

Abstract
The influenza virus PA endonuclease, which cleaves capped cellular pre-mRNAs to prime viral mRNA synthesis, is a promising target for novel anti–influenza virus therapeutics. The catalytic center of this enzyme resides in the N-terminal part of PA (PA-Nter) and contains two (or possibly one or three) Mg2+ or Mn2+ ions, which are critical for its catalytic function. There is great interest in PA inhibitors that are optimally designed to occupy the active site and chelate the metal ions. We focused here on a series of β-diketo acid (DKA) and DKA-bioisosteric compounds containing different scaffolds, and determined their structure-activity relationship in an enzymatic assay with PA-Nter, in order to build a three-dimensional pharmacophore model. In addition, we developed a molecular beacon (MB)–based PA-Nter assay that enabled us to compare the inhibition of Mn2+ versus Mg2+, the latter probably being the biologically relevant cofactor. This real-time MB assay allowed us to measure the enzyme kinetics of PA-Nter or perform high-throughput screening. Several DKA derivatives were found to cause strong inhibition of PA-Nter, with IC50 values comparable to that of the prototype L-742,001 (i.e., below 2 μM). Among the different compounds tested, L-742,001 appeared unique in having equal activity against either Mg2+ or Mn2+. Three compounds (10, with a pyrrole scaffold, and 40 and 41, with an indole scaffold) exhibited moderate antiviral activity in cell culture (EC99 values 64–95 μM) and were proven to affect viral RNA synthesis. Our approach of integrating complementary enzymatic, cellular, and mechanistic assays should guide ongoing development of improved influenza virus PA inhibitors.
Introduction
Influenza viruses cause significant morbidity and mortality and vaccination provides only partial protection in some populations, such as the elderly (Treanor et al., 2012). Antiviral therapy comprises the M2 blockers amantadine and rimantadine and the neuraminidase inhibitors oseltamivir and zanamivir (Vanderlinden and Naesens, 2014). The global spread of amantadine-resistant influenza viruses and growing resistance to neuraminidase inhibitors (in particular oseltamivir) underline the need for novel therapeutics with a different mode of action.
An attractive target is the influenza virus RNA polymerase complex (Ruigrok et al., 2010) composed of three proteins: PA, PB1, and PB2. The RNA-polymerizing activity resides in the PB1 subunit. PB2 and PA carry out the first step of the viral transcription process, referred to as “cap-snatching.” This reaction appears unique for Orthomyxo-, Arena- and Bunyaviridae (Morin et al., 2010; Reguera et al., 2010; Guilligay et al., 2014). In the case of influenza virus, the capped 5′ end of a cellular pre-mRNA is bound by PB2 and subsequently cleaved by PA at 10–13 nucleotides from the cap to serve as primer for viral mRNA synthesis (Bouloy et al., 1978; Plotch et al., 1981; Dias et al., 2009; Yuan et al., 2009). The catalytic site of the PA endonuclease, residing in its N-terminal part (PA-Nter; residues 1–256 or less), is highly conserved in influenza A and B viruses. Several crystallographic studies have revealed the protein structure of PA-Nter, which contains two (or possibly one or three) Mn2+ or Mg2+ ions in its catalytic core, although it is still unresolved which metal ions are present in native enzyme (Dias et al., 2009; Yuan et al., 2009; Zhao et al., 2009; DuBois et al., 2012; Kowalinski et al., 2012; Parhi et al., 2013). In biochemical assays, the enzyme’s activity appears higher with manganese, but magnesium appears more biologically relevant since the intracellular concentration of free Mg2+ is >1000-fold higher than that of Mn2+ (Maret, 2010). For unrelated nucleases, reduced substrate specificity was observed when tested with manganese instead of magnesium. This is probably attributable to the less rigid coordination requirements of manganese compared with magnesium (Yang et al., 2006).
The available crystal structures of PA-Nter enable rational design of PA inhibitors (PAIs). The first class of influenza endonuclease inhibitors (Tomassini et al., 1994; Hastings et al., 1996) are 4-substituted-2,4-dioxobutanoic acids with a characteristic β-diketo acid (DKA) motif. Among this series, L-742,001 (1 in Table 1) was identified as a particularly potent inhibitor, both in enzyme- and cell-based antiviral assays (Hastings et al., 1996; Nakazawa et al., 2008). Several other classes of endonuclease inhibitors have been reported (Cianci et al., 1996; Tomassini et al., 1996; Singh and Tomassini, 2001; Iwai et al., 2010; Baughman et al., 2012; Sagong et al., 2013; Chen et al., 2014). All of these compounds bear chelating motifs able to bind the bivalent metal ions in the catalytic core of PA-Nter (Rogolino et al., 2012). A similar concept has led to clinical development of HIV integrase (HIV IN) inhibitors, and is also explored to design inhibitors of HIV RNase H or HCV polymerase.
TABLE 1.
Inhibition of PA-Nter by different DKA or DKA-bioisosteric compounds as determined in the plasmid-based enzymatic assay
One microgram of PA-Nter was incubated with 1 μg of M13mp18 plasmid substrate and the compounds. After 2 hours incubation, cleavage was assessed by gel electrophoresis.
| Scaffold | Compound | R 1 | R 2 | IC50a |
|---|---|---|---|---|
| μM | ||||
Piperidine scaffold 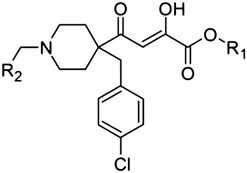
|
1 (L-742,001) | H | Ph | 0.5 |
| 2 | Me | Ph | 0.5 | |
| 3 | H | p-F-Ph | 1.4 | |
| 4 | Me | p-F-Ph | 0.4 | |
| 5 | H | Cy | 0.5 | |
| 6 | Me | Cy | 0.6 | |
Dihydroxybutenedioic acid 
|
7 | — | — | >500 |
1,3-Dioxo-1-phenylpropane scaffold and triazole bioisostere 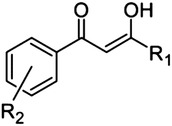
|
8 |
|
H | 77 |
| 9 | Ph | o-OH | >500 | |
Pyrrole scaffold 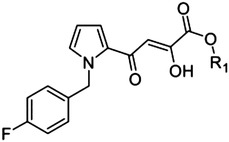
|
10 (L-731,988) | H | — | 0.8 |
| 11 | Me | — | 5.5 | |
2,4-Dioxo-4-phenylbutanoic acid (DPBA) scaffold 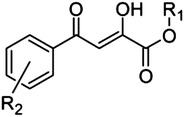
|
12 | H | H | 2.7 |
| 13 | Me | H | 14 | |
| 14 | H | o-OMe | 2.2 | |
| 15 | Me | o-OMe | 11 | |
| 16 | H | p-OMe | 2.0 | |
| 17 | H | p-Cl | 9.8 | |
| 18 | H | o-Cl | 9.8 | |
| 19 | Me | o-Cl | 15 | |
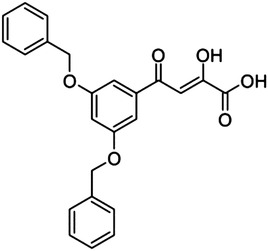
|
20 (L-708,906) | — | — | 137 |
2,4,6-Trioxo-6-phenylhexanoic acid scaffold 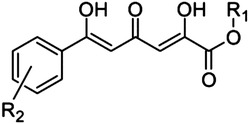
|
21 | H | H | >500 |
| 22 | Me | H | >500 | |
| 23 | H | o-OMe | >500 | |
| 24 | H | o-Cl | >500 | |
Quinolone scaffold 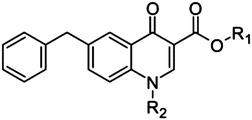
|
25 | H | H | >500 |
| 26 | H | CH2CH2OH | >500 | |
| 27 | H | Bn | >500 | |
| 28 | Et | Bn | >500 | |
Hydroxypyrimidine- carboxamide scaffold 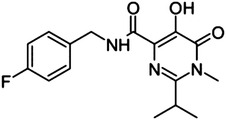
|
29 | — | — | >500 |
IC50, 50% inhibitory concentration. For each compound the IC50 value was calculated using nonlinear regression analysis. Values are the mean of at least three independent experiments.
