

Abstract
Purpose
The aim of the study was to identify immunogenic HLA-A*0201-binding epitopes derived from a number of classical midgut carcinoid-associated proteins. CD8+ T cells recognizing tumor-associated antigen (TAA) epitopes are of great interest for the establishment of immunotherapy as a novel treatment for this type of malignancy.
Experimental design
Midgut carcinoid tumor specimens were microdissected and expression levels of potential TAAs were investigated by quantitative real time PCR. HLA-A*0201-binding motifs were selected using HLA peptide binding prediction algorithms and stabilization of HLA-A*0201 was verified using TAP-deficient T2 cells. Peripheral blood of midgut carcinoid patients was analyzed for peptide epitope recognition and the feasibility of generating peptide-reactive CD8+ T cells in healthy blood donors was examined by an in vitro stimulation protocol using mature DCs. Activation of patient and healthy donor CD8+ T cells was analyzed by intracellular flow cytometry staining of interferon γ.
Results
Chromogranin A (CGA), tryptophan hydroxylase 1 (TPH-1), vesicular monoamine transporter 1 (VMAT-1), caudal type homeobox transcription factor 2 (CDX-2), and islet autoantigen 2 (IA-2) are properly expressed by midgut carcinoid tumor cells, with CGA mRNA expressed to greatest level. Midgut carcinoid patients have increased frequencies of peripheral blood CD8+ T cells recognizing a pool of HLA-A*0201 peptides derived from these proteins compared to healthy age-matched individuals. Activated peptide-specific CD8+ T cells could also be generated in healthy blood donors by in vitro stimulation.
Conclusion
We have identified a number of immunogenic midgut carcinoid-associated peptide epitopes recognized by CD8+ T cells. We show that midgut carcinoid patients display immune recognition of their tumors. Memory CD8+ T cells in patient blood are of great interest when pursuing an immunotherapeutic treatment strategy.
Keywords: Midgut carcinoid, Antigens, HLA-A*0201, CD8+ T cells, IFNγ
Introduction
Neuroendocrine tumors of the gastroenteropancreatic tract (GEP-NETs) represent a rare and heterogenous group of tumors often associated with hypersecretion of hormones, bioactive peptides and amines [21]. Although a novel classification by the WHO in 2000 uses the notions neuroendocrine tumor and neuroendocrine carcinoma [25, 31], the term carcinoid is still often used for these malignancies. Classical midgut carcinoids are often well-differentiated neuroendocrine tumors arising from lower jejunum, ileum, caecum and ascending colon and are associated mainly with serotonin overproduction [1]. The tumor has generally metastasized to regional lymph nodes and liver at the time of diagnosis and patients with liver metastases are at risk of developing the so called carcinoid syndrome characterized by flushes, diarrhea, and endocardial fibrosis due to the excessive hormonal secretion [36]. Current treatment options for metastasized midgut carcinoids involve palliative surgery and medical treatment with somatostatin analogues and interferon α (IFNα), options that often alleviate symptoms of hormone overproduction and might give a temporary stabilization of disease progression, but not provide a complete cure [21].
A growing tumor may cause activation of the immune system due to the disruption of tissue homeostasis; T cells directed against tumor-associated antigens (TAAs) can spontaneously develop in cancer patients. The functionality of these usually low-avidity T cells and their influence on the clinical course of the disease has, however, been questioned [20]. The tumor area is a hostile and suppressive milieu for cytotoxic T cells (CTLs) and TAA-directed T cells often demonstrate an anergized phenotype [38]. Regulatory T cells and inhibitory cytokines present in the tumor area are important factors that contribute to down-regulation of a possible TAA-directed immune response [40]. TAA-responses against malignant melanoma have been extensively investigated and patients are often described as having T cells recognizing several different TAAs present in peripheral blood and infiltrating the tumor area. These T cells are used for anti-cancer treatment by different immune-enhancing strategies. Clinical trials involving adoptive T cell transfer [7] and transfer of genetically engineered T cells [18] have shown encouraging results. Previous immunotherapeutic attempts for neuroendocrine malignancies have been performed by in vivo activation of T cells. Dendritic cell vaccinations using crude tumor lysates or the polypeptide hormones parathyroid hormone or calcitonin as tumor antigens have generated anti-tumor responses [27-29] and partial tumor regression in patients [32]. Several clinical trials involving DC vaccinations for various malignancies, including malignant melanoma [35], renal cell carcinoma [39], gastrointestinal cancer [19], and prostate cancer [30] have been conducted up to date.
We have previously proposed a number of potential midgut carcinoid antigens based on tissue restriction following a large screening of tumor material and normal tissues [37]. In this paper, we make a more extensive analysis of the potential antigens chromogranin A (CGA), tryptophan hydroxylase 1 (TPH-1), vesicular monoamine transporter 1 (VMAT-1), caudal type homeobox transcription factor 2 (CDX-2), islet autoantigen 2 (IA-2), and survivin. We assure tumoral antigen expression by microdissection of malignant cells and make a relative quantitative analysis of the mRNA expression by real time PCR. Peripheral blood from midgut carcinoid patients was investigated for CD8+ T cell recognition of computer-predicted HLA-A*0201-binding peptide epitopes derived from these proteins. The possibility of generating activated peptide-specific CD8+ T cells in blood from healthy donors was also examined by in vitro stimulations. Identification of immunogenic epitopes from midgut carcinoid-associated antigens could provide a first step towards the establishment of immunotherapy as novel treatment for this neuroendocrine tumor.
