

Abstract
Background
DNA methylation is an important epigenetic mode of genomic DNA modification and plays a vital role in maintaining epigenetic content and regulating gene expression. Cytosine-5 DNA methyltransferase (C5-MTase) are the key enzymes in the process of DNA methylation. However, there is no systematic analysis of the C5-MTase in cotton so far, and the function of DNMT2 genes has not been studied.
Methods
In this study, the whole genome of cotton C5-MTase coding genes was identified and analyzed using a bioinformatics method based on information from the cotton genome, and the function of GhDMT6 was further validated by VIGS experiments and subcellular localization analysis.
Results
33 C5-MTases were identified from three cotton genomes, and were divided into four subfamilies by systematic evolutionary analysis. After the protein domain alignment of C5-MTases in cotton, 6 highly conserved motifs were found in the C-terminus of 33 proteins involved in methylation modification, which indicated that C5-MTases had a basic catalytic methylation function. These proteins were divided into four classes based on the N-terminal difference, of which DNMT2 lacks the N-terminal regulatory domain. The expression of C5-MTases in different parts of cotton was different under different stress treatments, which indicated the functional diversity of cotton C5-MTase gene family. Among the C5-MTases, the GhDMT6 had a obvious up-regulated expression. After silencing GhDMT6 with VIGS, the phenotype of cotton seedlings under different stress treatments showed a significant difference. Compared with cotton seedlings that did not silence GhDMT6, cotton seedlings silencing GhDMT6 showed significant stress resistance.
Conclusion
The results show that C5-MTases plays an important role in cotton stress response, which is beneficial to further explore the function of DNMT2 subfamily genes.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12870-024-04985-x.
Keywords: C5-MTase, Gossypium raimondii, Gossypium arboreum, Gossypium hirsutum, Abiotic stress
Background
DNA methylation is the process of transferring a methyl (-CH3) group to a specific base of a DNA molecule and is catalyzed by DNA methyltransferase, with S-adenosine methionine (SAM) as a methyl donor [1]. It widely occurs in the epigenetics of bacteria, plants, and animals and is involved in transposons, the suppression of gene silencing, genomic imprinting, X chromosome inactivation, cell differentiation and embryo development [2–5]. DNA methylation is a dynamic process. When plants are subjected to stress, DNA methylation breaks through the inherent stability and limitation of their genome and makes a rapid response to adversity [6]. This induces the expression of some genes associated with stress to maintain plant growth, development and evolutionary process. Therefore, epigenetic modification precedes genomic evolution in response to adversity, and DNA methylation is considered the molecular response mechanism of plants in the face of adverse stress.
DNA methylation mostly take place in CHG at carbon 5 in cytosine (C5) [7] and primarily occurs in symmetric sequence CHG but also in CHG and CHH (H = A, C or T) sequences [5, 8]. There are two DNA methylation methods in plants: maintenance methylation and denovo methylation [9]. The former means that double-stranded DNA molecule through semi-reserved replication, which is distinguished by whether it is methylated. Parental methylation patterns are passed to the offspring [10]. The latter is a type of DNA methylation that happens when different C5-Mtases catalyze two strands of DNA without methylation. C5-Mtases in plants cotain four categories, which respectively are methyltransferase (MET), chromo methylase (CMT), domains rearranged methylase (DRM), and (DNA methyltransferase homologue 2)DNMT2 [11, 12]. METs are mainly used in methylation of the heterochromatin region of the CHG site of the symmetric sequence, which is an indispensable part of the methyltransferase [13]. CMTs are a special type of C5-Mtases that maintain methylation at CHG and CHH sites, and play a role in stabilizing the heterochromatin state of the genome [14]. DRM is homologous to the Dnmt3 of animals, and the function of them is to catalyze the methylation of cytosine and to maintain the cytosine methylation of non-CHG sites under the guidance of RNA [15, 16]. In previous study, after knockout of OsDRM2 gene, de novo methylation defect appeared in rice, which affected the DNA methylation level of rice genome, and finally led to abnormal reproductive and vegetative growth of rice [17]. The proteins encoded by the plant DNMT2 subfamily are very similar to those of mice, bacteria and yeast, and researches has shown that the DNMT2 subfamily genes may play a role in RNA methylation [18].
Abiotic stress can threaten the vegetative and reproductive growth of plants, accelerate the growth period and reduce the yield and quality of crops. Many studies have reported that the methylation level of plants also changes in response to abiotic stress [19, 20]. When subjected to different stresses, different DNA methylation in plant will active in the genome, which can regulate the expression of different stress response genes, thus opening up different regulatory pathways and improving plant tolerance [21]. DNA methylation is a very important regulatory pathway throughout the life cycle of organisms and plays a crucial role in the process of biological evolution, which is involved not only in growth and development but also in secondary metabolism [22–25]. DNA methylation is involved in the regulation of biological clock, photoperiod, stress resistance, metabolism, growth and development in many plant processes, including arabidopsis thaliana [26], solanaceae [27], legumes [21], maize [28], solanum lycopersicum [29]. However, studies on DNA methylation of cotton, which is an very important cash crop, have not been reported.
With the rapid change of global climate, the plant of cotton have been severely tested, especially the salt stress and drought stress seriously affect the yield and fiber quality of cotton. Many studies have reported that there is a close relationship between methylation levels and stress resistance in plants [30, 31], and indicated that C5-MTase is a key factor triggering methylation and can directly affect methylation levels [32].
However, there has been no relevant identification of C5-MTase in the cotton genome, and there has been no relevant study on how C5-MTase functions in response to salt stress and drought stress in cotton. With the completion of cotton genome sequencing [33–35], C5-MTase in cotton can be analyzed by genome-wide identification. In addition, the transcriptional abundance of GhDMT6 under salt stress and drought stress was analyzed. Our results provide a reference for the role of C5-MTase in abiotic stress resistance in cotton.
Results
Identification of C5-MTase family members
A total of 33 C5-MTase members were identified from the whole genome of cotton. Group A contained 9 C5-MTases and group D contained 8 C5-MTases, which were respectively named GaDMT1-GaDMT9 and GrDMT1-GrDMT8 according to their sequence on the chromosomes. Similarly, 16 C5-MTases were identified in the AD group, named GhDMT1-GhDMT16. Most of the C5-MTases are located on the chromosome in three cotton species. They consist of 344–1585 different amino acids, most of which contain 400–1000 amino acid residues. Because of the N-terminal difference in the gene structure, GaDMT3 contains up to 1585 amino acids, while GhDMT4 contains only 344 amino acids. The isoelectric point (PI) of C5-MTases ranges from 4.54 to 8.71. The predicted subcellular localization shows that most C5-MTases are located in the nucleus, but some are located in the outer chloroplast. Specially, GaDMT7 and GhDMT6 are located in the cell membrane (Table 1).
Table 1.