In this research, we investigated a series of DKAs (and DKA-bioisosteric compounds) incorporating different chemical scaffolds that were synthesized as specific PAIs (1–6, Table 1) or were originally designed toward HIV IN (7–47, Tables 1 and 2) (Maurin et al., 2004; Sechi et al., 2004, 2005, 2006; Bacchi et al., 2008, 2011; Zeng et al., 2008; Reddy et al., 2011). Although these compounds contain relevant scaffolds for PAI development, information on their activity against PA-Nter is lacking. Hence, their structure-activity relationship toward PA-Nter was analyzed to build a plausible pharmacophore model. A selection of compounds was also evaluated against untruncated PA.
TABLE 2.
Inhibition of PA-Nter by different DKA or DKA-bioisosteric indole derivatives as determined in the plasmid-based enzymatic assay
One microgram of PA-Nter was incubated with 1 μg of M13mp18 plasmid substrate and the compounds. After 2 hours incubation, cleavage was assessed by gel electrophoresis.
| Indole scaffold | Compound | R 1 | R 2 | IC50 |
|---|---|---|---|---|
| μM | ||||
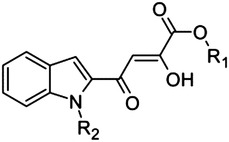
|
30 | H | Me | 3.4 |
| 31 | H | Bn | 22 | |
| 32 | Me | Me | 10 | |
| 33 | Me | Et | 4.1 | |
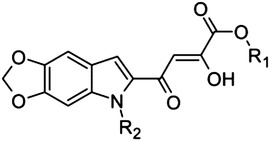
|
34 | H | Me | 0.6 |
| 35 | H | Et | 5.5 | |
| 36 | H | Bn | 8.9 | |
| 37 | Me | Bn | 204 | |
| 38 | Me | Me | 21 | |

|
39 | H | Me | 0.6 |
| 40 | H | Et | 0.8 | |
| 41 | H | Bn | 1.6 | |
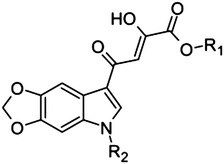
|
42 | H | Me | 3.5 |
| 43 | Me | Me | 22 | |
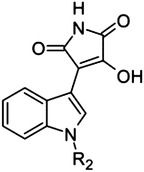
|
44 | — | Et | >500 |
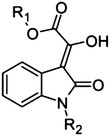
|
45 | H | H | >500 |
| 46 | Me | H | >500 | |
| 47 | Me | Ph | >500 |
In the case of HIV IN, the potency of DKAs in enzymatic assays was found to depend on which bivalent metal ion (Mg2+ or Mn2+) was used (Marchand et al., 2003). This prompted us to investigate whether a similar metal ion dependency may occur when evaluating PAIs in an enzymatic assay, such as the novel molecular beacon (MB) assay presented here. This real-time MB method, applicable for high-throughput screening, also enabled comparison of the kinetic parameters of PA-Nter for different substrates or metal ions.
To validate the inhibition data for PA-Nter, we used two cell culture methods: an influenza virus ribonucleoprotein (vRNP) reconstitution assay and a virus yield replication assay. A basic mechanistic study was also performed to verify that the antivirally active DKAs indeed act upon viral RNA synthesis.
Materials and Methods
Compounds.
The synthetic route to obtain 1–6 (i.e., L-742,001 and derivatives) can be found in the Supplemental Data and Supplemental Methods. Compound 7 was obtained from Sigma-Aldrich (St. Louis, MO). The procedures for chemical synthesis of 8–11 and 30–47 were reported elsewhere (Tomassini et al., 1994; Sechi et al., 2004, 2005, 2006; Maurin et al., 2004; Zeng et al., 2008; Bacchi et al., 2008, 2011; Reddy et al., 2011). Ribavirin (Virazole; ICN Pharmaceuticals, Costa Mesa, CA) and chloroquine diphosphate salt (Sigma-Aldrich) were included as reference compounds. The test compounds were stored as a 20–100 mM stock solution in dimethyl sulfoxide. In all cellular assays, the final dimethyl sulfoxide concentration was kept below 0.8%, a concentration that was free of aspecific effects on cell viability or luciferase activity.
Pharmacophore Building.
Twelve of the more active compounds (1–6, 10, 16, 34, 39–41) were chosen for generating the pharmacophore model. For each compound a full minimized model was built in Molecular Operating Environment software package (MOE, version 2010.11; Chemical Computing Group Inc., Montreal, QC, Canada) using MMFF94x force field with standard bond length and angles. The obtained database was used as starting point for the pharmacophore elucidation procedure implemented in MOE. The selected search scheme was PPCH_All (Planar-Polarity-Charge-Hydrophobicity), and all settings were kept as default with the exception of conformations (set to stochastic search). Among the proposed pharmacophore queries the best one was chosen on the basis of statistical parameters, i.e., high correlation coefficient and lower root-mean-square deviation value.
Production of Recombinant Influenza Virus PA-Nter Protein.
The coding sequence for PA-Nter (i.e., residues 1–217 from the PA protein of influenza virus strain A/X-31) was cloned in the pET28a(+) plasmid (Merck KGaA, Darmstadt, Germany) with an N-terminal 6xHis-tag, and this bacterial expression plasmid was transformed into Escherichia coli BL21-CodonPlus cells (Agilent Technologies, Santa Clara, CA). The plasmid was subjected to site-directed mutagenesis (QuikChange II; Agilent Technologies) to prepare the K134A-substituted PA-Nter enzyme.
The bacteria were grown to an optical density of 0.6, when isopropylthio-β-galactoside was added at a final concentration of 1 mM to induce expression of recombinant proteins for 5 hours at 37°C. The bacterial cells were ruptured using a French press, and the proteins were purified by 6xHis-Ni-NTA chromatography (Qiagen, Valencia, CA), followed by buffer exchange using PD-10 desalting columns (GE Healthcare, Diegem, Belgium) to keep the proteins in storage buffer (50 mM Tris-HCl pH 8, 100 mM NaCl, 10 mM β-mercaptoethanol, 50% glycerol). Protein purity was verified by SDS-PAGE with Coomassie Blue staining, and protein concentration was determined by Bradford assay. Finally, the purified proteins were divided in aliquots and stored at –80°C.
Plasmid-Based Endonuclease Assay.
This enzymatic endonuclease activity assay was performed according to a previously published method (Carcelli et al., 2014) with minor modifications. One microgram of recombinant PA-Nter was incubated with 1 μg (16.7 nM) of single-stranded circular DNA plasmid M13mp18 (Bayou Biolabs, Metairie, Louisiana) in the presence of the test compounds and at a final volume of 25 μl. The assay buffer contained 50 mM Tris-HCl pH 8, 100 mM NaCl, 10 mM β-mercaptoethanol, and 1 mM MnCl2 (unless stated otherwise). The reaction was incubated at 37°C for 2 hours and then stopped by heat inactivation (80°C, 20 minutes). The endonucleolytic digestion of the plasmid was visualized by gel electrophoresis on a 1% agarose gel with ethidium bromide staining. The amount of remaining intact plasmid was quantified by ImageQuant TL software (GE Healthcare). The percentage inhibition of PA endonuclease activity was plotted against the compound concentration on a semilogarithmic plot, using GraphPad Prism software (GraphPad Software, La Jolla, CA). Values were the mean ± S.E.M. of three independent experiments. The 50% inhibitory concentrations (IC50) were obtained by nonlinear least-squares regression analysis.
Molecular Beacon-Based Endonuclease Assay.
The sequences of the eight MB substrates are given in Table 3. MB-C15-DFO (Dragonfly Orange), MB-A15-DFO, MB-U6A2U7-DFO, MB-OH-DFO, and MB-het2-DFO were purchased from Eurogentec (Seraing, Belgium), whereas MB-het1-FAM, MB-het2-FAM, and MB-het3-FAM were synthesized by S.A.E. Marras. The molecular beacons were prepared by automated solid-phase DNA synthesis on an Applied Biosystems (Foster City, CA) ABI 394 DNA/RNA synthesizer. A controlled-pore glass column (Biosearch Technologies, Novato, CA) was used to incorporate a quencher moiety (dabcyl) at the 3′, end and a fluorescein phosphoramidite (Glen Research, Sterling, VA) was used to incorporate a fluorophore moiety at the 5′ end of the oligodeoxyribonucleotides. The molecular beacons were then purified by high-pressure liquid chromatography on a Beckman Coulter System Goldchromatograph (Indianapolis, IN) through a C-18 reverse-phase column (Waters Corporation, Milford, MA). Concentrations were determined on a Nanodrop 1000 Spectrophotometer (Thermo Scientific, Wilmington, DE).
