

Abstract
Post-translational modifications (PTMs) on intact histones play a major role in regulating chromatin dynamics and influence biological processes such as DNA transcription, replication, and repair. The nature and position of each histone PTM is crucial to decipher how this information is translated into biological response. In the present work, the potential of a novel tandem top-“double-down” approach –ultraviolet photodissociation followed by mobility and mass selected electron capture dissociation and mass spectrometry (UVPD-TIMS-q-ECD-ToF MS/MS)– is illustrated for the characterization of HeLa derived intact histone H4 proteoforms. Comparison between q-ECD-ToF MS/MS spectra and traditional FT-ICR-ECD MS/MS spectra of a H4 standard showed similar sequence coverage (~75%) with significant faster data acquisition in the ToF MS/MS platform (~3 min vs. ~15 min). Multiple mass shifts (e.g., 14 and 42 Da) were observed for the HeLa derived H4 proteoforms, for which the top-down UVPD and ECD fragmentation analysis were consistent in detecting the presence of acetylated PTMs at the N-terminus, Lys5, Lys8, Lys12 and Lys16 residues, as well as methylated, dimethylated, and trimethylated PTMs at the Lys20 residue with a high sequence coverage (~90%). The presented top-down results are in good agreement with bottom-up timsTOF MS/MS experiments and allowed for additional description of PTMs at the N-terminus. The integration of a 213 nm UV laser in the present platform allowed for UVPD events prior to the ion mobility-mass precursor separation for CID/ECD TOF MS. Selected c305+ UVPD fragments, from different H4 proteoforms (e.g., Ac+Me2, 2Ac+Me2 and 3Ac+Me2), exhibited multiple IMS bands, for which similar CID/ECD fragmentation patterns per IMS band pointed toward the presence of conformers, adopting the same PTM distribution, with a clear assignment of the PTM localization for each of the c305+ UVPD fragment H4 proteoforms. These results were consistent with the biological “zip” model, where acetylation proceeds in the Lys16 to Lys5 direction. This novel platform further enhances the structural toolbox with alternative fragmentation mechanisms (UVPD, CID and ECD) in tandem with fast, high resolution mobility separations and shows great promises for global proteoform analysis.
Graphical Abstract
1. INTRODUCTION
Histones (H2A, H2B, H3, and H4) are highly basic intrinsically disordered proteins1 that package deoxyribonucleic acid (DNA) into nucleosomes within the nuclei of eukaryotic cells.2, 3 The structure of a nucleosome consists of a DNA segment wrapped around an octameric core histones, containing a tetrameric H3(x2)/H4(x2) together with two pairs of H2A/H2B dimers (Figure S1).2, 3 The nucleosome is the fundamental subunit of chromatin, where the chromatin fiber can be further folded and condensed to produce chromosomes.4 A core histone is composed of a central helix fold and a flexible and highly basic N-terminal tail, which protrudes from the nucleosome (Figure S1) and are the main target for post-translational modifications (PTMs).5 These histone tails are highly subjected to acetylation (Arg and Lys residues), methylation (Arg and Lys residues), phosphorylation (Ser, Thr and Tyr residues) among others, at diverse and/or multiple positions.6–11 Combinatorial PTMs result in a histone code12 that are of particular interest due to their essential role in gene expression by regulating chromatin dynamics5, 13, 14 as well as DNA transcription, replication and repair.15–18 However, the enzymatic machinery that establishes the histone code can be deregulated in diseases such as, cancer leading to alterations in the PTM patterns having crucial functions in diverse cancer development and progression.19–22 Therefore, the position and nature of each PTM must be elucidated to decipher how the histone code is translated into biological response. The elucidation of such a code is the great challenge for proteomics given the stunning isobaric/isomeric PTM content in at the histone level.
Traditional proteomic approaches have become fundamental tools for the characterization of histone PTMs in biological systems.23–26 In particular, middle- and top-down proteomic methods are gaining momentum over bottom-up strategies with the emergence and advancement of electron-based fragmentation (ExD)27–29 and ultraviolet photodissociation (UVPD).30–33 These fragmentation techniques have demonstrated significant advances in elucidating isobaric and isomeric histone PTMs with much more detailed and confident characterization by preserving the labile PTMs and increasing sequence coverage.34–38 The introduction of the electromagnetostatic (EMS)39, 40 cell in 2008 has opened new avenues for proteomic analysis by making more straightforward and affordable the use of electron capture dissociation (ECD) over ECD performed on traditional FT-ICR mass spectrometer. The EMS cell is capable of performing ECD without the need for long reaction times or ultrahigh vacuum and has been implemented into widespread quadrupole,41–43 q-ToF44, 45 and Orbitrap46–49 mass spectrometers.
Ion mobility spectrometry coupled to tandem mass spectrometry (IMS-MS/MS) has gained impetus in proteomics due to superior speed and selectivity over traditional condensed-phase separations (e.g., liquid chromatography).50–53 The benefit of the EMS cell in combination with IMS has been recently reported into commercially available Agilent DTIMS-q-ToF MS (e.g., Agilent 6560),54 Waters q-TWIMS-ToF MS (e.g., Synapt G2-Si)55 and Bruker TIMS-q-ToF MS (e.g., Maxis Impact II)56, 57 platforms. In particular, the recent implementation in 2021 of the EMS cell into a TIMS-q-ToF MS instrument exhibited great promises in separating and discriminating isomeric/isobaric histone tail proteoforms.56 With the introduction of UVPD into IMS-MS platforms,58–60 several groups have reported the potential of UVPD prior and/or after the IMS separation step. The advantages of TIMS-UVPD-ToF MS were recently showcase for the mobility separation of histone tail proteoforms followed by the localization of the PTM locations based on mobility-selected-UVPD MS.61
In the present work, the potential of top-“double-down” ultraviolet photodissociation, ion mobility and mass -selected electron capture dissociation with mass spectrometry (UVPD-TIMS-q-ECD-ToF MS/MS) is illustrated for the characterization of histone H4 proteoforms. Proof of concept data is presented for an intact H4 (recombinant standard) and HeLa derived H4 proteoforms. Comparison between the top-down (UVPD and ECD) results and traditional bottom-up as well as between ECD-ToF MS/MS and FT-ICR-ECD MS/MS platforms are discussed based on PTM assignments and ECD fragmentation efficiency and sequence coverage, respectively. This UVPD-TIMS-q-ECD-ToF MS/MS platform enables top-down UVPD (213 nm) capabilities followed by a second top-down high resolution mobility separations (i.e., 2x-3x higher resolving power compared to previous IMS-UVPD implementations) and mass-selected ECD (EMS cell) MS/MS experiments.In the following discussion, a special emphasis is placed on the capabilities of performing top-“double-down” analysis for a more comprehensive characterization of H4 proteoforms.
2. EXPERIMENTAL SECTION
2.1. Materials and Reagents.
Recombinant human histone H4 (accession number: P62805, 11.3 kDa) was purchased from EpiCypher (Durham, NC). The H4 standard was extensively dialyzed (desalting) against 10 mM aqueous ammonium acetate (NH4Ac), obtained from Fisher Scientific (Pittsburgh, PA), and was analyzed at a concentration of 10 μM in 50:50 water/methanol. Details on the histone extraction workflow from human cells can be found elsewhere.62, 63 Briefly, the first step consisted of extracting the nucleus from HeLa cells, followed by an histone extraction step in acidic conditions and finally the histones were precipitated using cold acetone and dissolved in water as illustrated in Figure S2. The same histone extraction workflow was used for untreated HeLa cells as well as for HeLa cells treated with 5 M sodium butyrate, which is known to be a strong histone deacetylase inhibitor. The histones were purified by reversed-phase liquid chromatography (RP-LC) on a XBridge peptide BEH C18 column (10 mm × 250 mm × 5 μm) using a Dionex UltiMate 3000 LC system under similar elution condition as previously described.62, 63 Mass spectrometry analysis were then performed on each LC fraction to assign the histone identity as depicted in Figure S3. For bottom-up purposes, the extracted histone H4 from HeLa cells were derivatized with propionic anhydride followed by digestion with trypsin to generate peptides of 5 – 15 aa in length.62, 63 After digestion, a propionylation step was carried out on the histones at the N-terminus to improve chromatographic retention. Solutions of HeLa derived histones were analyzed at a concentration of 10 μM in 50:50 water/methanol. Low concentration Tuning Mix standard (G1969–85000), obtained from Agilent Technologies (Santa Clara, CA), was used to externally calibrate the TIMS instrument.
2.2. UVPD-TIMS-q-ECD-ToF MS Instrumentation.
The UVPD, TIMS and ECD capabilities were integrated into a Bruker Maxis Impact II ToF MS (Bruker Daltonics Inc., Billerica, MA) instrument, equipped with a nESI source, as depicted in Figure 1. nESI emitters were pulled in-house from quartz capillaries (O.D. = 1.0 mm and I.D. = 0.70 mm) using Sutter Instruments Co. P2000 laser puller (Sutter Instruments, Novato, CA). Protein solutions were loaded in a pulled-tip capillary, housed in a mounted custom built XYZ stage in front of the MS inlet, and sprayed at 1000 V via a tungsten wire inserted inside the nESI emitters.