Basic characteristics of C5-MTases in the cotton genome
| gene name | Type | Locus ID | Location (chromosome) | Position (domain) | CDS (bp) | AA | PI | Predicted subcellular localization |
|---|---|---|---|---|---|---|---|---|
| GaDMT1 | CMTc | Ga02G1310 | Chr02:91041776-91047696- | 442–816 | 2724 | 851 | 5.66 | Nucleus |
| GaDMT2 | DRM | Ga04G1492 | Chr04:86372417-86376133- | 511–630 | 4638 | 640 | 4.88 | Chloroplast |
| GaDMT3 | MET | Ga04G1657 | Chr04:90780693-90787721- | 1150–1579 | 1179 | 1585 | 5.51 | Nucleus |
| GaDMT4 | CMTd | Ga07G0535 | Chr07:5708914–5,715,570 + | 499–871 | 2082 | 907 | 5.09 | Nucleus |
| GaDMT5 | CMTa | Ga08G1800 | Chr08:111527760-111536767- | 589–729 | 2580 | 730 | 7.69 | Nucleus |
| GaDMT6 | CMTb | Ga08G1801 | Chr08:111537181-111547908- | 769–1145 | 1914 | 1182 | 8.64 | Nucleus |
| GaDMT7 | DNMT2 | Ga08G2785 | Chr08:127956353–127,959,973+ | 19–395 | 1992 | 398 | 6.41 |
Cell membrane, Endoplasmic reticulum |
| GaDMT8 | DRM | Ga09G0343 | Chr09:9182888–9,188,986+ | 569–687 | 1494 | 693 | 5.14 | Chloroplast |
| GaDMT9 | CMTd | Ga13G0623 | Chr13:9274883-9284481- | 431–799 | 2421 | 835 | 5.35 | Cytoplasm |
| GrDMT1 | CMTd | Gorai.001G052000 | Chr01 : 4,942,493–4,949,993 | 507–867 | 2718 | 905 | 4.84 | Nucleus |
| GrDMT2 | CMTc | Gorai.002G216500 | Chr02 : 56,627,700–56,634,321 | 454 ~ 812 | 2556 | 851 | 5.93 | Nucleus |
| GrDMT3 | CMTa | Gorai.004G180200 | Chr04 : 49,072,088–49,079,643 | 587–945 | 2952 | 983 | 7.4 | Nucleus |
| GrDMT4 | CMTb | Gorai.004G180300 | Chr04 : 49,082,203–49,092,215 | 789–1145 | 3558 | 1185 | 8.22 | Nucleus |
| GrDMT5 | DNMT2 | Gorai.004G274400 | Chr04 : 60,871,759–60,876,114 | 20–391 | 1221 | 406 | 6.05 | Nucleus |
| GrDMT6 | DRM | Gorai.006G031000 | Chr06 : 8,112,196–8,117,039 | 513–627 | 1911 | 636 | 4.54 | Chloroplast |
| GrDMT7 | DRM | Gorai.012G062900 | Chr12 : 8,876,628–8,881,084 | 526–640 | 1965 | 654 | 4.72 | Chloroplast |
| GrDMT8 | MET | Gorai.012G048000 | Chr12 : 6,069,727–6,077,197 | 1144–1570 | 4734 | 1577 | 5.36 | Nucleus |
| GhDMT1 | DRM | CotAD_37635 | At_chr06:11311066.11315214- | 512–630 | 1911 | 636 | 4.8 | Nucleus |
| GhDMT2 | MET | CotAD_51709 | At_chr9:63179674.63185543- | 1076–1505 | 4536 | 1511 | 6.34 | Nucleus |
| GhDMT3 | DRM | CotAD_46796 | At_chr9:65748738.65752451+ | 511–630 | 1923 | 640 | 4.85 | Chloroplast |
| GhDMT4 | CMTd | CotAD_10542 | Dt_chr1:40856014.40859213- | 1-308 | 1035 | 344 | 7.76 | Nucleus |
| GhDMT5 | CMTc | CotAD_49037 | Dt_chr2:8190450.8196335- | 496–781 | 2451 | 816 | 5.62 | Nucleus |
| GhDMT6 | DNMT2 | CotAD_04205 | Dt_chr5:13787674.13791315+ | 13–389 | 1206 | 401 | 5.82 | Cell membrane, Endoplasmic reticulum |
| GhDMT7 | DRM | CotAD_13275 | Dt_chr6:40651384.40655909- | 512–611 | 1878 | 625 | 4.72 | Chloroplast |
| GhDMT8 | CMTd | CotAD_24264 | Dt_chr7:37416068.37421133+ | 230–632 | 2007 | 668 | 4.81 | Nucleus |
| GhDMT9 | CMTb | CotAD_00990 | Dt_chr10: 6477855.6487242+ | 789–1157 | 3585 | 1194 | 8.51 | Nucleus |
| GhDMT10 | CMTa | CotAD_00992 | Dt_chr10:6495924.6502242+ | 299–680 | 2094 | 697 | 6.75 | Nucleus |
| GhDMT11 | CMTc | CotAD_14980 | scaffold39.1:602870.611908+ | 667–846 | 2646 | 881 | 6.94 | Nucleus |
| GhDMT12 | DRM | CotAD_18652 | scaffold71.1:1581335.1591239- | 547–666 | 2031 | 676 | 4.91 | Cytoplasm |
| GhDMT13 | CMTa | CotAD_41398 | scaffold294.1:1053646.1057162- | 319–500 | 1533 | 510 | 5.45 | Cytoplasm |
| GhDMT14 | CMTb | CotAD_41399 | scaffold294.1:1063401:1074111- | 780–1161 | 3597 | 1198 | 8.71 | Nucleus |
| GhDMT15 | MET | CotAD_46012 | scaffold1041.1:189137.194922+ | 1112–1541 | 4644 | 1547 | 5.57 | Nucleus |
| GhDMT16 | CMTd | CotAD_40093 | scaffold2005.1:29096.35764+ | 453–828 | 2595 | 864 | 5.02 | Nucleus |
Chromosomal localization analysis
The distribution map of the gene on the chromosome was constructed according to the gene locus information (Fig. 1). The 9 genes in group A were unevenly distributed on the 6 chromosomes of Chr02, Chr04, Chr07, Chr08, Chr09, and Chr13. Among them, Chr08 contained 3 genes on chromosome, which was the most one. GaDMT5 and GaDMT6 were a pair of tandem repeats (Fig. 1A). On the other hand, there were 8 genes in group D unevenly distributed on the 5 chromosomes of Chr01, Chr02, Chr04, Chr06, and Chr12. Among them, Chr06 has the most genes on chromosome, with 3 genes. GrDMT7 and GrDMT8 were a pair of tandem repeats (Fig. 1B). The AD group only mapped genes located on chromosomes (Fig. 1C). In the AD group, GhDMT9 and GhDMT10 were repeated in series. Among the 16 genes in AD group, 10 genes were located on chromosomes At_chr6、At_chr9、Dt_chr1、Dt_chr2、Dt_chr5、Dt_chr6、Dt_chr7、Dt_chr10, respectively, while another 6 genes were mapped to the scaffold.
Fig. 1.

Chromosome distribution and syntenic analysis of C5-MTases from cotton. (A) Positions of C5-MTase gene family members on A group chromosomes. (B) Positions of C5-MTase gene family members on D group chromosomes. (C) Positions of C5-MTase gene family members on AD group chromosomes
Conserved motifs and exon-intron analysis
Visualized analysis of the C5-MTase gene structure and conserved motifs was shown in Fig. 2. Gene structure analysis is an important method in the study of genetic evolution. The number of introns and exons in C5-MTase family members in groups D, A, and AD were analyzed. The C5-MTases structures for cotton were created (Fig. 2B). The results showed that the numbers of exons in different C5-MTases in cotton varied greatly. GrDMT8, GhDMT9, GhDMT11 and GhDMT14 gene had the most exons, with 24 exons, while the DNMT2, MET, CMTa, CMTb, CMTc, CMTd, DRM subfamily contained 10, 11–12, 13–19, 23–24, 21–24, 10–21, 10–13 exons, respectively.
Fig. 2.

Conserved motifs and exon-intron analysis of C5-MTases in cotton. (A) Phylogenetic tree of C5-MTases. (B) Exon-intron structures of C5-MTases. (C) Conservative motif analysis
Conserved motifs of C5-MTases members in three cotton genomes were analyzed (Fig. 2C), and the protein sequences of 33 C5-MTases members were entered into the MEME website and set 6specific motifs. The C-terminal catalytic region had 6 highly conserved motifs. Among these motifs, motif 4 and motif 6 were used for SAM-binding sites, and motif 1, motif 2, motif 3, and motif 5 were the C5-Mtase functional catalytic sites. To be specific, motif 3 was the active site, motif 5 was the target cytosine binding site, motif 2 was the DNA neutralization region, motif 1 was the target sequence location identification area. In this study, it was found that all C5-MTases members contain motif 4 except GhDMT4. The motifs of C5-MTases members within the same subfamily were approximately same, however, it was quite different between different subfamilies, but. For example, almost all MET contained motifs 2, 3, 4 and 5, butthe vast majority of C5-MTases members in DRM included motifs 3, 4, 5 and 6, and the DNMT2 subfamily usually contained motif 4 and motif 1 only.