TABLE 3.
Endonucleolytic cleavage by PA-Nter of different MB substrates
| MB | Sequence | Kma ± S.E.M. | Signal-to-noise ratiob ± S.E.M. |
|---|---|---|---|
| μM | |||
| MB-C15-DFO | 5′‐[DFO]‐GCAGG CCCCCCCCCCCCCCC CCTGC‐[BHQ-2]‐3′ | 0.09 ± 0.004 | 8.2 ± 1.3 |
| MB-A15-DFO | 5′‐[DFO]‐GCAGG AAAAAAAAAAAAAAA CCTGC‐[BHQ-2]‐3′ | 0.36 ± 0.01 | 9.4 ± 2.3 |
| MB-U6A2U7-DFO | 5′‐[DFO]‐GCAGG dUdUdUdUdUdUAAdUdUdUdUdUdUdU CCTGC‐[BHQ-2]‐3′ | 0.23 ± 0.02 | 11.7 ± 0.6 |
| MB-het1-FAM | 5′-[FAM]-GCGAGC TAGGAAACACCAAAGATGATATTT GCTCGC-[DABCYL]-3′ | 0.64 ± 0.03 | 4.4 ± 0.3 |
| MB-het2-FAM | 5′-[FAM]-CGCACG TTATGCTAAGCAAGTAAC CGTGCG-[DABCYL]-3′ | 0.15 ± 0.01 | 5.5 ± 0.4 |
| MB-het3-FAM | 5′-[FAM]-CGAGCG GCTGGTAAGGGTTTCCATATAA CGCTCG-[DABCYL]-3′ | 0.46 ± 0.06 | 7.0 ± 0.6 |
| MB-het2-DFO | 5′-[DFO]‐CGCACG TTATGCTAAGCAAGTAAC CGTGCG‐[BHQ-2]‐3′ | 0.46 ± 0.01 | 5.7 ± 0.8 |
| MB-OH-DFO | 5′‐[DFO]‐dUdUdUdUdU GGGGGGGGGGGGGGCCCCCCCCCCCCCC‐[BHQ2]‐3′ | 0.22 ± 0.01 | 1.8 ± 0.1 |
BHQ2, Black Hole Quencher 2 dye (Biosearch Technologies).
For each MB, the Michaelis–Menten equation was fitted to the initial reaction rate to estimate the Km values by nonlinear regression analysis. Values shown are the averages ± S.E.M. of at least two independent experiments.
Fold increase at 60 minutes using 100 nM MB.
The PA-Nter assay was performed at 37°C in a buffer consisting of 50 mM Tris-HCl pH 8, 100 mM NaCl, and 10 mM β-mercaptoethanol, to which either 1 mM MnCl2, 1 mM MgCl2, or no metal was added.
To determine the kinetic parameters for the different MB substrates, the substrate concentrations were varied from 25 nM to 5 μM, whereas the enzyme concentration was kept constant at 0.04 μg per microliter. These tests were performed in the presence of 1 mM MnCl2 and in 10-μl reaction volumes in a 384-well plate format. MB-het1-FAM, MB-het2-FAM, and MB-het3-FAM (FAM-labeled) were excited at 495 nm and emission was recorded at 520 nm, whereas the DFO-labeled MB molecules MB-C15-DFO, MB-A15-DFO, MB-U6A2U7-DFO, MB-OH-DFO, and MB-het2-DFO were read at 554-nm excitation and 576-nm emission wavelength. The increasing fluorescence signal was measured every 14 seconds for 60 minutes using a spectrofluorometer (Safire2; Tecan, Männedorf, Switzerland). All reactions were performed in duplicate. The initial reaction velocity V0 (determined at concentrations at which no substrate inhibition occurred) was calculated as the slope for the 5- to 15-minute time interval. To estimate the Km values, the Michaelis-Menten equation was fitted to these V0 data, on the basis of nonlinear regression analysis and using GraphPad Prism software.
Similar conditions were used for the inhibition assays. Serial dilutions of the test compounds were added to the reaction mixture containing buffer (with or without added metal ions) and 20 or 100 nM MB, and the reaction was started by adding 0.4 μg of PA-Nter. All reactions were performed in duplicate. The initial cleavage velocity was plotted against the compound concentration on a semilogarithmic scale, and for each compound the IC50 value was calculated using nonlinear regression analysis (GraphPad Prism). Values were the mean of at least two independent experiments.
The cleavage pattern of the molecular beacons was studied in the same buffer conditions, but the reaction was stopped at the indicated time points by heat inactivation (80°C, 20 minutes). The samples were loaded on 15% Tris/borate/EDTA–urea polyacrylamide gels (Criterion; Bio-Rad, Hercules, CA) for denaturing nucleic acid PAGE. The bands were visualized by fluorescent gel imaging using an Ettan difference gel electrophoresis imager apparatus (GE Healthcare).
Cells and Media.
Madin-Darby canine kidney (MDCK) cells (a kind gift from M. Matrosovich, Marburg, Germany) and human embryonic kidney 293T (HEK293T) cells (purchased from Thermo Fisher Scientific, Waltham, MA) were grown in Dulbecco’s modified Eagle medium supplemented with 10% fetal calf serum, 1 mM sodium pyruvate, and 0.075% sodium bicarbonate. Virus experiments were performed in MDCK infection medium, consisting of Ultra MDCK medium (Lonza, Basel, Switzerland) supplemented with 0.0225% sodium bicarbonate, 2 mM l-glutamine, and 2 μg/ml tosylphenylalanylchloromethylketone-treated trypsin (Sigma-Aldrich). The cells were incubated in a 5% CO2 humidified atmosphere.
vRNP Reconstitution Assay.
The assay to determine the inhibitory effect of the compounds on reconstituted influenza virus vRNPs is described in more detail elsewhere (Meneghesso et al., 2012; Stevaert et al., 2013). Four sets of vRNP reconstituting plasmids derived from different influenza virus (sub)types were used: 1) four reverse genetics plasmids derived from influenza A/PR/8/34 (encoded pVP-PB1, pVP-PB2, pVP-PA, and pVP-NP, which contain the cDNAs in the bidirectional expression cassette of pHH21), kindly given by M. Kim (Korea Research Institute of Chemical Technology, Daejeon) (Kim et al., 2013); 2) four plasmids derived from the avian influenza A/turkey/England/50-92/1991 (H5N1) virus, containing the cDNA sequences in a pol-II–driven expression cassette, generously given by W. Barclay (Imperial college London, United Kingdom) (Moncorgé et al., 2010; in this set, the PB2 protein contains the avian influenza Glu627 residue); 3) identical to the previous set, with the exception that the PB2-expressing plasmid was mutated to encode the human influenza Lys627 residue (Moncorgé et al., 2010); 4) four reverse genetics plasmids derived from influenza B/Yamanashi/166/98 [designated pAB251-PB1, pAB252-PB2, pAB253-PA, and pAB255-NP, which contain the cDNAs in a bidirectional expression cassette, generously donated by J. McCullers (St. Jude Children’s Research Hospital, Memphis, TN) (Hoffmann et al., 2002)]. Set 1 was used in combination with an influenza A–specific firefly luciferase (fluc) reporter plasmid (Kim et al., 2013) that was also given by M. Kim. For sets 2 and 3, the fluc reporter was a kind gift from W. Barclay. For set 4, an influenza B–specific fluc reporter plasmid was used, kindly donated by K. Nagata, University of Tsukuba, Tsukuba, Ibaraki, Japan (Wakai et al., 2011).