Figure 1.
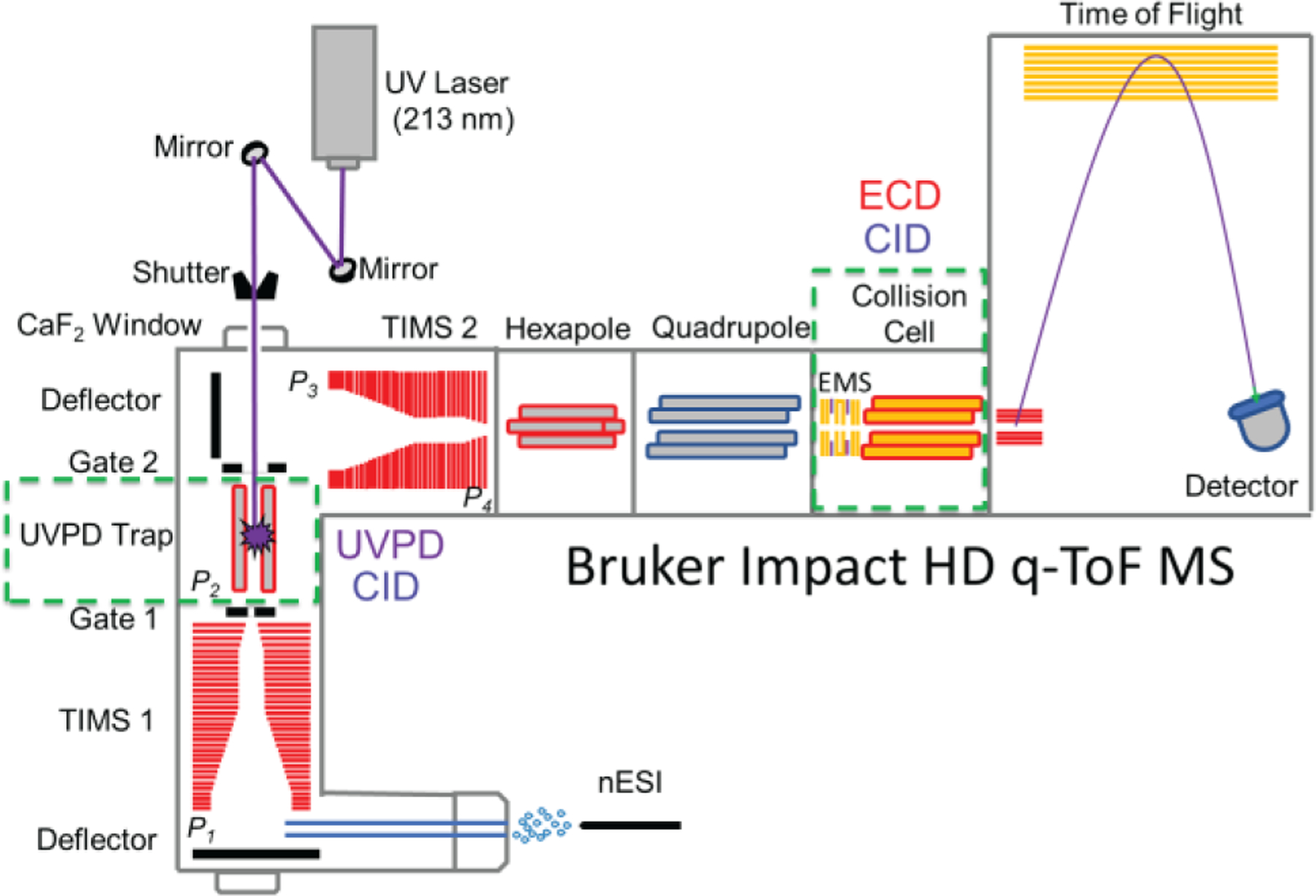
Schematic of the nESI-UVPD-TIMS-q-ECD-ToF MS/MS instrument. The green dashed lines represent the possibility for MS/MS events, for which UVPD/CID and ECD/CID can be performed in the UVPD trap and collision cell, respectively.
A 203 mm long UVPD linear ion trap with a quadrupolar design (d0 = 3.5 mm constructed of 4.0 mm round rods) was incorporated prior to the TIMS 2 analyzer and equipped with an entrance (gate 1) and end (gate 2) lens system (∼5 mm i.d. apertures). A smaller inner diameter lens (∼2 mm i.d. aperture) located between the TIMS and the trap region allows for maximum UVPD fragmentation in the trap region and minimal UV light transmission to the TIMS region. Potential UVPD product ions from the TIMS analyzer (<50% efficiency) are excluded by their ion mobilities to enter the trap region. A 213 nm laser beam, generated from the fifth harmonic of a Nd:YAG laser (NL204, EKSPLA, Vilnius, Lithuania), was aligned with the UVPD trap and operated at a repetition rate of 1 kHz with an energy of ∼0.2 mJ per pulse. The TIMS analyzers and the UVPD trap were controlled by a modular intelligent power source (MIPS, GAA Custom Electronics, WA), consisting of 16 channels with a 250 V output range and two radiofrequency (rf) drivers, and synchronized with the ToF-MS platform controls. Additional details on the synchronisation and timing sequences occurring during TIMS/UVPD acquisition can be found elsewhere.61 During UVPD and TIMS operation, precursor ions were UV irradiated with ∼155 laser pulses in the UVPD trap while the fragment ions were separated in the ion mobility domain (155 ms trap time) and mass domain (quadrupolar isolation window of 5 m/z) prior to ECD-TOF MS (Figure 1).
A custom-built 19 mm long EMS (e-MSion Inc., Corvallis, OR) cell was attached to a custom-built collision cell and mounted between the quadrupole exit and the pulsing plates of the ToF MS instrument. Electrons are generated at the center of the cell, through a heated rhenium filament (Scientific Instrument Services, Ringoes, NJ), and confined along the ion longitudinal axis. The filament was operated at a current of 2.5 A. The collision cell was operated using high purity argon (oxygen free) to enhance the cooling of the ions. Additional details on the ECD operation are described elsewhere.56, 57 ECD spectra were collected on quadrupole isolated (mass window of 3–5 Da) UVPD product ions, where each of the ECD events were synchronized with the ion mobility scan step allowing for precursor-fragments ion mobility alignment (Figure 1). A typical data acquisition lasted for ~5 min. UVPD/ECD spectra were deconvoluted using UniDec64 v4.4.0 and assignments were performed using ProSight Lite v1.4. The fragment ions were annotated with a mass error of ~20 ppm average with S/N >3–4 in the UVPD/ECD spectra.
The general fundamentals of TIMS as well as the calibration procedure have been described in the literature.65–68 TIMS experiments were carried out using nitrogen (N2) as buffer gas, at ambient temperature (T) with a gas velocity defined by the funnel entrances (P1 = 3.9 mbar / P3 = 2.1 mbar) and exits (P2 = 2.6 mbar / P4 = 0.74 mbar) pressure differences (Figure 1). TIMS 1 was operated in transmission mode (rf voltage of 160 Vpp at 755 kHz). Mobility separation of the UVPD fragment ions was performed in TIMS2 (rf voltage of 250Vpp at 880 kHz). The UPVD ion trap (rf voltage of 170 Vpp at 675 kHz) was operated in tandem with the TIMS2 separation. A deflector voltage of 300 V, a TIMS 1 voltage of 170 V, a TIMS exit lens (gate 1) of 169 V, a multipole exit lens (gate 2) of 135 V, as well as a TIMS 2 ramp voltage of −150 to −25 V were used for all the experiments. The scan rate (Sr = ΔVramp/tramp) was selected to trap all UVPD fragments in a single experiment and optimized for fast ion mobility acquisition.
2.3. FT-ICR-ECD MS/MS Instrumentation.
Complementary ECD experiments were conducted on a Solarix 7T FT-ICR mass spectrometer (Bruker, Billerica, MA) equipped with an Infinity cell and a nESI source (similar to the ToF platform) operated in positive ion mode. The high voltage, capillary exit, and skimmer I were set to 1500 V, 140 V, and 30 V respectively. Precursor ions were isolated in the quadrupole with a mass window of 10 Da, accumulated for 0.8 s in the collision cell, and further injected into the ICR cell. ECD experiments were performed with a heated hollow cathode operating at a current of 1.5 A. The ECD pulse length, ECD Bias and ECD Lens were set at 0.095 s, 0.95 V, and 10 V respectively. A total of 600 scans (m/z range 200–3000) were co-added with a data acquisition size of 512k words. A typical data acquisition lasted for ~15 min.
