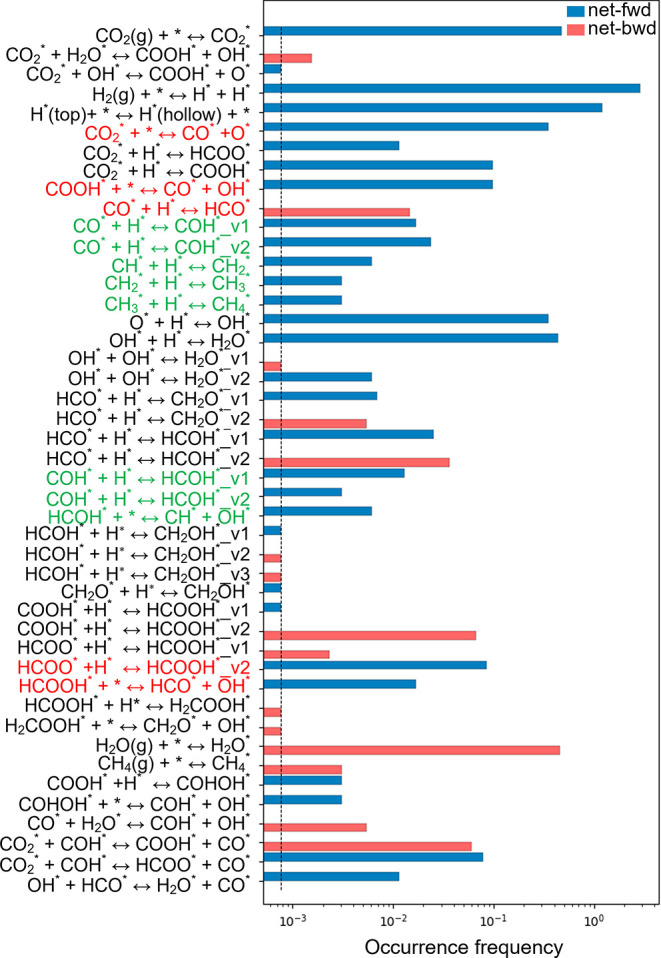
Occurrence frequency of the elementary steps (excluding events with zero frequency) during the time interval of 0–1300 s at a temperature of 473.15 K and a pressure of 1 bar (PH2 = 0.8 bar, PCO2 = 0.2 bar). Net rates of the reversible events are calculated by subtracting the reverse rates from the forward rates. The positive net rates are denoted as “net-fwd”, while the negative ones are labeled as “net-bwd”. Pathways for the RWGS reaction are highlighted in red, while pathways leading to methane formation are marked in green. Labels v1 and v2 represent the sets of neighboring sites with different types of site connectivity defined in kMC simulations, on which the same elementary process takes place.
The emergence of HCOOH evolution as illustrated in the kMC product distribution after ∼1300 s (Figure 5c) can be understood in terms of CO coverage. By comparing the event frequency between both time intervals (i.e., before and after 1300 s), it can be seen that desorption of gaseous HCOOH after 1300 s correlates with a greater formation and subsequent dissociation of HCO (as shown in Figure S9), which results in a higher surface coverage of CO species. The adlayer configurations in Figure S8 also show that more CO species occupy the surface at the steady-state time of 5900 s, compared with that at 500 s in Figure 7. Hence, at longer kMC simulation times, larger numbers of surface CO species can ultimately block the surface sites, thus preventing further HCOOH decomposition on the surface; HCOOH therefore desorbs to the gas phase when the surface coverage of CO is sufficiently high. This is furthermore supported by the exploration of the effect of increasing the ratio of H2/CO2 gas mixtures, which will be discussed in more detail later. Experimental results also reported that the presence of CO in the gas stream markedly inhibited the HCOOH decomposition reaction.108 Moreover, the process of HCOO hydrogenation to HCOOH becomes more pronounced at the time interval of 1300–2600 s (Figure S9), compared with that during 0–1300 s (Figure 6). Hence, formic acid slowly desorbs to the gas phase after 1300 s with a moderate desorption energy of 0.79 eV.
Figure 7.

Adlayer configurations for the 50 × 50 lattice at a time of 500 s under the different temperatures: (a) T = 473.15 K, (b) T = 573.15 K. The occurrence frequency of the elementary steps (excluding events with zero frequency) over the time interval of 0–500 s under the different temperatures: (c) T = 473.15 K, (d) T = 573.15 K. The partial pressures for H2 and CO2 are 0.8 and 0.2 bar, respectively. Net-fwd and net-bwd are defined as earlier mentioned, as well as the labels v1 and v2.
As discussed in the previous section, it is likely that the DFT-calculated CO adsorption energy represents an overestimation; hence, it is instructive to test the sensitivity of the kMC product selectivity with respect to CO binding energy. Additional simulations were performed with less exothermic CO adsorption energies to assess the impact of possible overestimation of the binding energy, as detailed in Section S7 of Supporting Information. These simulations reveal that the methane formation mechanism remains largely unchanged, while CO desorption is somewhat accelerated, leading to a higher fraction of CO being desorbed to the gas phase, as would be expected. Hence, the additional simulations validate the key features of the model applied in this study.
Temperature Effects
The impact of temperature on reaction mechanism and selectivity has also been considered by performing the kMC simulations at two different temperatures, 473.15 and 573.15 K, both under a pressure of 1 bar. Increasing temperature results in the production of gaseous CO as shown in Figure 5, in agreement with previous experiment,109 implying that elevated temperatures are required to facilitate CO desorption from the surface. Methane selectivity decreases within the time interval of 0–2055 s, whereas the selectivity of CO increases noticeably. To gain further insight into the product distribution at elevated temperatures, the adlayer configurations during the reaction process are visualized and compared at the temperatures of 473.15 and 573.15 K. Figure 7 shows the adlayer configurations after running the kMC simulation for 500 s. At 473.15 K, the lattice is predominantly covered with adsorbed H atoms, while adsorbed CO species tend to form islands, presenting a high ratio of H to CO coverages on the lattice. In contrast, higher concentrations of adsorbed CO species are observed on the surface at 573.15 K, and the H atoms adsorbed at the top sites are surrounded by at least two adsorbed CO molecules. This arrangement can hinder the H diffusion from the top sites to the hollow sites, further preventing the H2 dissociation at the top sites.
Our findings are in good agreement with experimental observations, which indicate that CO species adsorbed at the metal sites limit H2 dissociation.110 This is further supported by the finding that the partial kinetic order of CO at relatively high CO concentrations is negative.54 The elevated temperature accelerates the formation of CO, resulting in an enhanced presence of CO species, both on the surface and in the gas phase. The higher coverage of the adsorbed CO species on the Rh surface can impede the hydrogen dissociation and adsorption, thereby hindering further CO hydrogenation processes, leading to CO accumulation on the surface and its slow desorption to the gas phase. Figure 7 shows that the elevated temperature accelerates CO formation via CO2 and COOH dissociation pathways (with one way to obtain COOH being HCOOH dehydrogenation). The most favorable pathway under elevated temperature is HCO dehydrogenation, along with HCO derived from the promoted dissociation of HCOO and HCOOH intermediates. The prohibitive CO coverage under reaction conditions has also been reported to account for the lower activity of smaller Rh particles in CO hydrogenation.37 It was also reported that CO2 methanation appeared to be inhibited by CO on Rh/γ-Al2O3 catalyst.28 Overall, an increased coverage of adsorbed CO species on the Rh surface can be detrimental to hydrogen dissociative adsorption, which is an essential prerequisite for CO2 methanation.