Materials and methods
Patient material
Metastatic tumor specimens from eight patients diagnosed with midgut carcinoid tumor at the Department of Endocrine Oncology, Uppsala University Hospital, Uppsala, Sweden were snap frozen in liquid nitrogen after surgical resection and stored in −80°C until further use. Permission to collect tumor specimens was approved by the regional ethical committee (ref. no. Ups 02-077). Blood samples were collected from midgut carcinoid patients during routine disease monitoring at the Clinic of Endocrine Oncology, Uppsala University Hospital, Uppsala, Sweden. Permission to collect blood was approved by the regional ethical committee (ref. no. 2005:241) and informed consent was obtained from each patient. All patients demonstrated metastatic disease and were divided into two groups based on their tumor burden. The distinguishing criteria for a high tumor burden were ≥5 liver metastases while patients with <5 liver metastases and/or local metastases in lymph nodes and mesentery and at extra-abdominal sites were considered to have a low tumor burden. Plasma CGA was measured by a radioimmunosorbent assay (RIA) (Euro-Diagnostica, Malmö, Sweden). Average plasma CGA was 38.1 nmol/l in the patient group with a high tumor burden and 18.7 nmol/l in the group with a low tumor burden. Blood from 12 HLA-A*0201 positive patients with a high tumor burden and 17 HLA-A*0201 positive patients with a low tumor burden was included in the analysis. HLA-A typing was performed by incubating samples with a mouse monoclonal anti-HLA-A*0201 antibody (BB7.2, kind gift from Dr. J. Berzofsky, NCI, Bethesda, MD, USA) and a secondary FITC-labeled rabbit anti-mouse antibody (DAKO Cytomation, Copenhagen, Denmark) before flow cytometry analysis (FACS Calibur, BD Biosciences, San Diego, CA, USA).
Laser-assisted microdissection of midgut carcinoid tumor cells and RNA isolation
Eight tumor specimens from midgut carcinoid metastases were cut in 8 μm sections by using a microtome cryostat (Microm, Walldorf, Germany) and adhered to polyethylene-naphtalate (PEN) membrane slides (Carl Zeiss AB, Stockholm, Sweden). Tumor cells were isolated using the PALM® Robot Microbeam Laser Microdissection System (P.A.L.M Microlaser Technologies AG, Bernried, Germany) according to a procedure previously described [12, 17]. Total RNA was extracted from ∼5,000 microdissected tumor cells from each tumor specimen using the Pico Pure RNA isolation kit (Arcturus, Mountain View, CA, USA) according to the manufacturer’s protocol. RNA quality and quantity were evaluated using the RNA 6000 pico kit (Agilent Technologies, Palo Alto, CA, USA) and the Agilent 2100 Bioanalyzer (Agilent Technologies).
Quantitative real time PCR
Two nanograms of total RNA from microdissected tumor cells were reverse transcribed into cDNA using Superscript II™ RNase H− Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA). The cDNA was analyzed for expression of CGA, TPH-1, VMAT-1, CDX-2, IA-2, and survivin using primer pairs described in Table 1. Primer pairs were designed to span 2 or more exons to avoid amplification of genomic DNA and were designed de novo unless otherwise stated. Gene-specific PCR products were continuously measured by the iCycler IQ real time detection system (Bio-Rad Laboratories, Hercules, CA, USA) during 40 cycles with iQ SYBR Green Supermix (Bio-Rad) at 60°C. The reaction setup included DNase and RNase free water, 5 pmol/μl of forward and reverse primers, iQ SYBR Green Supermix and cDNA template. Data were evaluated using the 2−ΔΔct method [14] using the mRNA level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from each individual sample for normalization. Average expression with standard deviation from triplicate samples was plotted in relation to average expression of GAPDH (set to 100).
Table 1.
Primers for quantitative real time PCR
| Target | Primer sequences (5′–3′) | Product size (bp) |
|---|---|---|
| Chromogranin A (CGA) | CCC CAC TGT AGT GCT GAA CC (S) | 154 |
| GGA GTG CTC CTG TTC TCC C (AS) | ||
| Tryptophan hydroxylase 1 (TPH-1) | GAA GAT GCA AAG GAG AAG ATG (S) | 175 |
| GAC CTT AGC AAG GGC ATC AC (AS) | ||
| Vesicular monoamine transporter 1 (VMAT-1) | GGT GGA TTC TTC TAT GAT GCC C (S) | 126 |
| GTG GAT GGA CCT ATA GCA AAG C (AS) | ||
| [8] | ||
| Caudal type homeobox transcription factor 2 (CDX-2) | ACTACAGTCGCTACATCACCA (S) | 244 |
| GAAGACACCGGACTCAAGGG (AS) | ||
| Primer bank http://www.pga.mgh.harvard.edu/primerbank/ | ||
| Islet autoantigen 2 (IA-2) | GTC CCA TAC CAT CGC AGA CT (S) | 109 |
| GTA GCG GTC ACA CTG CTT GA (AS) | ||
| Survivin | GCA GTT TGA AGA ATT AAC CCT TGG (S) | 157 |
| TCA ATC CAT GGC AGC CAG CTG (AS) | ||
| Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | CCCATGTTCGTCATGGGTGT (S) | 145 |
| TGGTCATGAGTCCTTCCACGATA (AS) | ||
| [9] |
Cell lines
The HLA-A*0201 positive T2 cell line (ATCC, Teddington, UK) was cultured in RPMI 1640 supplemented with 10% foetal bovine serum (FBS) and 1% penicillin-streptomycin (PeSt). T2 cells are deficient in transporters associated with antigen processing (TAP) and are therefore unable to present endogenous peptides through the proteasome degradation pathway [3]. The EBV-transformed B lymphoblastoid cell line C1R.A2 (kind gift from Dr. J. Berzofsky) does not express endogenous HLA-A or B molecules but is stably transfected with a genomic clone of HLA-A*0201 [11]. C1R.A2 was cultured as described above with the addition of 200 μg/ml geneticin to ensure HLA-A*0201 transgene expression. All cell culture reagents were from Invitrogen.
Epitope prediction and peptide synthesis
Potential HLA-A*0201 binding motifs from CGA (AAH06459), TPH-1 (NM_004179), VMAT-1 (U39905), CDX-2 (NM_001265), IA-2 (L18983), were selected based on their score using the HLA peptide binding prediction algorithms available online: http://www-bimas.cit.nih.gov/molbio/hla_bind/ [22] and http://www.syfpeithi.de/[23]. A total of 16 HLA-A*0201-restricted peptides were chosen. The survivin peptide was previously published [2, 26] as was one of the IA-2 peptides [33]. All peptides were synthesized by Sigma-Genosys Ltd., Haverhill, UK with a purity of 70–99% verified by high performance liquid chromatography (HPLC).