Structural domain and promoter analysis
The phylogenetic results of C5-MTase and the protein domain results of C5-MTase protein were analyzed (Fig. 3A). It was found that the same subfamily of genes had similar protein structure. After analyzing the domains of 33 C5-MTase proteins of cotton (Fig. 3B), it was found that all C5-MTase proteins contained Cyt-C5-DNA-methylase domains. The Cyt-C5-DNA-methylase domain was the conservative protein domain of the C5-MTase family. Genes contained different domains and were subdivided into different subfamilies. Genes contained similar domains and could be subdivided into the same subfamily. For example, the CMT subfamily contained the CD-CMT3-like domain except for GhDMT16, the MET subfamily contains 2 DNMT1-RFD domains and 2 BAH-DCM domains, and the DNMT2 subfamily contained only one Cyt-C5-DNA-methylase domain.
Fig. 3.

Structural domain and promoters of C5-MTases from three cotton species. (A) Phylogenetic tree of C5-MTases. (B) Structural domain of C5-MTases proteins. (C) Promoters of C5-MTases.Collinear analysis
As a knob in the process of gene expression, promoters played a crucial role in plant growth and development. The promoters of 33 C5-MTases were analyzed in our study (Fig. 3C). The results showed that all C5-MTases promoters contained hormone response elements, but the distribution of these elements was not confirmed. It is also proved that that all the C5-MTases promoters contain one or more photo-responsive elements, which indicated that C5-MTases may be involved in plant circadian rhythm, photoperiod and other processes. Most of the C5-MTases promoters contained anaerobic induction elements and MeJA regulatory elements, and they also contained many different types of promoter elements.
In order to further investigate the evolutionary relationship between G.arboreum(A), G.raimondii(D) and G.hirsutum(AD) genomes, a collinear analysis between the two genomes was performed (Fig. 4). 56 linear gene pairs and 3 tandem repeat gene pairs were identified by collinearity analysis (Fig. 1). GaDMT5 and GaDMT6, GrDMT3 and GrDMT4, GhDMT9 and GhDMT10 were three tandem repeat gene pairs, respectively. Ga(A)-Ga(A), Gh(AD)-Gh(AD) and Gr(D)-Gr(D) had 1,10 and 1 co-linear gene pair, respectively. 18,9, and 17 linear gene pairs in Ga(A)-Gh(AD), Ga(A)-Gr(D), and Gh(AD)-Gr(D) were identified, respectively (supplementary table S3). A total of 44 genes were replicated genome-wide. Some of the C5-MTase genes in the AD group were mapped to scaffold, which caused that the gene pairs shown in the image were different from the actual ones. For example, a total of 10 homologous gene pairs were identified, but only 2 pairs were marked on the chromosome and 8 pairs were not.
Fig. 4.

Syntenic relationship of C5-MTases duplicated gene pairs in A, D, and AD
The cotton C5-MTase gene was multicollinearity and a collinear map was furtherly constructed between AD and A, D (Fig. 5). The finding showed that most of the C5-MTases were located on synlinear chromosomal segments, indicating that group A and group D had high similarity. CMT subfamily, DRM subfamily, MET subfamily, and DNMT2 subfamily all had orthologous genes in group A, group D, and group AD, indicating that the C5-MTases family was highly conserved in the course of evolution. The above characteristics were inherited in the process of doubling the hybridization between group A and group D to form the AD group. It is found that the CMTa subfamily has no orthologous genes. The reason was guessed that the function of this subfamily was similar to the other subfamily, which lead to it was lost or was replaced by other subfamily with evolution. All these results proved that the doubling of group A and group D to form group AD was an incomplete inheritance process.
Fig. 5.

Multiple collinear analysis of C5-MTase genes from three cotton species. The CMT subfamily genes are linked by purple lines. The DRM subfamily genes are linked by green line. The MET subfamily genes are linked by blue line. The DNMT2 subfamily genes are linked by red lines
Selection pressure and interspecies evolution analysis
Genomes are selected to response to the environment pressure as species evolve. For purpose of understanding the selection process of the C5-MTase gene family during evolution, selection stress analysis of the three C5-MTase genomes were performed. The ratio between no-nsynonymous (Ka) and synonymous (Ks) was calculated using software (Fig. 6A). It was considered that there was a positive selection effect when Ka/Ks was greater than 1, a neutral selection occurs when equal 1, and a negative selection effect (purification effect or purification selection) when less than 1.The results showed that there were 34 gene pairs with a KA/Ks ratio less than 0.5 for C5-MTase. The Ka/Ks ratio of 8 pairs was greater than 0.5 but less than 0.99(Fig. 6B). The results strongly proved that the C5-MTases had negative selection effect during evolution and experienced strong purification selection.
Fig. 6.

Analysis of non-synonymous (Ka) to synonymous (Ks) ratio. (A) non-synonymous (Ka) and synonymous (Ks) divergence values for Ga–Ga, Ga-Gr, Ga-Gh, Gh-Gh Gh-Gr and Gr-Gr are shown in circular chart. (B) Predicted logarithm of genes
Analysis of inter-species evolution
Phylogenetic trees can reveal the homologous and evolutionary relationships among different species. In order to investigate the evolutionary relationship between the members of the cotton C5-MTases family and those of arabidopsis thaliana, cocoa, glycine max, oryza sativa, chlamydomonas reinhardtii and other crops, the biomolecular structure of C5-MTases in 13 species were compared to construct a phylogenetic tree (Fig. 7). As with other crop divisions, the C5-MTases members of the cotton genome were divided into four subfamilies, CMT, DRM, MET, and DNMT2. Among these subfamilies, the CMT had the largest number of genes, which was divided into CMTa, CMTb, CMTc, and CMTd. There was only CMTc types existed in the monocots but more than two types of CMT existed in the dicotyledones. This suggested that the differentiation of the CMT subfamily may occur after cotyledon and dicotyledones evolution. The CMT subfamily plays an important role in the development of vascular plant, while there was no CMT in chlamydomonas reinhardtii, which was suspected that the CMT subfamily genes were unique to vascular plant. Interestingly, it was found that GhDMT numbers were lower in some subfamilies of the tetraploid species AD (G.hirsutum) than in AtDMT. For example, the MET subfamily in arabidopsis thaliana consisted of 3 members, while AD had only 2. Combined with this phenomenon, i.e. was speculated that GhDMTs in the MET subfamily played a greater important role in cotton than AtDMTs in Arabidopsis thaliana. DNMT2 subfamily members were present in all 13 species, indicating that the DNMT2 subfamily was highly conserved and did not differentiate during species evolution. Cocoa was closely related to cotton in the evolutionary branch, indicating that the C5-MTases in cotton was closely related to that in cacao.
Fig. 7.

Phylogenetic analysis of C5-MTase family from among species. Ricinus communis(Rc), Vitis vinifera L (Vv), Oryza sativa(Os), Cacao(Co), Chlamydomonas reinhardtii(Cr), Medicago truncatula(Mt), Arabidopsis thaliana(At) Solanum lycopersicum (Sl) Solanum tuberosum (St) and Glycine max (Gly)
Expression analysis
Gene function is closed related with its expression.
To study the expression patterns of C5-MTases in different cotton tissues under salt and drought stress, the raimondii, Shixiya 1, and TM-1 were developed to the trefoil stage, and real-time quantitative PCR was performed (Fig. 8). The results showed that the three cotton species had different expression patterns under different stress conditions. Through cluster analysis, it was found that GrDMT3, GhDMT6, GhDMT8, GhDMT9, and GhDMT14 had higher expression in response to salt and drought stress. Because of GhDMT8, GhDMT9, and GhDMT14 belong to the CMT subfamily, it is speculated that CMTa, CMTb, CMTc, and CMTd subfamily regulated a certain type of genes and played a role together in response to drought and salt stress. Raimondii, Shixiya 1, and TM-1 had obvious tissue differences when treated with drought and salt stress. Raimondii and Shixiya 1 had more genes up-regulated in leaf parts than in rhizome parts, TM-1 had more genes down-regulated in leaf parts than in rhizome parts. However, the responses time to stress of C5-MTases in raimondii, Shixiya 1, and TM-1 were kept in consistent. The expression level of C5-MTases within 12 h under drought or salt stress was high, but it was low within 12 to 24 h.