Impact of H:CO Ratio
The coverages of H and CO species can influence the pathways controlling the selectivity of methane and CO, which is evident from the occurrence frequencies of all of the elementary events obtained from the kMC simulations. Figure 5d shows an increase in methane selectivity accompanied by a decrease in CO selectivity during the period of 2055 and 2450 s at 573.15 K. To analyze the selectivity trend, event frequencies for the time interval of 1660–2055 and 2055–2450 s (representing the opposite selectivity trends) are compared and shown in Figure S10. This can be attributed to the difference in the occurrence frequency for CO hydrogenation to COH, which occurs more frequently between 2055 and 2450 s. As discussed above, CH species result from the dissociation of the adsorbed HCOH, the formation of which in turn depends on the rate of COH formation. Hence, the higher rate of methane formation between 2055 and 2450 s is correlated with the increasing frequency of COH formation. Indeed, between 1660 and 2055 s, the net rate (defined as the forward rate minus the reverse rate) for the HCOH dissociation is close to the dashed vertical line (corresponding to the occurrence of a single event in the entire duration of the simulation), meaning that only a few events occur. However, from 2055 to 2450 s, the dissociation of HCOH occurs more frequently, as the concentration of HCOH species increases due to the higher COH formation frequency. Meanwhile, Figure 8a shows a decline in hydrogen coverage at hollow sites from 2055 to 2450 s at 573.15 K. Conversely, the coverage of the adsorbed CO increases, which is shown by the adlayer configurations, with a higher substantial ratio of H to CO coverages at 2055 s than 2450 s (Figure S11). Hence, the relative coverages of the H and CO species on the Rh(111) surface can play a decisive role in the CO hydrogenation to COH, with higher COH formation frequencies being observed for optimal H:CO surface coverages. The H:CO surface coverage ratio can also explain the different selectivities observed at different temperatures. The ratio of hydrogen adsorbed at the hollow sites and the adsorbed CO is 0.15 at the initial state and decreases to 0.06 at the steady state at 573.15 K (Figure 8a). However, at 473.15 K, the H:CO coverage ratio is significantly different, starting at 4.62 and sharply declining to 0.75 at the steady state (Figure 8b), which is considerably greater than the H:CO coverage ratio at steady state at 573.15 K. Hence, CO2 methanation is favored at 473.15 K, and gaseous CO is produced at 573.15 K, due to the higher surface H coverage at the steady state at 473.15 K (Figure S8). Additionally, the production of gaseous H2O under 573.15 K decreases over time (Figure 5b). This is because the H2O species can desorb to the gas phase once they are formed via RWGS reaction, while the evolution of gaseous CO occurs until the surface coverage of CO is sufficiently high due to the high desorption energy of CO. Along with CO species accumulated on the surface slowly desorb to the gas phase, gaseous H2O constitutes a decreasing fraction of the total gaseous species.
Figure 8.

Evolution of the coverages of surface species over time at the temperatures of (a) 573.15 K and (b) 473.15 K, with a pressure of 1 bar (PH2 = 0.8 bar, PCO2 = 0.2 bar). Herein, the coverage is defined as the fraction of a specific surface species over the total species, independent of the number of the sites, since the different types of active sites are occupied by the multidentate species.
Based on the effects of the relative coverages of H and CO species discussed above, we considered the effect of the gaseous H2 to CO2 ratio by performing simulations with 3:1, 4:1, and 9:1 H2/CO2 gas mixtures under typical experimental conditions of 473.15 K and 1 bar. Under these conditions, methane and formic acid were the only carbon-contained species evolving to the gas phase in these simulations. The selectivity to methane was promoted with a higher H2 to CO2 ratio in the mixture, which is in agreement with previously reported thermodynamic analysis.111,112 This trend is confirmed, as shown in Table 3, which demonstrates that as the H2/CO2 gas mixture ratio increases, the steady-state coverage of the CO species decreases, whereas the steady-state coverage of the H species adsorbed at the hollow sites increases (θCO = 0.410, θH = 0.129 for 3:1 H2/CO2 mixture, θCO = 0.319, θH = 0.210 for 4:1 H2/CO2 mixture, and θCO = 0.180, θH = 0.348 for 9:1 H2/CO2 mixture).
Table 3. Selectivity of the Carbon-Based Gas Products, and the Coverage of the Surface Species over the Rh(111) Surface under Various Reaction Conditionsa.
| T = 473.15 K, PH2 = 0.75 bar, PCO2 = 0.25 bar | T = 473.15 K, PH2 = 0.8 bar, PCO2 = 0.2 bar | T = 473.15 K, PH2 = 0.9 bar, PCO2 = 0.1 bar | |
|---|---|---|---|
| CH4 selectivity | 0.654 | 0.759 | 0.882 |
| HCOOH selectivity | 0.346 | 0.241 | 0.118 |
| θCO | 0.410 | 0.319 | 0.180 |
| θCO2 | 0.021 | 0.009 | 0.006 |
| θCOOH | 0.001 | 0.007 | 0.004 |
| θHCOO | 0.009 | 0.005 | 0.004 |
| θO | 0.004 | 0.004 | 0.001 |
| θH (hollow) | 0.129 | 0.210 | 0.348 |
| θOH | 0.001 | 0.002 | 0.001 |
| θH2O | 0.047 | 0.026 | 0.014 |
| θH (top) | 0.367 | 0.417 | 0.440 |
Coverage is defined as earlier mentioned.
In addition, Table S12 shows that higher pressures can enhance methane selectivity at a given reaction temperature with a higher ratio of H/CO coverage, in agreement with previous reports.111−113 Furthermore, the rates of CO2 hydrogenation, expressed as the turnover frequency (TOF), were obtained through simulations at varied temperatures with a pressure of 1 bar and a 4:1 H2/CO2 gas mixture. The apparent activation energy for the overall process was derived via the Arrhenius equation. Figure S13 shows that the calculated apparent activation energy is 19.94 kcal/mol between temperatures of 473.15 and 573.15 K, which is comparable with experimental measurements. Bell and co-workers107 obtained an apparent activation energy of 16.6 kcal/mol for CO2 hydrogenation on Rh/SiO2 catalyst under a fixed pressure of 608 Torr for H2 and 152 Torr for CO2. In addition, Bell, Somorjai, and co-workers114 reported a value of 17.0 kcal/mol by investigating methane formation for both the bare Rh surface and titania-promoted Rh surface at atmospheric pressure with a gaseous H2/CO2 ratio of 3. Meanwhile, other studies reported 16.2, 17.3, and 19.4 kcal/mol for the apparent activation energy of CO2 hydrogenation over Rh catalyst supported by alumina, silica, and titania, respectively,115 confirming that our calculated activation energy is in close agreement with experiments.52,107,114,115
Comparison of CO2 Hydrogenation Performance over Rh and Cu Catalysts
Our computational results show that CO2 chemisorption on the Rh(111) surface is exothermic, with −0.33 eV of adsorption energy, which contrasts with the adsorption energies reported for the same species over different metal surfaces. DFT calculations by Higham et al.93 showed that the bent CO2 adsorbate is only metastable on low-index Cu surfaces, with endothermic adsorption energies of 0.10 and 0.05 eV reported for Cu(110) and (100) surfaces, respectively, whereas no such adsorption mode was identified for the Cu(111) facet. Kowalec et al.92 similarly reported a chemisorption energy of 0.09 eV for the bent CO2 species on Pd(111), corroborating experiments that demonstrated the absence of CO2 chemisorption on this surface facet. The difference in stability of the bent CO2 species on different metal surfaces may lie in the different stabilities of the surfaces; the calculated surface energies for Cu(111) and Pd(111) are 1.29 J/m2116 and 1.72 J/m2,92 respectively, which are lower than our calculated value of 2.85 J/m2 for Rh(111). Higher surface energies may promote stronger adsorption interactions due to the inherent instability of the surface facet, resulting in exothermic chemisorption of CO2 on the Rh(111) surface.
