

Abstract
In order to engage their students in a core methodology of the new genomics era, an ever-increasing number of faculty at primarily undergraduate institutions are gaining access to microarray technology. Their students are conducting successful microarray experiments designed to address a variety of interesting questions. A next step in these teaching and research laboratory projects is often validation of the microarray data for individual selected genes. In the research community, this usually involves the use of real-time polymerase chain reaction (PCR), a technology that requires instrumentation and reagents that are prohibitively expensive for most undergraduate institutions. The results of a survey of faculty teaching undergraduates in classroom and research settings indicate a clear need for an alternative approach. We sought to develop an inexpensive and student-friendly gel electrophoresis-based PCR method for quantifying messenger RNA (mRNA) levels using undergraduate researchers as models for students in teaching and research laboratories. We compared the results for three selected genes measured by microarray analysis, real-time PCR, and the gel electrophoresis-based method. The data support the use of the gel electrophoresis-based method as an inexpensive, convenient, yet reliable alternative for quantifying mRNA levels in undergraduate laboratories.
Keywords: cDNA, gene expression, microarrays, mRNA, quantitative PCR, real-time PCR, undergraduate
INTRODUCTION
In this article, we present a student-friendly method for validation of gene expression microarray data that uses equipment and supplies readily available at primarily undergraduate institutions. The method is inexpensive, reliable, and adaptable to the timing of undergraduate teaching laboratories. As an undergraduate research group at Missouri Western State College, three of us (T.E., S.F., and W.B.) have been studying the effects on gene expression of a class of antitumor drugs. We are addressing several hypotheses about the sequence and structural requirements for the binding of these compounds to the DNA minor groove. One of our approaches has been to use DNA microarrays to examine the effects on yeast cells of minor groove binding compounds. The results pointed us to several genes that were affected, and we were excited to be able to examine their upstream sequences in light of our hypotheses. However, we soon realized that we needed to validate and confirm the microarray results with an independent method of measuring the messenger RNA (mRNA) levels of selected individual genes. The literature taught us that the current method of choice is automated real-time polymerase chain reaction (PCR) (Wall and Edwards, 2002; Bijwaard et al., 2001; Gibson et al., 1996; Heid et al., 1996), but the costs of an instrument and its operation were beyond our budget. Earlier published reports led us to consider several different gel electrophoresis-based methods of using PCR quantitatively (Gilliland et al., 1990; Noonan et al., 1990; Vu et al., 2000). We chose to develop one of these in our research laboratory as an inexpensive alternative to automated real-time PCR. The gel electrophoresis-based experiments were conducted by the undergraduate coauthors of this report (S.F. and W.B.) in order to test the ease with which they can be adapted for use in undergraduate research and teaching environments.
As instructors at a National Science Foundation-sponsored Genome Consortium for Active Teaching (GCAT) workshop at Georgetown University in July 2004, two of us (L.H. and T.E.) worked closely with a diverse group of 18 faculty from undergraduate institutions across the country (http://www.bio.davidson.edu/Biology/GCAT/workshop2.html). The participants learned to conduct microarray experiments and analyze microarray data. In the course of the workshop, the gel electrophoresis-based method used at Missouri Western State College for measuring complementary DNA (cDNA) levels was presented to the group and it generated a large amount of interest. We began to consider that the need to make this type of measurement without an automated real-time PCR instrument is widespread in undergraduate settings. A three-part plan was developed to address this issue. First, faculty at undergraduate institutions would be surveyed to gauge the need for a reliable and affordable alternative to real-time PCR and determine whether the level of interest in the workshop group is reflected in a larger group. Second, two of us (L.H. and L.C.) would conduct an experiment to evaluate the reliability of gel electrophoresis-based measurements made by undergraduate researchers in comparison to real-time data from the same samples. Third, dissemination to the undergraduate educational community would be accomplished in the form of this article.
VALIDATING MICROARRAY DATA WITH REVERSE TRANSCRIPTION-PCR
Since exploding onto the scene 10 years ago (Schena et al., 1995), microarrays have been used for a great variety of applications (Brewster et al.,2003; Hakak et al., 2001; Jost et al., 2001). However, the data derived from them has undergone increasing scrutiny with regard to dependability and reproducibility (Bolufer et al., 2001). In addition, individual transcript data can be unreliable (Townsend, 2004; Asyali et al., 2004; Jin et al., 2001). This problem emerges from the massive nature of the data sets generated. At p < .05, there can be five false gene expression identifications per 100 genes, amounting to 300 genes misidentified as differentially transcribed in an experiment using Saccharomyces cerevisiae. Even at p < .001, six misidentified genes are expected from the yeast genome. As a result, the research community has come to expect that microarray measurements of relative mRNA levels be validated by an independent method (Bustin and Dorudi, 2002). The widespread choice of real-time PCR for this purpose is apparent in the literature. For example, the microarray measurement of changes in gene expression during pancreatic tumor cell growth (Bai et al., 2004) and modified vaccinia virus Ankara (MVA) infection (Guerra et al., 2004) were confirmed by real-time PCR. The uses of microarrays to study biodegradation genes in soil microbes (Rhee et al., 2004), to measure differences in gene expression in patients with acute and chronic multiple sclerosis (Tajouri et al., 2003), and to compare gene expression patterns in dogs and wolves (Saetre et al., 2004) were also complemented by real-time PCR. These and many other examples illustrate that the measurement of mRNA levels of individual genes is an important companion to the observation of global patterns of gene expression.
SURVEY OF EDUCATORS
In order to assess the degree of need among undergraduate educators for a convenient and inexpensive method to measure mRNA levels, we sent an e-mail survey instrument to all subscribers to the listservs for the Council on Undergraduate Research (http://www.cur.org/) and the Genome Consortium for Active Teaching (http://www.bio.davidson.edu/Biology/GCAT/GCAT.html). Thirty faculty members responded, representing a diversity of institutions from small, private liberal arts colleges to large, public universities. As shown in Table 1, half of the responding faculty reported having used microarrays in undergraduate courses or in their undergraduate research laboratories, reflecting the increasing role that microarrays are playing in undergraduate education. The faculty and their students are addressing a variety of interesting problems, such as observation of the yeast host response to expression of components of a virus, determination of the gene expression effects of insect herbivory on tomatoes and maize, and identification of temperature-regulated genes in Escherichia coli.
Table 1. Survey of undergraduate educators.

Of the responding faculty, 77% reported that they have reason to measure cDNA levels of individual genes. Examples of applications described in response to the survey include the measurement of changes in cDNA levels when bacteria are exposed to compounds that induce or repress heme precursors, the study of genes expressed during sexual development in a basidiomycete fungus, and the examination of the impact of changing chromosome copy number on levels of gene expression in Tetrahymena. Despite the widespread interest among the faculty in measuring individual mRNA levels, only three of the respondents have performed real-time PCR experiments, and only six have a real-time PCR machine at their institution. Six others have plans to purchase a real-time instrument once funds have been obtained through grants or institutional sources, and eight reported that they have a budget for real-time PCR reagents. However, all but one of the respondents have access to the thermal cycler and gel electrophoresis equipment needed to implement a gel electrophoresis-based quantitative PCR method. Of the responding faculty, 80% replied that they would find a gel electrophoresis-based method for quantifying mRNA useful in a laboratory course and 87% reported that they would be able to use it in an undergraduate research setting. These results reinforced our initial observation from the GCAT workshop that the need for a convenient, inexpensive, and reliable alternative to real-time PCR is widespread at undergraduate institutions.