Fig. 8.

Expression analysis of C5-MTases from cotton under drought and salt stress
Interaction network of GhDMT6 protein analysis
The qRT-PCRassays results showed that the GhDMT6 was highly expressed from TM-1 under drought and salt stress. GhDMT6 is a gene of the DNMT2 subfamily. To investigate the function of the DNMT2 subfamily, protein comparison was used to obtain orthologs of GhDMT6 in arabidopsis thaliana. Interaction network analysis of GhDMT6 was performed with proteins by sequence program using STRING data (Fig. 9). The function of the DNMT2 subfamily could be inferred from the in-depth study of AtDMT. GhDMT6 interacted with Retinoblastoma-related protein 1(RBR1) and Transcription factor-like protein DPB (DPB). MET1 gene interacts with member of the E2F transcription factors (E2F1, ATE2F2, E2F3). RBR1, DPB, E2F1, ATE2F2 and E2F3 were all genes related to cell division. It is inferred that C5-MTase is involved in cell division.
Fig. 9.
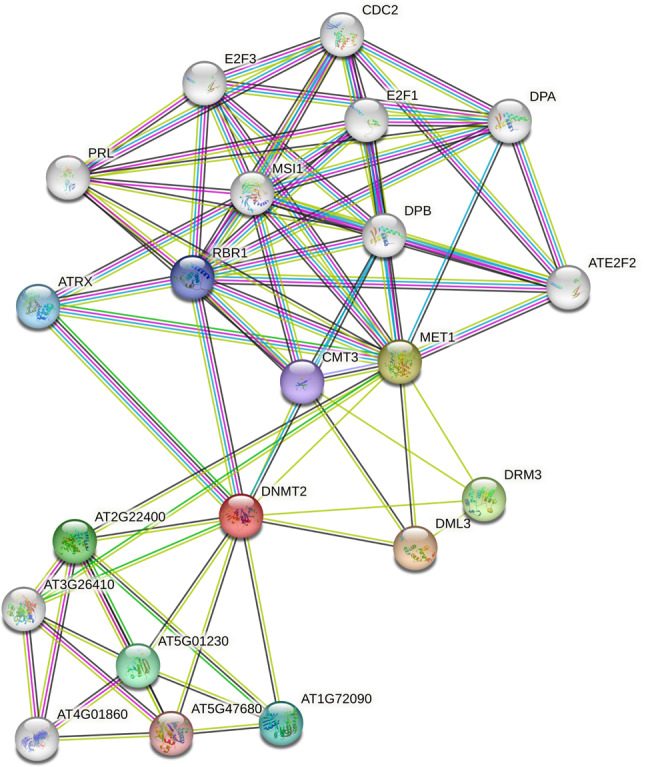
Interaction network of GhDMT6 protein
Subcellular localization analysis of GhDMT6
To further explore the function of GhDMT6, subcellular localization was used to determine the GhDMT6 protein location. The location of red fluorescence was observed after GhDMT6-RFP vector was injected into tobacco leaves. The result showed GhDMT6 protein located in the cell membrane and endoplasmic reticulum. Fluorescence of GhDMT6 found by confocal microscopy indicated that it was mainly distributed within the cell and formed a hollow ring enclosing the nucleus with characteristic endoplasmic reticulum expression. This result demonstrated that GhDMT6 may be localized to the ER (Fig. 10).
Fig. 10.

Subcellular localization of GhDMT6 protein
Expression and silencing analysis of GhDMT6 under drought and salt stress in cotton
To understand the role of GhDMT6 in response to drought and salt stress, phenotypic changes in cotton were observed by specifically silencing the GhDMT6 gene. The leaf bleaching illustrated that the GhDMT6 gene was specifically silenced. The fluorescent quantitative analysis showed the expression levels of GhDMT6 in roots, stems and leaves of plants injected with pYL156:GhDMT6 were significantly lower than control (Fig. 11B and D). After 6 days of natural drought, there were significant differences in the phenotypes of cotton seedlings. The cotyledons of CK and pYL156 seedlings were shed, and the new true leaves were yellow, withered and severely dehydrated (Fig. 11A), but the symptoms of pYL156:GhDMT6 seedlings were milder. Similarly, after 2 days of 200 mM NaCl stress treatment, there was also significant difference in the phenotype of cotton seedlings. CK and pYL156 seedlings had shed leaves and yellowed true leaf margins, while pYL156: GhDMT6 cotton had milder symptoms (Fig. 11C).
Fig. 11.

Specifically silencing GhDMT6 gene. A Phenotypic differences of cotton under drought stress. B Expression of GhDMT6 after drought stress. C Phenotypic differences of cotton after salt stress. D Expression of GhDMT6 after salt stress
Discussion
Cotton is one of the pioneer plants in saline-alkali lands because of its unique ability to resist salt and drought [36]. After the completion of the genome project of cotton [37], the identification and study of gene family classifications, evolutionary features and function prediction at the whole-genome level becomes a hotspot of cotton functional gene research, which also provides a method to study the mechanism of stress tolerance in cotton for us. Many reports have shown that DNA methylation is a response to biological adversity and regulates the expression of downstream genes on the basis of maintaining its genome unchanged [38, 39]. The methylation level in plants is a dynamic process, which initiates different genes downstream and then participates in different regulatory pathways [40]. C5-MTases are key enzymes in DNA methylation, which is closely related to resistance to stress [41]. However, studies of the C5-MTases in cotton have been largely absent. Therefore, the study of genome-wide C5-MTases in cotton is strongly necessary, which would have great significance of cotton breeding, the identification of functional genes and the mechanism of cotton resistance.
In this study, the classification of C5-MTases in different crops were quantitatively compared, which indicated that the number of DNMT2 family genes showed a big difference. G.hirsutum (tetraploid) had only one gene, while arabidopsis thaliana (diploid) had two genes. DNMT2 family members decreased as the genome increases, which may be due to the functional differentiation or loss of the DNMT2 family during evolution. The gene structure of the CMT subfamily was similar to the MET subfamily, except that there was a conserved region (chromodomain) of approximately 60 amino acid residues between motif 3 and motif 4 of the CMT gene. There was no CMTb subfamily gene in arabidopsis thaliana, but there were 3 genes in the MET subfamily in arabidopsis thaliana which were significantly more than in cotton. We infer that CMT and MET have the same or synergetic effect. The above results are consistent with the conjecture that the CMT subfamily may originate from the MET subfamily which is the closest to CMT subfamily [42, 43]. The N-terminal of CMT subfamily and MET subfamily both have BAH domains, but CMT has only one BAH domain while MET1 has two. In arabidopsis thaliana, the function of AtCMT1 is lost after the BAH domain of AtCMT1 is mutated or destroyed, and arabidopsis thaliana, CMT subfamily to maintain its own function [44].
DNA methylation in the non-CG context is widespread in the plant kingdom. Non-CG methylation in arabidopsis thaliana is coordinately regulated by DRM subfamily and CMT subfamily proteins. CMT may play an important role during the abiotic stress response via non-CG methylation. It was reported that OsCMT3b was subfunctionalized to accommodate a distinct cluster of non-CG-methylated sites at highly GC-rich regions in the rice genome [45]. Chen Zhu et al. found that CsCMT1 and CsCMT2 genes in camellia sinensis were up-regulation under drought stress and cold stress [46]. Vijay gahlaut found that TaCMT3-6B in triticum aestivum showed up-regulation when subjected to drought stress and heat stress [42]. In this study, the expression of C5-MTases in cotton under drought and salt stress was analyzed and it was found that CMT subfamily (GhDMT8, GhDMT9 and GhDMT14) were up-regulated after drought and salt stress. Different from G.hirsutum, the DRM family members, GrDMT2 and GrDMT4, were highly expressed in G.raimondii under drought and salt stress. It was hypothesized that non-CG site methylation occurred mainly in response to drought and salt stress in G.raimondii. DNMT2 is the original C5-MTases family [47]. How DNMT2 subfamily members play a role in response to abiotic stresses is unknown. Although the structure of DNMT2 is highly similar to the other C5-MTases, DNMT2 has only a C-terminal catalytic region and no complete N-terminal regulatory region. It has long been thought that the C5-MTases activity of DNMT2 may be very weak. Interestingly, the DNMT2 members, GhDMT6, GrDMT3, and GaDMT7, were up-regulated after PEG6000 and 200mM NaCl treatment in our study. These results indicated that DNMT2 subfamily might play an important role during the abiotic stress response via DNA methylation. It will be worth in-depth study to test this hypothesis in the future by genetic.