The combined DFT and kMC simulation results presented predict that methane is a significant CO2 hydrogenation product instead of methanol over the Rh surface. In terms of reaction mechanisms explored, the adsorbed CO is an essential intermediate, which facilitates further hydrogenation into COH. HCOH derived from COH hydrogenation can be dissociated into CH, which undergoes further hydrogenation to yield methane. In contrast, Cu catalysts are reported as the dominant active constituent for effective synthesis of methanol from CO2 hydrogenation and have been extensively studied.117,118 For unsupported Cu catalysts, methanol desorption is less activated than methanol dissociation, as shown by DFT calculations of CO2 hydrogenation on Cu(100) and (110) surfaces.93 Furthermore, the mechanisms of methanol synthesis on Cu catalysts have been discussed via either formate pathway119,120 or carboxyl pathway.121,122 Yang et al.123 proposed CO produced by the fast RWGS reaction did not undergo subsequent hydrogenation to methanol over Cu catalysts, but instead simply accumulated as a product, which was demonstrated by both experiments and calculations. The desorption energy of 0.79 eV for CO from Cu(111) also indicates that CO potentially undergoes desorption.124 In the kinetic regime of CO2 hydrogenation, an inverse kinetic isotope effect of H/D substitution on Cu/ZnO/Al2O3 catalyst was observed, which is stronger for methanol synthesis than for CO formation, suggesting that the two reactions do not share a common intermediate.125 Hence, methanol synthesis on Cu catalysts can be achieved without CO subsequent hydrogenation and dissociation, whereas CO is a significant intermediate for methane formation on Rh catalysts, which is indicated by the kMC results in the present work. In contrast, previous studies focusing on CO2 hydrogenation over Cu(100)83 suggested that the key process involved CO2 hydrogenation to formate, leading to the formation of formic acid and methanol, with few CO2 species undergoing dissociation into CO; COH derived from CO hydrogenation was found to be unstable on Cu(100) surface, and consequently minimal HCOH was expected to be present, unlike that for Rh(111) where HCOH is a key intermediate leading to the formation of CH species, and ultimately methane. In addition, HCOOH readily desorbs from Cu(100) and is only weakly bound at low surface coverages, whereas for Rh(111), HCOOH dissociates to HCO, which undergoes further dehydration to CO, thus limiting methanol production via HCOOH hydrogenation and favoring methane formation via HCOH as discussed above.
Summary and Conclusions
Our DFT simulations have shown that the Rh catalyst promotes CO2 activation and dissociation, as well as H2 dissociation, as demonstrated by the geometric and electronic-structure analysis of the adsorption structures for CO2 and H2 molecules, along with low activation energies for H2 dissociation. The RWGS reaction can proceed via three possible mechanisms: the redox mechanism, the carboxyl mechanism, and the formate mechanism, via formic acid. Analysis of the DFT results suggests that methane formation is favored by CO2 direct hydrogenation to formate, with subsequent hydrogenation to formic acid, which dissociates to HCO. Subsequently, the dissociation of HCOH derived from HCO hydrogenation can yield CH, which undergoes further hydrogenation to yield the final product methane. However, kMC simulations demonstrate that adsorbed CO is a crucial intermediate for methane formation, undergoing hydrogenation into COH and subsequently HCOH, which itself then subsequently undergoes dissociation to CH and then hydrogenation to methane formation as indicated by DFT simulations.
The reaction temperature was found to have a profound effect on the reaction mechanism and product selectivity. Higher methane production via the Sabatier reaction was observed to take place on the surfaces with a higher H/CO ratio at 473.15 K, with no gaseous CO production observed; however, the evolution of gaseous CO starts to occur at 573.15 K, as evident from the adlayer configurations under these conditions. This finding highlights the crucial role of the H/CO ratio in controlling the product distribution in CO2 hydrogenation over Rh-based catalysts. The elevated temperature accelerates CO formation via the three mechanisms. Subsequently, the higher coverage of the adsorbed CO species on the Rh surface can impede hydrogen dissociation and adsorption, thereby hindering further CO hydrogenation processes. This mechanism leads to CO accumulation on the surface and eventually slow desorption to the gas phase at the elevated temperature. Hence, a higher ratio of H to CO coverage on the Rh(111) surface enhances methane formation, with the key steps being CO hydrogenation to COH and the dissociation of HCOH. The contrast with the CO2 hydrogenation over copper, where the selectivity toward methanol is observed, can be largely attributed to lower CO surface coverages, the instability of the COH intermediate, and thus the minimal presence of the HCOH species (a key intermediate for the Sabatier reaction, as illustrated by the present work); instead, copper favors direct hydrogenation to formate, leading to methanol formation.
In summary, the present work not only provides new insights into the mechanism of CO2 methanation on Rh(111) surfaces but also illustrates the value of combining different computational techniques to provide a multiscale analysis of reaction mechanisms. While DFT simulations can provide valuable insights from reaction profiles with static energies, kMC simulations can provide a deeper insight into the exploration of the reaction mechanism with a statistical and dynamical method based on the DFT-calculated reaction profile, elucidating mechanistic subtleties arising from competing processes and intermediates.
Acknowledgments
The authors acknowledge the use of YOUNG and ARCHER2 UK National Supercomputing Service (http://www.archer2.ac.uk) via membership of UK’s HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202/1). This work used the UK Materials and Molecular Modelling Hub for computational resources, MMM Hub, which is partially funded by EPSRC (EP/T022213/1, EP/W032260/1, and EP/P020194/1). The authors also acknowledge the use of the UCL Kathleen and Myriad High Performance Computing Facility (Kathleen@UCL, Myriad@UCL), and associated support services, in the completion of this work. The authors are grateful to Professor Michael Stamatakis for the use of the Zacros software package. M.D.H. acknowledges EPSRC/UKRI (EP/T028629/1) for financial support and the UK Catalysis Hub Consortium (funded by EPSRC (Grants EP/R026815/1)) for the provision of additional resources. S.J. acknowledges China Scholarship Council (CSC) for the financial support.
Supporting Information Available
The Supporting Information is available free of charge. The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c05939.