OPTIONS FOR MEASURING mRNA LEVELS
Most of the methods used today to measure steady state mRNA levels are based on the production of cDNA by reverse transcription and the use of quantitative PCR to measure cDNA levels for individual genes. To interpret the results of such experiments appropriately, it is important to have a clear understanding of the PCR process itself. In the reactions used for this purpose, a very small amount of cDNA is targeted by primers specific for a given gene. The reaction product accumulates exponentially during the early part of the reaction, but once reactants such as deoxynucleotides and primers become limiting, and inhibitors such as pyrophosphate accumulate, the efficiency of the reaction decreases. As a result, the curve of product versus number of cycles reaches an inflection point, and eventually a plateau (Nakayama et al.,1992). Before the inflection point, in the exponential part of the curve, the amount of PCR product accumulating is described by P = I (1 + E)C, where I is the initial amount of cDNA at the start of PCR; E is the efficiency of amplification, varying between 0 and 1.0; and C is the number of cycles. One measure of the relative initial amounts of cDNA in two samples is the ratio of the amounts of product for a given number of cycles, assuming the efficiency of the two reactions is the same. That is, P1/P2 = I1/I2. Another measure of the ratio of initial cDNA levels derives from the equation I1/I2 = (1 + E)ΔC, where ΔC is the difference in the number of cycles required to yield the same amount of product in the two reactions.
These considerations led to reports within the past 15 yr of several different methods for the measurement of cDNA levels by quantitative PCR (Bustin, 2002; Halford et al., 1999). The methods vary widely with respect to the cost of the required equipment and reagents and also as to the ease with which they can be adapted for use in undergraduate settings. The most popular method of late is automated real-time PCR (Bijwaard et al., 2001; Gibson et al., 1996; Heid et al., 1996). A real-time PCR instrument is usually a combination of a thermal cycler and a fluorimeter that enables the investigator to monitor the products of a reaction while it is proceeding. The ability to measure DNA sensitively and quantitatively allows the determination of the point in a reaction when the amount of the amplification product of cDNA exceeds a user-defined threshold. With proper controls, the cycle at threshold (Ct) gives a measure of the initial cDNA level. The least expensive and most straightforward way of measuring product is through the use of a fluorescent dye specific for double-stranded DNA, such as SYBR Green I. After each cycle of PCR, the reaction mixture is exposed to light at the absorption wavelength for the dye, and emission is used as a measure of the amount of product. However, this method detects not only the specific product, but also products resulting from nonspecific priming and primer-dimer formation. Therefore, optimization of reactions is critical, and real-time PCR machines use melting profiles after a reaction has been conducted to determine how much of the product is specific. The issue of specificity can be bypassed with more expensive gene-specific probe systems. Molecular beacons contain a reporter fluorophore and a quencher on their 5′ and 3′ ends that produce fluorescence only when the central part of the oligonucleotide is bound through specific base pairing to the PCR product to be measured (Abravaya et al., 2003). TaqMan probes (http://www.appliedbiosystems.com) bind specifically to the cDNA template in an area central to the region being amplified, and emit fluorescence only after a quencher is removed from the 5′ end by the action of Taq polymerase (Pongers-Willemse et al., 1998). Real-time PCR is a sensitive and accurate method, but it involves expensive equipment and reagents.
Before the advent of real-time PCR, the literature described several gel electrophoresis-based PCR methods developed to measure mRNA levels. Each of these uses reverse transcription to produce cDNA and quantitative PCR to measure the level of cDNA for selected genes. One such method, competitive reverse transcription-PCR, controls for RNA stability and differences in the efficiency of reverse transcription (Gilliland et al., 1990; Vu et al., 2000). Control RNA of a different size than the mRNA to be measured is produced by in vitro transcription of the coding sequence cloned into the appropriate plasmid. The control RNA is added in known amounts to the RNA sample and cDNA is produced. PCR with primers that amplify cDNA templates from both the control RNA and the specific mRNA is then conducted. Gel electrophoresis of the products and densitometry of band intensities give a measure of the absolute level of mRNA for the selected gene in the original RNA sample by calibration against the known standards. Competitive reverse transcription-PCR is a sensitive and accurate method, but it requires time-consuming procedures for cloning and transcribing a control RNA for each gene to be studied.
Another gel electrophoresis-based method, primer dropping, employs amplification within the same reaction of cDNA from both the gene to be measured and a control gene (Wall and Edwards, 2002). If the mRNA from either of these two genes is present at substantially higher levels than the other, one cDNA amplification product accumulates at levels past the exponential phase of the reaction before the other reaches the threshold of detection. Primer dropping allows one of the two amplifications to proceed for a small number of cycles before primers for the other amplification are added, ensuring that both genes are in the exponential phase of the reaction at the same time. In this way, band intensities can be used to measure the mRNA levels of a gene when they are substantially different from the mRNA levels for the control gene. This method did not meet our need for a flexible and convenient method that could be easily and quickly applied to any gene of interest from a microarray experiment.
We chose to adapt a method of quantitative amplification of cDNA that is based on the exponential relationship between the amount of accumulated product and the initial template level (Noonan et al., 1990). Identical amplifications are conducted with primers for a given gene and cDNA template derived from two RNA populations to be compared. Before the inflection point of the curve of product versus cycle number, the ratio of product amounts for the two reactions is proportional to the ratio of initial cDNA templates present. The amount of product present at several points in the exponential phase of the reactions can be measured by densitometry of the intensities of bands after electrophoresis. In this way, the relative amounts of cDNA in two samples can be measured. Before making measurements with this method, it is important to consider several sources of experimental error. Because RNA samples can vary in quality, and the efficiencies of reverse transcription and PCR are not constant, Noonan et al. (1990) employed an internal control. Primers for the amplification of the cDNA for a ubiquitously expressed gene were included in the reaction along with primers for cDNA from the gene to be measured. This approach lacks adaptability because it is difficult to optimize the annealing temperature for a duplex reaction, and unwanted products can appear. An alternative approach is to use parallel PCR amplifications to measure the cDNA levels from an experimental gene relative to those of a reference gene (Lesur and Campbell, 2004). Because this method is better suited to undergraduate settings, we chose to develop it in our research laboratory and to compare it with microarray analysis and real-time PCR.