Briefly, the four relevant plasmids (i.e., the expression plasmids for PB1, PB2, PA and NP) were combined with the corresponding fluc reporter plasmid, and cotransfected into HEK293T cells using Lipofectamine 2000 (Meneghesso et al., 2012; Stevaert et al., 2013). The cells were transferred to a 96-well plate containing serial dilutions of the test compounds, and after 24 hours incubation at 37°C, luciferase activity was determined using the ONE-Glo assay system from Promega (Madison, WI). The 50% effective concentration (EC50) was defined as the compound concentration causing 50% reduction in the vRNP-driven firefly luciferase signal, compared with cells receiving medium instead of compound. These EC50 values were calculated by interpolation assuming a semi-log dose-response effect. In parallel, compound cytotoxic activity was determined in untransfected HEK293T cells that had been incubated with serial dilutions of the compounds for 24 hours, using the MTS cell viability assay (CellTiter 96 AQueous One Solution Cell Proliferation Assay; Promega). These spectrophotometric data were used to calculate the 50% cytotoxic concentration (CC50), i.e., the concentration reducing cell viability by 50%, compared with the wells receiving medium instead of compound. Ribavirin was included as the reference compound.
Virus Yield Assay.
To determine anti–influenza virus activity in infected cell cultures, we performed a virus yield assay (Stevaert et al., 2013). One day prior to infection, MDCK cells were seeded into 96-well plates at 25,000 cells per well. At day 0, serial dilutions of the test compounds were added, immediately followed by infection with influenza A/PR/8/34 virus. The multiplicity of infection was 150 CCID50 per well [50% cell culture infectious dose, determined by the method of Reed and Muench]. After 24 hours incubation at 35°C, the supernatants were collected and stored at –80°C. The virus amount in these samples was estimated by determining the viral genome copy number in a one-step quantitative real-time reverse transcription (RT-PCR) assay (CellsDirect One-Step qRT-PCR kit; Invitrogen/Life Technologies, Gent, Belgium), with influenza virus M1–specific primers and probe [see Vanderlinden et al. (2010) for all details]. Absolute quantification of viral RNA (vRNA) copies was performed by including an M1-plasmid standard. The EC99 and EC90 values were calculated by interpolation from data of at least three experiments and defined as the compound concentration causing, respectively, a 2-log10 and 1-log10 reduction in vRNA copy number, compared with the virus control receiving no compound. In parallel, the CC50 values after 24 hours incubation with compounds were determined in uninfected MDCK cells, using the MTS cell viability assay (CellTiter 96 AQueous One Solution Cell Proliferation Assay; Promega). Ribavirin was included as the reference compound.
One-Cycle Virus Replication Assay.
A simplified time-of-addition experiment was performed as previously described in more detail (Stevaert et al., 2013). The compounds were added to confluent MDCK cells at either 30 minutes before or 1 hour after virus infection with influenza virus A/PR/8/34, and incubated at 35°C. At 8 hours postinfection, the supernatant was removed and total cellular RNA extracts were prepared with the RNeasy Mini Kit (Qiagen). To quantify the negative-sense vRNA, the samples were analyzed by two-step real-time RT-PCR (Vanderlinden et al., 2010). cDNA synthesis was performed on 0.5 μg of total cellular RNA using Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen/Life Technologies) and 80 nM M1-FOR primer. Real-time PCR was then performed, using influenza virus M1-specific primers and probe (Vanderlinden et al., 2010), and quantitative PCR MasterMix (Eurogentec). All samples were analyzed in duplicate. The vRNA copy number was quantified by including an M1-plasmid standard, and the fold increase in vRNA copies was calculated relative to the viral copy number added at time zero. Chloroquine was included as the reference compound.
Results
Selection of DKAs and Derivatives.
Tables 1 and 2 show the chemical structures of the different series of DKA derivatives that we evaluated for inhibitory activity against PA-Nter. In the known PAI, L-742,001 (1), included as the reference, the β-diketo acid motif is attached to a piperidine moiety, which carries two cyclic substituents. These two “wings” occupy opposite hydrophobic pockets around the catalytic center of PA-Nter and are considered crucial for achieving strong binding to PA-Nter (DuBois et al., 2012; Stevaert et al., 2013). To acquire more information about the structural features of this prototype, we prepared some closely related analogs (3 and 5), N-substituted at the piperidine ring, and also considered the diketo methylesters (2, 4, and 6).
Moreover, on the basis of the assumption that the DKA moiety is essential for anti-PA activity, our aim was to explore this structural motif and the related chemical space. First, we selected a series of DKA analogs carrying an indole scaffold (30–43; Table 2) in which major modifications were made to determine the effect of different substituents on activity. Second, we included other series of compounds where: 1) the β-diketo acid group was been replaced with a bioisostere (44–47); 2) the aromatic scaffold had been replaced with a second DKA functionality (7); 3) a triazole bioisostere has been incorporated in place of the carboxyl group (8); 4) an additional phenyl group had been introduced on the 1,3-dioxo-phenylpropane framework (9); and 5) the DKA motif had been connected to a different heterocycle (10, 11). Third, since 2,4-dioxo-4-phenylbutanoic acid (DPBA, 12; Table 1) represents a suitable platform to examine structural and biologic features of DKAs, several substitutions on the phenyl (13–20), as well as on the diketo moiety with an additional keto group (21–24), were evaluated.
Finally, inspired by studies on HIV IN (Bacchi et al., 2011), we considered two additional chemotypes as putative DKA-related compounds: the 4-quinolone-3-carboxylic acids 25–27 (and ethyl ester 28), which bear a monoketo acid motif, and the bioisosteric compound 29, where the DKA moiety was replaced by a 4,5-dihydroxypyrimidine-4-carboxamide scaffold.
Activity in the Plasmid-Based PA Endonuclease Assay.
We used a PA endonuclease assay based on end-point gel electrophoretic analysis of substrate cleavage (Fig. 1). Among the various PA-Nter substrates that were reported to work (Dias et al., 2009; DuBois et al., 2012; Kowalinski et al., 2012; Parhi et al., 2013; Bauman et al., 2013; Datta et al., 2013; Chen et al., 2014), we chose the M13mp18 single-stranded DNA plasmid since it is commercially available. The active site substitution K134A in the PA-Nter enzyme completely abolished the endonucleolytic cleavage, in agreement with previous reports (Hara et al., 2006; Yuan et al., 2009; Crépin et al., 2010).
Fig. 1.

Illustration of the activity of DKAs in the plasmid-based PA-Nter assay. Recombinant PA-Nter (1 μg) was incubated with 1 μg (16.7 nM) of single-stranded circular DNA plasmid M13mp18 in the presence of the test compounds. The endonucleolytic digestion of the plasmid was visualized by gel electrophoresis, and the amount of remaining intact plasmid was quantified. For L-742,001 and 10 (L-731,988), the IC50 values were 0.5 μM and 0.8 μM, respectively. Wild-type (WT) enzyme showed complete cleavage with Mn2+ but was inactive when Mn2+ was omitted from the reaction mixture or replaced by Mg2+. The endonuclease activity was also completely abolished by the active site substitution K134A.
At the time of its discovery (Hastings et al., 1996) L-742,001 (1) was reported to have an IC50 value of 0.43 μM in an enzymatic endonuclease assay with a capped mRNA substrate and influenza virus ribonucleoprotein complexes, which contain the entire PA protein within the heterotrimeric viral polymerase. We here obtained the same IC50 value (i.e., 0.5 μM; Table 1) against isolated PA-Nter. This supports the view that the endonuclease catalytic site has the same structure in isolated PA-Nter as in native polymerase, and implicates that enzymatic assays with PA-Nter are relevant for developing catalytic site inhibitors (Crépin et al., 2010; Noble et al., 2012). Regarding the direct analogs of L-742,001 that we tested (Table 1), the IC50 data demonstrate that the activity is unchanged when the phenyl moiety of L-742,001 is replaced by a cyclohexyl group (5), and only slightly higher when this phenyl carries a p-fluoro substituent (3). Also, methyl-esterification of the terminal carboxyl in the DKA part did not affect the inhibitory activity (2, 4, and 6).
A prototypic inhibitor that has been used in several enzymatic studies with PA-Nter is 2,4-dioxo-4-phenylbutanoic acid (DPBA, 12). The IC50 value for DPBA in our plasmid assay was 2.7 μM, which is comparable to the data reported by others (Dias et al., 2009; Kowalinski et al., 2012; Noble et al., 2012). This value was unchanged upon addition of a methoxy in ortho (14) or para position (16). Addition of an ortho-chlorine had no effect (18), but a chlorine in para position (17) reduced the potency by a factor of 4. We also included three methyl esters (13, 15, and 19); their IC50 values were consistently 4- to 5-fold higher than the corresponding carboxylic acids. Also, replacement of the carboxylic functionality with a triazole ring bioisostere (8) or a phenyl moiety (9) caused a dramatic reduction in inhibitory activity. The length of the DKA arm seems optimal in DPBA, since analogs carrying an extra keto group (21–24) were devoid of activity. The critical role for the phenyl ring of DPBA is evident from the fact that unsubstituted dihydroxybutenedioic acid (7) was inactive. Introduction of two large aromatic substituents (benzyloxygroups) onto this phenyl ring [20; known as the HIV IN inhibitor L-708,906 (Hazuda et al., 2000)] led to a dramatic (50-fold) reduction in activity.