T2 stabilization assays
Peptide binding and stabilization of HLA-A*0201 were verified using T2 cells. T2 cells were stripped in 0.131 M citric acid, 0.066 M Na2HPO4 (pH 3.3) for 45 s, washed and resuspended in serum-free culture media. Cells (2 × 105) were incubated with 3 μg/ml β2 microglobulin (Sigma Chemicals, St. Louis, MO, USA) and 100 μg/ml peptide in a total volume of 500 μl for 4 h at 37°C. Cells were washed and stained with the HLA-A*0201 monoclonal antibody BB7.2 and a secondary FITC labeled rabbit anti-mouse antibody (DAKO Cytomation) before FACS evaluation (FACSCalibur, BD Biosciences). Stabilization ratios were calculated by dividing mean fluorescence for peptide-pulsed T2 cells with mean fluorescence for T2 cells pulsed with a negative control peptide (MLLRYIGKK) with no predicted binding affinity to HLA-A*0201.
Isolation of PBMCs from patients and healthy blood donors
Heparinized patient blood was subjected to Ficoll–Paque density centrifugation (Amersham Biosciences, Uppsala, Sweden) and peripheral blood mononuclear cells (PBMCs) were either analyzed directly or cryopreserved in pooled human AB serum with 10% dimethylsulfoxide (DMSO) (Apoteket AB, Umeå, Sweden). Cryopreserved PBMCs were cultured in RPMI 1640 supplemented with 1% pooled human AB serum, 0.5% l-glutamine, 1% HEPES, 0.2% 2-mercaptoethanol, 1% PeSt, and 0.1% fungizone (all cell culture reagents from Invitrogen) with the addition of 10 U/ml human IL-2 (Proleukin® Chiron, Emeryville, CA, USA) for 24 h prior to analysis. Buffy coats from peripheral blood of HLA-A*0201 positive age-matched healthy blood donors obtained from Uppsala University Hospital Blood Center, Uppsala, Sweden were used as controls and were prepared in the same way.
Detection of activated CD8+ T cells by intracellular staining of interferon γ
Interferon γ (IFNγ) production against predicted peptide epitopes was measured by intracellular cytokine staining after blocking of cellular secretion. C1R.A2 cells were pulsed individually with each peptide (50 μg/ml) for 2 h before the addition of PBMCs in a 1:1 ratio. A HLA-A*0201 peptide from the cytomegalovirus pp65 (NLVPMVATV) and CD4+ T cell epitopes from tetanus toxin [6] served as positive controls. After 2 h cellular secretion was blocked by the addition of Brefeldin A (8 μg/ml) and the incubation was allowed to continue for 5 h. Cells were permeabilized (BD Perm, BD Biosciences) and stained with CD3-APC, CD8-PE, and IFNγ-FITC antibodies for 30 min at 4°C and analyzed by flow cytometry (FACS Calibur, BD Biosciences). Evaluation was based on a minimum of 10,000 events and evaluated cells were gated on the lymphocyte population in a forward and side scatter dot plot and on positive CD3 expression. The FL3 channel was used to exclude autofluorescent cells. A well-defined population of at least 0.10% IFNγ-secreting cells within the CD3+CD8+ T cell population, after subtraction of occasional background against unpulsed C1R.A2, was considered positive recognition.
Statistical analysis
Statistical analysis of data was performed by Statisticon AB, Uppsala, Sweden. Fischer’s exact test and a multiple permutation test with Fischer’s omnibus combination function were used to establish statistical relevance between patients and healthy controls.
Generation of peptide-specific T cells from healthy donors
Peripheral blood mononuclear cells of HLA-A*0201 positive healthy blood donors were washed and separated by plastic adherence for 2 h at 37°C. Non-adherent cells were collected and CD8+ T cells were purified using MACS CD8 microbeads (Miltenyi Biotech, Auburn, CA, USA) according to manufacturer’s instructions. CD8+ T cells were cryopreserved until further use. Negative fractions (non-adherent, non-CD8+ cells) were cryopreserved in aliquots for restimulations. Adherent cells were differentiated into immature dendritic cells by culture in RPMI 1640 supplemented with 1% pooled human AB serum, 0.5% l-glutamine, 1% HEPES, 0.2% 2-mercaptoethanol, 1% PeSt, and 0.1% fungizone (Invitrogen) for 6 days with the addition of 50 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) (Leukine®, Berlex Oncology, Wayne, NJ, USA) and 25 ng/ml IL-4 (Gentaur, Brussels, Belgium) every 2 days. Immature dendritic cells were harvested and transduced with an adenovirus vector encoding CD40L (AdCD40L) [15] with a multiplicity of infection (MOI) of 150. The cells were cultured at a density of up to 5 × 105 cells/cm2 for 2 days after which a phenotypic evaluation based on surface expression of HLA-ABC, HLA-DR, CD14, CD80, CD86, CD83, and CCR7 (BD Biosciences) was performed. Mature dendritic cells were pulsed for 4 h at 37°C with β2 microglobulin (0.3 μg/ml) and peptide (50 μg/ml). Autologous CD8+ T cells were added in a ratio of 10:1 and the cells were cultured with a cell density of ∼3 × 106 cells/cm2 for 12 days with the addition of IL-7 (Nordic Biosite, Täby, Sweden) (10 ng/ml) on the day of stimulation and IL-2 (20 U/ml) on day 2. Three restimulations were performed weekly when indicated using irradiated (40 Gy) peptide-pulsed autologous cells and with the addition of IL-7 and IL-2 as described. Peptide-specific activation was detected by intracellular staining of IFNγ using the assay described above.