MATERIALS AND METHODS
Growth of Yeast and RNA Isolation
Overnight cultures of Saccharomyces cerevisiae strain S288C grown at 30°C in yeast extract/peptone/dextrose (YPD) with shaking at 175 rpm were used to seed two 5-ml cultures. One culture remained untreated and the other was treated with 4′,6-diamidino-2-phenylindole (DAPI) at a final concentration of 10 μM. The cultures were incubated for 4 h after which both A660 readings were 2.0. Total RNA was isolated using the RiboPure-Yeast kit from Ambion, Inc. (Austin, TX). The procedure uses zirconia beads for cell disruption, phenol, and sodium dodecyl sulfate (SDS) to reduce RNase activity, and filter cartridges to bind and wash precipitated RNA. We chose to collect the RNA with two elutions of 50 μl each, and added 1 μl of RNasin Plus RNase inhibitor to each sample (Promega Corporation, Madison, WI).
Measuring RNA Quantity and Quality
Elimination of contaminating yeast genomic DNA in RNA preparations is crucial before cDNA synthesis and PCR. We tested each RNA sample by amplifying with primers for the housekeeping gene TDH1. Although the RNA isolation procedure includes a DNase I digestion, about half of our samples showed a product, indicating DNA contamination. We therefore performed a second DNase I treatment with twice the enzyme recommended by the RNA isolation kit and for twice as long, then performed the PCR check again. Spectrophotometric readings at 260 and 280 nm were then made for the RNA samples. The yield of RNA varied from 20 to 30 μg from each 5-ml yeast culture and the A260–A280 ratios were between 1.8 and 2.1. As a further quality check, 1 μg of each RNA sample was subjected to denaturing agarose gel electrophoresis. The presence of 28S and 18S ribosomal RNA over a background smear of mRNA and the absence of both degraded RNA and high molecular weight DNA indicated good quality RNA preparations.
Microarray Procedure
Total RNA from DAPI-treated and untreated yeast was used as starting material for the Genisphere 3DNA Array 900 Expression Array Detection Kit (http://www.genisphere.com/). The RNA was reverse transcribed with Superscript II (Invitrogen Life Technologies, http://www.invitrogen.com) using primers that include a capture sequence for fluorescent dendrimers. Hybridization of the cDNA to an Institute for Systems Biology 70 mer oligonucleotide DNA chip provided by GCAT was performed with 40 μl of hybridization buffer (0.5 M NaPO4, 1% SDS, 2 mM ethylenediamine tetraacetic acid, 2× saline-sodium citrate, and 4× Denhardt) at 58°C overnight. The 3DNA Cy3 and Cy5 fluorescent dendrimers were hybridized in hybridization buffer for 4 h at 58°C. Dithiothreitol (DTT) at a level of 1 mM was added to the wash solutions. The chip was scanned at Davidson College by Dr. Malcolm Campbell. We used Scanalyze (http://rana.lbl.gov/index.htm?software/) to grid the microarray data and collect intensities from the two channels.
Reverse Transcription
Reverse transcription of total RNA for the preparation of cDNA to be used in PCR was also performed using Superscript II as the reverse transcriptase, but with oligo(dT)15 primers. A solution of 5 μl RNA (0.2 μg/μl), 1 μl deoxynucleotide triphosphate (dNTP) mix (10 mM each), and 5 μl dH2O was heated to 65°C for 5 min and then chilled on ice. To this mixture was added 4 μl of 5× first-strand buffer, 2 μl of 0.1 M DTT, and 1 μl RNasin Plus RNase inhibitor. The contents were gently mixed, briefly centrifuged, and allowed to incubate for 2 min at 42°C. Reverse transcriptase was added (1 μl), and the reaction was incubated for 50 min at 42°C. The reaction was stopped by heating at 70°C for 15 min.
Polymerase Chain Reaction
Polymerase chain reaction was performed in 40-μl reactions containing 84 pmol of each of two primers, 4 μl of 10 mM dNTP mix, 4 μl of Mg-free 10× buffer (100 mM Tris pH 9.0 at 25°C, 500 mM KCl, 1% Triton X-100), 4 μl 25 mM MgCl2, and 2 units Taq DNA Polymerase. The PCR reagents were assembled as a master mix that was split into reactions that received 1 μl of cDNA from either treated or untreated cells as template. PCR was performed with cycles of 94°C for 15 s, 58°C to 62°C for 15 s, and 74°C for 30 s. Aliquots of 7 μl of the reactions were removed after a determined number of cycles and two-cycle intervals thereafter until four samples were taken from each 40-μl reaction. We designed primers for the amplification of cDNA from each of three yeast genes. The forward primer 5′-ATGTCTGAATCAGTGGCCATTATAGGTGC-3′ and reverse primer 5′-ATCAATGCCACGAGCAGAAATAGCC-3′ yielded a 180-base pair (bp) product from BNA4 using an annealing temperature of 62°C. For OLI1, a 124-bp product was produced by the forward primer 5′-GAGCAGGTATGGTATTGCTATCGT-3′ and the reverse primer 5′-CCTGTAGCTTCTGATAAGGCGAAACC-3′, with annealing at 58°C. Forward primer 5′-ATCGATGTCGCTGTTGACTCCACT-3′ and reverse primer 5′-CAATGGAGCCAAACAGTTGGTGGT-3′ gave a 206-bp product for TDH1 at an annealing temperature of 60°C. Although the TDH1 reverse primer is also predicted to bind to the paralogs TDH2 and TDH3, the forward primer is specific for TDH1.
Gel Electrophoresis and Data Acquisition
Aliquots taken from reactions at increasing cycles were electrophoresed on 7% polyacrylamide gels at 20 volts/cm for 30 min and stained with 1 μg/ml ethidium bromide for 15 min. Gel electrophoresis images were captured with a Kodak DC120 digital camera, and the densitometry feature of Kodak Digital Science 1D version 2.0.3 was used to measure band intensities. The relative intensities of specific bands were calculated by reference to the 100-bp band from the molecular weight marker.
Real-Time PCR
An ABI Prism 7000 was used for real-time PCR analysis of the three selected genes. Amplification reactions were assembled as described above but with ABI 2× SYBR Green I Master Mix comprising half the total volume, and with the total volume per reaction adjusted to 50 μl. The cDNA templates were diluted two-, four- and eight-fold, and three replicate reactions were performed for each.
RESULTS AND DATA ANALYSIS
In order to evaluate the gel electrophoresis-based method by comparison to microarray and real-time PCR analyses, we chose to measure the effect of experimental treatment on the mRNA levels for one gene in each of the three categories of induced, repressed, and unaffected. Of particular importance was our choice of an unaffected housekeeping gene, because we wished to offer it as a reference for use in the gel electrophoresis-based method. We chose TDH1 because it is the yeast ortholog of the gene for G3PDH (glyceraldehyde 3-phosphate dehydrogenase), widely used in other species (Ceol et al., 2001; Halford et al., 1999). Our choice of the induced and repressed genes was guided by the fact that one of our undergraduate research groups (T.E., W.B., and S.F.) is interested in compounds that bind the DNA minor groove and affect gene expression. Although the upstream sequence of TDH1 is devoid of the features we hypothesize are needed for interaction with minor groove binders, OLI1, a mitochondrial gene coding for subunit 9 of the F0 ATP synthase, and BNA4, coding for kynurenine 3-mono-oxygenase have upstream regions that contain the sequence and structural features that we hypothesize are important for binding of the compounds we are studying.