The same DKA fragment as in DPBA and L-742,001 is present in the extended series of indole derivatives that we tested (Table 2). Several of these had strong activity against PA-Nter with IC50 values below 1 μM, which is comparable to the value for L-742,001. The following parameters were found to influence the activity: 1) The N-substituent of the indole increased the IC50 value in the order methyl < ethyl < benzyl (cf., 30–31, 34–36, and 39–41). 2) The location of the DKA arm on the indole moiety (i.e., at position 2 or 3) had variable impact in relationship to the size of the indole moiety. Position 3 represents the optimum geometry in the case of the simple indole ring (compare 39 and 41 with 30 and 31, respectively). 3) When the indole part was enlarged by adding an extra dioxole ring, this extension led to increased potency for the 2-diketo derivatives (compare 34 and 36 with 30 and 31, respectively). However, for the compounds bearing the diketo motif in 3-position, this extra dioxole ring appeared to reduce the activity (cf., 42 and 39). 4) The DKA-methyl esters of these indole derivatives (37 and 43) were markedly less active than the corresponding analogs containing a free carboxyl group (36 and 42, respectively).
A favorable IC50 value (0.8 μM; Fig. 1; Table 1) was also noted for the pyrrole derivative 10 (known as the Merck compound L-731,988), which was previously discovered as a potent inhibitor of HIV IN (Hazuda et al., 2000). As with the data above, its methyl ester proved to be sevenfold less active. This pyrrole derivative can be viewed as an analog of indole 31, in which the ring system is “size-reduced” to a pyrrole, and the benzyl is substituted with a p-fluoro atom.
Finally, we evaluated several compounds in which the two-metal–chelating moiety is incorporated into a cyclic system. This strategy proved highly successful for creating a potent and selective HIV IN inhibitor (Agrawal et al., 2012). In the case of PA-Nter, neither of the cyclic diketo analogs tested, i.e., the indole derivatives 44–47, as well as the other DKA bioisosteres, i.e., the quinolones 25–28 and the hydroxypyrimidine carboxamide 29, had any activity in the enzymatic PA-Nter assay.
Pharmacophore Model Generation.
To elucidate the structural requirements for novel PAIs, a three-dimensional pharmacophore model was built (Fig. 2) on the basis of the efficacy data of a selection of tested compounds; namely, the training set contained twelve molecules with an IC50 value against PA-Nter of 2 μM or less, i.e., the β-diketo acids or esters 1–6, 10, 16, 34, and 39–41, and these were submitted to a pharmacophore generation protocol using the Molecular Operating Environment software package. With the exception of 16, 34, and 39, all compounds also showed antiviral activity in cell culture (see below). Fifteen PAI pharmacophore models were generated and the best hit was evaluated in terms of statistical parameters, such as correlation coefficient and root-mean-square deviation value. This model featured five pharmacophore elements, including three metal ligator (ML) features, one aromatic or hydrophobic element (Hyd-Aro), and one cyclic aromatic region (Aro) (Fig. 2, A and B). This five-point pharmacophore was then validated by screening our in-house database of about 150 compounds, chosen on the basis of their previously evaluated chelating ability toward metal cofactors of other metalloenzymes, and considering compounds having molecular diversity. All selected compounds were retrieved from the database and fitted nicely with the chemical features of our hypothesis, thus confirming the reliability of the proposed model.
Fig. 2.

Three-dimensional arrangement of five pharmacophore features in the β-diketo acid motif. (A) Twelve active compounds 1–6, 10, 16, 34, 39–41 (i.e., β-diketo acids or esters with IC50 values ≤ 2 μM) were aligned and mapped to generate a five-point pharmacophore. (B) The pharmacophore features are depicted as metal ligators (ML1, ML2, and ML3) in cyan, aromatic (Aro), and hydrophobic or aromatic (Hyd-Aro) features in pink. (C) Interfeature distances are given in angstroms (Å). (D) These distances are in agreement with both proposed models for interaction between the β-diketo acid motif and the metal ions. Left: in one model [which is based on the cocrystal structure of PA-Nter in complex with L-742,001 (DuBois et al., 2012)], the carboxylate and α-hydroxyl functionalities of the DKA motif coordinate one ion, and the third coplanar oxygen at the γ-position chelates the second metal ion together with the α-hydroxyl group. Middle: in an alternative model [previously proposed by us on the basis of L-742,001 docking within the holo form of PA-Nter, and assuming that L-742,001 mainly binds in its monodeprotonated form (Stevaert et al., 2013)], the γ-oxygen of the DKA motif is not involved. Instead, one metal is coordinated by the lone oxygen pair of the α-hydroxyl group and one oxygen atom of the carboxylate group, whereas the second metal ion interacts with both oxygens of the carboxylate moiety. Right: overlay of both models showing the intermetal distances.
As shown in Fig. 2A, the overall mapping results indicate that almost all compounds aligned in a similar spatial arrangement within the model, highlighting important structural similarities between compounds bearing piperidine and indole scaffolds. A good fitting was shown for the coplanar diketo acid/ester fragment of all compounds within three points, whereas the orientation of the substituted heterocyclic portion mapped into the remaining pharmacophore elements, with the exception of the para-F-benzyl moiety of the pyrrole scaffold (10), which has a different orientation. Moreover, the N-methylcyclohexyl substituent of the diketo methyl ester 6 is located in an opposite conformation with respect to the same moiety in the corresponding acid 5. In L-742,001, the two aromatic moieties attached to the piperidine ring are oriented in opposite directions and perpendicularly to the DKA motif. Tolerance for various orientations of the aromatic and hydrophobic moieties agrees with structural data showing that the catalytic core of PA-Nter is surrounded by different hydrophobic pockets well suited for inhibitor binding (DuBois et al., 2012; Kowalinski et al., 2012). Hence, it is relevant to design DKA derivatives containing hydrophobic substituents in an opportune topological disposition, so as to occupy these alternative pockets in PA-Nter.
With regard to the metal chelating fragment, the three-dimensional spatial arrangement and distance constraints between the chemical features are in agreement with a previously published minimal pharmacophore model (Parkes et al., 2003). Both features ML1-ML2 and ML2-ML3 (i.e., the donor triad atoms chemotype; Fig. 2B) are able to coordinate the two metal ions in the PA-Nter active site. In our pharmacophore model, the interfeature distances between ML2 and ML1, and between ML2 and ML3, are 2.64 and 2.77 Å, respectively (Fig. 2C; distance ML1–ML3 is 4.74 Å). The other two additional features (Aro, Hyd/Aro) would be involved in favorable contacts with the hydrophobic binding pockets around the active center. The distances between the key ML1, ML2, and ML3 features and the aromatic feature Aro are 4.41, 6.12, and 6.46 Å, respectively. The distance between the optimally oriented Aro and Hyd/Aro features is 2.55 Å, whereas that between ML1 and Hyd/Aro is 2.98 Å.
In Fig. 2D, two proposed models for interaction of the β-diketo acid motif with the metal ions are shown (see figure legend for all details). Importantly, the ML1-ML2-ML3 interfeature distances in our pharmacophoric model are coherent with favorable ligand-metal interactions and in agreement with both binding models (DuBois et al., 2012; Stevaert et al., 2013). Namely, the Mn2+ to Mn2+ distance is 3.82 Å in the crystal-based and 3.31 Å in the docking-based model (Fig. 2D). In thermodynamic models using Mg2+ as the cofactor, the Mg2+ to Mg2+ distance was predicted to decrease from 4.91 to 3.85 Å upon RNA binding (Xiao et al., 2014). These changes in the intermetal distance along the catalytic pathway were already demonstrated for Bacillus halodurans RNase H (Yang et al., 2006) and EcoRV (Horton and Perona, 2004).