Results
Expression of potential TAAs by microdissected metastatic carcinoid tumor cells
The PALM® Robot Microbeam Laser Microdissection System was used to specifically select tumor cells from frozen tissue sections of midgut carcinoid specimens. Microdissected cells were subjected to total RNA isolation. Total RNA quality and quantity were evaluated using Agilent 2100 Bioanalyzer and then subjected to cDNA synthesis. Quantitative real time PCR was run on a total of eight metastases using primers for TAAs CGA, TPH-1, VMAT-1, CDX-2, IA-2, and survivin. The relative expression level of these genes was normalized evaluating the expression of GAPDH (set to 100), Fig. 1. CGA served as a marker for neuroendocrine cell origin. All metastases expressed immense amounts of CGA mRNA, a majority of metastases expressed CGA mRNA to a level over a 100-fold the level of GAPDH. The tumor-associated proteins TPH-1, VMAT-1, CDX-2, and IA-2 were also well expressed by all metastases. However, CDX-2 and IA-2 were always expressed to a lesser extent than TPH-1 and VMAT-1. Surprisingly, only one mesentery metastasis expressed survivin and at a level lower than one tenth the level of GAPDH. Therefore, we are presently further exploring its expression by microarray analyses.
Fig. 1.

Expression of potential TAAs by microdissected metastatic midgut carcinoid tumor cells. Four liver and four mesentery metastases were analyzed for gene expression of potential TAAs by quantitative real time PCR. Average expression of CGA, TPH-1, VMAT-1, CDX-2, IA-2, and survivin was plotted with standard deviation from triplicate samples in relation to average expression of GAPDH (set to 100) for each sample
Peptide prediction and stabilization of HLA-A*0201 on T2 cells
The proposed midgut carcinoid TAAs CGA, TPH-1, VMAT-1, CDX-2, IA-2, and survivin were analyzed for potential HLA-A*0201 peptides using the BIMAS and SYFPEITHI computer algorithms available online. Peptides with high binding prediction scores were chosen for T cell recognition studies. In the case of VMAT-1, all peptides chosen were non-identical with sequences derived from the homologous isoform VMAT-2 and in the case of TPH-1, all peptides except TPH1307–315 were non-identical with sequences derived from the homologous isoform TPH-2. Stabilization assays using TAP-deficient T2 cells verified binding of all peptides to HLA-A*0201 in comparison to a control peptide with no predicted binding. Selected peptides with their binding prediction scores and stabilization ratios are shown in Table 2.
Table 2.
Potential midgut carcinoid antigens with peptide locations and sequences, and stabilization ratios
| Protein | Peptide | Sequence | BIMAS score | SYFPEITI score | Stabilization ratio on T2 cells |
|---|---|---|---|---|---|
| Chromogranin A (CGA) | CGA10–19 | LLCAGQVTAL | 84 | 27 | 1.7 |
| CGA74–82 | NLLKELQDL | 182 | 26 | 1.7 | |
| CGA209–217 | GLVDREKGL | 88 | 22 | 1.8 | |
| Tryptophan hydroxylase 1 (TPH-1) | TPH1241–249 | FLSGLAFRV | 1,856 | 24 | 3.3 |
| TPH1248–256 | RVFHCTQYV | 124 | 17 | 2.1 | |
| TPH1307–315 | KLATCYFFT | 1,248 | 17 | 2.8 | |
| TPH1333–341 | GLLSSISEL | 182 | 29 | 2.8 | |
| TPH1364–372 | CLITTFQDV | 132 | 22 | 2.1 | |
| Vesicular monoamine transporter 1 (VMAT-1) | VMAT132–40 | LLDNMLFTV | 630 | 28 | 2.7 |
| VMAT148–57 | FLYDMEFKEV | 7,975 | 24 | 2.7 | |
| Caudal type homeobox transcription factor 2 (CDX-2) | CDX25–13 | YLLDKDVSM | 347 | 24 | 2.0 |
| CDX223–32 | GLNLAPQNFV | 383 | 23 | 1.8 | |
| CDX2224–232 | GLSERQVKI | 43 | 25 | 1.8 | |
| Islet autoantigen 2 (IA-2) | IA2277–285 | GLLYLAQEL | 79 | 25 | 2.8 |
| IA2797–805 | MVWESGCTV | 352 | 19 | 2.4 | |
| Survivin (Surv) | Surv95–104 | ELTLGEFLKL | 3 | 19 | 1.5 |
Memory CD8+ T cells recognizing TAA-derived peptides can be detected in peripheral blood of carcinoid patients after short-term re-activation
To identify immunogenic epitopes derived from our proposed TAAs and to investigate whether midgut carcinoid patients have had immune activation against their tumor the HLA-A*0201-binding peptides were tested in short-term stimulation assays with intracellular staining of IFNγ. PBMCs isolated from patients and healthy blood donors were mixed with peptide-pulsed C1R.A2 in a 1:1 ratio. C1R.A2 can efficiently present HLA-A*0201 binding peptides to T cells and will induce an IFNγ response by memory T cells similar to peptide-pulsed dendritic cells (O. Forsberg et al., submitted manuscript). T cells were stimulated for a total of 7 h with blocking of cellular secretion after 2 h. T cells were then assayed for IFNγ production by intracellular cytokine staining and FACS analysis. A well-defined population of at least 0.10% IFNγ secreting cells within the CD3+CD8+ population was considered positive recognition. IFNγ production is believed to be peptide-specific and not due to an allogenic response against C1R.A2 due to the short incubation time. Patient T cells were always assayed for IFNγ production against unpulsed C1R.A2. Background was in all cases limited and was subtracted from the results with peptide-pulsed C1R.A2. In addition, none of the patients displayed positive IFNγ against all peptides tested, indicating peptide-specific responses. The results show that patients have a higher frequency of activated CD8+ T cells against midgut carcinoid-derived peptides compared to a group of age-matched healthy controls, 14.5% (17/117) of assays using PBMCs from patients carrying a high tumor burden and 17.1% (25/146) of assays using PBMCs from patients with a low tumor burden were positive compared to 5.6% (16/288) in healthy controls, as shown in Table 3. Percentages were based on 117 peptide assays using PBMCs from 12 high tumor burden patients, 146 peptide assays using PBMCs from 17 low tumor burden patients and 288 peptide assays using PBMCs from 40 healthy blood donors. Statistical analysis was performed to establish relevant differences in CD8+ T cell recognition frequency between patients and healthy donors. Fischer’s exact test was used to calculate P-values for each individual peptide and a joint P-value for the two patient groups and the healthy donors was obtained from the separate P-values by a multiple permutation test with Fischer’s omnibus combination function. Peptides not yielding any IFNγ responses in patients or healthy controls were excluded from the statistical analysis (TPH1241–249). The analysis shows a significant difference in recognition frequency between the two patient groups relative to healthy blood donors (P = 0.0284 for high burden patients, P = 0.0048 for low burden patients). All patients combined reached close to statistical relevance (P = 0.0506). Patients also reach higher frequencies of IFNγ-secreting cells within the CD3+CD8+ population 0.10–1.47% compared to healthy blood donors 0.10–0.51%, shown in Fig. 2. IFNγ against CGA-derived peptides was frequently detected in patients and to a lesser extent also in healthy controls indicating the immunogenicity of this highly expressed tumor-associated protein. As shown in Table 3, 21.7% (10/46) of patient assays showed positive recognition of CGA-derived peptides compared to 9.3% (5/54) of assays using PBMCs from healthy controls, Table 3. The plasma CGA levels in patients tested for T cell recognition of CGA are demonstrated in Fig. 3. Patients who displayed positive recognition of one or more of the 3 CGA-derived epitopes are indicated in the + column and patients who did not show recognition of any CGA-derived epitope are displayed in the − column.