Further details on ZPE-corrected adsorption energies, surface energies, determining of rate constants, surface lateral interactions, electronic analysis (including density of states and crystal orbital Hamilton population), impact of the magnitude of CO adsorption energy, adlayer configurations, event frequency, pressure effects, apparent activation energy, and configurations used in cluster expansion model (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Aresta M.; Dibenedetto A.; Angelini A. Catalysis for the valorization of exhaust carbon: from CO2 to chemicals, materials, and fuels. Technological use of CO2. Chem. Rev. 2014, 114 (3), 1709–1742. 10.1021/cr4002758. [DOI] [PubMed] [Google Scholar]
- Hu B.; Guild C.; Suib S. L. Thermal, electrochemical, and photochemical conversion of CO2 to fuels and value-added products. J. CO2 Util. 2013, 1, 18–27. 10.1016/j.jcou.2013.03.004. [DOI] [Google Scholar]
- Steinfeld A. Solar thermochemical production of hydrogen-a review. Sol. Energy 2005, 78 (5), 603–615. 10.1016/j.solener.2003.12.012. [DOI] [Google Scholar]
- Ye R.-P.; Ding J.; Gong W.; Argyle M. D.; Zhong Q.; Wang Y.; Russell C. K.; Xu Z.; Russell A. G.; Li Q.; et al. CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Commun. 2019, 10 (1), 5698 10.1038/s41467-019-13638-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattel S.; Liu P.; Chen J. G. Tuning selectivity of CO2 hydrogenation reactions at the metal/oxide interface. J. Am. Chem. Soc. 2017, 139 (29), 9739–9754. 10.1021/jacs.7b05362. [DOI] [PubMed] [Google Scholar]
- Götz M.; Lefebvre J.; Mörs F.; Koch A. M.; Graf F.; Bajohr S.; Reimert R.; Kolb T. Renewable Power-to-Gas: A technological and economic review. Renewable Energy 2016, 85, 1371–1390. 10.1016/j.renene.2015.07.066. [DOI] [Google Scholar]
- Ozturk M.; Dincer I. A comprehensive review on power-to-gas with hydrogen options for cleaner applications. Int. J. Hydrogen Energy 2021, 46 (62), 31511–31522. 10.1016/j.ijhydene.2021.07.066. [DOI] [Google Scholar]
- Li S.; Ahmed R.; Yi Y.; Bogaerts A. Methane to methanol through heterogeneous catalysis and plasma catalysis. Catalysts 2021, 11 (5), 590. 10.3390/catal11050590. [DOI] [Google Scholar]
- Guo X.; Fang G.; Li G.; Ma H.; Fan H.; Yu L.; Ma C.; Wu X.; Deng D.; Wei M.; et al. Direct, nonoxidative conversion of methane to ethylene, aromatics, and hydrogen. science 2014, 344 (6184), 616–619. 10.1126/science.1253150. [DOI] [PubMed] [Google Scholar]
- Morejudo S. H.; Zanón R.; Escolástico S.; Yuste-Tirados I.; Malerød-Fjeld H.; Vestre P. K.; Coors W. G.; Martínez A.; Norby T.; Serra J. M.; Kjølseth C. Direct conversion of methane to aromatics in a catalytic co-ionic membrane reactor. Science 2016, 353 (6299), 563–566. 10.1126/science.aag0274. [DOI] [PubMed] [Google Scholar]
- Rönsch S.; Schneider J.; Matthischke S.; Schlüter M.; Götz M.; Lefebvre J.; Prabhakaran P.; Bajohr S. Review on methanation–From fundamentals to current projects. Fuel 2016, 166, 276–296. 10.1016/j.fuel.2015.10.111. [DOI] [Google Scholar]
- Su X.; Xu J.; Liang B.; Duan H.; Hou B.; Huang Y. Catalytic carbon dioxide hydrogenation to methane: A review of recent studies. J. Energy Chem. 2016, 25 (4), 553–565. 10.1016/j.jechem.2016.03.009. [DOI] [Google Scholar]
- Miao B.; Ma S. S. K.; Wang X.; Su H.; Chan S. H. Catalysis mechanisms of CO2 and CO methanation. Catal. Sci. Technol. 2016, 6 (12), 4048–4058. 10.1039/C6CY00478D. [DOI] [Google Scholar]
- Albeladi N.; Alsulami Q. A.; Narasimharao K. Recent Progress in Nickel and Silica Containing Catalysts for CO2 Hydrogenation to CH4. Catalysts 2023, 13 (7), 1104. 10.3390/catal13071104. [DOI] [Google Scholar]
- Shen L.; Xu J.; Zhu M.; Han Y.-F. Essential role of the support for nickel-based CO2 methanation catalysts. ACS Catal. 2020, 10 (24), 14581–14591. 10.1021/acscatal.0c03471. [DOI] [Google Scholar]
- Jalama K. Carbon dioxide hydrogenation over nickel-, ruthenium-, and copper-based catalysts: Review of kinetics and mechanism. Catal. Rev. 2017, 59 (2), 95–164. 10.1080/01614940.2017.1316172. [DOI] [Google Scholar]
- Heine C.; Lechner B. A.; Bluhm H.; Salmeron M. Recycling of CO2: probing the chemical state of the Ni (111) surface during the methanation reaction with ambient-pressure X-ray photoelectron spectroscopy. J. Am. Chem. Soc. 2016, 138 (40), 13246–13252. 10.1021/jacs.6b06939. [DOI] [PubMed] [Google Scholar]
- Karelovic A.; Ruiz P. CO2 hydrogenation at low temperature over Rh/γ-Al2O3 catalysts: Effect of the metal particle size on catalytic performances and reaction mechanism. Appl. Catal., B: Environ. 2012, 113-114, 237–249. 10.1016/j.apcatb.2011.11.043. [DOI] [Google Scholar]
- Karelovic A.; Ruiz P. Mechanistic study of low temperature CO2 methanation over Rh/TiO2 catalysts. J. Catal. 2013, 301, 141–153. 10.1016/j.jcat.2013.02.009. [DOI] [Google Scholar]
- Deleitenburg C.; Trovarelli A. Metal-support interactions in Rh/CeO2, Rh/TiO2, and Rh/Nb2O5 catalysts as inferred from CO2 methanation activity. J. Catal. 1995, 156 (1), 171–174. 10.1006/jcat.1995.1244. [DOI] [Google Scholar]
- Dong T.; Liu X.; Tang Z.; Yuan H.; Jiang D.; Wang Y.; Liu Z.; Zhang X.; Huang S.; Liu H.; et al. Ru decorated TiOx nanoparticles via laser bombardment for photothermal co-catalytic CO2 hydrogenation to methane with high selectivity. Appl. Catal., B: Environ. 2023, 326, 122176 10.1016/j.apcatb.2022.122176. [DOI] [Google Scholar]
- Guo Y.; Mei S.; Yuan K.; Wang D.-J.; Liu H.-C.; Yan C.-H.; Zhang Y.-W. Low-temperature CO2 methanation over CeO2-supported Ru single atoms, nanoclusters, and nanoparticles competitively tuned by strong metal–support interactions and H-spillover effect. ACS Catal. 2018, 8 (7), 6203–6215. 10.1021/acscatal.7b04469. [DOI] [Google Scholar]