Using DAPI as a model for minor groove binding compounds, we cultured yeast in its presence and absence. We then isolated RNA from the two cultures for use in microarray analysis. Figure 1 shows the microarray results from the three selected genes. The cDNA derived from untreated yeast was labeled green, and that from DAPI-treated yeast was labeled red. The spot for TDH1 is yellow, with a ratio of treated to untreated of 1.1, indicating that its mRNA is present at equal levels in the two samples. The spot for BNA4 is red, with a treated to untreated ratio of 4.5, indicating a higher level of mRNA from DAPI-treated cells. OLI1 mRNA levels are lower in the DAPI-treated sample, because it is green, with an untreated to treated ratio of 8.3. These results are indicated in Table 2. In addition, the spot intensities give an indication of the relative abundance of mRNA species. The mRNA for BNA4 appears to be present at a lower level than that for the other two genes.
Figure 1.
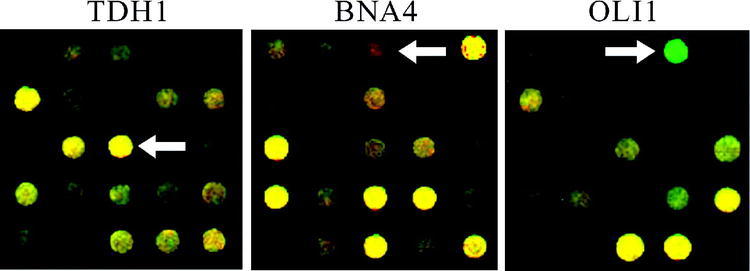
Microarray data for TDH1, BNA4, and OLI1. Total RNA isolated from yeast grown in the presence of DAPI was used to prepare cDNA that gives a red fluorescent signal, while that derived from untreated yeast gave a green signal. TDH1 is unaffected (yellow), BNA4 is at higher levels after treatment (red), and OLI1 is at lower levels after treatment (green)
Table 2. Comparison of ratios for selected genesa.
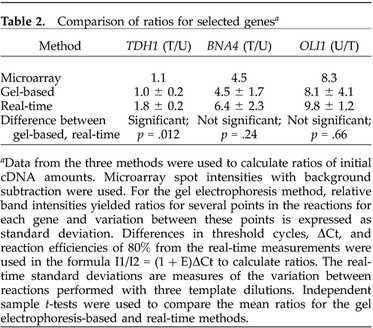
aData from the three methods were used to calculate ratios of initial cDNA amounts. Microarray spot intensities with background subtraction were used. For the gel electrophoresis method, relative band intensities yielded ratios for several points in the reactions for each gene and variation between these points is expressed as standard deviation. Differences in threshold cycles, ΔCt, and reaction efficiencies of 80% from the real-time measurements were used in the formula I1/I2 = (1 + E)ΔCt to calculate ratios. The real-time standard deviations are measures of the variation between reactions performed with three template dilutions. Independent sample t-tests were used to compare the mean ratios for the gel electrophoresis-based and real-time methods
We subjected the very same RNA samples used for microarray analysis to the gel electrophoresis-based method for measuring cDNA levels. Accordingly, mRNA from DAPI-treated and untreated yeast cultures was converted into cDNA and subjected to PCR with primers for the three selected genes. The polyacrylamide gel electrophoresis of reaction samples taken at two-cycle intervals from 16 to 22 cycles for TDH1 and OLI1 and from 20 to 26 cycles for BNA4 is shown in Figure 2. The images show that the specific PCR product for TDH1 accumulates at an equal rate in the treated and untreated cDNA reactions. For BNA4, the product appears much earlier in the treated reaction, while the opposite is true for OLI1. The intensities of these bands were measured by using a digital camera and the densitometry feature of Kodak Digital Science 1D version 2.0.3. The values were used to calculate relative intensities compared with the 100-bp band of the molecular weight marker on each gel.
Figure 2.
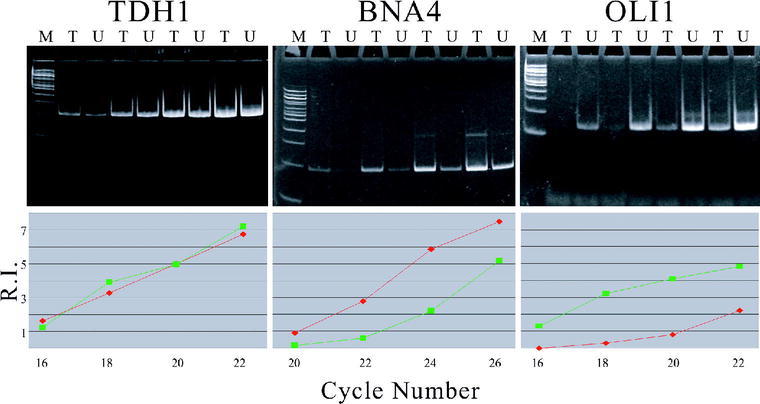
Gel electrophoresis of amplifications specific for TDH1, BNA4, and OLI1, with treated (T) and untreated (U) cDNA templates. Aliquots were taken at 16, 18, 20, and 22 cycles for TDH1 and OLI1, and at 20, 22, 24, and 26 cycles for BNA4. M is a 100-bp ladder used as a molecular weight standard. Graphs of relative band intensities (R.I.) versus cycles are shown for each gel, where diamond = treated and square = untreated.
The graphs in Figure 2 of relative intensities (R.I.) versus number of cycles provide information about the relative initial amounts of cDNA in the treated and untreated samples. For TDH1, equal intensities of PCR product bands for treated and untreated samples are apparent throughout the reaction. These data support the conclusion that TDH1 mRNA is present at equal levels in the two samples. For BNA4, the results indicate a higher initial level of mRNA in the DAPI-treated sample than in the untreated sample, while the opposite is true for OLI1. When considering which band intensity data to use for calculating ratios of treated to untreated cDNA, we adopted two criteria. We chose to eliminate data points that were past the inflection point and therefore past the exponential phase of the reaction. The graphs in Figure 2 reveal that only the data points for treated BNA4 at 26 cycles and untreated OLI1 at 22 cycles fell into this category. We also established 0.25 as a level of relative band intensity below which data acquisition by the digital camera is unreliable. Use of this criterion resulted in the elimination of data points for untreated BNA4 at 20 cycles and treated OLI1 at 16 cycles. Relative intensities of the remaining data points were used to calculate ratios of initial cDNA levels for the three genes. As shown in Table 2, the ratios of treated to untreated initial cDNA levels are 1.0 for TDH1 and 4.5 for BNA4, and the untreated to treated ratio is 8.1 for OLI1. The standard deviations are measures of the variation between ratios derived from different numbers of cycles. The ratios are in good accord with the microarray data, indicating no effect of DAPI on the steady state mRNA levels for TDH1, an increase for BNA4, and a decrease for OLI1. In addition, the results support the use of TDH1 as a reference gene in the gel electrophoresis-based method.