The twelve compounds selected for pharmacophore generation were also submitted to basic physicochemical calculations to try to predict their membrane-permeating capability, which is required to achieve antiviral activity in cell culture. The calculated atom-based values (Supplemental Table 1) for almost all selected compounds (1–6, 10, 16, 34, and 39–41) fall within the desirable range for good absorption and membrane permeability (i.e., logP < 5, molecular weight < 500, H-bond acceptor and donor counts of <5 and <10, respectively). However, three compounds (i.e., 16, 34, and 39) showed unfavorable predicted logP values, which may (at least partially) explain their inactivity in cellular assays (see below). Other studies suggest that compounds with a polar surface area (PSA) < 60 Å2 would probably be well absorbed (>90%), whereas compounds with PSA > 140 Å2 are predicted to be poorly absorbed (<10%) (Lipinski et al., 2001). All twelve compounds had an intermediate PSA value well below 140 Å2.
Development of the Molecular Beacon–Based Endonuclease Assay.
The plasmid-based endonuclease method has the disadvantages of being discontinuous and time consuming, which makes it less suitable for screening large series of compounds. We therefore developed an alternative real-time assay that uses a molecular beacon as the enzyme substrate and is amenable to high-throughput format. An MB is a single-stranded DNA probe that contains a fluorophore moiety at one end and a nonfluorescent quencher moiety at the other. When intact, the MB forms a stem-and-loop structure, bringing the fluorophore and quencher in very close proximity, which results in superior quenching efficiency compared with a linear dual-labeled probe. MB cleavage by recombinant PA-Nter (depicted in Fig. 3A) separates the fluorophore from the quencher, and the evolving fluorescence reveals the cleavage process in real-time (Marras et al., 2006).
Fig. 3.
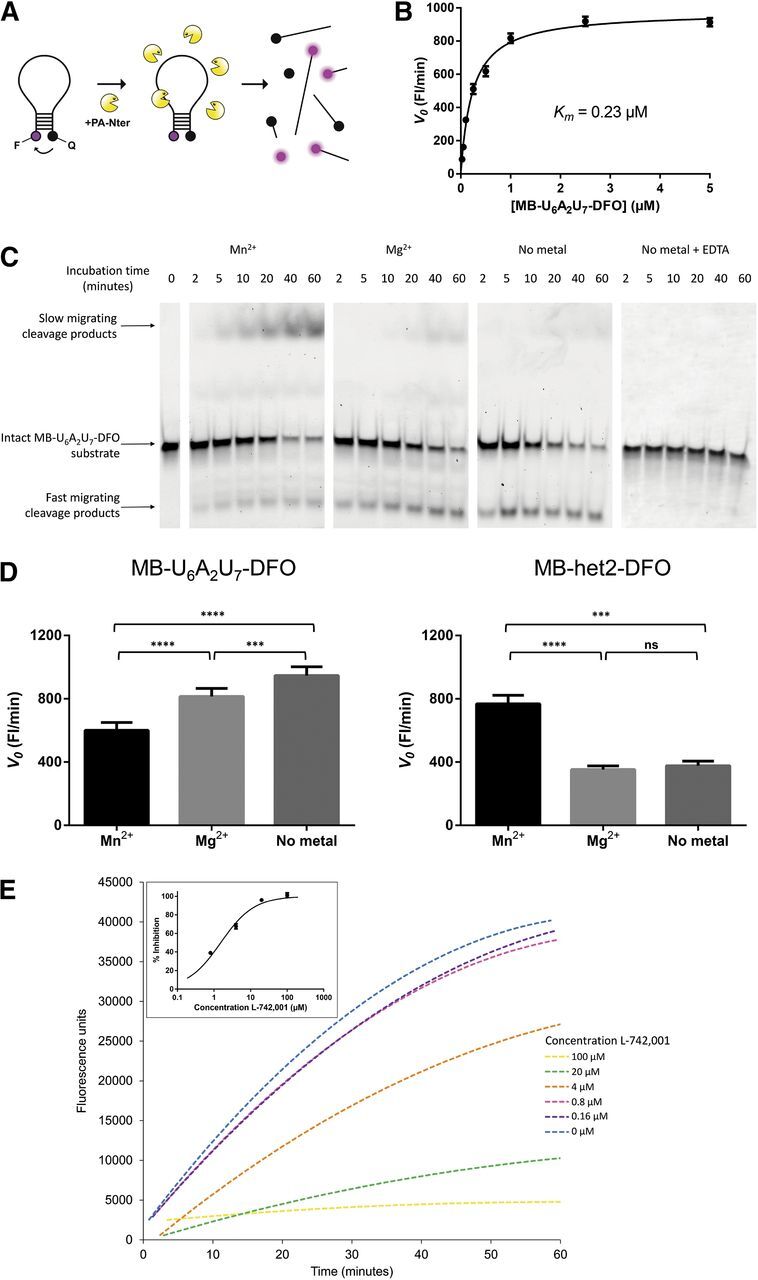
Utility of the MB assay for determination of PA-Nter enzyme properties and activity of PA inhibitors. (A) Schematic representation of the MB assay. MB cleavage by recombinant PA-Nter (which can occur at different sites) separates the fluorophore (F) from the quencher (Q). The evolving fluorescence, indicating enzymatic activity, can be monitored in real-time. (B) Michaelis-Menten curve for MB-U6A2U7-DFO. This MB was incubated with PA-Nter and the evolving fluorescence was recorded as a function of time and MB substrate concentration. The initial cleavage rate (V0) was obtained from the slope (fluorescence units per minute) of the best-fit line derived from the 5–15-minute interval of the reaction. Values shown are the averages ± S.E.M. of three independent experiments. (C) Gel electrophoretic analysis of the MB cleavage pattern. The amount of intact MB-U6A2U7-DFO substrate decreased over time when incubated with PA-Nter. The first-appearing cleavage products migrated faster than the intact MB substrate, whereas the late-cleavage products showed slower migration through the gel. When using Mn2+ as a cofactor, PA-Nter appears able to cleave shorter oligonucleotides than in the presence of Mg2+ or without addition of a bivalent metal (i.e., in the presence of the copurified metal). Addition of 100 mM EDTA completely abrogated the endonuclease activity. (D) Influence of the metal ion on the initial cleavage rate of MB-U6A2U7-DFO and MB-het2-DFO. The initial cleavage rate (V0; on the basis of the real-time fluorographs) for MB-U6A2U7-DFO (left graph) was 1.4-fold and significantly higher with Mg2+ than with Mn2+, and still increased when no metal ion was added. An opposite effect was seen with MB-het2-DFO (right graph): In this case, the reaction rate was 2.2-fold higher in the presence of Mn2+ compared with Mg2+. The statistical significance (****P < 0.0001; ***P < 0.001; ns, not significant) was calculated by one-way analysis of variance followed by a Tukey’s multiple comparison test using GraphPad Prism. (E) Dose-dependent inhibition of MB cleavage by L-742,001. MB-U6A2U7-DFO (at 20 nM) was incubated with 1 μg PA-Nter and increasing concentrations of L-742,001. The initial cleavage rate (V0) was obtained from the slope (fluorescence units per minute) of the best-fit line derived from the 5–15-minute interval of the reaction. These V0-values were used to calculate the percentage of inhibition. The inset shows the resulting dose-response curve.
First, the real-time MB method was used to determine the kinetic parameters of PA-Nter versus Mn2+ or Mg2+, and eight different oligonucleotide substrates (i.e., an MB with a heterogeneous sequence, or containing oligo-C, -A, or -U). The Km values determined for the eight different MB substrates were in the range of 0.09–0.64 μM (see Fig. 3B and Table 3, which also contains the signal-to-noise ratio at 100 nM MB). This indicates that, under the conditions of this MB assay, PA-Nter does not possess much preference for specific dinucleotide linkages. In this context, it is noteworthy that the MB-het2-DFO substrate, which contains two 5′-GC-3′ motifs, did not prove to be a better substrate than the dU-rich MB-U6A2U7-DFO. On the contrary, we noted a 6-times higher Vmax with MB-U6A2U7-DFO than with MB-het2-DFO. This contrasts with the observation by another group that PA-Nter has a preference for 5′-GC-3′ in RNA substrates (Datta et al., 2013). However, efficient cleavage of U-rich RNA substrates was already demonstrated by Dias et al. (2009). In these two studies, PA-Nter was evaluated against RNA substrates, whereas our MB assay uses DNA-oligonucleotides (which were preferred because of their higher chemical stability). Other groups have successfully developed fluorescent assays for PA endonuclease using totally different DNA or RNA probes (Kowalinski et al., 2012; Noble et al., 2012; Bauman et al., 2013; Datta et al., 2013; Chen et al., 2014), confirming that PA-Nter can cleave different DNA or RNA sequences in enzymatic assays. From a technical viewpoint, our MB assay is superior in having a higher signal-to-noise ratio than fluorescent assays using nonbent oligonucleotide substrates.