- Wang X.; Hong Y.; Shi H.; Szanyi J. Kinetic modeling and transient DRIFTS–MS studies of CO2 methanation over Ru/Al2O3 catalysts. J. Catal. 2016, 343, 185–195. 10.1016/j.jcat.2016.02.001. [DOI] [Google Scholar]
- Abe T.; Tanizawa M.; Watanabe K.; Taguchi A. CO2 methanation property of Ru nanoparticle-loaded TiO2 prepared by a polygonal barrel-sputtering method. Energy Environ. Sci. 2009, 2 (3), 315–321. 10.1039/b817740f. [DOI] [Google Scholar]
- Wang X.; Shi H.; Szanyi J. Controlling selectivities in CO2 reduction through mechanistic understanding. Nat. Commun. 2017, 8 (1), 513 10.1038/s41467-017-00558-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Shi H.; Kwak J. H.; Szanyi J. Mechanism of CO2 hydrogenation on Pd/Al2O3 catalysts: kinetics and transient DRIFTS-MS studies. ACS Catal. 2015, 5 (11), 6337–6349. 10.1021/acscatal.5b01464. [DOI] [Google Scholar]
- Schild C.; Wokaun A.; Baiker A. Surface species in CO2 methanation over amorphous palladium/zirconia catalysts. J. Mol. Catal. 1991, 69 (3), 347–357. 10.1016/0304-5102(91)80115-J. [DOI] [Google Scholar]
- Beuls A.; Swalus C.; Jacquemin M.; Heyen G.; Karelovic A.; Ruiz P. Methanation of CO2: Further insight into the mechanism over Rh/γ-Al2O3 catalyst. Appl. Catal., B: Environ. 2012, 113-114, 2–10. 10.1016/j.apcatb.2011.02.033. [DOI] [Google Scholar]
- Martin N. M.; Hemmingsson F.; Schaefer A.; Ek M.; Merte L. R.; Hejral U.; Gustafson J.; Skoglundh M.; Dippel A.-C.; Gutowski O.; et al. Structure–function relationship for CO2 methanation over ceria supported Rh and Ni catalysts under atmospheric pressure conditions. Catal. Sci. Technol. 2019, 9 (7), 1644–1653. 10.1039/C8CY02097C. [DOI] [Google Scholar]
- Erdőhelyi A. Hydrogenation of carbon dioxide on supported Rh catalysts. Catalysts 2020, 10 (2), 155. 10.3390/catal10020155. [DOI] [Google Scholar]
- Yang N.; Medford A. J.; Liu X.; Studt F.; Bligaard T.; Bent S. F.; Nørskov J. K. Intrinsic selectivity and structure sensitivity of rhodium catalysts for C2+ oxygenate production. J. Am. Chem. Soc. 2016, 138 (11), 3705–3714. 10.1021/jacs.5b12087. [DOI] [PubMed] [Google Scholar]
- Ulissi Z. W.; Medford A. J.; Bligaard T.; Nørskov J. K. To address surface reaction network complexity using scaling relations machine learning and DFT calculations. Nat. Commun. 2017, 8 (1), 14621 10.1038/ncomms14621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y.; Liu P. Mechanism of ethanol synthesis from syngas on Rh(111). J. Am. Chem. Soc. 2009, 131 (36), 13054–13061. 10.1021/ja903013x. [DOI] [PubMed] [Google Scholar]
- Köhler L.; Kresse G. Density functional study of CO on Rh(111). Phys. Rev. B 2004, 70 (16), 165405 10.1103/PhysRevB.70.165405. [DOI] [Google Scholar]
- Beutl M.; Lesnik J.; Rendulic K. Adsorption dynamics for CO, CO-clusters and H2 (D2) on rhodium (111). Surf. Sci. 1999, 429 (1–3), 71–83. 10.1016/S0039-6028(99)00340-4. [DOI] [Google Scholar]
- Solymosi F.; Kiss J.; Kovács I. Adsorption of HCOOH on Rh(111) and its reaction with preadsorbed oxygen. Surf. Sci. 1987, 192 (1), 47–65. 10.1016/S0039-6028(87)81161-5. [DOI] [Google Scholar]
- Schumann M.; Nielsen M. R.; Smitshuysen T. E.; Hansen T. W.; Damsgaard C. D.; Yang A.-C. A.; Cargnello M.; Grunwaldt J.-D.; Jensen A. D.; Christensen J. M. Rationalizing an unexpected structure sensitivity in heterogeneous catalysis-CO hydrogenation over Rh as a case study. ACS Catal. 2021, 11 (9), 5189–5201. 10.1021/acscatal.0c05002. [DOI] [Google Scholar]
- Jiang B.; Li C.; Dag Ö.; Abe H.; Takei T.; Imai T.; Hossain M. S. A.; Islam M. T.; Wood K.; Henzie J.; Yamauchi Y. Mesoporous metallic rhodium nanoparticles. Nat. Commun. 2017, 8 (1), 15581 10.1038/ncomms15581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arandiyan H.; Kani K.; Wang Y.; Jiang B.; Kim J.; Yoshino M.; Rezaei M.; Rowan A. E.; Dai H.; Yamauchi Y. Highly selective reduction of carbon dioxide to methane on novel mesoporous Rh catalysts. ACS Appl. Mater. Interfaces 2018, 10 (30), 24963–24968. 10.1021/acsami.8b06977. [DOI] [PubMed] [Google Scholar]
- Jangam A.; Das S.; Dewangan N.; Hongmanorom P.; Hui W. M.; Kawi S. Conversion of CO2 to C1 chemicals: catalyst design, kinetics and mechanism aspects of the reactions. Catal. Today 2020, 358, 3–29. 10.1016/j.cattod.2019.08.049. [DOI] [Google Scholar]
- Peebles D. E.; Goodman D.; White J. Methanation of carbon dioxide on nickel (100) and the effects of surface modifiers. J. Phys. Chem. A 1983, 87 (22), 4378–4387. 10.1021/j100245a014. [DOI] [Google Scholar]
- Weatherbee G. D.; Bartholomew C. H. Hydrogenation of CO2 on group VIII metals: II. Kinetics and mechanism of CO2 hydrogenation on nickel.. J. Catal. 1982, 77 (2), 460–472. 10.1016/0021-9517(82)90186-5. [DOI] [Google Scholar]
- Kattel S.; Yan B.; Chen J. G.; Liu P. CO2 hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of synergy between Pt and oxide support. J. Catal. 2016, 343, 115–126. 10.1016/j.jcat.2015.12.019. [DOI] [Google Scholar]
- Silveri F.; Quesne M. G.; Viñes F.; Illas F.; Catlow C. R. A.; de Leeuw N. H. Catalytic reduction of carbon dioxide on the (001), (011), and (111) surfaces of TiC and ZrC: A computational study. J. Phys. Chem. C 2022, 126 (11), 5138–5150. 10.1021/acs.jpcc.1c10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akamaru S.; Shimazaki T.; Kubo M.; Abe T. Density functional theory analysis of methanation reaction of CO2 on Ru nanoparticle supported on TiO2 (1 0 1). Appl. Catal., A 2014, 470, 405–411. 10.1016/j.apcata.2013.11.016. [DOI] [Google Scholar]
- Schild C.; Wokaun A.; Baiker A. On the mechanism of CO and CO2 hydrogenation reactions on zirconia-supported catalysts: a diffuse reflectance FTIR study: Part II. Surface species on copper/zirconia catalysts: implications for methanol synthesis selectivity. J. Mol. Catal. 1990, 63 (2), 243–254. 10.1016/0304-5102(90)85147-A. [DOI] [Google Scholar]