2.4. Bottom-up MS Analysis.
The digested peptides were separated in a 75 μm ID x 17 cm Reprosil-Pur C18-AQ (3 μm; Dr. Maisch HPLC GmbH, Germany) nano-column fitted on an EASY-nLC system (Thermo Scientific, San Jose, Ca) using the following gradient at a flow-rate of 300 nL/min: 2% to 28% solvent B (A = 0.1% formic acid; B = 95% MeCN, 0.1% formic acid) over 45 minutes, from 28% to 80% solvent B in 5 minutes, 80% B for 10 minutes. This nLC was coupled online to an QExactive-Orbitrap mass spectrometer (Thermo Scientific) and data-independent acquisition (DIA) was used to acquire data. Briefly, full scan MS (m/z 300−1100) was acquired in the Orbitrap with a resolution of 70,000 and an AGC target of 1x10e6. MS/MS experiments were performed in centroid mode in the ion trap with sequential isolation windows of m/z 24 with an AGC target of 2x10e5, a CID collision energy of 30 eV and a maximum injection time of 50 msec. Data were analyzed using the in-house software, EpiProfile,69 wherein the precursor and fragment extracted ion chromatography was used to accurately determine the chromatographic profile and to discriminate isobaric forms of peptides.
3. RESULTS AND DISCUSSION
3.1. Cross-platform Comparison of H4 Histone Top-Down ECD: q-ECD-ToF MS/MS vs. FT-ICR-ECD MS/MS.
The nESI-MS analysis of the intact H4 standard (11.3 kDa), acquired using the UVPD-TIMS-q-EMS-ToF MS/MS platform, resulted in a broad charge state distribution, ranging from [M + 6H]6+ to [M + 17H]17+ molecular ions, under denaturing solution conditions (Figure S4a). This broad charge state distribution was rationalized as a structural change in the folded protein (6+ to 8+) towards more extended conformations (9+ to 17+), due to the exposure of the basic residues which is typical of an intrinsically disordered protein. The nESI-MS distribution obtained using the FT-ICR configuration revealed a similar charge state distribution (7+ to 17+, Figure S4b).
The ECD MS/MS spectra of the quadrupole isolated [M + 11H]11+ (blue, m/z 1022.4) and [M + 13H]13+ (red, m/z 865.3) molecular ions of intact H4 standard (inserts in Figure S4), obtained from the q-ToF MS/MS and FT-ICR MS/MS platforms, are illustrated in Figures 2a and 2b, respectively. Inspection of the ECD spectra showed similar fragmentation patterns between the two platforms. Typical product ions were obtained across the protein, for which a fragmentation efficiency of ~45% was obtained in the q-ToF MS/MS platform while being higher (~65%) using the FT-ICR MS/MS instrument. This may be explained by the differences in the ECD speed between the two platforms, for which faster ECD events (~10 μs) occurred in the q-ToF MS/MS platform as compared to the FT-ICR MS/MS instrument (~100 ms). Nevertheless, the assigned ECD fragment ions accounted for over 70% sequence coverage in the q-ToF MS/MS platform (Figure 2a), which was comparable (~75%) with the sequence coverage obtained from the FT-ICR MS/MS instrument (Figure 2b). In addition, the EMS cell presents the advantage of performing ECD without the need for long reaction times or ultra-high vacuum and can be easily implemented with trapped ion mobility spectrometry.56, 57 In particular, significantly faster ECD data acquisition were obtained in the q-ToF MS/MS platform (~3 min) as compared to the FT-ICR-MS/MS instrument (~15 min).
Figure 2.
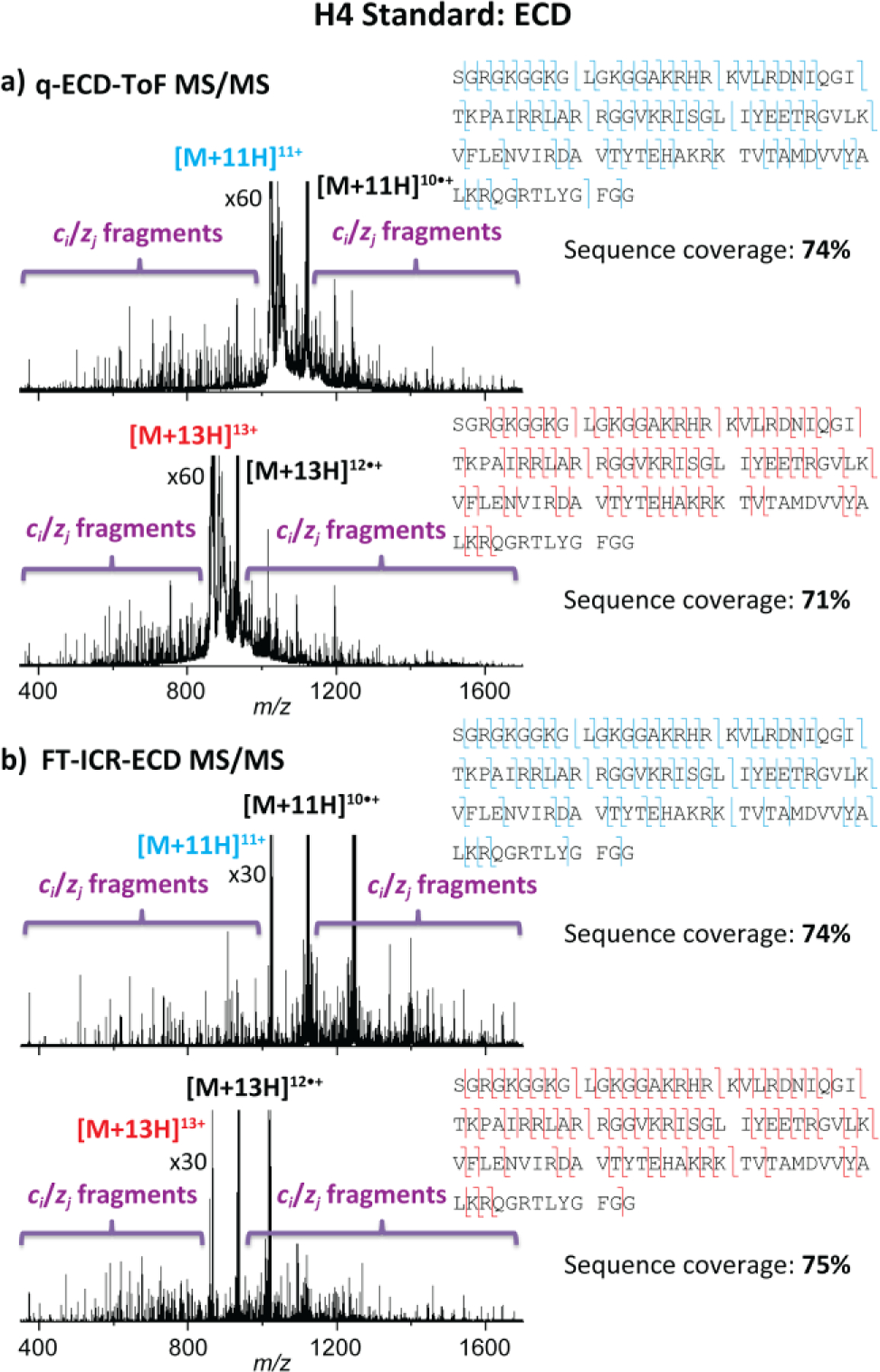
ECD spectra of the [M + 11H]11+ (blue, m/z 1022.4) and [M + 13H]13+ (red, m/z 865.3) species of intact H4 standard within (a) q-ECD-ToF MS/MS and (b) FT-ICR-ECD MS/MS platforms. The sequence coverages are denoted on the top right of each panel.
3.2. Top-down UVPD, CID and ECD MS/MS Fragmentation of HeLa Derived H4 proteoforms.
Combinatorial PTMs on histones play an important role in many biological processes.12 Different epigenetic mechanisms, involving histone acetyl transferases (HATs) and histone deacetylases (HDACs), lead to histone lysine acetylation and deacetylation on histone tails, respectively, resulting in changes in chromatin structure and transcription levels.70 During inhibition of HDAC activity (HDACi), HAT activity continues, which results in histone hyperacetylation. In particular, we previously demonstrated the utility of the TIMS-q-EMS-ToF MS/MS platform for discriminating acetylation / trimethylation PTM histone tail proteoforms.56 Here, intact H4 extracted from HeLa cells (H4 HeLa) containing wild-type PTMs, as well as from HeLa cells treated with sodium butyrate (H4 HeLa HDACi), which is known to be a strong HDAC inhibitor were studied.71 The nESI-MS analysis of the H4 HeLa and H4 HeLa HDACi exhibited a similar charge state distribution as compared to the full H4 standard, ranging from [M + 7H]7+ to [M + 17H]17+ molecular ions, under the same starting solution denaturing conditions (Figure 3a). Figure 3b illustrates the nESI-MS spectra in the m/z 1020–1047 range, which corresponds to the [M + 11H]11+ region, for the three H4 protein conditions. At each charge state, additional MS peaks at higher m/z values were observed in H4 HeLa and H4 HeLa HDACi and assigned to different PTMs. In particular, multiple shifts of 14 Da and 42 Da were attributed to methylation and acetylation or trimethylation, respectively (Figure 3b). The significant increase in PTM abundances for H4 HeLa HDACi was consistent with histone hyperacetylation. The MS analysis allowed to identify the nature and number of PTMs present in H4 HeLa and H4 HeLa HDACi (Table S1).