By using gel electrophoresis with fluorescence detection (Fig. 3C), we confirmed that the fluorescence enhancement observed by real-time fluorometry was attributable to MB cleavage and not to mere opening of the stem structure upon binding of the MB to the active site of PA-Nter. An intriguing and unexpected finding relates to the impact of the bivalent metal ion on cleavage efficiency by PA-Nter. While using the 7-kb plasmid substrate (M13mp18), we observed no endonuclease activity when Mn2+ was omitted from the reaction mixture or replaced by Mg2+ (second and third from last lanes in Fig. 1), in agreement with a previous report (Dias et al., 2009). In contrast, PA-Nter was found to efficiently cleave the MB substrate when magnesium instead of manganese was added, and even without addition of a metal ion, which is probably attributable to copurification of a metal ion (probably Mg2+) (Xiao et al., 2014) during preparation of the PA-Nter enzyme. Significant substrate cleavage in the absence of added salt was also observed by Noble et al. (2012) in experiments with untruncated PA. The effect of the metal ion depended on which MB sequence was used, since one MB was more rapidly cleaved in the presence of Mg2+ than Mn2+, whereas for another sequence, the opposite was seen (see Fig. 3D for all details).
When analyzing the MB cleavage by gel electrophoresis (Fig. 3C), we observed a difference in the cleavage pattern, depending on which metal ion was used. This is in accordance with the results obtained by Noble et al. (2012). When using Mn2+ as a cofactor, PA-Nter appears able to cleave shorter oligonucleotides than in the presence of Mg2+ or without addition of a bivalent metal (i.e., in the presence of the copurified metal). The Mn2+ condition gave rise to cleavage products which increased in function of time and appeared as an extra band on the gel, which migrated more slowly than the intact beacon. Most probably, migration of the shorter oligonucleotides is slowed down by the presence of the positively charged fluorophore. The endonuclease activity resulting from the copurified metal was completely abrogated upon addition of EDTA to the reaction mixture (Fig. 3C, right). The observation that the metal cofactor changes the cleavage pattern suggests that the structure of the active site depends on which metal ions are present. Since this may influence the binding properties of PAIs, we used the MB assay to evaluate a series of representative DKA inhibitors with activity toward PA-Nter in the plasmid-based assay. The MB assay with these compounds was performed in the presence of Mn2+, Mg2+, or without addition of a bivalent metal ion, and using either MB-U6A2U7-DFO or MB-het2-DFO as the substrate. As shown in Table 4, the metal ion and MB substrate had very little, if any, impact on the IC50 values for L-742,001 (1). In Fig. 3E, the fluorographs for this compound and derived dose-response curve are shown for the experiments with Mn2+; similar curves were obtained in the Mg2+ or no-added-metal conditions. In contrast, other PAIs, i.e., 10, 31, 34, 39, 40, and 41, had up to 61-fold lower IC50 values in the presence of Mn2+ compared with Mg2+ (Table 4). When the data obtained in the plasmid and molecular beacon assay (both with Mn2+) are being compared, both assays gave the same IC50 values for L-742,001, 5, 10, 34, 40, and 41. For some compounds, we observed markedly higher IC50 values when using the MB instead of the plasmid substrate (i.e., factor increase of 35 for 39 and 57 for 12). On the other hand, for 31, a sevenfold lower IC50 value was obtained in the MB assay than in the plasmid assay.
TABLE 4.
Inhibitory activity of a selection of DKA derivatives in the MB versus plasmid assay with PA-Nter, or an assay using untruncated PA
IC50 (μM), 50% inhibitory concentration. For each compound the IC50 value was calculated using nonlinear regression analysis. Values are the mean of at least two independent experiments.
| Compound | PA-Nter, molecular beacon assay |
Plasmid assayc Mn2+ | Untruncated PAd Mn2+ | |||||
|---|---|---|---|---|---|---|---|---|
| MB-U6A2U7-DFOa | MB-het2-DFOb |
|||||||
| Mn2+ | Mg2+ | No metal | Mn2+ | Mg2+ | No metal | |||
| 1 | 1.5 | 3.1 | 2.5 | 3.9 | 7.1 | 3.5 | 0.5 | 0.5 |
| 5 | 3.9 | 6.3 | 9.1 | 0.5 | ||||
| 10 | 4.2 | 177 | 100 | 2.4 | >200 | >200 | 0.8 | |
| 12 | 154 | >200 | >200 | 2.7 | ||||
| 30 | 26 | 41 | 41 | 3.4 | 1.1 | |||
| 31 | 3.2 | 44 | 54 | 22 | ||||
| 34 | 3.6 | 59 | 180 | 0.6 | ||||
| 39 | 21 | >200 | >200 | 0.6 | 1.1 | |||
| 40 | 5.2 | >200 | 174 | 0.8 | ||||
| 41 | 1.4 | 86 | 98 | 1.6 | ||||
| 42 | 23 | 65 | 72 | 3.5 | ||||
MB-U6A2U7-DFO, 5′‐[DFO]‐GCAGG (dU)6A2(dU)6 CCTGC‐[BHQ2]‐3′.
MB-het2-DFO, 5′-[DFO]‐CGCACG TTATGCTAAGCAAGTAACC GTGCG‐[BHQ2]‐3′.
See Noble et al. (2012) for a description of the fluorescence-based enzymatic assay with untruncated PA expressed in insect cells.
Finally, we verified that assays using truncated PA-Nter are adequate to investigate PAIs interacting with the catalytic site. Namely, the inhibitory activity of a few DKA compounds was determined in an enzymatic assay with untruncated PA and Mn2+ (Noble et al., 2012). The IC50 values for compounds 1, 30, and 39 (Table 4, right column) were very similar to the values obtained with PA-Nter using the plasmid/Mn2+ assay.
Anti–Influenza Virus Activity in Cell Culture.
Besides evaluation in the PA-Nter enzymatic assay, all DKA analogs were examined for anti–influenza virus activity in cell culture. We first performed the influenza vRNP reconstitution assay in HEK293T cells. Using L-742,001, we previously demonstrated that this method is well suited for determining the activity and selectivity of PAIs in cell culture (Stevaert et al., 2013). In the present study, we further expanded this vRNP assay by including four different vRNP complexes, i.e., derived from a human influenza A or B virus, an avian H5N1 virus containing the PB2-627Glu avian signature, or the PB2-627Lys human adaptation residue (Moncorgé et al., 2010). As shown in Table 5, L-742,001 and its close analogs 2–6 had strong and consistent activity against all four vRNP complexes, which agrees with our previous analysis that the key residues for binding of L-742,001 in PA-Nter show little variation among influenza A (human or avian) or B viruses (Stevaert et al., 2013).
TABLE 5.