The Ka/Ks ratio for a total of 42 gene pairs were calculated to analyze the effects on C5-MTases during species evolution. It was found that the Ka/Ks ratio for all 42 gene pairs was less than 1, indicating that the C5-MTases family in the three cotton genomes underwent strong purification selection. In addition, this study predicted that 56 gene pairs played an important role in the expansion of C5-MTases during evolution (Supplemental table S1). There were fewer repetitions between Ga (A)-Ga (A), Gr (D)-Gr (D), Gh (AD)-Gh (AD), but more between Ga (A)-Gh (AD), Ga (A)-Gr (D), Gr (D)-Gh (AD). These results further confirmed that G.hirsutum (AD) derived from interspecific hybridization between G.arboreum (A) and G.raimondii (D) [48, 49]. In addition, we found 3 tandem repeat gene pairs that promote the evolution of C5-MTases function. During the evolution of cotton, the differentially expressed repetitive genes may undergo functional differentiation, and the function of C5-MTases can be stably preserved, indicating that the C5-MTases family plays a crucial role in the growth of cotton.
The promoters of C5-MTases in three cotton genomesand the protein-protein interaction of C5-MTases were detailedly analyzed. The C5-MTase promoter regions in three cotton species identified four stress responsiveness elements, such as defense and stress responsiveness, low-temperature responsiveness, anaerobic induction, and drought-inducibility. Most of the C5-MTase promoters identified plant hormone regulatory elements, such as abscisic acid responsiveness、MeJA-responsiveness、gibberellin-responsive element、salicylic acid responsiveness and auxin-responsive element, The promoter of C5-MTase has also found circadian control and light responsive element. All these findings demonstrated that C5-MTase participated in multiple signaling pathways and played an important role on defense responses, growth and development in cotton. In present study, several cell cycle genes were proved that interacted with GhDMTs protein, such as RBR1, DPB, E2F1, ATE2F2 and E2F3 [50–52]. Previous studies have shown that the cyclin-dependent kinase (CDK)-RB-E2F regulatory axis constitutes the core transcription mechanism that drives cell cycle progression, determines the timing and accuracy of genome replication, and ensures the accurate delivery of genetic material [53]. The related components regulating the CDK-RB-E2F pathway have been identified in almost all the human malignancies [54]. So GhDMTs are considered as key factor involved in cell cycle process. The growth stress mechanism of GhDMTs under abiotic stress needs further study.
Cell division, differentiation and apoptosis in plants are dynamic equilibrium processes. When plants are stressed, this balance is broken, establishing a new balance in response to stress [55]. The cell cycle is an important part of this rebalancing process. The cell cycle of eukaryotic cells refers to the cycle of continuous division of eukaryotic cells from the end of secondary mitosis to the end of the next division [56, 57]. Cell cycle is not a fixed valueor individual state, but cell cycle length is different, which reflects the state of the cell [58]. The length of cell cycle is mainly determined by the G1 phase of interphase, and the time is affected by both the organism itself and the environment [59, 60]. E2F plays an important role in regulating the cell cycle. E2F transcription factor is a member of the cell cycle gene, a key component of cyclin and retinoblastoma E2F pathway [61, 62]. Transcription factor-like protein DPB can be involved in regulating G1/S conversion. It can improve the DNA-binding activity of E2F protein after heterodimerization [63]. After the GhDMT6 was silenced under drought and salt stress, the activity of DPB gene was enhanced and the DNA binding activity of E2F protein after heterodimerization was increased to inhibit cell division. Cell division requires a lot of nutrients and energy reserves [64]. Therefore, GhDMT6 silenced plants showed stronger drought and salt tolerance than the control. At the same time, compared with CK plants, GhDMT6 silenced plants after stress enhanced their resistance by prolonging cell cycle, reducing the number of cell division, and further reducing the loss of nutrition and energy, resist coercion.
Conclusion
Based on the analysis of C5-MTase family in three cotton genomes, the characteristics of C5-MTase family were studied. In addition, we silenced the GhDMT6, and studied the expression of GhDMT6 under drought and salt stress. GhDMT6 gene is very conservative in the evolution of species. After silencing GhDMT6 by VIGS, cotton seedlings showed enhanced stress resistance. These results provide a basis for further studies on the response of C5-MTase gene to abiotic stresses by delaying the number of cell divisions during plant development, which would have great significance of great significance to cotton breeding, the identification of functional genes and the mechanism of cotton resistance.
Methods
Identification of C5-MTase family in cotton
The cotton genome information was downloaded from COTTONGEN (https://www.cottongen.org/) [65]. The DNA-methylase structure domain (PF00145) was downloaded from the Pfam (http://pfam.xfam.org/) database [66] (IPR001525). The DNA-methylase.hmm hidden markov model was constructed with DNA-methylase.hmm as the reference HMMER3.0 (http://hmmer.org/ download.html). The cotton genome database was queried to obtain the gene location and name of candidate protein family members containing DNA-methylase structure domains in cotton and to obtain GFF (general feature format) files from genome annotation files. Then, the gene position was obtained on the chromosome and used local BLAST 2.2.31 + to obtain the CDS sequence and protein sequence of the corresponding gene and obtained the whole sequence of the gene corresponding to the genome based on its position on the chromosome. The gene protein sequence was downloaded in the SMART software(http://smart.embl-heidelberg.de/). The Pfam30.0 database was analyzed to ensure that each candidate gene contains a DNA-methylase structure domain. Subcellular location prediction was performed on cello [67](http://cello.life.nctu.edu.tw/). The ProtParam(https://web.expasy.org/protparam/) was obtained by protein analysis.
Chromosomal locations of C5-MTases from cotton
The annotated and genomic files of the three cotton genomes were downloaded from COTTONGEN (https://www.cottongen.org/). TBtools software [68, 69] was used to map the positions of C5-MTases on chromosomes in three cotton genomes.
Analysis of conserved motifs and gene structure of C5-MTases in cotton
The protein sequence of C5-MTases in cotton was input into MEME website (http://meme-suite.org/) for conservative sequence recognition and obtains MAST profile. Gene sequences and genomic sequences were entered for C5-MTases at the Gene Structure Display Server 2.0 website (http://gsds.gao-lab.org/) site for intron and exon analysis to obtain the tab profile. Enter the obtained files into TBtools software to draw composite images [68, 69].
Analysis of structural domain and promoter regions
NCBI-CDD website (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi) was use to identify the domain contained in C5-MTases. DNA sequence of 2000 bp upstream of C5-MTases was extracted as promoter sequence. The C5-MTase promoters sequence was entered into PlantCare website [70] (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) for analysis to predict which cis-elements contained in the C5-MTases promoter.
Collinearity and selective pressure calculation analysis
The genomic sequences and GFF files of the three cotton genomes were input into MCScanX software [71] to conduct the same-line analysis among the duplicated gene pairs from G.arboreum(A), G.raimondii(D) and G.hirsutum(AD). The homologous gene information was entered into the Kaks_Calculator 2.0 program for selection stress analysis [72].
Phylogenetic analysis
Cotton DNA-methylase (PF00145, IPR001525) was used as the key word in Phytozome v12.1 [73] (https://phytozome.jgi.doe.gov/pz/portal.html) in the database rather than the homologous sequences of other species (supplementary table S2). Clustal W software was used to analyze the amino acid sequence alignment. MEGA7.0 software [74] was used to construct the phylogenetic tree with the neighbor-joining method. The number of bootstraps was 1000.