The cDNA preparations from the DAPI-treated and untreated RNA isolations were subjected by L.C. and L.H. to real-time PCR analysis using the same primers and PCR conditions as for the gel electrophoresis-based method. An ABI Prism 7000 was used to determine the fluorescence of DNA-bound SYBR Green I as a function of the number of cycles. Because SYBR Green I measures the total double-stranded DNA that accumulates in the reaction tube, it is important to determine how much of the PCR product is specific and how much arises from nonspecific priming and primer dimer formation. For this reason, each reaction was subjected to a dissociation protocol after the cycles were complete. The expected result of a well-optimized reaction is a product that melts at a single, reproducible melting temperature characteristic of that DNA sequence. Other PCR products will usually display lower melting temperatures. For the three genes selected, the dissociation protocols indicated the production of only minor amounts of products that melted at temperatures 10–20 degrees below the main peak, in agreement with the gel results above. However, for BNA4, the dissociation peak was broader, covering almost 10 degrees, while more typical product melts, including those for the other two genes in this study, cover 2–3 degrees. This observation is in accord with the appearance of a slight array of background bands on the electrophoresis gel loaded with BNA4 PCR products. We therefore decided to check whether the BNA4 product from the gel electrophoresis experiments was specific by digesting it with four different restriction endonucleases. The 180-bp product was cut at position 164 by HaeIII, position 105 by MwoI, position 111 by TaqI, and positions 135 and 165 by Tsp509I, as predicted by the DNA sequence. These results verify the identity of the specific BNA4 product that was quantified by the gel electrophoresis method. In this case, measuring a specific band with the gel electrophoresis-based method gives a cleaner result than SYBR Green I real-time PCR, because only the correctly sized products are quantified.
Output from the real-time PCR instrument also included plots of SYBR Green I fluorescence versus the number of cycles, shown in Figure 3. In order to make the plots more intelligible by reducing the total number of colored lines plotted, only one of each replicate set is shown; the others in each set were almost identical. The treated and untreated cDNA were used to prepare three different dilutions for use as templates with primers for each of the three genes. The TDH1 plot shows that all of the reactions enter the exponential phase of the reaction at approximately the same number of cycles, indicating little effect of the treatment on mRNA levels. For BNA4, the three lines for treated cDNA are shifted to the left, indicating that the treatment raised the mRNA levels. Because the treated cDNA lines are shifted to the right for OLI1, the data support the conclusion that the treatment lowered mRNA levels. The threshold level was set at 10 standard deviations above the baseline fluorescence and is indicated in the plots by a blue horizontal line. Table 3 shows values for the threshold number of cycles, Ct, for each of the three genes. Table 3 also includes threshold cycle data from the gel electrophoresis-based method. Each value is the number of cycles required for the appearance of a specific PCR product at a band intensity equal to the 100-bp marker band.
Figure 3.
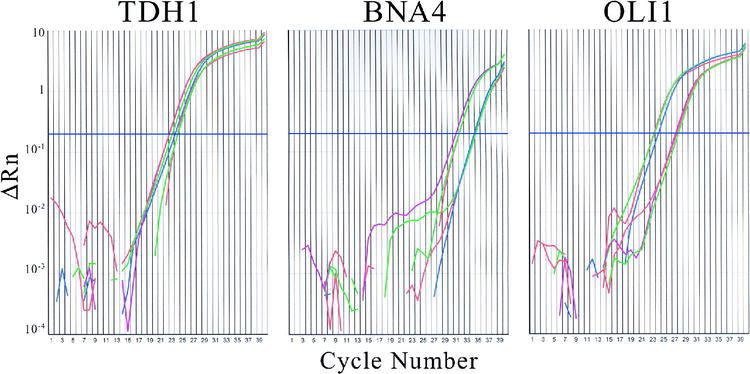
Real-time plots of fluorescence (ΔRn) versus number of cycles for treated and untreated cDNA amplified with primers for TDH1, BNA4, and OLI1. Lines for two-, four- and eight-fold dilutions of untreated cDNA are green, red, and blue, respectively. Corresponding lines for treated cDNA are red, purple, and green.
Table 3. Threshold cycle data for gel electrophoresis-based and real-time methodsa.
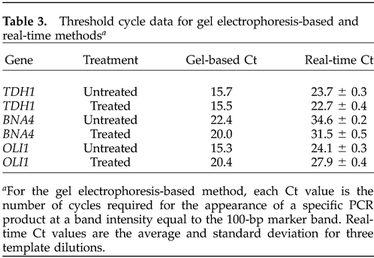
aFor the gel electrophoresis-based method, each Ct value is the number of cycles required for the appearance of a specific PCR product at a band intensity equal to the 100-bp marker band. Real-time Ct values are the average and standard deviation for three template dilutions
There is a good relative correspondence between the threshold numbers of cycles required for detection with the gel electrophoresis-based and real-time PCR methods. The data indicate that BNA4 mRNA levels are higher in the treated than in the untreated sample, while the opposite is true for OLI1. The larger number of cycles for BNA4 product detection indicates the lower level of its cDNA, as was shown with the microarray analysis.
Real-time Ct values were also used to produce ratios of initial cDNA levels. Ct values from the three replicates of each dilution of cDNA template were averaged and the differences, ΔCt, between reactions with treated and untreated cDNA, were calculated. Considering that the accumulation of product is given by P = (1 + E)C, where P is the amount of product, E is the efficiency of amplification, and C is the number of cycles, the plots of fluorescence versus number of cycles were used to determine the efficiency for each of the reactions. The efficiencies varied only slightly, from 80% to 82%, and were used with the ΔCt values in the formula I1/I2 = (1 + E)ΔCt to calculate ratios of treated to untreated initial cDNA levels. The relative levels of initial cDNA present in untreated versus treated samples are reported in Table 2. The standard deviations reflect variation between measurements made with three dilutions of the template. The ratios of treated to untreated mRNA levels are 1–8 for TDH1 and 6–4 for BNA4, and the ratio of untreated to treated mRNA levels is 9–8 for OLI1.
We used an independent samples t-test to compare the mean ratios reported in Table 2 for the gel electrophoresis-based and real-time methods. For the induced gene BNA4, the two methods produced the same conclusion because there was no statistically significant difference between the means (p = .24). Similarly, means for the repressed gene OLI1 were not statistically different (p = .66). However, the analysis did reveal a difference between the mean ratios for TDH1 (p = .012). This may mean that that the real-time method is more sensitive, or that proprietary components in the SYBR Green I Master Mix provide improved consistency between reactions. The measurement of a slight induction of TDH1 as a result of our experimental treatment indicates that it may not be the ideal choice of a housekeeping reference gene under all conditions. However, the difference in TDH1 results from the microarray and gel electrophoresis-based methods to real-time PCR does not disallow the conclusion that BNA4 is induced and OLI1 is repressed. Although this analysis reveals differences in the results from the two methods of quantifying individual mRNA levels, they are both in good general accord with the microarray results. The encouraging conclusion with regard to the development of a reliable gel electrophoresis-based method is that all three methods revealed the same trends for the effects of treatment on the three genes studied. The treatment increased the steady state mRNA level for BNA4, decreased it for OLI1, and had little effect on TDH1. Considered as a whole, the results support the conclusion that the gel electrophoresis-based method is a convenient and economical way to reliably quantify steady state mRNA levels.