Table 3.
Memory CD8+ T cells in peripheral blood of midgut carcinoid patients and healthy blood donors detected by intracellular staining of IFNγ
| Peptide | Patients with high tumor burden (n = 12) | Patients with low tumor burden (n = 17) | Healthy controls (n = 40) |
|---|---|---|---|
| CGA10–19 | 0/7 | 3/8 | 1/18 |
| CGA74–82 | 2/8 | 3/8 | 4/18 |
| CGA209–217 | 1/7 | 1/8 | 0/18 |
| TPH1241–249 | 0/8 | 0/10 | 0/18 |
| TPH1248–256 | 3/8 | 0/10 | 1/18 |
| TPH1307–315 | 2/8 | 5/10 | 4/18 |
| TPH1333–341 | 2/8 | 1/10 | 0/18 |
| TPH1364–372 | 0/8 | 1/10 | 0/18 |
| VMAT132–40 | 0/8 | 1/9 | 2/18 |
| VMAT148–57 | 3/8 | 0/9 | 0/18 |
| CDX25–13 | 0/5 | 2/8 | 1/18 |
| CDX223–32 | 0/5 | 3/8 | 1/18 |
| CDX2224–232 | 0/5 | 2/8 | 0/18 |
| IA2277–285 | 2/8 | 2/10 | 0/18 |
| IA2797–805 | 0/8 | 0/10 | 1/18 |
| Surv95–104 | 2/8 | 1/10 | 1/18 |
| Total | 17/117 = 14.5% | 25/146 = 17.1% | 16/288 = 5.6% |
Fig. 2.
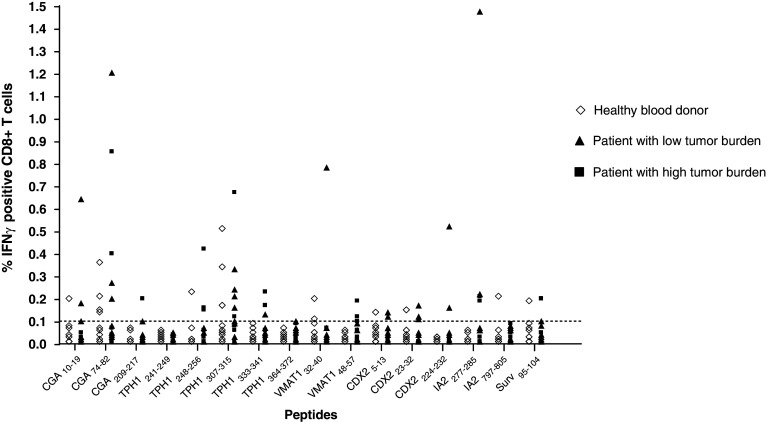
CD8+ T cell IFNγ production in response to peptide-pulsed C1R.A2. PBMCs from healthy blood donors (n = 40) and midgut carcinoid patients (n = 12 with a high tumor burden and n = 17 with a low tumor burden) were tested for CD8+ T cell recognition of potential TAA-derived HLA-A*0201-binding peptides. CD8+ T cell recognition was detected by intracellular FACS staining of IFNγ following a short-term re-activation assay. Frequencies of IFNγ-secreting cells within the CD3+CD8+ population were plotted for each peptide after subtraction of occasional background against unpulsed C1R.A2. The threshold for positive recognition was set to 0.10% IFNγ-secreting cells within the CD3+CD8+ T cell population (dotted line)
Fig. 3.

Presence of CGA-directed CD8+ T cells in patients with intermediate plasma CGA levels. Plasma CGA levels, measured by RIA, are demonstrated in patients tested for CD8+ T cell recognition of CGA. Plasma levels of patients with positive recognition of any of the three CGA peptide epitopes (≥0.10% IFNγ-secreting cells within the CD3+CD8+ population) are indicated in the +column, while patients with no recognition of any CGA peptide epitope are indicated in the −column
Generation of IFNγ-secreting, TAA-specific CD8+ T cells from healthy donors
Peripheral blood mononuclear cells from healthy blood donors were stimulated with autologous DCs pulsed with midgut carcinoid tumor-derived peptides in order to generate a population of TAA-reactive CD8+ T cells. The DCs were genetically modified to express CD40L, an important molecule for DC maturation and a T helper type-1 response. Cultured CD40L-expressing DCs display a mature phenotype based on FACS evaluation of surface markers (data not shown) and generate robust IL-12 secretion [16]. Stimulated T cells were analyzed by intracellular cytokine staining of IFNγ and FACS analysis. PBMCs from two healthy blood donors with relatively high frequencies IFNγ-secreting CD8+ T cells against the epitopes CGA74–82 and Surv95–104 were stimulated once with mature DCs and IFNγ secretion was assayed before stimulation and 12 days post-stimulation. This stimulation enriched IFNγ-secreting CD8+ T cells 4.3-fold and 10-fold, respectively, FACS analysis dot plots are shown in Fig. 4. Epitopes from the TAAs TPH-1, VMAT-1, and IA-2 with no or very low CD8+ T cell recognition in healthy blood donors could generate a distinct population of IFNγ-secreting CD8+ T cells specifically recognizing these epitopes. An extended stimulation protocol involving an initial stimulation with mature DCs and three rounds of restimulations with peptide-pulsed autologous blood cells was used and these CD8+ T cells reached frequencies of 0.16–1.22% of the total CD3+CD8+ population. FACS analysis dot plots are shown in Fig. 5.