Expression nalysis of cotton C5-MTase under stresses
The phytotron sand culture cultivation method was used for planting three species of cotton, TM-1 (AD), raimondii (D)and Shixiya 1 (A) under16h light/8 h dark, day 28 °C, night 25 °C. Cotton seedlings were dealing with salt (200mM NaCl) and drought (20% PEG6000) stress at the three-leaf stage. The root, stem, and leaf samples of cotton were taken at 0 h, 1 h, 3 h, 6 h, 12 h and 24 h after experimental treatment. Total RNA was extracted from root, stem, and leaf samples by RN38 EASYspin Plus rapid plant RNA extraction kit and reversed transcribed into cDNA by TransScript II All-in-One First-Strand cDNA Synthesis SuperMix for qPCR (One-Step gDNA Removal) kit. The primers for the real-time fluorogenic quantitative PCR were designed with the NCBI-line primer design tool primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) (supplementary table S3). The RNA was reverse-transcribed into cDNA samples and used as a template for quantitative PCR experiments. QRT-PCRassays were performed on the Bio-Rad 7500 rapid fluorescence quantitative PCR platform, and the gene expression level of C5-MTase was detected by 2−ΔΔCt method.
Interaction network of GhDMT proteins
GhDMT6 protein sequence was used as target sequence to obtain arabidopsis thaliana homologous gene. The homologous gene sequence of arabidopsis thaliana was input into STRING software (https://string-db.org/) to further analyze the interaction between GhDMT proteins. The confidence parameter was 0.15 thresholds.
Subcellular localization analysis of GhDMT6
GhDMT6 gene was ligated into pBI121 vector containing red fluorescent protein to construct GhDMT6-RFP subcellular localization vector. The positive control, no-load control and GhDMT6-RFP vector were transferred into agrobacterium tumefaciens (LBA4404). Tobacco leaves were transfected with agrobacterium tumefaciens at 6 weeks after incubation in an indoor growth chamber, and instantaneously transformed tobacco leaves were used to visualize and locate RFP proteins under confocal laser scanning microscopy [75].
Virus‑induced gene silencing (VIGS) of GhDMT6
A 423 bp DNA fragment was extracted from GhDMT6 gene by restriction enzyme (EcoRI and XmaI) and ligated to the pYL156 vector to construct the pYL156: GhDMT6 vector. TM-1 was planted in an artificial climate incubator and prepared the injection when the two cotyledons were flattened. pYL156:GhDMT6, pYL156 Vector, pYL156:PDS were injected into cotton cotyledons and continued to culture cotton seedlings until the albino phenotype appeared, indicating successful gene silencing. Cotton seedlings were subjected to drought (natural drought) and salt stress (200 mM NaCl/24 h), and observed the changes of cotton phenotype. The roots, stems and leaves of plants were took as samples, and further analyzed the relative expression levels of GhDMT6.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We would like to thank all the teachers for their guidance and technical assistance.
Author contributions
XY: Conceptualization, Methodology, Software, Writing -original draft, Writing - review & editing. YH and NW: Conceptualization, Methodology, Writing - review & editing. ZB: VIGS and qPCR experiments, Writing - review & editing. YL, ZY, XL: Methodology, Writing- review & editing. XW, BZ, MH and XC: VIGS and qPCR experiments, Methodology. LS and JW: Methodology. DW and SW: Software, Writing - review & editing. LG and CC: Writing - review & editing. WY and KF: Conceptualization, Supervision.
Funding
This work was funded by China Project of Cotton Biological Breeding (NO.2023ZD04040), Modern Agricultural Industrial Technology System Construction Special Project of Jiangxi Province (NO. JXARS-22), The technology integration and demonstration promotion project of “Zhi-Mi-AI” simplified and efficient cultivation of cotton in Jiangxi Province (No. CCRI 2023-17) and Guiding project of Agriculture, animal husbandry and fishery in Jiangxi Province(NO.2023-14).
Data availability
All data supporting the conclusions of this article are provided within the article and its additional files.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no conflict of interest to declare.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Xiaomin Yang, Zhigang Bai, Yunxin He and Ning Wang contributed equally to this work.
Contributor Information
Keyun Feng, Email: fengkeyun@126.com.
Wuwei Ye, Email: yew158@163.com.
References
- 1.Edwards JR, Yarychkivska O, Boulard M, Bestor TH. DNA methylation and DNA methyltransferases. Epigenetics Chromatin. 2017;10:10–23. doi: 10.1186/s13072-017-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–20. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bender J. DNA methylation and epigenetics. Annu Rev Plant Biol. 2004;55:1186–91. doi: 10.1146/annurev.arplant.55.031903.141641. [DOI] [PubMed] [Google Scholar]
- 4.Heard E, Disteche CM. Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev. 2006;20:1848–67. doi: 10.1101/gad.1422906. [DOI] [PubMed] [Google Scholar]
- 5.Arthur B, Qiang H, Pooja N, Liam S, Hannah G, Matthew M, Qi H, Jacob P, Tzung-Fu H, An YQ. Dynamic DNA methylation in Plant Growth and Development. Int J Mol Sci. 2018;19:2144–61. doi: 10.3390/ijms19072144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang PJ. Plant genomic DNA methylation in response to stresses: potential applications and challenges in plant breeding. Prog Nat Sci. 2009;19:1037–45. doi: 10.1016/j.pnsc.2008.10.014. [DOI] [Google Scholar]
- 7.Lister R, OMalley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–36. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–9. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aufsatz W, Mette M, Matzke A, Matzke M. The role of MET1 in RNA-directed de novoand maintenance methylation of CG dinucleotides. Plant Mol Biol. 2004;54:793–804. doi: 10.1007/s11103-004-0179-1. [DOI] [PubMed] [Google Scholar]
- 10.Schumacher A. Epigenetic and genotype-specific effects on the stability of de novo imposed methylation patterns in transgenic mice. J Biol Chem. 2014;17:A673–673. doi: 10.1074/jbc.M004839200. [DOI] [PubMed] [Google Scholar]
- 11.Pavlopoulou A, Kossida S. Plant cytosine-5 DNA methyltransferases: structure, function, and molecular evolution. Genomics. 2007;90:530–41. doi: 10.1016/j.ygeno.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 12.Finnegan EJ, Kovac KA. Plant DNA methyltransferases. Plant Mol Biol. 2000;43:189–201. doi: 10.1023/A:1006427226972. [DOI] [PubMed] [Google Scholar]
- 13.Zubko E, Gentry M, Kunova A, Meyer P. De novo DNA methylation activity of methyltransferase 1 (MET1) partially restores body methylation in Arabidopsis thaliana. Plant J Cell Mol Biology. 2012;71:1029–37. doi: 10.1111/j.1365-313X.2012.05051.x. [DOI] [PubMed] [Google Scholar]