Figure 3.
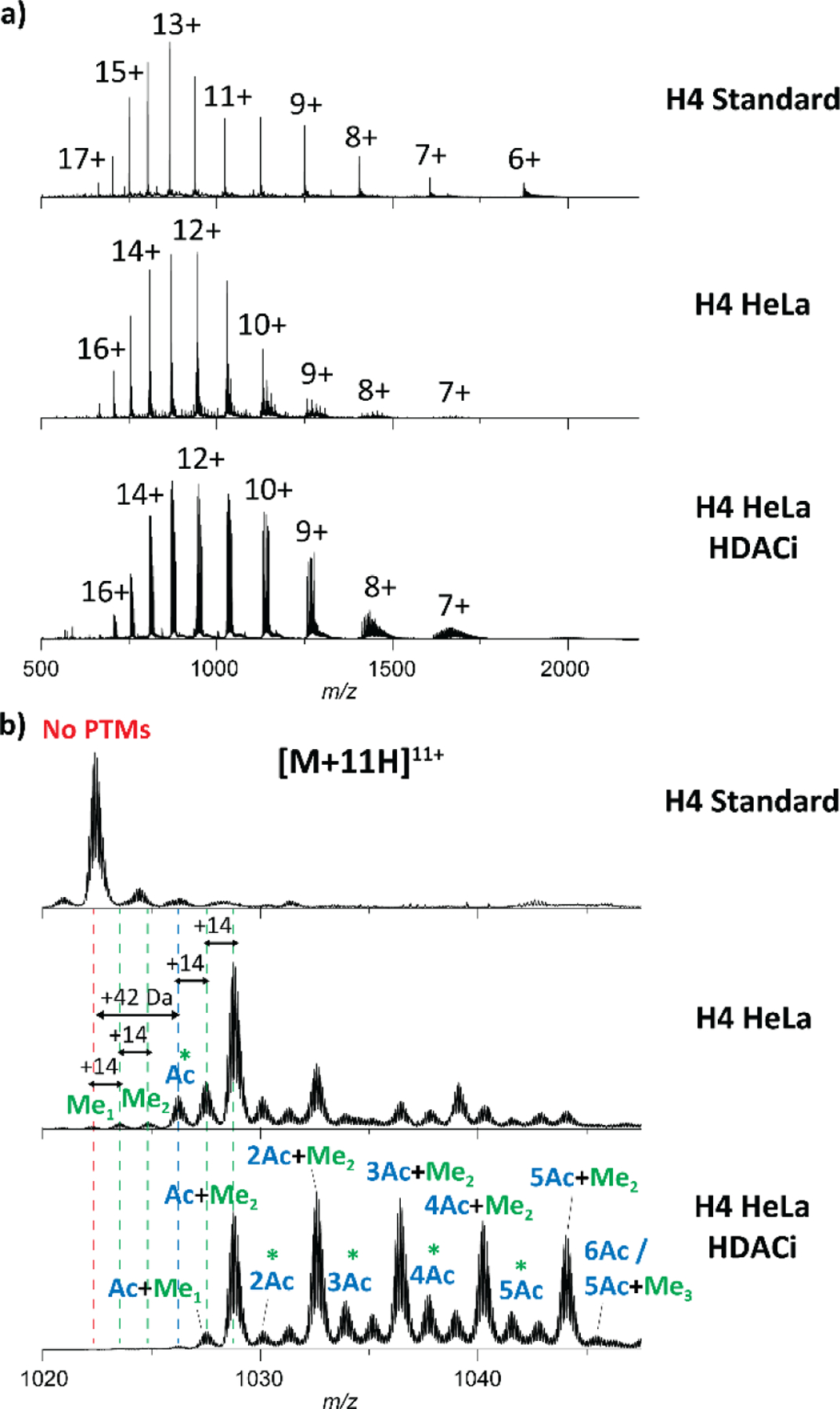
H4 nESI-MS spectra (a) and zoom in the [M + 11H]11+ region (b) for H4 standard, H4 HeLa and H4 HeLa HDACi. PTMs containing acetylation and/or methylations are colored in blue and green, respectively. Note that the green stars (*) highlight for potential isobaric trimethylation species.
The PTM localization on H4 HeLa were determined from top-down MS/MS experiments. Note that for proof-of-concept, H4 from HDACi treated HeLa cells was selected due to the higher abundance of the PTMs relative to H4 HeLa. The ECD MS/MS spectrum of the quadrupole isolated [M + 11H]11+ ion regions, comprising all PTMs (wide m/z isolation window), for H4 HeLa HDACi is illustrated in Figure 4a. Typical product ions were obtained across the protein with a fragmentation efficiency of ~85% and sequence coverage of ~89% (Figure 4a). In addition, similar ECD fragmentation patterns were observed between H4 standard and H4 HeLa HDACi for [z8• to z81•] product ions, indicating that the Lys31, Lys44, Lys59, Lys77, Lys79 and Lys91 residues are not carrying any PTMs. However, specific product ions were observed for the H4 HeLa HDACi, where multiple shifts of 42 Da were obtained for [c2′ to c19′] fragment ions, involving the presence of acetylated PTMs at the N-ter, Lys5, Lys8, Lys12 and Lys16 residues (highlighted in blue in Figure 4a). Moreover, multiple shifts of 14 Da in addition to the multiple shifts of 42 Da were observed for [c20′ to c101′] and [z83• to z101•] product ions, suggesting the presence of methylated, dimethylated and trimethylated PTMs at the Lys20 residue (highlighted in green in Figure 4a) with the dimethylated distribution being the most abundant.
Figure 4.
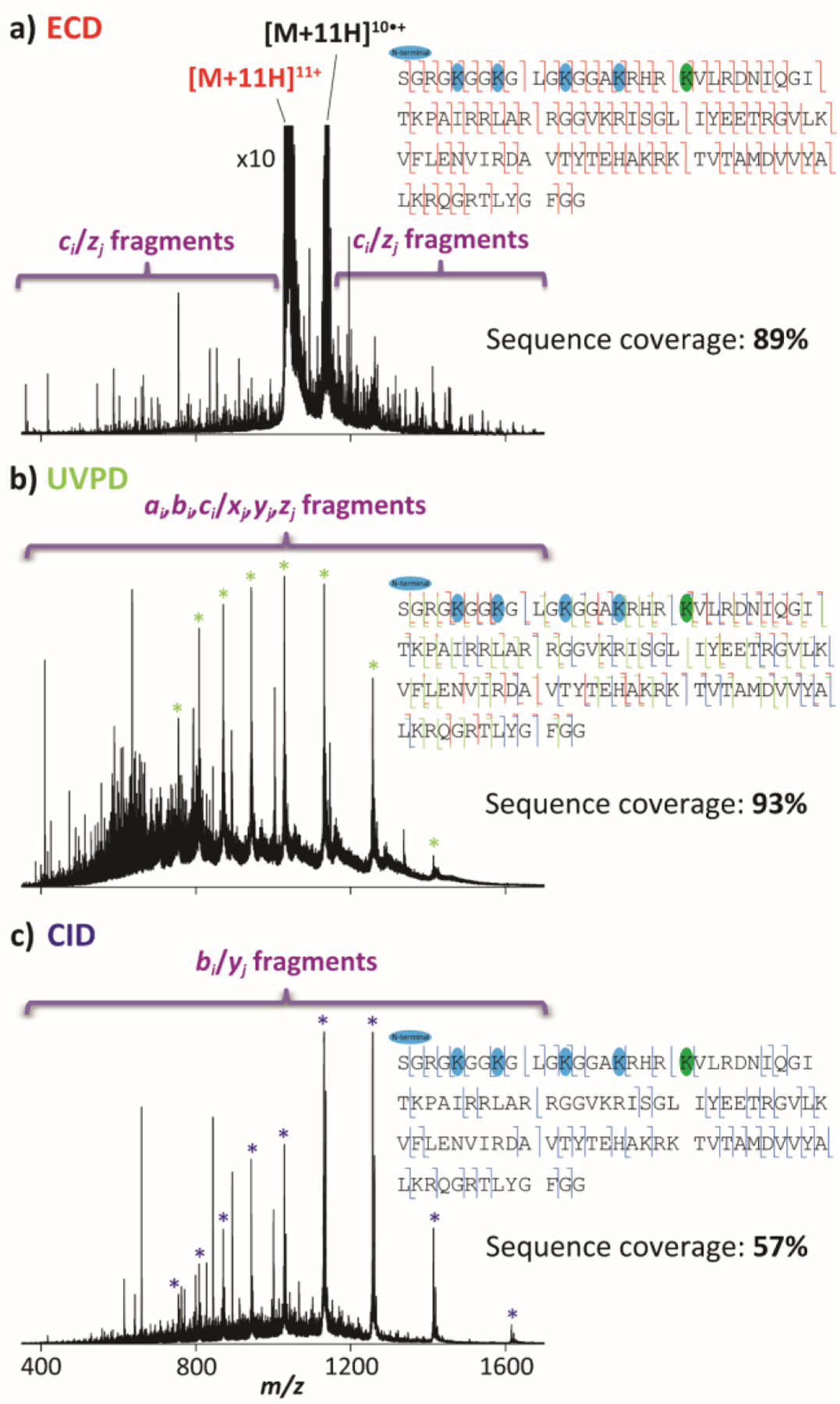
Top-down (a) ECD spectrum of the [M + 11H]11+ species, (b) UVPD spectrum without mass isolation and (c) CID spectrum without mass isolation of intact H4 HeLa HDACi within UVPD-TIMS-q-EMS-ToF MS platform. The sequence coverages are denoted on the top right of each panel. PTMs containing acetylation and methylations are colored in blue and green, respectively.