- Upham D. C.; Derk A. R.; Sharma S.; Metiu H.; McFarland E. W. CO2 methanation by Ru-doped ceria: the role of the oxidation state of the surface. Catal. Sci. Technol. 2015, 5 (3), 1783–1791. 10.1039/C4CY01106F. [DOI] [Google Scholar]
- Aldana P. U.; Ocampo F.; Kobl K.; Louis B.; Thibault-Starzyk F.; Daturi M.; Bazin P.; Thomas S.; Roger A. Catalytic CO2 valorization into CH4 on Ni-based ceria-zirconia. Reaction mechanism by operando IR spectroscopy. Catal. Today 2013, 215, 201–207. 10.1016/j.cattod.2013.02.019. [DOI] [Google Scholar]
- Liu X.; Sun L.; Deng W.-Q. Theoretical investigation of CO2 adsorption and dissociation on low index surfaces of transition metals. J. Phys. Chem. C 2018, 122 (15), 8306–8314. 10.1021/acs.jpcc.7b12660. [DOI] [Google Scholar]
- Van Tol M.; Gielbert A.; Nieuwenhuys B. The adsorption and dissociation of CO2 on Rh. Appl. Surf. Sci. 1993, 67 (1–4), 166–178. 10.1016/0169-4332(93)90309-Y. [DOI] [Google Scholar]
- Castner D.; Sexton B.; Somorjai G. Leed and thermal desorption studies of small molecules (H2, O2, CO, CO2, NO, C2H4, C2H2 AND C) chemisorbed on the rhodium (111) and (100) surfaces. Surf. Sci. 1978, 71 (3), 519–540. 10.1016/0039-6028(78)90444-2. [DOI] [Google Scholar]
- Sexton B.; Somorjai G. The hydrogenation of CO and CO2 over polycrystalline rhodium: Correlation of surface composition, kinetics and product distributions. J. Catal. 1977, 46 (2), 167–189. 10.1016/0021-9517(77)90198-1. [DOI] [Google Scholar]
- Kim J.; Ha H.; Doh W. H.; Ueda K.; Mase K.; Kondoh H.; Mun B. S.; Kim H. Y.; Park J. Y. How Rh surface breaks CO2 molecules under ambient pressure. Nat. Commun. 2020, 11 (1), 5649 10.1038/s41467-020-19398-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solymosi F.; Tombacz I.; Kocsis M. Hydrogenation of CO on supported Rh catalysts. J. Catal. 1982, 75 (1), 78–93. 10.1016/0021-9517(82)90123-3. [DOI] [Google Scholar]
- McCarty J.; Wise H. Hydrogenation of surface carbon on alumina-supported nickel. J. Catal. 1979, 57 (3), 406–416. 10.1016/0021-9517(79)90007-1. [DOI] [Google Scholar]
- Ekerdt J. G.; Bell A. T. Synthesis of hydrocarbons from CO and H2 over silica-supported Ru: reaction rate measurements and infrared spectra of adsorbed species. J. Catal. 1979, 58 (2), 170–187. 10.1016/0021-9517(79)90255-0. [DOI] [Google Scholar]
- Rabo J.; Risch A.; Poutsma M. Reactions of carbon monoxide and hydrogen on Co, Ni, Ru, and Pd metals. J. Catal. 1978, 53 (3), 295–311. 10.1016/0021-9517(78)90102-1. [DOI] [Google Scholar]
- Yates J. Jr; Williams E.; Weinberg W. Does chemisorbed carbon monoxide dissociate on rhodium?. Surf. Sci. 1980, 91 (2–3), 562–570. 10.1016/0039-6028(80)90351-9. [DOI] [Google Scholar]
- Solymosi F.; Erdöhelyi A. Dissociation of CO on supported Rh. Surf. Sci. Lett. 1981, 110 (2), L630–L633. 10.1016/0167-2584(81)90473-4. [DOI] [Google Scholar]
- Claver C.Rhodium Catalysis; Springer: 2018. [Google Scholar]
- Vanzan M.; Marsili M.; Corni S. Study of the Rate-Determining Step of Rh Catalyzed CO2 Reduction: Insight on the Hydrogen Assisted Molecular Dissociation. Catalysts 2021, 11 (5), 538. 10.3390/catal11050538. [DOI] [Google Scholar]
- Jacquemin M.; Beuls A.; Ruiz P. Catalytic production of methane from CO2 and H2 at low temperature: Insight on the reaction mechanism. Catal. Today 2010, 157 (1–4), 462–466. 10.1016/j.cattod.2010.06.016. [DOI] [Google Scholar]
- Nolen M. A.; Tacey S. A.; Kwon S.; Farberow C. A. Theoretical assessments of CO2 activation and hydrogenation pathways on transition-metal surfaces. Appl. Surf. Sci. 2023, 637, 157873 10.1016/j.apsusc.2023.157873. [DOI] [Google Scholar]
- Kraus P.; Frank I. Constrained chemical dynamics of CO dissociation/hydrogenation on Rh surfaces. Chem. – Eur. J. 2018, 24 (28), 7188–7199. 10.1002/chem.201705867. [DOI] [PubMed] [Google Scholar]
- Ouyang M.; Papanikolaou K. G.; Boubnov A.; Hoffman A. S.; Giannakakis G.; Bare S. R.; Stamatakis M.; Flytzani-Stephanopoulos M.; Sykes E. C. H. Directing reaction pathways via in situ control of active site geometries in PdAu single-atom alloy catalysts. Nat. Commun. 2021, 12 (1), 1549 10.1038/s41467-021-21555-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S.; Sun G.; Pei C.; Zhao Z.-J.; Gong J. Origin of Performances of Pt/Cu Single-Atom Alloy Catalysts for Propane Dehydrogenation. J. Phys. Chem. C 2021, 125 (34), 18708–18716. 10.1021/acs.jpcc.1c04295. [DOI] [Google Scholar]
- Piccinin S.; Stamatakis M. CO Oxidation on Pd(111): A First-Principles-Based Kinetic Monte Carlo Study. ACS Catal. 2014, 4 (7), 2143–2152. 10.1021/cs500377j. [DOI] [Google Scholar]
- Lozano-Reis P.; Prats H.; Gamallo P.; Illas F.; Sayós R. Multiscale Study of the Mechanism of Catalytic CO2 Hydrogenation: Role of the Ni(111) Facets. ACS Catal. 2020, 10 (15), 8077–8089. 10.1021/acscatal.0c01599. [DOI] [Google Scholar]
- Chutia A.; Thetford A.; Stamatakis M.; Catlow C. R. A. A DFT and KMC based study on the mechanism of the water gas shift reaction on the Pd(100) surface. Phys. Chem. Chem. Phys. 2020, 22 (6), 3620–3632. 10.1039/C9CP05476F. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54 (16), 11169–11186. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6 (1), 15–50. 10.1016/0927-0256(96)00008-0. [DOI] [Google Scholar]
- Kresse G.; Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 59 (3), 1758. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77 (18), 3865. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132 (15), 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Owen E.; Yates E. XLI. Precision measurements of crystal parameters. London, Edinburgh, Dublin Philos. Mag. J. Sci. 1933, 15 (98), 472–488. 10.1080/14786443309462199. [DOI] [Google Scholar]