- 14.Wendte JM, Zhang Y, Ji L, Shi X, Schmitz RJ. Epimutations are associated with CHROMOMETHYLASE 3-induced de novo DNA methylation. eLife Sci. 2019;8:1–22. doi: 10.7554/eLife.47891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong X, Du J, Hale C, Gallego-Bartolome J, Feng S, Vashisht A, Chory J, Wohlschlegel J, Patel D, Jacobsen S. Molecular Mechanism of Action of Plant DRM De Novo DNA methyltransferases. Cell. 2014;157:1050–60. doi: 10.1016/j.cell.2014.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao X, Springer NM, Muszynski MG, Phillips RL, Kaeppler S, Jacobsen SE. Conserved plant genes with similarity to mammalian de novo DNA methyltransferases. Proc Natl Acad Sci USA. 2000;25:4979–81. doi: 10.1073/pnas.97.9.4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moritoh S, Eun CH, Ono A, Asao H, Terada R. Targeted disruption of an orthologue of DOMAINS REARRANGED METHYLASE 2, OsDRM2, impairs the growth of rice plants by abnormal DNA methylation. Plant J Cell Mol Biology. 2012;71:1–14. doi: 10.1111/j.1365-313X.2012.04974.x. [DOI] [PubMed] [Google Scholar]
- 18.Albert J, Ann E-M. Tomasz, Jurkowski, Frank, Lyko, Gunter: mechanism and biological role of Dnmt2 in nucleic acid methylation. RNA Biol. 2016;27:1547–55. doi: 10.1080/15476286.2016.1191737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ganguly D, Crisp PA, Eichten SR, Pogson BJ. The Arabidopsis DNA methylome is stable under Transgenerational Drought stress. Plant Physiol. 2017;6:1–56. doi: 10.1104/pp.17.00744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guizhen G, Jun, Li, Hao, Feng, Kun, Xu, Guixin Y. Comparison of the heat stress induced variations in DNA methylation between heat-tolerant and heat-sensitive rapeseed seedlings. Breed Sci. 2014;64:125–33. doi: 10.1270/jsbbs.64.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rohini G, Romika K, Sneha T, Shweta G, Wenqing X. Genomic Survey, Gene expression analysis and structural modeling suggest diverse roles of DNA methyltransferases in Legumes. PLoS ONE. 2014;9:e88947–8. doi: 10.1371/journal.pone.0088947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu R, How-Kit A, Stammitti L, Teyssier E, Rolin D, Mortain-Bertrand A, Halle S, Liu M, Kong J, Wu C. A DEMETER-like DNA demethylase governs tomato fruit ripening. Proc Natl Acad Sci. 2015;112:10804–9. doi: 10.1073/pnas.1503362112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mei-Qing X, Zhang Y-J. Xue-Ting, Wu: global analysis reveals the crucial roles of DNA methylation during rice seed development. Plant Physiol. 2015;168:1–62. doi: 10.1104/pp.15.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poonam B, Monika M, Ajay K. Vishwakarma, Jyoti, Bhardwaj, Sudesh: AtROS1 overexpression provides evidence for epigenetic regulation of genes encoding enzymes of flavonoid biosynthesis and antioxidant pathways during salt stress in transgenic tobacco. J Exp Bot. 2015;66:5959–69. doi: 10.1093/jxb/erv304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bo Z, Tieman DM, Chen J, Xu Y, Klee HJ. Chilling-induced tomato flavor loss is associated with altered volatile synthesis and transient changes in DNA methylation. Proc Natl Acad Sci U S A. 2016;113:12580–5. doi: 10.1073/pnas.1613910113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogneva ZV, Dubrovina AS, Kiselev KV. Age-associated alterations in DNA methylation and expression of methyltransferase and demethylase genes in Arabidopsis thaliana. Biol Plant. 2016;60:628–34. doi: 10.1007/s10535-016-0638-y. [DOI] [Google Scholar]
- 27.Kumar R, Chauhan PK, Khurana A. Identification and expression profiling of DNA methyltransferases during development and stress conditions in Solanaceae. Funct Integr Genom. 2016;16:1–16. doi: 10.1007/s10142-016-0502-3. [DOI] [PubMed] [Google Scholar]
- 28.Qian Y, Xi Y, Cheng B, Zhu S. Genome-wide identification and expression profiling of DNA methyltransferase gene family in maize. Plant Cell Rep. 2014;33:1661–72. doi: 10.1007/s00299-014-1645-0. [DOI] [PubMed] [Google Scholar]
- 29.Cao D, Ju Z, Gao C, Mei X, Fu D, Zhu H, Luo Y, Zhu B. Genome-wide identification of cytosine-5 DNA methyltransferases and demethylases in Solanum lycopersicum. Gene. 2014;550:230–7. doi: 10.1016/j.gene.2014.08.034. [DOI] [PubMed] [Google Scholar]
- 30.Ashapkin VV, Kutueva LI, Aleksandrushkina NI, Vanyushin BF. Epigenetic Mechanisms of Plant Adaptation to Biotic and Abiotic stresses. Int J Mol Sci. 2020;21:7457–90. doi: 10.3390/ijms21207457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bai J. In response to Abiotic Stress, DNA methylation confers EpiGenetic changes in plants. Plants (Basel Switzerland) 2021;10:1096–101. doi: 10.3390/plants10061096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du J, Zhong X, Bernatavichute Y, Stroud H, Feng S, Caro E, Vashisht A, Terragni J, Chin HG, Tu A. Dual Binding of Chromomethylase Domains to H3K9me2-Containing nucleosomes directs DNA methylation in plants. CELL -CAMBRIDGE MA. 2012;151:167–80. doi: 10.1016/j.cell.2012.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du X, Huang G, He S, Yang Z, Sun G, Ma X, Li N, Zhang X, Sun J, Liu M. Resequencing of 243 diploid cotton accessions based on an updated a genome identifies the genetic basis of key agronomic traits. Nat Genet. 2018;10:1–11. doi: 10.1038/s41588-018-0116-x. [DOI] [PubMed] [Google Scholar]
- 34.Repeated polyploidization Of Gossypium genomes and the evolution of spinnable cotton fibres. Nature. 2012;492:423–7. doi: 10.1038/nature11798. [DOI] [PubMed] [Google Scholar]
- 35.Genome sequence of cultivated Upland Cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotechnol. 2015;33:524–30. doi: 10.1038/nbt.3208. [DOI] [PubMed] [Google Scholar]
- 36.Binglei Z, Xiugui C, Xuke L, Na S, Xiaoge W, Xiaomin Y, Shuai W, Junjuan W, Lixue G, Delong W. Transcriptome analysis of Gossypium hirsutum L. reveals different mechanisms among NaCl, NaOH and Na2CO3 stress tolerance. Entific Rep. 2018;8:13527–31. doi: 10.1038/s41598-018-31668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuguang G, Cairui X, Guanghui, Changsong, Kohel RJ, Zhiying. Shang: Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nature biotechnology 2015; 33:524–530. [DOI] [PubMed]
- 38.Szyf M. DNA methylation mediating long-term genome responses to the Environment - ScienceDirect. Ref Module Biomedical Sci. 2014;1:1–8. [Google Scholar]
- 39.Song Q, Zhang T, Stelly DM, Chen ZJ. Epigenomic and functional analyses reveal roles of epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons. Genome Biol. 2017;18:99–113. doi: 10.1186/s13059-017-1229-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shiba H, Takayama S. RNA silencing systems and their relevance to allele-specific DNA methylation in plants. Biosci Biotechnol Biochem. 2007;71:2632–46. doi: 10.1271/bbb.70339. [DOI] [PubMed] [Google Scholar]
- 41.Zhou L, Cheng X, Connolly BA, Dickman MJ, Hurd PJ, Hornby DP. Zebularine: a novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J Mol Biol. 2002;321:591–9. doi: 10.1016/S0022-2836(02)00676-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gahlaut V, Samtani H, Khurana P. Genome-wide identification and expression profiling of cytosine-5 DNA methyltransferases during drought and heat stress in wheat (Triticum aestivum) Genomics. 2020;112:4796–807. doi: 10.1016/j.ygeno.2020.08.031. [DOI] [PubMed] [Google Scholar]