The integration of the UVPD trap prior to the TIMS 2 allowed for additional top-down UVPD and CID fragmentation capabilities without mass selection (Figure 1). The UVPD and CID spectra for H4 HeLa are illustrated in Figures 4b and 4c, respectively. Typical ai,bi,ci/xj,yj,zj and bi/yj product ions were obtained across the protein with a sequence coverage of ~93% and ~57% in UVPD and CID, respectively. Both fragmentation techniques were consistent with the top-down ECD experiments in term of PTM assignments. Although acetylation and trimethylation PTMs are not labile enough to be cleaved in CID, the lower sequence coverage together with the lower abundance of fragments containing PTMs as compared to UVPD and ECD, make the later fragmentation techniques more suitable for a better identification and assignment of the PTM sites at the protein level.
Complementary bottom-up experiments were conducted on H4 HeLa HDACi, for which Gly4-Arg17, Lys20-Arg23, Asp24-Arg35, Arg40-Arg45, Ile46-Arg55, Gly56-Arg67, Asp68-Arg78 and Lys79-Arg92 H4 fragments were observed (red rectangles in Figure S5). The Asp24-Arg35, Arg40-Arg45, Gly56-Arg67, Asp68-Arg78 and Lys79-Arg92 H4 fragments did not show any PTMs at lysine residues while the Gly4-Arg17 and Lys20-Arg23 H4 fragments exhibited acetylation at Lys5, Lys8, Lys12 and Lys16 residues as well as methylations at Lys20 residues, in good agreement with the top-down MS/MS experiments (Figure S5). However, the PTM at the N-terminal was not identified by the bottom-up approach, due to the absence of H4 fragments comprising the N-terminal part, making the top-down MS/MS approach better-suited and faster for the comprehensive characterization of full histone proteoforms. However, the top-down approach usually has limitations in term of sensitivity when compared to the bottom-up workflow.
3.3. Top-“Double-Down” Mass Spectrometry of Histone H4 Proteoforms.
The top-down UVPD and ECD approaches showed useful for identifying and localizing the number and type of PTMs. The added ion mobility pre-separation of proteoforms, posterior to UVPD fragmentation and prior to top-down ECD MS/MS, can be very effective for the discrimination of potential isomeric/isobaric interferences. While the top-down and bottom-up experiments were carried out on H4 HeLa cells treated with sodium butyrate to facilitate the elucidation of the PTM positions for proof-of-concept purposes, the novel top-“double-down” mass spectrometry experiments were performed on the untreated H4 HeLa cells to evaluate the potential of this approach on real life sample. The overall ion mobility profiles for H4 standard and H4 HeLa exhibited a large structural heterogeneity across the charge state distribution in agreement with an intrinsically disordered protein (Figure S6a). In addition, the conformational space of both intact H4 proteins occupied a wide ion mobility range (1/K0 ~0.9–1.55 V.s/cm2), indicative of compact to extended structural diversity through the exposure of the basic residues that significantly increase the electrostatic interactions (i.e., coulombic repulsions) with the net charge. Comparison between the ion mobility profiles of H4 standard and H4 HeLa exhibited less conformational flexibility for H4 containing PTMs (Figure S6a). This suggests that the presence of PTMs at lysine residues disturbs the electrostatic interaction network by preventing protonation at these lysine residues, re-defining the accessible conformational space of the H4 protein. This is supported by comparing the ion mobility profiles of individuals PTMs in H4 HeLa, for which a conformational collapse was observed when increasing the number of PTMs (Figure S6b). As most of the H4 PTMs occur at the N-terminal (tail), results suggest that this segment drive the conformational changes observed for H4 in the presence of PTMs.
The integration of a 213 nm UV laser in the present platform, prior to the TIMS 2, permitted fast ion mobility separations of the UVPD fragments, as illustrated in Figure 5a. Since all the PTMs were localized in the N-terminal part of the HeLa derived H4 samples, the ion mobility analysis were focused on one selected c305+ UVPD product ions (high intensity) which comprise all the identified PTMs. In particular, the ion mobility profiles of the c305+ fragment ions were investigated in H4 standard (m/z 621.0) as well as in H4 HeLa, for which the c305+ product ions carried distinct PTM distributions, including Ac+Me2 (m/z 635.0), 2Ac+Me2 (m/z 643.4) and 3Ac+Me2 (m/z 651.8, Figure 5). These UVPD fragment of interests were then mass isolated in the quadrupole and filtered in the ion mobility domain in order to remove potential isobaric interferences as illustrated in Figures 5b and 5c (top left of each panel). The integration of the EMS cell in the present platform, posterior to the TIMS 2, allowed for fast ion mobility-selected ECD MS/MS acquisitions. As the ion mobility separation step of the UVPD fragments occurs, ECD MS/MS spectra of the selected c305+ UVPD product ions can be synchronized with the ion mobility scans for precursor-fragments ion mobility alignment as depicted in Figures 5b and 5c (bottom left of each panel). Note that similar experiments involving CID fragmentation can be performed in the collision cell of the instrument (Figure S7). Proof of concept UVPD-TIMS-q-ECD/CID-ToF MS/MS analysis were carried out on the c305+ H4 standard, where the assigned ECD and CID fragment ions accounted for a high sequence coverage of ~90% and ~87%, respectively (Figures 5b and S7b). The TIMS distribution of the c305+ Ac+Me2, 2Ac+Me2 and 3Ac+Me2 product ions in H4 HeLa, for which multiple PTM combinations can occur within the N-terminus and five available Lys residues, exhibited two IMS bands for each of the PTM combinations (Figures 5c and S7c, bottom left of each panel). The PTM localization of these fragment ions were determined from top-“double-down” mass spectrometry experiments (Figures S8-S10). The ECD and CID MS/MS spectra per IMS band of the quadrupole isolated c305+ Ac+Me2, 2Ac+Me2 and 3Ac+Me2 product ions are illustrated in Figures 5c and S7c (right of each panel), respectively. The assigned ECD fragment ions of the peptides accounted for ~90% sequence coverage while being lower (~70%) using top-“double-down” mass spectrometry with CID. In addition, similar ECD and CID fragmentation patterns were observed across the two IMS bands for each of the c305+ PTM combination, attesting for the presence of conformers sharing the same PTM distributions (single proteoform). Furthermore, these top-“double-down” MS/MS experiments clearly pointed toward the presence of an acetylation at the N-term together with a dimethylation at Lys20 for c305+ Ac+Me2, acetylations at the N-term and Lys16 together with a dimethylation at Lys20 for c305+ 2Ac+Me2 and acetylations at the N-term, Lys16 and Lys12 together with a dimethylation at Lys20 for c305+ 3Ac+Me2 (Figures 5c and S7-S10). In summary, this workflow was able to highlight the presence of conformers sharing similar PTM distributions (single proteoform), for which acetylation proceeded in the Lys16 to Lys5 direction in agreement with the previously introduced biological ‘zip’ model (Figure S11).72 The present approach has also the potential to separate proteoforms without the need for pre-separation/cleaning techniques (e.g., LC system) by directly injecting the protein of interest. Further top- “double-down” mass spectrometry analysis can take advantage of the ECD implementation to investigate more labile PTMs, such as phosphorylation and/or increase the PTM abundances for potential isomeric proteoforms for a more comprehensive (or blind) global histone proteoform analysis.
Figure 5.