Fig. 4.

Activation of TAA-directed memory CD8+ T cells from healthy blood donors. PBMCs from two healthy blood donors (I and II) with relatively high frequencies IFNγ-secreting CD8+ T cells against the epitopes CGA74–82 and Surv95–104 were stimulated once with peptide-pulsed CD40L-matured DCs and IFNγ secretion was assayed before stimulation and 12 days post-stimulation. The percentages of IFNγ-producing cells are given in the upper right quadrant. Background against C1R.A2 pulsed with an irrelevant HLA-A*0201-binding peptide, indicated within brackets, was subtracted. This stimulation enriched IFNγ-secreting CD8+ T cells 4.3-fold and 10-fold for CGA74–82 and Surv95–104, respectively
Fig. 5.

Generation of IFNγ-secreting CD8+ T cells specific for TAA-derived peptides from peripheral blood of healthy donors. PBMCs from four healthy blood donors (III and VI) with undetectable levels of CD8+ T cells against epitopes from the TAAs: TPH-1, VMAT-1, and IA-2 were stimulated once with peptide-pulsed CD40L-matured DCs and three times with peptide-pulsed autologous blood cells. These CD8+ T cells reached IFNγ-secreting frequencies of 0.16–1.22% of the total CD3+CD8+ population, shown in the upper right quadrant. Background against C1R.A2 pulsed with an irrelevant HLA-A*0201-binding peptide are shown and have been subtracted from percentages indicated for relevant peptides
Discussion
Metastasized midgut carcinoid tumors remain a therapeutic challenge despite improvements regarding medical treatment and surgery. Immunotherapy in the form of a DC vaccine or adoptive T cell therapy could imply a novel addition to current treatments. We have previously proposed a number of proteins with preferential expression in midgut carcinoid tumors by reverse transcription PCR and immunohistochemistry analyses [37]. By performing microdissection of tumor specimens, we were able to synthesize cDNA from malignant cells exclusively and a relative quantitation of potential antigen mRNA expression levels was made by quantitative real time PCR. CGA was used as a marker for neuroendocrine differentiation and was found to be immensely expressed: over a 100-fold the level of the housekeeping gene GAPDH. The tumor-associated proteins TPH-1, VMAT-1, CDX-2, and IA-2 were well expressed in all metastases examined. However expression of survivin, a protein considered over-expressed in many tumors [13], was detected in only one metastasis. This is considerably less than our previous analyses when we examined bulk tumor material [37]. A possible explanation could be that survivin is differentially expressed in various tumor specimens, suggesting specific variation in the gene profiling of these tumors at different stages of malignancy. A more detailed gene profiling of these tumors are ongoing and might verify this aspect. However, since survivin is a broad TAA, it is of general interest to analyze the occurrence and activation potential of survivin-reactive T cells.
Immune responses against TAAs have been described in many cancers [4, 5, 20]. This response can help to define proteins and epitopes with potential use in immunotherapy. Pioneering immunotherapy trials for neuroendocrine tumors performed by Schott et al. [27–29] used crude tumor lysates, hormone proteins and the carcinoembryonic antigen (CEA) with delayed type hypersensitivity and T cell proliferative responses as read-outs. More recently, Stift et al. [32] reported clinical responses following dendritic cell vaccinations using tumor lysate for medullary thyroid carcinoma. We have identified a number of defined CD8+ T cell epitopes derived from midgut carcinoid-associated proteins and analyzed specific CD8+ T cell activation against these epitopes in the form of IFNγ production. A selection of 16 HLA-A*0201-restricted peptides derived from six proposed TAAs was investigated for recognition by CD8+ T cells in patient blood compared to blood from an age-matched group of healthy blood donors. The frequency of IFNγ production in response to these peptide epitopes was 14.5% in a group of patients with high tumor burden and 17.1% in a group of patients with a low tumor burden. The frequency in healthy blood donors was merely 5.6%. These differences proved statistically significant after establishing a joint P-value for all peptide assays performed in each group. The frequencies of IFNγ-positive cells within the CD3+CD8+ T cell population are also higher in the patient groups indicating greater amplification of TAA-reactive CD8+ T cells in patients. The patient group with a low tumor burden displayed a slightly higher recognition frequency compared to the patient group with a high tumor burden and this could be explained by the fact that a high tumor burden to a larger extent anergize or delete tumor-reactive T cells [24]. We have with our analysis detected functional IFNγ-producing T cells, a tetramer analysis that merely detects T cell recognition might have yielded higher frequencies in the patient group with a higher tumor load. The protein with highest overall CD8+ T cell recognition in patients was the highly expressed neuroendocrine marker CGA. CGA might therefore be an attractive target for immunotherapy despite its expression in several non-malignant tissues [34]. CGA is secreted in large amounts in midgut carcinoid patients and plasma levels are often measured for monitoring tumor mass. Plasma CGA levels in patients analyzed for CD8+ T cell recognition of CGA show positive recognition in patients with an intermediate plasma CGA level. Patients with no CGA recognition were found to have a low or an extremely high plasma CGA level. Although the number of included patients are few, this could be interpreted as that a certain level of progressive tumor growth are needed to activate T cells and that these T cells might eventually submit to anergy or deletion when the tumor mass is too great.
The TAA-derived peptides were also used in stimulation assays to generate activated CD8+ T cells in healthy blood donors. We obtained well-defined populations of peptide-reactive cells. Such populations can be sorted using tetramers and expansion protocols together with cytotoxicity studies can pave the way for adoptive T cell therapy. Generating a substantial population IFNγ-secreting CD8+ T cells is more easily achieved when pre-activated memory CD8+ T cells are available. T cells recognizing the CGA74–82 epitope was brought up to a frequency of 1.56% IFNγ-secreting cells within the CD3+CD8+ T cell population by a single round of stimulation using mature peptide-pulsed DCs. Similarly, the Surv95–104 epitope could generate a population of 2.07% IFNγ-secreting cells. It was also possible to detect IFNγ-secreting CD8+ T cells specifically recognizing epitopes from the TAAs TPH-1, VMAT-1, and IA-2 in blood from healthy donors. Generating tumor-reactive, high avidity CTLs from low frequency precursors requires an optimal stimulation protocol [10] and identified epitopes will be further investigated for cytolytic activity after in vitro stimulations. This requires more and larger volumes of patient blood compared to the current investigation.