EDUCATIONAL EFFECT ON UNDERGRADUATE RESEARCHERS
When our Missouri Western State College research team set out to quantify mRNA levels, we discussed several objectives for the educational impact it would have on the undergraduate researchers. First, we wanted the students to improve their understanding of fundamental molecular techniques used in the study of gene expression. Undergraduate researcher and coauthor William “Dan” Bradford was asked whether performing the gel-based measurements improved his understanding of reverse transcription and quantitative PCR. Dan replied that “performing the gel electrophoresis-based measurements did improve my understanding of reverse transcription. I learned firsthand that after we reverse transcribe the mRNA, we can measure gene expression using PCR. There was value in learning about using PCR in a quantitative way since it is usually used to create a large amount of product from a very small amount of template. Using this method, we can detect very small differences in template cDNA, and use that to measure gene expression.” A second objective was to stimulate the undergraduates to learn about and think critically about methods for measuring changes in mRNA levels. When asked whether performing the gel-based measurements caused her to think more critically about the reliability of microarray measurements, undergraduate researcher and coauthor Sara Freel said, “even though we would like to trust the data from the microarray, whatever data you get must be supported with other types of measurements to be reliable. You can never have too many controls.” Dan Bradford commented that “the gel-based method gave me a better understanding of real-time PCR instruments. Working with our collaborators, we had to invest the time to understand how the method worked.”
The students were also asked to comment on the overall objective of this study to develop a method that is student friendly and appropriate for use in undergraduate teaching and research settings. Sara said, “it was not very difficult to learn how to use the method, and I had only minor questions throughout the procedure. I think this will provide a tremendous amount of learning opportunities for undergraduate students.” According to Dan, “the method is appropriate for use in undergraduate teaching laboratories, and its implementation into undergraduate curricula will reinforce the central dogma of molecular biology. The ease with which the method can be performed and the fact that it was developed by undergraduates illustrate its fitness for undergraduate teaching and research laboratories.”
QUANTIFYING mRNA LEVELS IN UNDERGRADUATE LABORATORIES
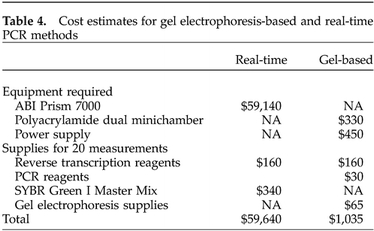
We have found the gel electrophoresis-based method of measuring mRNA levels to be convenient and student friendly, and suggest that it can be easily adapted to undergraduate teaching and research environments. As supported by the cost comparison in Table 4, the method is also a good fit to the budgetary constraints of most undergraduate institutions. The pieces of equipment required for the gel electrophoresis-based method, a thermal cycler and agarose gel electrophoresis equipment, are commonly found in undergraduate teaching and research laboratories. The power supply and dual minichamber for polyacrylamide gel electrophoresis that were used in the current study could be purchased for less than $800. By contrast, real-time PCR instruments range from $15,000 to $60,000. The cost of the supplies needed to make 20 measurements of mRNA levels with SYBR Green I real-time PCR is estimated to be twice as high as for the gel electrophoresis-based method (Table 4). Using a molecular beacon or TaqMan probe would be even more expensive, because for a single gene, the detector oligonucleotide alone costs more than $300. As supported by the survey responses reported in Table 1, these costs are prohibitive for most undergraduate institutions. Although GCAT (http://www.bio.davidson.edu/Biology/GCAT/GCAT.html) has addressed this problem for microarray analysis through inexpensive arrays and free scanning via its National Science Foundation-supported scanners, no such resource is available for real-time PCR.
Table 4. Cost estimates for gel electrophoresis-based and real-time PCR methods.
A recommended time line for using the gel electrophoresis-based method in a series of four weekly 3-h laboratory class periods is shown in Figure 4. The first day could begin with a discussion of the value of measuring mRNA levels for individual genes, the merits of various methods to do so, and the design of specific experiments to address hypotheses developed by the class. This discussion would probably not require the entire class time. The next period could be devoted to the isolation of RNA. We estimate that the RiboPure-Yeast procedure takes about 2 h to perform. During the third class period, students could measure the quantity and quality of the isolated RNA by spectrophotometry and denaturing gel electrophoresis. We estimate 1.5 h for these experiments, leaving the remainder of the period for reverse transcription reactions. The final laboratory period could be spent performing DNA amplifications with the cDNA, conducting gel electrophoresis, and analyzing the data with regard to the original hypotheses. A more detailed description of the time line for using the method in an undergraduate laboratory course is posted on the GCAT Web site (http://www.bio.davidson.edu/Biology/GCAT/GCAT.html). In a course using microarray analysis, the use of the gel electrophoresis-based method would add only two laboratory periods, because the RNA preparation and assessment of quality and quantity experiments would serve both purposes.
Figure 4.

Time line for use of the gel electrophoresis-based method in an undergraduate laboratory course that meets for weekly 3-h periods. Missouri Western State College students Bart Phillips, William Bradford, and Steven Hart discuss the measurement of mRNA levels and Sara Freel isolates RNA; denaturing agarose gel electrophoresis of RNA samples; PCR products measured by densitometry of a polyacrylamide gel.
RECOMMENDATIONS
Our experience in developing a gel electrophoresis-based method for measuring cDNA levels as part of an undergraduate research project has led us to offer several recommendations for its use. First, the method we have described is subject to several types of errors. The preparation and quantitation of quality RNA is essential. It is important to purify RNA that passes both the UV spectrophotometry and denaturing gel electrophoresis quality control checks. The A260–A280 ratio should be between 1.8 and 2.1. Both 28S and 18S ribosomal RNA bands should be visible with a background smear of mRNA and the absence of both degraded RNA and high-molecular-weight DNA, as seen in the denaturing agarose gel in the third panel of Figure 4. A critical negative control experiment is to use some of the prepared RNA as template in a standard PCR amplification with primers that will amplify cDNA or genomic DNA. If a product appears, then contaminating genomic DNA is present, and the DNase I treatment needs to be repeated. It is also important that the RNA samples to be compared are included in the reverse transcription reaction in the same amounts and that they will be converted into cDNA with the same efficiency. Because it involves PCR, the amplification of the cDNA is sensitive to variations in reagent concentrations as well as to template quantity and quality. We advise the use of a master mix whenever possible, and experimental designs that avoid pipetting very small volumes. In addition, the usual safeguards against contamination of pre-PCR reagents with reaction products are essential. The genes we selected for testing were not dramatically affected by the treatment. If a larger difference between the cDNA levels in two samples occurs, the amplification curve of one sample may pass the exponential phase before the other reaches the threshold of detection. This problem is easily solved by diluting the first sample until both amplifications are in the exponential phase at approximately the same number of cycles. In this case, gel electrophoresis band intensities need to be corrected by the dilution factor. Another key point is that it is critical that the PCR product measured is derived from the gene to be analyzed. As an alternative to the melting point analysis conducted with real-time PCR, we advise the use of restriction digests to verify the identity of the specific PCR product. When the above recommendations are implemented, we have found the gel electrophoresis-based method to be reliable and reproducible.