- Monkhorst H. J.; Pack J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13 (12), 5188. 10.1103/PhysRevB.13.5188. [DOI] [Google Scholar]
- Henkelman G.; Jónsson H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113 (22), 9978–9985. 10.1063/1.1323224. [DOI] [Google Scholar]
- Henkelman G.; Uberuaga B. P.; Jónsson H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113 (22), 9901–9904. 10.1063/1.1329672. [DOI] [Google Scholar]
- Henkelman G.; Jónsson H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 1999, 111 (15), 7010–7022. 10.1063/1.480097. [DOI] [Google Scholar]
- Heyden A.; Bell A. T.; Keil F. J. Efficient methods for finding transition states in chemical reactions: Comparison of improved dimer method and partitioned rational function optimization method. J. Chem. Phys. 2005, 123 (22), 224101 10.1063/1.2104507. [DOI] [PubMed] [Google Scholar]
- Stamatakis M.; Vlachos D. G. A graph-theoretical kinetic Monte Carlo framework for on-lattice chemical kinetics. J. Chem. Phys. 2011, 134 (21), 214115 10.1063/1.3596751. [DOI] [PubMed] [Google Scholar]
- Nielsen J.; d’Avezac M.; Hetherington J.; Stamatakis M. Parallel kinetic Monte Carlo simulation framework incorporating accurate models of adsorbate lateral interactions. J. Chem. Phys. 2013, 139 (22), 224706 10.1063/1.4840395. [DOI] [PubMed] [Google Scholar]
- Ravipati S.; Savva G. D.; Christidi I. A.; Guichard R.; Nielsen J.; Reocreux R.; Stamatakis M. Coupling the Time-Warp Algorithm with the Graph-Theoretical Kinetic Monte Carlo Framework for Distributed Simulations of Heterogeneous Catalysts. Comput. Phys. Commun. 2022, 270, 108148. 10.1016/j.cpc.2021.108148. [DOI] [Google Scholar]
- Lombardo S. J.; Bell A. T. Monte Carlo simulations of the effect of pressure on isothermal and temperature-programmed desorption kinetics. Surf. Sci. 1991, 245 (1–2), 213–224. 10.1016/0039-6028(91)90480-G. [DOI] [Google Scholar]
- Chatterjee A.; Voter A. F. Accurate acceleration of kinetic Monte Carlo simulations through the modification of rate constants. J. Chem. Phys. 2010, 132 (19), 194101 10.1063/1.3409606. [DOI] [PubMed] [Google Scholar]
- Dybeck E. C.; Plaisance C. P.; Neurock M. Generalized Temporal Acceleration Scheme for Kinetic Monte Carlo Simulations of Surface Catalytic Processes by Scaling the Rates of Fast Reactions. J. Chem. Theory Comput. 2017, 13 (4), 1525–1538. 10.1021/acs.jctc.6b00859. [DOI] [PubMed] [Google Scholar]
- Vignola E.; Steinmann S. N.; Vandegehuchte B. D.; Curulla D.; Stamatakis M.; Sautet P. A machine learning approach to graph-theoretical cluster expansions of the energy of adsorbate layers. J. Chem. Phys. 2017, 147 (5), 054106 10.1063/1.4985890. [DOI] [PubMed] [Google Scholar]
- Ko J.; Kim B.-K.; Han J. W. Density functional theory study for catalytic activation and dissociation of CO2 on bimetallic alloy surfaces. J. Phys. Chem. C 2016, 120 (6), 3438–3447. 10.1021/acs.jpcc.6b00221. [DOI] [Google Scholar]
- Peng G.; Sibener S.; Schatz G. C.; Ceyer S. T.; Mavrikakis M. CO2 hydrogenation to formic acid on Ni (111). J. Phys. Chem. C 2012, 116 (4), 3001–3006. 10.1021/jp210408x. [DOI] [Google Scholar]
- Favaro M.; Xiao H.; Cheng T.; Goddard W. A. III; Yano J.; Crumlin E. J. Subsurface oxide plays a critical role in CO2 activation by Cu (111) surfaces to form chemisorbed CO2, the first step in reduction of CO2. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (26), 6706–6711. 10.1073/pnas.1701405114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huš M.; Kopač D.; Štefančič N. S.; Jurković D. L.; Dasireddy V. D.; Likozar B. Unravelling the mechanisms of CO2 hydrogenation to methanol on Cu-based catalysts using first-principles multiscale modelling and experiments. Catal. Sci. Technol. 2017, 7 (24), 5900–5913. 10.1039/C7CY01659J. [DOI] [Google Scholar]
- Kowalec I.; Kabalan L.; Catlow R.; Logsdail A. A Computational Study of Direct CO2 Hydrogenation to Methanol on Pd Catalysts. 2022. Phys. Chem. Chem. Phys. 2022, 24 (16), 9360–9373. 10.1039/D2CP01019D. [DOI] [PubMed] [Google Scholar]
- Higham M. D.; Quesne M. G.; Catlow C. R. A. Mechanism of CO2 conversion to methanol over Cu (110) and Cu (100) surfaces. Dalton Trans. 2020, 49 (25), 8478–8497. 10.1039/D0DT00754D. [DOI] [PubMed] [Google Scholar]
- Quesne M. G.; Roldan A.; de Leeuw N. H.; Catlow C. R. A. Carbon dioxide and water co-adsorption on the low-index surfaces of TiC, VC, ZrC and NbC: a DFT study. Phys. Chem. Chem. Phys. 2019, 21 (20), 10750–10760. 10.1039/C9CP00924H. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Liu F.; Zhao X.; Wang B.; Ling L. First-principles study about the effect of coverage on H2 adsorption and dissociation over a Rh (100) surface. J. Phys. Chem. C 2015, 119 (19), 10355–10364. 10.1021/jp512504b. [DOI] [Google Scholar]
- Heyl D.; Rodemerck U.; Bentrup U. Mechanistic study of low-temperature CO2 hydrogenation over modified Rh/Al2O3 catalysts. ACS Catal. 2016, 6 (9), 6275–6284. 10.1021/acscatal.6b01295. [DOI] [Google Scholar]
- Huai L.-y.; Su T.; Wen H.; Jin X.; Liu J.-y. NO reduction by H2 on the Rh(111) and Rh (221) surfaces: A mechanistic and kinetic study. J. Phys. Chem. C 2016, 120 (10), 5410–5419. 10.1021/acs.jpcc.5b10740. [DOI] [Google Scholar]
- Wellendorff J.; Silbaugh T. L.; Garcia-Pintos D.; Nørskov J. K.; Bligaard T.; Studt F.; Campbell C. T. A benchmark database for adsorption bond energies to transition metal surfaces and comparison to selected DFT functionals. Surf. Sci. 2015, 640, 36–44. 10.1016/j.susc.2015.03.023. [DOI] [Google Scholar]
- Dulaurent O.; Chandes K.; Bouly C.; Bianchi D. Heat of adsorption of carbon monoxide on a Pd/Rh three-way catalyst and on a Rh/Al2O3 solid. J. Catal. 2000, 192 (2), 262–272. 10.1006/jcat.2000.2844. [DOI] [Google Scholar]