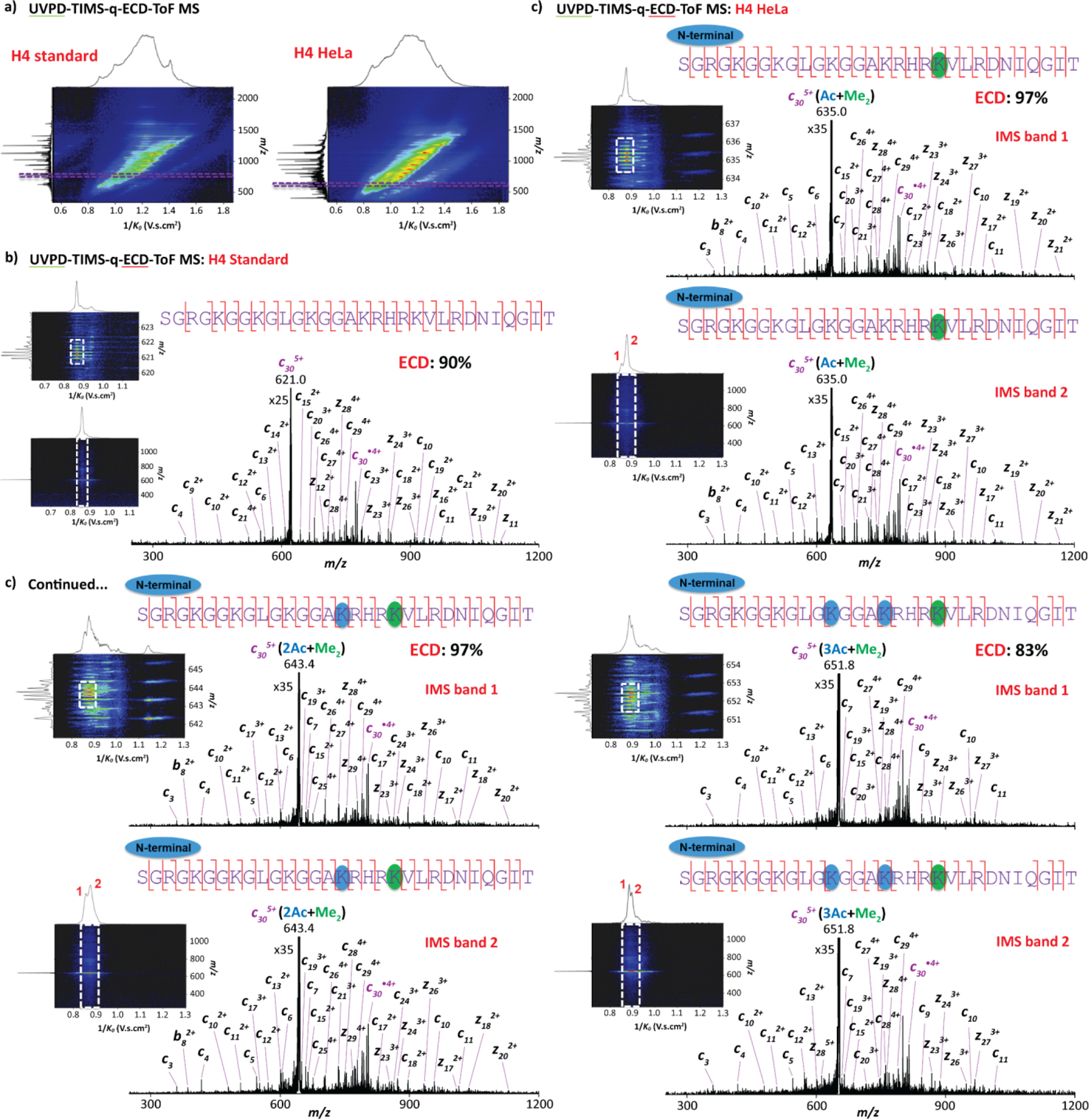
Top-“double-down” mass spectrometry analysis showing (a) 2D UVPD-TIMS-ToF MS contour maps for both H4 proteins and 2D UVPD-TIMS-q-ECD-ToF MS contour maps of the c305+ fragments (left panel) together with the ECD spectra per IMS band (right panel) for the (b) H4 standard and (c) H4 HeLa.
4. CONCLUSIONS
The potential of top-“double-down” mass spectrometry in a UVPD-TIMS-q-ECD-ToF MS/MS platform was effectively demonstrated for the comprehensive characterization of intact H4 standard (no PTMs) as well as HeLa cell acid-extracted H4 containing various PTM combinations. The comparison between the top-down ECD MS/MS spectra obtained from the q-ToF MS and FT-ICR MS configurations showed similar sequence coverage with significant faster data acquisition in the q-ToF MS platform. The histone PTMs were successfully detected in H4 HeLa/HDACi with high sequence coverage (~90%) using top-down UVPD and ECD MS/MS at the protein level and validated using traditional bottom-up histone analysis.
The ion mobility analysis showed that the H4 conformational state varies with the type and number of PTMs. As the number of PTMs increases, a shift towards more compact structures was observed. Complementary mobility separation after the UVPD events permitted the removal of potential isobaric/isomeric interferences prior to the second fragmentation step. Mobility-selected ECD/CID ToF MS/MS spectra of selected UVPD product ions (c305+ with different PTM distribution) permitted a clear H4 proteoform identification, for which the presence of conformers sharing similar PTM distribution (single proteoform) were highlighted. The present results are in agreement the “zip” model, where acetylation proceeds in the Lys16 to Lys5 direction. The higher throughput of the tims-q-ECD ToF MS/MS effectively translates in the possibility to interrogate several UVPD fragments in a single experiment (~12–24 from different proteoforms over a 1–2h spray event). This novel platform further enhances the structural toolbox with alternative fragmentation mechanisms (UVPD, CID and ECD) in tandem with fast, high resolution mobility separations and shows great promises for global histone proteoform analysis.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
The authors acknowledge the financial support from NIH grants 1R01GM134247-01, 5R01AI118891-08 and 1P01CA196539-01; and from the National Science Foundation Division of Chemistry, under CAREER award CHE-1654274 and grant CHE-2127882, with co-funding from the Division of Molecular and Cellular Biosciences to FFL.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Supporting Information contain additional Figures that illustrate the three-dimensional structure of a nucleosome, histone extraction workflow from HeLa cells, LC profiles obtained from the acid-extracted histones of HeLa cells, nESI-MS spectra of H4 standard generated from UVPD-TIMS-q-EMS-ToF MS and FT-ICR MS platforms, H4 sequence map showing the fragments observed using bottom-up experiments in HeLa HDACi, TIMS distributions for H4 standard and H4 HeLa per PTMs, top- “double-down” UVPD-TIMS-q-CID-ToF MS of the c305+ fragment ions, ECD sequence coverage including all acetylation modification possibilities, summary of the top- “double-down” mass spectrometry analysis and table showing the theoretical and measured monoisotopic m/z values for each PTM. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Hansen JC; Lu X; Ross ED; Woody RW Intrinsic protein disorder, amino acid composition, and histone terminal domains. J. Biol. Chem. 2006, 281 (4), 1853–1856. DOI: 10.1074/jbc.R500022200 From NLM. [DOI] [PubMed] [Google Scholar]
- (2).Davey CA; Sargent DF; Luger K; Maeder AW; Richmond TJ Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J. Mol. Biol. 2002, 319 (5), 1097–1113. DOI: 10.1016/s0022-2836(02)00386-8 From NLM. [DOI] [PubMed] [Google Scholar]
- (3).Bowman GD; Poirier MG Post-Translational Modifications of Histones That Influence Nucleosome Dynamics. Chem. Rev. 2015, 115 (6), 2274–2295. DOI: 10.1021/cr500350x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).McGinty RK; Tan S Nucleosome Structure and Function. Chem. Rev. 2015, 115 (6), 2255–2273. DOI: 10.1021/cr500373h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bannister AJ; Kouzarides T Regulation of chromatin by histone modifications. Cell Res. 2011, 21 (3), 381–395. DOI: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Jung HR; Pasini D; Helin K; Jensen ON Quantitative mass spectrometry of histones H3.2 and H3.3 in Suz12-deficient mouse embryonic stem cells reveals distinct, dynamic post-translational modifications at Lys-27 and Lys-36. Mol. Cell. Proteom. 2010, 9 (5), 838–850. DOI: 10.1074/mcp.M900489-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wang Y; Wysocka J; Sayegh J; Lee YH; Perlin JR; Leonelli L; Sonbuchner LS; McDonald CH; Cook RG; Dou Y; Roeder RG; Clarke S; Stallcup MR; Allis CD; Coonrod SA Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 2004, 306 (5694), 279–283. DOI: 10.1126/science.1101400. [DOI] [PubMed] [Google Scholar]
- (8).Turner BM Defining an epigenetic code. Nat. Cell. Biol. 2007, 9 (1), 2–6. DOI: 10.1038/ncb0107-2. [DOI] [PubMed] [Google Scholar]
- (9).Sidoli S; Garcia BA Middle-down proteomics: a still unexploited resource for chromatin biology. Exp. Rev. Proteom. 2017, 14 (7), 617–626. DOI: 10.1080/14789450.2017.1345632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mosammaparast N; Shi Y Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Biochem. 2010, 79, 155–179. DOI: 10.1146/annurev.biochem.78.070907.103946. [DOI] [PubMed] [Google Scholar]
- (11).Tvardovskiy A; Schwammle V; Kempf SJ; Rogowska-Wrzesinska A; Jensen ON Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Res. 2017, 45 (16), 9272–9289. DOI: 10.1093/nar/gkx696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jenuwein T; Allis CD Translating the histone code. Science 2001, 293 (5532), 1074–1080. DOI: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- (13).Wisniewski JR; Zougman A; Mann M Nepsilon-formylation of lysine is a widespread post-translational modification of nuclear proteins occurring at residues involved in regulation of chromatin function. Nucleic Acids Res. 2008, 36 (2), 570–577. DOI: 10.1093/nar/gkm1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kouzarides T Chromatin modifications and their function. Cell 2007, 128 (4), 693–705. DOI: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- (15).van Attikum H; Gasser SM The histone code at DNA breaks: a guide to repair? Nat. Rev. Mol. Cell. Biol. 2005, 6 (10), 757–765. DOI: 10.1038/nrm1737 From NLM. [DOI] [PubMed] [Google Scholar]
- (16).Wolffe AP Histone deacetylase: a regulator of transcription. Science 1996, 272 (5260), 371–372. DOI: 10.1126/science.272.5260.371 From NLM. [DOI] [PubMed] [Google Scholar]
- (17).Kimura H; Sato Y DNA Replication and Histone Modification. In DNA Replication, Recombination, and Repair, 2016; pp 469–488. [Google Scholar]