Identification of immunogenic epitopes derived from midgut carcinoid-associated proteins is a pivotal step towards the establishment of immunotherapy as a novel treatment for this type of neuroendocrine tumor. To our knowledge this is the first study to detect and characterize a TAA-specific CD8+ T cell response against midgut carcinoid tumor antigens. Immunogenic midgut carcinoid-associated proteins will constitute essential components in specific DC vaccines. Alternatively, in vitro generation of tumor-reactive CD8+ T cells or expansion of a memory CD8+ T cell pool from patients are of great interest for an adoptive T cell transfer approach.
Acknowledgments
The authors thank research nurses Monica Hurtig and Lena Olsson at the Clinic of Endocrine Oncology, Uppsala University Hospital for providing the blood samples used in this study. They also thank Dr. Mohammad Alimohammadi, Dept. of Medical Sciences, Uppsala University for sharing valuable peptides. The authors express their gratitude to Verto Institute (Stamford, CT, USA), Dr. Raymond and Beverly Sackler for financial and scientific support.
References
- 1.Åkerström G, Hellman P, Hessman O, Osmak L. Management of midgut carcinoids. J Surg Oncol. 2005;89:161–169. doi: 10.1002/jso.20188. [DOI] [PubMed] [Google Scholar]
- 2.Andersen MH, Østergaard Pedersen L, Becker JC, Straten PT. Identification of a cytotoxic T lymphocyte response to the apoptosis inhibitor protein survivin in cancer patients. Cancer Res. 2000;61:869–872. [PubMed] [Google Scholar]
- 3.Anderson KS, Alexander J, Wei M, Cresswell P. Intracellular transport of class I MHC molecules in antigen processing mutant cell lines. J Immunol. 1993;157:3407–3419. [PubMed] [Google Scholar]
- 4.Boon T, Coulie PG, Van den Eynde BJ, Van der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- 5.Chakraborty NG, Stevens RL, Mehrotra S, Laska E, Taxel P, Sporn JR, Schauer P, Albertsen PC. Recognition of PSA-derived peptide antigens by T cells from prostate cancer patients without any prior stimulation. Cancer Immunol Immunother. 2003;52:497–505. doi: 10.1007/s00262-003-0377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diethelm-Okita BM, Raju R, Okita DK, Conti-Fine BM. Epitope repertoire of human CD4+ T cells on tetanus toxin: identification of immunodominant sequence segments. J Infect Dis. 1997;175:382–391. doi: 10.1093/infdis/175.2.382. [DOI] [PubMed] [Google Scholar]
- 7.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, Rogers LJ, Gracia GJ, Jones SA, Mangiameli DP, Pelletier MM, Gea-Banacloche J, Robinson MR, Berman DM, Filie AC, Abati A, Rosenberg SA. Adoptive cell transfer following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Essand M, Vikman S, Grawe J, Gedda L, Hellberg C, Öberg K, Totterman TH, Giandomenico V. Identification and characterization of a novel splicing variant of vesicular monoamine transporter 1. J Mol Endocrinol. 2005;35(3):489–501. doi: 10.1677/jme.1.01875. [DOI] [PubMed] [Google Scholar]
- 9.Giandomenico V, Simonsson M, Gronroos E, Ericsson J. Coactivator-dependent acetylation stabilizes members of the SREBP family of transcription factors. Mol Cell Biol. 2003;23(7):2587–2599. doi: 10.1128/MCB.23.7.2587-2599.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ho WY, Nguyen HN, Wolfl M, Kuball J, Greenberg PD. In vitro methods for generating CD8+ T-cell clones for immunotherapy from the naïve repertoire. J Immunol Methods. 2006;310:40–52. doi: 10.1016/j.jim.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 11.Hogan KT, Shimojo N, Walk SF, Engelhard VH, Maloy WL, Coligan JE, Biddison WE. Mutations in the α2 helix of HLA-A2 affect presentation but do not inhibit binding of influenza virus matrix peptide. J Exp Med. 1988;168:725–736. doi: 10.1084/jem.168.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leja J, Dzojic H, Gustafson E, Öberg K, Giandomenico V, Essand M. A novel chromogranin-a promoter-driven oncolytic adenovirus for midgut carcinoid therapy. Clin Cancer Res 15. 2007;13(8):2455–2462. doi: 10.1158/1078-0432.CCR-06-2532. [DOI] [PubMed] [Google Scholar]
- 13.Li F. Role of survivin and its splice variants in tumorigenesis. Br J Cancer. 2005;92:212–216. doi: 10.1038/sj.bjc.6602340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔct) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 15.Loskog A, Dzojic H, Vikman S, Ninalga C, Essand M, Korsgren O, Totterman TH. Adenovirus CD40 ligand gene therapy counteracts immune escape mechanisms in the tumor microenvironment. J Immunol. 2004;172(11):7200–7205. doi: 10.4049/jimmunol.172.11.7200. [DOI] [PubMed] [Google Scholar]
- 16.Loskog A, Ninalga C, Totterman T. Dendritic cells engineered to express CD40L continuously produce IL12 and resist negative signals from Tr1/Th3 dominated tumors. Cancer Immunol Immunother. 2006;55:588–597. doi: 10.1007/s00262-005-0051-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Micke P, Ostman A, Lundeberg J, Ponten F. Laser-assisted cell microdissection using the PALM System. Methods Mol Biol. 2005;293:151–166. doi: 10.1385/1-59259-853-6:151. [DOI] [PubMed] [Google Scholar]