We recommend that care be taken when choosing a housekeeping gene for use as a reference. This point is illustrated by the fact that although the TDH1 gene we used as a reference appeared to be unaffected as measured by microarray analysis and the gel electrophoresis-based method, the real-time PCR data showed a slight effect of the treatment. A better choice may be the yeast gene TUB1, because its transcript was found to be stable and unregulated in a series of experiments involving aging in yeast cells (Lesur and Campbell, 2004). Although a one-sample t-test showed expression ratios for TDH1 from 18 microarray experiments from our laboratories to be statistically different from unity (p < .01), the same analysis showed TUB1 expressed to be unchanged (p = .39).
We would also like to point out some alternatives to the procedures we used. There are several choices available for RNA isolation kits. Because the isolation of good-quality RNA is key, we suggest looking at the Ambion Web site for “Ten Ways to Improve Your RNA Isolation” (http://www.ambion.com/techlib/tn/91/9113.html). The negative control experiment of using RNA samples in an amplification to test for contaminating DNA could be omitted if the primers span an intron. In this case, genomic DNA would reveal a larger size product in the amplification of cDNA. Our selected genes, and 95% of yeast genes in general, do not contain introns. PCR products could be measured using agarose gel electrophoresis instead of polyacrylamide gel electrophoresis. Although we have found that polyacrylamide gels produce tighter bands that are more easily measured with densitometry, agarose gels may be easier to use in a teaching laboratory setting. If needed, a fluorophore that detects smaller amounts of DNA, such as SYBR Green I, could be used in place of ethidium bromide to stain gels. If a digital camera and software system designed for gel analysis are unavailable, densitometry could be performed using a normal digital camera and a software package such as NIH Image (http://rsb.info.nih.gov/nih-image/) or ImageJ (http://rsb.info.nih.gov/ij/).
We also recommend that time be invested in the optimization of reactions before an attempt to use either real-time PCR or the gel electrophoresis-based method is made. This process begins with primer design; we used the PrimerQuest tool at Integrated DNA Technologies (http://scitools.idtdna.com/Primerquest/). We used an annealing temperature of 4 degrees below the Tm reported by the manufacturer and conducted four reactions with cDNA template and MgCl2 concentrations varying from 1.5 mM to 4.5 mM. Using the MgCl2 concentration that yielded the fewest nonspecific products and least primer-dimer, we then optimized the annealing temperature. We suggest using “hot start” PCR as a next step, if needed (Santoyo et al. 1997). Although it is possible to exclude from measurement nonspecific products that migrate differently during gel electrophoresis than the specific product, we urge caution in this approach. It is also possible that the nonspecific and specific products have the same mobility. The use of a reaction that is not completely optimized can also be problematic in real-time PCR employing SYBR Green I, as opposed to a gene-specific probe system. We recommend that the specific product be verified by restriction digestion whenever possible.
A final important consideration is that even though the gel electrophoresis-based method agreed favorably with microarray and real-time data for three selected genes, it is not the most reliable method available. That real-time PCR holds this position is well accepted. We recommend that the gel electrophoresis-based method be used as a convenient and inexpensive alternative in undergraduate teaching and research settings where real-time PCR is impractical. It could also be used in a transitional role between microarrays and real-time measurements. For example, microarray experiments could be used in an undergraduate research project to generate a list of genes, and the gel electrophoresis-based method could be used to determine which of these measurements are the most reliable. Data of this sort would be suitable for local or even national presentations, or publication in an undergraduate journal. The mRNA levels for selected genes should then be further validated by real-time PCR before data are used in a professional publication.
The results of this study demonstrate that the gel electrophoresis-based method can produce data for quantifying mRNA levels that are in accord with microarray analysis and real-time PCR. The results validate the use of the method in our undergraduate research group at Missouri Western State College, allowing us to address important questions in an economical, yet reliable way. The dissemination of the method also provides opportunities for the surveyed faculty and many others to engage their undergraduate students, tomorrow's scientists and teachers, in technology that is fundamental to the new genomics era.
Acknowledgments
Thanks to the Genome Consortium for Active Teaching for access to the microarrays and to the participants of the Georgetown GCAT workshop (supported by National Science Foundation RUI grant DBI 0408386) for encouragement. Thanks also to the survey responders, to Kendy Jones for figure preparation, to Bart Phillips and Dr. Malcolm Campbell for critical reading of the manuscript, to Dr. Johanna Hardin and Dr. David Ashley for help with statistical analysis, and to Dr. John N. Anderson for invaluable advice. This work was supported by National Institutes of Health AREA grants 1R15CA096723–01 to Missouri Western State College and 1R15AG21907–01A1 to Pomona College, and by National Science Foundation RUI grants MCB 0113837 and MRI DBI 0318944 to Pomona College.