- (18).Williamson EA; Wray JW; Bansal P; Hromas R Overview for the histone codes for DNA repair. Prog. Mol. Biol. Transl. Sci. 2012, 110, 207–227. DOI: 10.1016/B978-0-12-387665-2.00008-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Seligson DB; Horvath S; Shi T; Yu H; Tze S; Grunstein M; Kurdistani SK Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435 (7046), 1262–1266. DOI: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- (20).Kurdistani SK Histone modifications as markers of cancer prognosis: a cellular view. Br. J. Cancer 2007, 97 (1), 1–5. DOI: 10.1038/sj.bjc.6603844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Audia JE; Campbell RM Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8 (4), a019521. DOI: 10.1101/cshperspect.a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yörüker EE; Holdenrieder S; Gezer U Potential of circulating nucleosome-associated histone modifications in cancer. Transl. Cancer Res. 2018, 7 (S2), S185–S191. DOI: 10.21037/tcr.2017.09.42. [DOI] [Google Scholar]
- (23).Benevento M; Tonge PD; Puri MC; Nagy A; Heck AJ; Munoz J Fluctuations in histone H4 isoforms during cellular reprogramming monitored by middle-down proteomics. Proteomics 2015, 15 (18), 3219–3231. DOI: 10.1002/pmic.201500031. [DOI] [PubMed] [Google Scholar]
- (24).Trelle MB; Jensen ON Functional proteomics in histone research and epigenetics. Exp. Rev. Proteomics 2007, 4 (4), 491–503. DOI: 10.1586/14789450.4.4.491. [DOI] [PubMed] [Google Scholar]
- (25).Sidoli S; Cheng L; Jensen ON Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. J. Proteom. 2012, 75 (12), 3419–3433. DOI: 10.1016/j.jprot.2011.12.029. [DOI] [PubMed] [Google Scholar]
- (26).Lu C; Coradin M; Porter EG; Garcia BA Accelerating the Field of Epigenetic Histone Modification Through Mass Spectrometry-Based Approaches. Mol. Cell. Proteom. 2020, 20, 100006. DOI: 10.1074/mcp.R120.002257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhurov KO; Fornelli L; Wodrich MD; Laskay UA; Tsybin YO Principles of electron capture and transfer dissociation mass spectrometry applied to peptide and protein structure analysis. Chem. Soc. Rev. 2013, 42 (12), 5014–5030. DOI: 10.1039/c3cs35477f. [DOI] [PubMed] [Google Scholar]
- (28).Kim MS; Pandey A Electron transfer dissociation mass spectrometry in proteomics. Proteomics 2012, 12 (4–5), 530–542. DOI: 10.1002/pmic.201100517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Riley NM; Coon JJ The Role of Electron Transfer Dissociation in Modern Proteomics. Anal. Chem. 2018, 90 (1), 40–64. DOI: 10.1021/acs.analchem.7b04810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Shaw JB; Li W; Holden DD; Zhang Y; Griep-Raming J; Fellers RT; Early BP; Thomas PM; Kelleher NL; Brodbelt JS Complete protein characterization using top-down mass spectrometry and ultraviolet photodissociation. J. Am. Chem. Soc. 2013, 135 (34), 12646–12651. DOI: 10.1021/ja4029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Fornelli L; Srzentic K; Toby TK; Doubleday PF; Huguet R; Mullen C; Melani RD; Dos Santos Seckler H; DeHart CJ; Weisbrod CR; Durbin KR; Greer JB; Early BP; Fellers RT; Zabrouskov V; Thomas PM; Compton PD; Kelleher NL Thorough Performance Evaluation of 213 nm Ultraviolet Photodissociation for Top-down Proteomics. Mol. Cell. Proteom. 2020, 19 (2), 405–420. DOI: 10.1074/mcp.TIR119.001638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Fort KL; Dyachenko A; Potel CM; Corradini E; Marino F; Barendregt A; Makarov AA; Scheltema RA; Heck AJ Implementation of Ultraviolet Photodissociation on a Benchtop Q Exactive Mass Spectrometer and Its Application to Phosphoproteomics. Anal. Chem. 2016, 88 (4), 2303–2310. DOI: 10.1021/acs.analchem.5b04162. [DOI] [PubMed] [Google Scholar]
- (33).Cintron-Diaz YL; Gomez-Hernandez ME; Verhaert M; Verhaert P; Fernandez-Lima F Spatially Resolved Neuropeptide Characterization from Neuropathological Formalin-Fixed, Paraffin-Embedded Tissue Sections by a Combination of Imaging MALDI FT-ICR Mass Spectrometry Histochemistry and Liquid Extraction Surface Analysis-Trapped Ion Mobility Spectrometry-Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2022, 33 (4), 681–687. DOI: 10.1021/jasms.1c00376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Brodbelt JS; Morrison LJ; Santos I Ultraviolet Photodissociation Mass Spectrometry for Analysis of Biological Molecules. Chem. Rev. 2020, 120 (7), 3328–3380. DOI: 10.1021/acs.chemrev.9b00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Greer SM; Sidoli S; Coradin M; Schack Jespersen M; Schwammle V; Jensen ON; Garcia BA; Brodbelt JS Extensive Characterization of Heavily Modified Histone Tails by 193 nm Ultraviolet Photodissociation Mass Spectrometry via a Middle-Down Strategy. Anal. Chem. 2018, 90 (17), 10425–10433. DOI: 10.1021/acs.analchem.8b02320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Siuti N; Roth MJ; Mizzen CA; Kelleher NL; Pesavento JJ Gene-specific characterization of human histone H2B by electron capture dissociation. J. Proteome Res. 2006, 5 (2), 233–239. DOI: 10.1021/pr050268v. [DOI] [PubMed] [Google Scholar]
- (37).Boyne MT 2nd; Pesavento JJ; Mizzen CA; Kelleher NL Precise characterization of human histones in the H2A gene family by top down mass spectrometry. J. Proteome Res. 2006, 5 (2), 248–253. DOI: 10.1021/pr050269n. [DOI] [PubMed] [Google Scholar]
- (38).Jung HR; Sidoli S; Haldbo S; Sprenger RR; Schwammle V; Pasini D; Helin K; Jensen ON Precision mapping of coexisting modifications in histone H3 tails from embryonic stem cells by ETD-MS/MS. Anal. Chem. 2013, 85 (17), 8232–8239. DOI: 10.1021/ac401299w. [DOI] [PubMed] [Google Scholar]
- (39).Voinov VG; Deinzer ML; Barofsky DF Electron capture dissociation in a linear radiofrequency-free magnetic cell. Rapid Commun. Mass Spectrom. 2008, 22 (19), 3087–3088. DOI: 10.1002/rcm.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Voinov VG; Beckman JS; Deinzer ML; Barofsky DF Electron-capture dissociation (ECD), collision-induced dissociation (CID) and ECD/CID in a linear radio-frequency-free magnetic cell. Rapid Commun. Mass Spectrom. 2009, 23 (18), 3028–3030. DOI: 10.1002/rcm.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Voinov VG; Deinzer ML; Barofsky DF Radio-frequency-free cell for electron capture dissociation in tandem mass spectrometry. Anal. Chem. 2009, 81 (3), 1238–1243. DOI: 10.1021/ac802084w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Voinov VG; Bennett SE; Beckman JS; Barofsky DF ECD of tyrosine phosphorylation in a triple quadrupole mass spectrometer with a radio-frequency-free electromagnetostatic cell. J. Am. Soc. Mass Spectrom. 2014, 25 (10), 1730–1738. DOI: 10.1007/s13361-014-0956-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Voinov VG; Bennett SE; Barofsky DF Electron-induced dissociation of peptides in a triple quadrupole mass spectrometer retrofitted with an electromagnetostatic cell. J. Am. Soc. Mass Spectrom. 2015, 26 (5), 752–761. DOI: 10.1007/s13361-014-1074-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Voinov VG; Deinzer ML; Beckman JS; Barofsky DF Electron capture, collision-induced, and electron capture-collision induced dissociation in Q-TOF. J. Am. Soc. Mass Spectrom. 2011, 22 (4), 607–611. DOI: 10.1007/s13361-010-0072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Voinov VG; Hoffman PD; Bennett SE; Beckman JS; Barofsky DF Electron Capture Dissociation of Sodium-Adducted Peptides on a Modified Quadrupole/Time-of-Flight Mass Spectrometer. J. Am. Soc. Mass Spectrom. 2015, 26 (12), 2096–2104. DOI: 10.1007/s13361-015-1230-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Fort KL; Cramer CN; Voinov VG; Vasil'ev YV; Lopez NI; Beckman JS; Heck AJR Exploring ECD on a Benchtop Q Exactive Orbitrap Mass Spectrometer. J. Proteome Res. 2018, 17 (2), 926–933. DOI: 10.1021/acs.jproteome.7b00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Shaw JB; Malhan N; Vasil'ev YV; Lopez NI; Makarov A; Beckman JS; Voinov VG Sequencing Grade Tandem Mass Spectrometry for Top-Down Proteomics Using Hybrid Electron Capture Dissociation Methods in a Benchtop Orbitrap Mass Spectrometer. Anal. Chem. 2018, 90 (18), 10819–10827. DOI: 10.1021/acs.analchem.8b01901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Shaw JB; Liu W; Vasil Ev YV; Bracken CC; Malhan N; Guthals A; Beckman JS; Voinov VG Direct Determination of Antibody Chain Pairing by Top-down and Middle-down Mass Spectrometry Using Electron Capture Dissociation and Ultraviolet Photodissociation. Anal. Chem. 2020, 92 (1), 766–773. DOI: 10.1021/acs.analchem.9b03129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zhou M; Liu W; Shaw JB Charge Movement and Structural Changes in the Gas-Phase Unfolding of Multimeric Protein Complexes Captured by Native Top-Down Mass Spectrometry. Anal. Chem. 2020, 92 (2), 1788–1795. DOI: 10.1021/acs.analchem.9b03469. [DOI] [PubMed] [Google Scholar]