- Stroppa A.; Kresse G. The shortcomings of semi-local and hybrid functionals: what we can learn from surface science studies. New J. Phys. 2008, 10 (6), 063020 10.1088/1367-2630/10/6/063020. [DOI] [Google Scholar]
- Gajdoš M.; Eichler A.; Hafner J. CO adsorption on close-packed transition and noble metal surfaces: trends from ab initio calculations. J. Phys.: Condens. Matter 2004, 16 (8), 1141. 10.1088/0953-8984/16/8/001. [DOI] [Google Scholar]
- Feibelman P. J.; Hammer B.; Nørskov J. K.; Wagner F.; Scheffler M.; Stumpf R.; Watwe R.; Dumesic J. The CO/Pt (111) puzzle. J. Phys. Chem. B 2001, 105 (18), 4018–4025. 10.1021/jp002302t. [DOI] [Google Scholar]
- Hammer B.; Hansen L. B.; Nørskov J. K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59 (11), 7413. 10.1103/PhysRevB.59.7413. [DOI] [Google Scholar]
- Dubois L.; Somorjai G. The chemisorption of CO and CO2 on Rh(111) studied by high resolution electron energy loss spectroscopy. Surf. Sci. 1980, 91 (2–3), 514–532. 10.1016/0039-6028(80)90348-9. [DOI] [Google Scholar]
- Hsing C.-R.; Chang C.-M.; Cheng C.; Wei C.-M. Quantum Monte Carlo studies of CO adsorption on transition metal surfaces. J. Phys. Chem. C 2019, 123 (25), 15659–15664. 10.1021/acs.jpcc.9b03780. [DOI] [Google Scholar]
- Krenn G.; Bako I.; Schennach R. CO adsorption and CO and O coadsorption on Rh(111) studied by reflection absorption infrared spectroscopy and density functional theory. J. Chem. Phys. 2006, 124 (14), 144703 10.1063/1.2184308. [DOI] [PubMed] [Google Scholar]
- Fisher I. A.; Bell A. T. A Comparative Study of CO and CO2 Hydrogenation over Rh/SiO2. J. Catal. 1996, 162 (1), 54–65. 10.1006/jcat.1996.0259. [DOI] [Google Scholar]
- Solymosi F.; Erdöhelyi A. Decomposition of formic acid on supported Rh catalysts. J. Catal. 1985, 91 (2), 327–337. 10.1016/0021-9517(85)90346-X. [DOI] [Google Scholar]
- Iizuka T.; Tanaka Y.; Tanabe K. Hydrogenation of CO and CO2 over rhodium catalysts supported on various metal oxides. J. Catal. 1982, 76 (1), 1–8. 10.1016/0021-9517(82)90230-5. [DOI] [Google Scholar]
- Bando K. K.; Ichikuni N.; Soga K.; Kunimori K.; Arakawa H.; Asakura K. Characterization of Rh Particles and Li-Promoted Rh Particles in Y Zeolite during CO2 Hydrogenatio-A New Mechanism for Catalysis Controlled by the Dynamic Structure of Rh Particles and the Li Additive Effect. J. Catal. 2000, 194 (1), 91–104. 10.1006/jcat.2000.2911. [DOI] [Google Scholar]
- Miguel C. V.; Soria M. A.; Mendes A.; Madeira L. M. Direct CO2 hydrogenation to methane or methanol from post-combustion exhaust streams–A thermodynamic study. J. Nat. Gas Sci. Eng. 2015, 22, 1–8. 10.1016/j.jngse.2014.11.010. [DOI] [Google Scholar]
- Gao J.; Wang Y.; Ping Y.; Hu D.; Xu G.; Gu F.; Su F. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Adv. 2012, 2 (6), 2358–2368. 10.1039/c2ra00632d. [DOI] [Google Scholar]
- Sun D.; Simakov D. S. Thermal management of a Sabatier reactor for CO2 conversion into CH4: Simulation-based analysis. J. CO2 Util. 2017, 21, 368–382. 10.1016/j.jcou.2017.07.015. [DOI] [Google Scholar]
- Williams K. J.; Boffa A. B.; Salmeron M.; Bell A. T.; Somorjai G. A. The kinetics of CO2 hydrogenation on a Rh foil promoted by titania overlayers. Catal. Lett. 1991, 9 (5), 415–426. 10.1007/BF00764834. [DOI] [Google Scholar]
- Solymosi F.; Erdöhelyi A.; Bánsági T. Methanation of CO2 on supported rhodium catalyst. J. Catal. 1981, 68 (2), 371–382. 10.1016/0021-9517(81)90106-8. [DOI] [Google Scholar]
- Fishman M.; Zhuang H. L.; Mathew K.; Dirschka W.; Hennig R. G. Accuracy of exchange-correlation functionals and effect of solvation on the surface energy of copper. Phys. Rev. B 2013, 87 (24), 245402 10.1103/PhysRevB.87.245402. [DOI] [Google Scholar]
- Kattel S.; Ramírez P. J.; Chen J. G.; Rodriguez J. A.; Liu P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 2017, 355 (6331), 1296–1299. 10.1126/science.aal3573. [DOI] [PubMed] [Google Scholar]
- Studt F.; Behrens M.; Kunkes E. L.; Thomas N.; Zander S.; Tarasov A.; Schumann J.; Frei E.; Varley J. B.; Abild-Pedersen F.; et al. The mechanism of CO and CO2 hydrogenation to methanol over Cu-based catalysts. ChemCatChem 2015, 7 (7), 1105–1111. 10.1002/cctc.201500123. [DOI] [Google Scholar]
- Grabow L. C.; Mavrikakis M. Mechanism of Methanol Synthesis on Cu through CO2 and CO Hydrogenation. ACS Catal. 2011, 1 (4), 365–384. 10.1021/cs200055d. [DOI] [Google Scholar]
- Chen Y.; Choi S.; Thompson L. T. Low-Temperature CO2 Hydrogenation to Liquid Products via a Heterogeneous Cascade Catalytic System. ACS Catal. 2015, 5 (3), 1717–1725. 10.1021/cs501656x. [DOI] [Google Scholar]
- Zhao Y.-F.; Yang Y.; Mims C.; Peden C. H. F.; Li J.; Mei D. Insight into methanol synthesis from CO2 hydrogenation on Cu(111): Complex reaction network and the effects of H2O. J. Catal. 2011, 281 (2), 199–211. 10.1016/j.jcat.2011.04.012. [DOI] [Google Scholar]
- Yang Y.; Mims C. A.; Mei D. H.; Peden C. H. F.; Campbell C. T. Mechanistic studies of methanol synthesis over Cu from CO/CO2/H2/H2O mixtures: The source of C in methanol and the role of water. J. Catal. 2013, 298, 10–17. 10.1016/j.jcat.2012.10.028. [DOI] [Google Scholar]
- Yang Y.; Evans J.; Rodriguez J. A.; White M. G.; Liu P. Fundamental studies of methanol synthesis from CO2 hydrogenation on Cu(111), Cu clusters, and Cu/ZnO(0001̅). Phys. Chem. Chem. Phys. 2010, 12 (33), 9909–9917. 10.1039/c001484b. [DOI] [PubMed] [Google Scholar]
- Greeley J.; Mavrikakis M. Methanol Decomposition on Cu(111): A DFT Study. J. Catal. 2002, 208 (2), 291–300. 10.1006/jcat.2002.3586. [DOI] [Google Scholar]
- Kunkes E. L.; Studt F.; Abild-Pedersen F.; Schlögl R.; Behrens M. Hydrogenation of CO2 to methanol and CO on Cu/ZnO/Al2O3: Is there a common intermediate or not?. J. Catal. 2015, 328, 43–48. 10.1016/j.jcat.2014.12.016. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.