REFERENCES
- Abravaya K., Huff J., Marshall R., Merchant B., Mullen C., Schneider G., Robinson J. Molecular beacons as diagnostic tools: technology and applications. Clin. Chem. Lab. Med. (2003).;41((4),):468–474.. doi: 10.1515/CCLM.2003.070. [DOI] [PubMed] [Google Scholar]
- Asyali M.H., Shoukri M.M., Demirkaya O., Khabar K.S. Assessment of reliability of microarray data and estimation of signal thresholds using mixture modeling. Nucl. Acids Res. (2004).;32((8),):2323–2335.. doi: 10.1093/nar/gkh544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J., Sata N., Nagai H., Wada T., Yoshida K., Mano H., Sata F., Kishi R. Genistein-induced changes in gene expression in Panc 1 cells at physiological concentrations of genistein. Pancreas. (2004).;29((2),):93–98.. doi: 10.1097/00006676-200408000-00002. [DOI] [PubMed] [Google Scholar]
- Bijwaard K.E., Aguilera N.S., Monczak Y., Trudel M., Taubenberger J.K., Lichy J.H. Quantitative real-time reverse transcription-PCR assay for cyclin D1 expression: utility in the diagnosis of mantle cell lymphoma. Clin. Chem. (2001).;47((2),):195–201.. [PubMed] [Google Scholar]
- Bolufer P., Lo Coco F., Grimwade D., Barragan E., Diverio D., Cassinat B., Chomienne C., Gonzalez M., Colomer D., Gomez M.T., Marugan I., Roman J., Delgado M.D., Garcia-Marco J.A., Bornstein R., Vizmanos J.L., Martinez B., Jansen J., Villegas A., de Blas J.M., Cabello P., Sanz M.A. Variability in the levels of PML-RAR alpha fusion transcripts detected by the laboratories participating in an external quality control program using several reverse transcription polymerase chain reaction protocols. Haematologica. (2001).;86((6),):570–576.. [PubMed] [Google Scholar]
- Brewster J.L., Beason B., Eckdahl T.T., Evans I. The microarray revolution: perspectives from educators. Biochem. Mol. Biol. Educ. (2003).;32((4),):217–227.. doi: 10.1002/bmb.2004.494032040362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin S.A. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J. Mol. Endocrinol. (2002).;29((1),):23–39.. doi: 10.1677/jme.0.0290023. [DOI] [PubMed] [Google Scholar]
- Bustin S.A., Dorudi S. The value of microarray techniques for quantitative gene profiling in molecular diagnostics. Trends Mol. Med. (2002).;8((6),):269–272.. doi: 10.1016/s1471-4914(02)02334-1. [DOI] [PubMed] [Google Scholar]
- Ceol M., Forino M., Gambaro G., Sauer U., Schleicher E.D., D'Angelo A., Anglani F. Quantitation of TGF-beta1 mRNA in porcine mesangial cells by comparative kinetic RT/PCR: comparison with ribonuclease protection assay and in situ hybridization. J. Clin. Lab. Anal. (2001).;15((4),):215–222.. doi: 10.1002/jcla.1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson U.E., Heid C.A., Williams P.M. A novel method for real-time quantitative RT-PCR. Genome Res. (1996).;6((10),):995–1001.. doi: 10.1101/gr.6.10.995. [DOI] [PubMed] [Google Scholar]
- Gilliland G., Perrin S., Blanchard K., Bunn H.F. Analysis of cytokine mRNA and DNA: detection and quantitation by competitive polymerase chain reaction. Proc. Natl. Acad. Sci. U.S.A. (1990).;87((7),):2725–2729.. doi: 10.1073/pnas.87.7.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra S., Lopez-Fernandez L.A., Conde R., Pascual-Montano A., Harshman K., Esteban M. Microarray analysis reveals characteristic changes of host cell gene expression in response to attenuated modified vaccinia virus Ankara infection of human HeLa cells. J. Virol. (2004).;78((11),):5820–5834.. doi: 10.1128/JVI.78.11.5820-5834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakak Y., Walker J.R., Li C., Wong W.H., Davis K.L., Buxbaum J.D., Haroutunian V., Fienberg A.A. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. U.S.A. (2001).;98((8),):4746–4751.. doi: 10.1073/pnas.081071198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford W.P., Falco V.C., Gebhardt B.M., Carr D.J. The inherent quantitative capacity of the reverse transcription-polymerase chain reaction. Anal. Biochem. (1999).;266((2),):181–191.. doi: 10.1006/abio.1998.2913. [DOI] [PubMed] [Google Scholar]
- Heid C.A., Stevens J., Livak K.J., Williams P.M. Real-time quantitative PCR. Genome Res. (1996).;6((10),):986–994.. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- Jin W., Riley R.M., Wolfinger R.D., White K.P., Passador-Gurgel G., Gibson G. The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat. Genet. (2001).;29((4),):389–395.. doi: 10.1038/ng766. [DOI] [PubMed] [Google Scholar]
- Jost J.P., Oakeley E.J., Zhu B., Benjamin D., Thiry S., Siegmann M., Jost Y.C. 5-Methylcytosine DNA glycosylase participates in the genome-wide loss of DNA methylation occurring during mouse myoblast differentiation. Nucl. Acids Res. (2001).;29((21),):4452–4461.. doi: 10.1093/nar/29.21.4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesur I., Campbell J.L. The transcriptome of prematurely aging yeast cells is similar to that of telomerase-deficient cells. Mol. Biol. Cell. (2004).;15((3),):1297–1312.. doi: 10.1091/mbc.E03-10-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H., Yokoi H., Fujita J. Quantification of mRNA by non-radioactive RT-PCR and CCD imaging system. Nucl. Acids Res. (1992).;20,(18):4939.. doi: 10.1093/nar/20.18.4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noonan K.E., Beck C., Holzmayer T.A., Chin J.E., Wunder J.S., Andrulis I.L., Gazdar A.F., Willman C.L., Griffith B., Von Hoff D.D., Roninson I.B. Quantitative analysis of MDR1 (multidrug resistance) gene expression in human tumors by polymerase chain reaction. Proc. Natl. Acad. Sci. U.S.A. (1990).;87((18),):7160–7164.. doi: 10.1073/pnas.87.18.7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongers-Willemse M.J., Verhagen O.J., Tibbe G.J., Wijkhuijs A.J., de Haas V., Roovers E., van der Schoot C.E., van Dongen J.J. Real-time quantitative PCR for the detection of minimal residual disease in acute lymphoblastic leukemia using junctional region specific TaqMan probes. Leukemia. (1998).;12((12),):2006–2014.. doi: 10.1038/sj.leu.2401246. [DOI] [PubMed] [Google Scholar]
- Rhee S.K., Liu X., Wu L., Chong S.C., Wan X., Zhou J. Detection of genes involved in biodegradation and biotransformation in microbial communities by using 50-mer oligonucleotide microarrays. Appl. Environ. Microbiol. (2004).;70((7),):4303–4317.. doi: 10.1128/AEM.70.7.4303-4317.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saetre P., Lindberg J., Leonard J.A., Olsson K., Pettersson U., Ellegren H., Bergstrom T.F., Vila C., Jazin E. From wild wolf to domestic dog: gene expression changes in the brain. Mol. Brain Res. (2004).;126((2),):198–206.. doi: 10.1016/j.molbrainres.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Santoyo J., Alcalde J., Mendez R., Pulido D., de Haro C. Cloning and characterization of a cDNA encoding a protein synthesis initiation factor-2alpha (eIF-2alpha) kinase from Drosophila melanogaster. J. Biol. Chem. (1997).;272((19),):12544–12550.. doi: 10.1074/jbc.272.19.12544. [DOI] [PubMed] [Google Scholar]
- Schena M., Shalon D., Davis R.W., Brown P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. (1995).;270((5235),):467–470.. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Tajouri L., Mellick A.S., Ashton K.J., Tannenberg A.E., Nagra R.M., Tourtellotte W.W., Griffiths L.R. Quantitative and qualitative changes in gene expression patterns characterize the activity of plaques in multiple sclerosis. Mol. Brain Res. (2003).;119((2),):170–183.. doi: 10.1016/j.molbrainres.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Townsend J.P. Resolution of large and small differences in gene expression using models for the Bayesian analysis of gene expression levels and spotted DNA microarrays. BMC Bioinformatics. (2004).;5((1),):54.. doi: 10.1186/1471-2105-5-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu H.L., Troubetzkoy S., Nguyen H.H., Russell M.W., Mestecky J. A method for quantification of absolute amounts of nucleic acids by (RT)-PCR and a new mathematical model for data analysis. Nucl. Acids Res. (2000).;28((7),):E18.. doi: 10.1093/nar/28.7.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall S.J., Edwards D.R. Quantitative reverse transcription-polymerase chain reaction (RT-PCR): a comparison of primer-dropping, competitive, and real-time RT-PCRs. Anal. Biochem. (2002).;300((2),):269–273.. doi: 10.1006/abio.2001.5458. [DOI] [PubMed] [Google Scholar]