- 43.Gianoglio S, Moglia A, Acquadro A, Comino C, Portis E. The genome-wide identification and transcriptional levels of DNA methyltransferases and demethylases in globe artichoke. PLoS ONE. 2017;12:e0181669–75. doi: 10.1371/journal.pone.0181669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mull L, Ebbs ML, Bender J. A histone methylation-dependent DNA methylation pathway is uniquely impaired by Deficiency in Arabidopsis S-Adenosylhomocysteine hydrolase. Genetics. 2006;174(3):1161–71. doi: 10.1534/genetics.106.063974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daoheng H, Yiming Y, Chun W, Yanping L, Yue L, Li F, Dongdong L, Bo L, Jinbu J, Rui X. Multiplex CRISPR-Cas9 editing of DNA methyltransferases in rice uncovers a class of non-CG methylation specific for GC-rich regions. Plant Cell. 2021;33:2950–64. doi: 10.1093/plcell/koab162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu QD, Xiuxin Characterization of DNA methylation variations during Fruit Development and Ripening of Sweet Orange. Plant Mol Biology Report. 2015;33:1–11. doi: 10.1007/s11105-014-0732-2. [DOI] [Google Scholar]
- 47.Newton ILG. Evidence of adaptive evolution in Wolbachia-regulated gene DNMT2 and its role in the Dipteran Immune Response and Pathogen Blocking. Viruses. 2021;13:1–25. doi: 10.3390/v13081464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salmon A, Flagel L, Ying B, Udall JA, Wendel JF. Homoeologous nonreciprocal recombination in polyploid cotton. New Phytol. 2010;186:123–34. doi: 10.1111/j.1469-8137.2009.03093.x. [DOI] [PubMed] [Google Scholar]
- 49.Guo WZ, Sang ZQ, Zhou BL, Zhang TZ. Genetic relationships of D-genome species based on two types of EST-SSR markers derived from G. Arboreum and G. Raimondii in Gossypium. Plant ence. 2007;172:808–14. [Google Scholar]
- 50.Timmers C, Sharma N, Opavsky R, Maiti B, Wu L, Wu J, Orringer D, Trikha P, Saavedra HI, Leone G. E2f1, E2f2, and E2f3 control E2F target expression and cellular proliferation via a p53-dependent negative feedback loop. Mol Cell Biol. 2007;27(1):65–78. doi: 10.1128/MCB.02147-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharma N, Timmers C, Trikha P, Saavedra HI, Obery A, Leone G. Control of the p53-p21CIP1 Axis by E2f1, E2f2, and E2f3 is essential for G1/S progression and Cellular Transformation. J Biol Chem. 2006;281:36124–31. doi: 10.1074/jbc.M604152200. [DOI] [PubMed] [Google Scholar]
- 52.Juan C, del Pozo SD-T, Nerea Cisneros, Gutierrez C. The balance between Cell Division and Endoreplication depends on E2FC-DPB, transcription factors regulated by the Ubiquitin-SCFSKP2A pathway in Arabidopsis. Plant Cell. 2006;18:2224–35. doi: 10.1105/tpc.105.039651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang K, Kumar R. Interferon-α inhibits cyclin E- and cyclin D1-Dependent CDK-2 kinase activity Associated with RB protein and E2F in Daudi Cells. Biochem Biophys Res Commun. 1994;200:522–8. doi: 10.1006/bbrc.1994.1479. [DOI] [PubMed] [Google Scholar]
- 54.Mei-Rong B, Hai-Bo W, Jin-Ping GU, Otorhinolaryngology DO. The roles of RB-E2F pathways in breast Cancer and its therapy. Chin J Biochem Mol Biology. 2017;33:572–8. [Google Scholar]
- 55.Parsons RL, Behringer FJ, Medford JI. The SCHIZOID gene regulates differentiation and cell division in Arabidopsis thaliana shoots. Planta. 2000;211:34–42. doi: 10.1007/s004250000255. [DOI] [PubMed] [Google Scholar]
- 56.Yanishevsky RM, Stein GH. Regulation of the cell cycle in eukaryotic cells. Int Rev Cytol. 1981;69:223–59. doi: 10.1016/S0074-7696(08)62324-4. [DOI] [PubMed] [Google Scholar]
- 57.Yuuta I. Yamato, Yoshida, Fumi, Yagisawa, Haruko, Kuroiwa, Tsuneyoshi: the cell cycle, including the mitotic cycle and organelle division cycles, as revealed by cytological observations. J Electron Microsc. 2011;60:S117–36. doi: 10.1093/jmicro/dfr034. [DOI] [PubMed] [Google Scholar]
- 58.Levy N, Yonish-Rouach E, Oren M, Kimchi A. Complementation by wild-type p53 of interleukin-6 effects on M1 cells: induction of cell cycle exit and cooperativity with c-myc suppression. Mol Cell Biol. 1993;13:7942–52. doi: 10.1128/mcb.13.12.7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsushime H, Roussel MF, Ashmun RA, Sherr CJ. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 1991;65:701–13. doi: 10.1016/0092-8674(91)90101-4. [DOI] [PubMed] [Google Scholar]
- 60.Massagué J. G1 cell-cycle control and cancer. Nature. 2004;432:298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- 61.Chaboute ME. Cell cycle regulation of the Tobacco Ribonucleotide Reductase Small Subunit Gene is mediated by E2F-like elements. Plant Cell. 2000;12:1987–2000. doi: 10.1105/tpc.12.10.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu M, Sheppard KA, Peng CY, Yee AS, Piwnica-Worms H. Cyclin A/CDK2 binds directly to E2F-1 and inhibits the DNA-binding activity of E2F-1/DP-1 by phosphorylation. Mol Cell Biol. 1994;14:8420–31. doi: 10.1128/mcb.14.12.8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Magyar Z, Atanassova A, De VL, Rombauts S. Characterization of two distinct DP-related genes from Arabidopsis thaliana. FEBS Lett. 2000;486:79–87. doi: 10.1016/S0014-5793(00)02238-9. [DOI] [PubMed] [Google Scholar]
- 64.Muller EB, Ananthasubramaniam B, Klanjšček T, Nisbet RM. Entrainment of cell division in phytoplankton with dynamic energy budgets. J Sea Res. 2011;66:447–55. doi: 10.1016/j.seares.2011.04.004. [DOI] [Google Scholar]
- 65.Yu J, Jung S, Cheng CH, Lee T, Zheng P, Buble K, Crabb J, Humann J, Hough H, Jones D. CottonGen: the Community database for Cotton Genomics, Genetics, and breeding research. Plants (Basel Switzerland) 2021;10:D1229–36. doi: 10.3390/plants10122805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sonnhammer ELL, Eddy SR, Ewan B, Alex B, Richard D. Pfam: multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res. 1998;10:320–2. doi: 10.1093/nar/26.1.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Andrea P, Luigi MP, Piero F, Rita C. BaCelLo: a balanced subcellular localization predictor. Bioinformatics. 2006;22:e408–16. doi: 10.1093/bioinformatics/btl222. [DOI] [PubMed] [Google Scholar]
- 68.Chen C, Xia R, Chen H, He Y. TBtools, a toolkit for biologists integrating various HTS-data handling tools with a user-friendly interface. Cold Spring Harbor Lab. 2018;14:14–48. [Google Scholar]
- 69.Chen C, Chen H, Zhang Y, Thomas HR, Xia R. TBtools: an integrative Toolkit developed for interactive analyses of big Biological Data. Mol Plant. 2020;13:1–26. doi: 10.1016/j.molp.2020.06.009. [DOI] [PubMed] [Google Scholar]
- 70.Lamarca A, Brunette W, Koizumi D, Lease M, Borriello G. PlantCare: an investigation in practical ubiquitous systems. Springer Verlag. 2002;2:1–19. [Google Scholar]
- 71.Yupeng W, Jingping, Li AH, Paterson MCScanX-transposed: detecting transposed gene duplications based on multiple colinearity scans. Bioinf (Oxford England) 2013;29:1458–60. doi: 10.1093/bioinformatics/btt150. [DOI] [PubMed] [Google Scholar]
- 72.Dapeng W, Zhang Y, Zhang Z. KaKs_Calculator 2.0: a Toolkit incorporating Gamma-Series methods and sliding window strategies. Genomics Proteom Bioinf. 2010;8:77–80. doi: 10.1016/S1672-0229(10)60008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Goodstein DM, Shengqiang S, Russell H, Rochak N, Hayes RD, Joni F, Therese M, William D, Uffe H, Nicholas P. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;D1:D1178–86. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2015;33:1870–4. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mravec J, Skpa P, Bailly A, Hoyerová K, Keek P, Bielach A, Petráek J, Zhang J, Gaykova V, Stierhof YD. Subcellular homeostasis of phytohormone auxin is mediated by the ER-localized PIN5 transporter. Nature. 2009;459:1136–40. doi: 10.1038/nature08066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the conclusions of this article are provided within the article and its additional files.