- (50).Burnum-Johnson KE; Zheng X; Dodds JN; Ash J; Fourches D; Nicora CD; Wendler JP; Metz TO; Waters KM; Jansson JK; Smith RD; Baker ES Ion Mobility Spectrometry and the Omics: Distinguishing Isomers, Molecular Classes and Contaminant Ions in Complex Samples. Trends Anal. Chem. 2019, 116, 292–299. DOI: 10.1016/j.trac.2019.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Zhong Y; Hyung SJ; Ruotolo BT Ion mobility-mass spectrometry for structural proteomics. Exp. Rev. Proteom. 2012, 9 (1), 47–58. DOI: 10.1586/epr.11.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Shliaha PV; Gorshkov V; Kovalchuk SI; Schwammle V; Baird MA; Shvartsburg AA; Jensen ON Middle-Down Proteomic Analyses with Ion Mobility Separations of Endogenous Isomeric Proteoforms. Anal. Chem. 2020, 92 (3), 2364–2368. DOI: 10.1021/acs.analchem.9b05011. [DOI] [PubMed] [Google Scholar]
- (53).Wei J; Wu J; Tang Y; Ridgeway ME; Park MA; Costello CE; Zaia J; Lin C Characterization and Quantification of Highly Sulfated Glycosaminoglycan Isomers by Gated-Trapped Ion Mobility Spectrometry Negative Electron Transfer Dissociation MS/MS. Anal. Chem. 2019, 91 (4), 2994–3001. DOI: 10.1021/acs.analchem.8b05283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Gadkari VV; Ramirez CR; Vallejo DD; Kurulugama RT; Fjeldsted JC; Ruotolo BT Enhanced Collision Induced Unfolding and Electron Capture Dissociation of Native-like Protein Ions. Anal. Chem. 2020, 92 (23), 15489–15496. DOI: 10.1021/acs.analchem.0c03372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Williams JP; Morrison LJ; Brown JM; Beckman JS; Voinov VG; Lermyte F Top-Down Characterization of Denatured Proteins and Native Protein Complexes Using Electron Capture Dissociation Implemented within a Modified Ion Mobility-Mass Spectrometer. Anal. Chem. 2020, 92 (5), 3674–3681. DOI: 10.1021/acs.analchem.9b04763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Jeanne Dit Fouque K; Kaplan D; Voinov VG; Holck FHV; Jensen ON; Fernandez-Lima F Proteoform Differentiation using Tandem Trapped Ion Mobility, Electron Capture Dissociation, and ToF Mass Spectrometry. Anal. Chem. 2021, 93 (27), 9575–9582. DOI: 10.1021/acs.analchem.1c01735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Jeanne Dit Fouque K; Wellmann M; Leyva Bombuse D; Santos-Fernandez M; Cintron-Diaz YL; Gomez-Hernandez ME; Kaplan D; Voinov VG; Fernandez-Lima F Effective discrimination of gas-phase peptide conformers using TIMS-ECD-ToF MS/MS. Anal. Methods 2021, 13 (43), 5216–5223. DOI: 10.1039/d1ay01461g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Simon AL; Chirot F; Choi CM; Clavier C; Barbaire M; Maurelli J; Dagany X; MacAleese L; Dugourd P Tandem ion mobility spectrometry coupled to laser excitation. Rev. Sci. Instrum. 2015, 86 (9), 094101. DOI: 10.1063/1.4930604. [DOI] [PubMed] [Google Scholar]
- (59).Theisen A; Black R; Corinti D; Brown JM; Bellina B; Barran PE Initial Protein Unfolding Events in Ubiquitin, Cytochrome c and Myoglobin Are Revealed with the Use of 213 nm UVPD Coupled to IM-MS. J. Am. Soc. Mass Spectrom. 2019, 30 (1), 24–33. DOI: 10.1007/s13361-018-1992-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Liu FC; Ridgeway ME; Winfred J; Polfer NC; Lee J; Theisen A; Wootton CA; Park MA; Bleiholder C Tandem-trapped ion mobility spectrometry/mass spectrometry coupled with ultraviolet photodissociation. Rapid Commun. Mass Spectrom. 2021, 35 (22), e9192. DOI: 10.1002/rcm.9192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Miller SA; Jeanne Dit Fouque K; Ridgeway ME; Park MA; Fernandez-Lima F Trapped Ion Mobility Spectrometry, Ultraviolet Photodissociation, and Time-of-Flight Mass Spectrometry for Gas-Phase Peptide Isobars/Isomers/Conformers Discrimination. J. Am. Soc. Mass Spectrom. 2022, 33 (7), 1267–1275. DOI: 10.1021/jasms.2c00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Sidoli S; Bhanu NV; Karch KR; Wang X; Garcia BA Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis. J. Vis. Exp. 2016, (111). DOI: 10.3791/54112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Bhanu NV; Sidoli S; Garcia BA A Workflow for Ultra-rapid Analysis of Histone Post-translational Modifications with Direct-injection Mass Spectrometry. Bio. Protoc. 2020, 10 (18), e3756. DOI: 10.21769/BioProtoc.3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Marty MT; Baldwin AJ; Marklund EG; Hochberg GK; Benesch JL; Robinson CV Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal. Chem. 2015, 87 (8), 4370–4376. DOI: 10.1021/acs.analchem.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Hernandez DR; Debord JD; Ridgeway ME; Kaplan DA; Park MA; Fernandez-Lima F Ion dynamics in a trapped ion mobility spectrometer. Analyst 2014, 139 (8), 1913–1921. DOI: 10.1039/c3an02174b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Ridgeway ME; Lubeck M; Jordens J; Mann M; Park MA Trapped ion mobility spectrometry: A short review. Int. J. Mass Spectrom. 2018, 425, 22–35. DOI: 10.1016/j.ijms.2018.01.006. [DOI] [Google Scholar]
- (67).Michelmann K; Silveira JA; Ridgeway ME; Park MA Fundamentals of trapped ion mobility spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26 (1), 14–24. DOI: 10.1007/s13361-014-0999-4. [DOI] [PubMed] [Google Scholar]
- (68).Silveira JA; Michelmann K; Ridgeway ME; Park MA Fundamentals of Trapped Ion Mobility Spectrometry Part II: Fluid Dynamics. J. Am. Soc. Mass Spectrom. 2016, 27 (4), 585–595. DOI: 10.1007/s13361-015-1310-z. [DOI] [PubMed] [Google Scholar]
- (69).Yuan ZF; Sidoli S; Marchione DM; Simithy J; Janssen KA; Szurgot MR; Garcia BA EpiProfile 2.0: A Computational Platform for Processing Epi-Proteomics Mass Spectrometry Data. J. Proteome Res. 2018, 17 (7), 2533–2541. DOI: 10.1021/acs.jproteome.8b00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Kim B; Hong J An overview of naturally occurring histone deacetylase inhibitors. Curr. Top. Med. Chem. 2015, 14 (24), 2759–2782. DOI: 10.2174/1568026615666141208105614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Davie JR Inhibition of histone deacetylase activity by butyrate. J. Nutr. 2003, 133 (7 Suppl), 2485S–2493S. DOI: 10.1093/jn/133.7.2485S. [DOI] [PubMed] [Google Scholar]
- (72).Zhang K; Williams KE; Huang L; Yau P; Siino JS; Bradbury EM; Jones PR; Minch MJ; Burlingame AL Histone acetylation and deacetylation: identification of acetylation and methylation sites of HeLa histone H4 by mass spectrometry. Mol. Cell. Proteom. 2002, 1 (7), 500–508. DOI: 10.1074/mcp.m200031-mcp200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.