- 18.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morse MA, Nair SK, Mosca PJ, Hobeika AC, Clay TM, Deng Y, Boczkowski D, Proia A, Neidzwiecki D, Clavien PA, Hurwitz HI, Schlom J, Gilboa E, Lyerly HK. Immunotherapy with autologous, human dendritic cells transfected with carcinoembryonic antigen mRNA. Cancer Invest. 2003;21(3):341–349. doi: 10.1081/CNV-120018224. [DOI] [PubMed] [Google Scholar]
- 20.Nagorsen D, Scheibenbogen C, Marincola FM, Letsch A, Keilholz U. Natural T cell immunity against cancer. Clin Cancer Res. 2003;9:4296–4303. [PubMed] [Google Scholar]
- 21.Öberg K. Neuroendocrine tumors of the gastrointestinal tract: recent advances in molecular genetics, diagnosis, and treatment. Curr Opin Oncol. 2005;17:386–391. doi: 10.1097/01.cco.0000167739.56948.a9. [DOI] [PubMed] [Google Scholar]
- 22.Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994;152(1):163–175. [PubMed] [Google Scholar]
- 23.Rammensee HG, Bachmann J, Emmerich NN, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 24.Redmond WL, Sherman LA. Peripheral tolerance of CD8 T lymphocytes. Immunity. 2005;22:275–284. doi: 10.1016/j.immuni.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 25.Rindi G, Klöppel G. Endocrine tumors of the gut and pancreas tumor biology and classification. Neuroendocrinology. 2004;80(Suppl 1):12–15. doi: 10.1159/000080733. [DOI] [PubMed] [Google Scholar]
- 26.Schmitz M, Diestelkoetter P, Weigle B, Schmachtenberg F, Stevanovic S, Ockert D, Rammensee HG, Rieber EP. Generation of survivin-specific CD8+ T effector cells by dendritic cells pulsed with protein or selected peptides. Cancer Res. 2000;60:4845–4849. [PubMed] [Google Scholar]
- 27.Schott M, Seissler J, Feldkamp J, von Schilling C, Scherbaum WA. Dendritic cell immunotherapy induces antitumor response in parathyroid carcinoma and neuroendocrine pancreas carcinoma. Horm Metab Res. 1999;31:662–664. doi: 10.1055/s-2007-978817. [DOI] [PubMed] [Google Scholar]
- 28.Schott M, Seissler J, Lettmann M, Fouxon V, Scherbaum WA, Feldkamp J. Immunotherapy for medullary thyroid carcinoma by dendritic cell vaccination. J Clin Endocrinol Metab. 2001;86:4965–4969. doi: 10.1210/jc.86.10.4965. [DOI] [PubMed] [Google Scholar]
- 29.Schott M, Feldkamp J, Klucken M, Kobbe G, Scherbaum WA, Seissler J. Calcitonin-specific antitumor immunity in medullary thyroid carcinoma following dendritic cell vaccination. Cancer Immunol Immunother. 2002;51:663–668. doi: 10.1007/s00262-002-0325-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Small EJ, Schellhammer PF, Higano CS, Redfern CH, Nemunaitis JJ, Valone FH, Verjee SS, Jones LA, Herschberg RM. Placebo-controlled phase III trial of immunologic therapy with Sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24:3089–3094. doi: 10.1200/JCO.2005.04.5252. [DOI] [PubMed] [Google Scholar]
- 31.Solcia E, Klöppel G, Sobin LH. Histological typing of endocrine tumors. WHO international histological classification of tumors. 2nd. Berlin: Springer; 2000. [Google Scholar]
- 32.Stift A, Sachet M, Yagubian R, Bittermann C, Dubsky P, Brostjan C, Pfragner R, Niederle B, Jakesz R, Gnant M, Friedl J. Dendritic cell vaccination in medullary thyroid carcinoma. Clin Cancer Res. 2004;10(9):2944–2953. doi: 10.1158/1078-0432.CCR-03-0698. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi K, Honeyman MC, Harrison LC. Cytotoxic T cells to an epitope in the islet autoantigen IA-2 are not disease-specific. Clinical Immunology. 2001;9(3):360–364. doi: 10.1006/clim.2001.5031. [DOI] [PubMed] [Google Scholar]
- 34.Taupenot L, Harper KL, O’Connor DT. The Chromogranin-Secretogranin family. N Engl J Med. 2003;348:1134–1149. doi: 10.1056/NEJMra021405. [DOI] [PubMed] [Google Scholar]
- 35.Tuettenberg A, Becker C, Huter E, Knop J, Enk AH, Jonuleit H. Induction of strong and persistent Melan A/MART-1-specific immune responses by adjuvant dendritic cell-based vaccination of stage II melanoma patients. Int J Cancer. 2006;118:2617–2627. doi: 10.1002/ijc.21679. [DOI] [PubMed] [Google Scholar]
- 36.Van der Horst-Schrivers ANA, Machteld Wymenga AN, Links TP, Willemse PHB, Kema IP, de Vries EGE. Complications of midgut carcinoid tumors and carcinoid syndrome. Neuroendocrinology. 2004;80(Suppl 1):28–32. doi: 10.1159/000080737. [DOI] [PubMed] [Google Scholar]
- 37.Vikman S, Essand M, Cunningham JL, de la Torre M, Öberg K, Tötterman TH, Giandomenico V. Gene expression in midgut carcinoid tumors: potential targets for immunotherapy. Acta Oncol. 2005;44:32–40. doi: 10.1080/02841860510007404. [DOI] [PubMed] [Google Scholar]
- 38.Whiteside TL. Immune responses to malignancies. J Allergy Clin Immunol. 2003;111:S677–S686. doi: 10.1067/mai.2003.90. [DOI] [PubMed] [Google Scholar]
- 39.Wierecky J, Muller MR, Wirths S, Halder-Oehler E, Dorfel D, Schmidt SM, Hantschel M, Brugger W, Schroder S, Horger MS, Kanz L, Brossart P. Immunologic and clinical responses after vaccinations with peptide-pulsed dendritic cells in metastatic renal cancer patients. Cancer Res. 2006;66(11):5910–5918. doi: 10.1158/0008-5472.CAN-05-3905. [DOI] [PubMed] [Google Scholar]
- 40.Zou W. Regulatory T cells, tumor immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]