Activity of compounds 1–29 in two influenza virus cell–based assays
Data shown are the mean ± S.E.M. of at least three independent tests.
| Compound |
Influenza virusa vRNP reconstitution assay in HEK293T cellsb |
Virus yield assay in MDCK cellsb |
||||||
|---|---|---|---|---|---|---|---|---|
| Antiviral activity
EC50c |
Cytotoxicity CC50d | Human A/PR/8/34
antiviral activitye
|
Cytotoxicity CC50 | |||||
| Human A/PR/8/34 | Human B/Yamanashi | Avian 50-92/ PB2-627E | Avian 50-92/ PB2-627K | EC99 | EC90 | |||
| μM | ||||||||
| 1 | 3.6 | 3.1 | 4.6 | 11 | ≥104 | 8.4 | 5.4 | 181 |
| 2 | 2.8 | 6.9 | 8.5 | 20 | ≥102 | 6.0 | 3.9 | >100 |
| 3 | 1.8 | 0.8 | 6.6 | 17 | ≥103 | 2.9 | 2.0 | >100 |
| 4 | 2.3 | 0.4 | 7.6 | 15 | ≥93 | 5.6 | 3.8 | >100 |
| 5 | 1.7 | 4.4 | 3.9 | 8.8 | ≥106 | 1.5 | 1.0 | >100 |
| 6 | 2.3 | 11 | 7.2 | 15 | ≥94 | 2.7 | 1.9 | >100 |
| 7 | >100 | NDf | ND | ND | >200 | ND | ND | ND |
| 8 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 9 | 57 | 25 | 105 | 29 | 117 | >100 | >100 | >200 |
| 10 | 84 | 116 | 88 | 95 | >200 | 74 | 58 | >200 |
| 11 | 132 | 49 | 47 | 65 | >200 | >100 | ≥90 | ≥184 |
| 12 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 13 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 14 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 15 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 16 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 17 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 18 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 19 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 20 | 72 | 97 | 32 | 37 | >200 | 79 | 55 | ≥194 |
| 21 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 22 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 23 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 24 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 25 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 26 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 27 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 28 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 29 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| Ribavirin | 16 | 3.1 | 50 | 45 | >200 | 10 | 6.7 | >200 |
Reconstituted vRNP complexes from: human influenza A virus, strain A/PR/8/34; human influenza B virus, strain B/Yamanashi/166/98; and avian influenza A virus, strain A/turkey/England/50-92/1991 with a PB2-627E or PB2-627K residue.
HEK293T cells, human embryonic kidney 293T cells; MDCK cells, Madin-Darby canine kidney cells.
EC50, 50% effective concentration (μM), i.e., compound concentration producing 50% reduction in vRNP-driven firefly reporter signal, estimated at 24 hours after transfection.
CC50, 50% cytotoxic concentration (μM) at 24 hours determined by MTS cell viability assay.
Compound concentration (μM) causing a 1-log10 (EC90) or 2-log10 (EC99) reduction in virus yield at 24 hours postinfection, as determined by real-time RT-PCR.
ND, Not done.
Among our series of DKA analogs with an indole scaffold (Tables 2 and 6), the following were shown to inhibit influenza vRNP activity: 33 and 40, which have an N-ethyl group; and 31, 36, 37, and 41, which carry an N-benzyl substituent (introduced to modulate lipophilicity, thus assumed to improve cellular uptake). Of these, 31, 36, 40, and 41 are unesterified DKAs, whereas 33 and 37 are methyl esters. The highest potency was obtained for 36. With the exception of 41, these indole derivatives were not toxic at the highest concentration tested (200 μM). Importantly, although these compounds displayed relatively low potency in the vRNP assay [i.e., average EC50 values ranging from 14 μM (36) to 118 μM (40)], the same values were obtained for all four vRNP complexes, whether derived from influenza A (human or avian) or B viruses.
TABLE 6.
Activity of compounds 30–47 in two influenza virus cell–based assays
| Compound | Influenza virusa vRNP reconstitution assay in HEK293T cells |
Virus yield assay in MDCK cellsb |
||||||
|---|---|---|---|---|---|---|---|---|
| Antiviral activity EC50c |
Cytotoxicity CC50d | Antiviral activitye
Human A/PR/8/34 |
Cytotoxicity CC50 | |||||
| Human A/ PR/8/34 | Human B/ Yamanashi | Avian 50-92/ PB2-627E | Avian 50-92/ PB2-627K | EC99 | EC90 | |||
| μM | ||||||||
| 30 | >100 | NDf | ND | ND | >200 | ND | ND | ND |
| 31 | 92 | 59 | >200 | 87 | >200 | 85 | 55 | 126 |
| 32 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 33 | 77 | 18 | 39 | 62 | >200 | >25 | >25 | ≥169 |
| 34 | >100 | ND | ND | ND | >100 | ND | ND | ND |
| 35 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 36 | 16 | 11 | 17 | 12 | >200 | >12.5 | >12.5 | >200 |
| 37 | 48 | 29 | 54 | 30 | >200 | >25 | >25 | ≥184 |
| 38 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 39 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 40 | 166 | 65 | 121 | 121 | >200 | 95 | 69 | ≥194 |
| 41 | 45 | 43 | 96 | 62 | 136 | 64 | 38 | >200 |
| 42 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 43 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 44 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 45 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 46 | >100 | ND | ND | ND | >200 | ND | ND | ND |
| 47 | >100 | ND | ND | ND | >200 | ND | ND | ND |
Reconstituted vRNP complexes from: human influenza A virus, strain A/PR/8/34; human influenza B virus, strain B/Yamanashi/166/98; and avian influenza A virus, strain A/turkey/England/50-92/1991 with a PB2-627E or PB2-627K residue.
HEK293T cells, human embryonic kidney 293T cells; MDCK cells, Madin-Darby canine kidney cells.
EC50, 50% effective concentration (μM), i.e., compound concentration producing 50% reduction in vRNP-driven firefly reporter signal, estimated at 24 hours after transfection.
CC50, 50% cytotoxic concentration (μM) at 24 hours determined by MTS cell viability assay.
Compound concentration (μM) causing a 1-log10 (EC90) or 2-log10 (EC99) reduction in virus yield at 24 hours postinfection, as determined by real-time RT-PCR.
ND, Not done.
Three other analogs in Table 1 had noticeable activity in the vRNP assay: 10 (L-731,988) and its methyl ester 11 (both are strong inhibitors in the enzymatic test) and 20 (L-708,906; a substituted DPBA derivative with weak activity in the enzymatic test). Several DKAs with potent activity in the PA-Nter enzymatic assay (i.e., IC50 values below 5 μM; Tables 1 and 2) were devoid of any activity in the vRNP cell culture assay. This might be explained by insufficient cellular uptake owing to the anionic charge of the DKA moiety. On the other hand, the activity of 9 in the vRNP assay appears unrelated to inhibition of the endonuclease since this compound was inactive in the enzymatic assay.
In a second stage, the compounds showing activity in the vRNP assay were submitted to a virus yield assay in influenza virus–infected MDCK cells. For L-742,001 and its close analogs (1-6), and 10, 20, 31, 40, and 41, the inhibitory activity noted in the vRNP reconstitution assay was confirmed in influenza virus–infected cells. Although their inhibitory potency was quite moderate (i.e., antiviral EC99 values of ∼80 μM), our cell culture data indicate that these compounds may represent some novel and cell-permeable PAIs. To validate this assumption, we performed a basic time-of-addition experiment (Fig. 4) in which viral RNA synthesis is measured after one virus replication cycle. We previously demonstrated (Stevaert et al., 2013) that the polyphenolic compound epigallocatechin gallate, which acts as a PAI in enzymatic assays (Kuzuhara et al., 2009; Kowalinski et al., 2012), has an unrelated antiviral mode of action in cell culture. Figure 4 shows that the reference compound chloroquine, which inhibits influenza virus entry by increasing the endosomal pH (Vanderlinden et al., 2012), was devoid of activity when added postentry, i.e., at 1 hour after virus addition. L-742,001 kept its full inhibitory activity when added at +1 hour, confirming that its action point is situated after virus entry. For 10, 40, and 41, the highest inhibition was seen (i.e., at least 500-fold reduction in viral RNA synthesis) when the compounds were added 30 minutes prior to the virus. However, the finding that these compounds still produced ∼60- to ∼300-fold reduction in viral RNA copy number when added at +1 hour, indicates that they do inhibit viral RNA synthesis quite potently, which fits with their designation as PAIs.
Fig. 4.

Compounds 10 (L-731,988), 40, and 41 inhibit viral RNA synthesis besides affecting the virus entry process. Light gray bars: the test compounds L-742,001 (20 μM), 10 (200 μM), 40 (150 μM), 41 (150 μM), or chloroquine (80 μM) were added to MDCK cells, and after 30 minutes incubation at 35°C, influenza virus A/PR/8/34 was added. Dark gray bars: virus was added first and allowed to enter during 1 hour incubation, after which the compounds were added at the same concentrations as above. In both conditions, total cellular RNA was extracted at 10 hours postinfection (VC, untreated virus control). The number of vRNA copies was quantified by two-step real-time RT-PCR. On the y-axis, the fold increase in vRNA copies is shown relative to the viral copy number added at time zero. L-742,001 remains fully effective when added after virus entry. In contrast, the reported entry inhibitor chloroquine (Vanderlinden et al., 2012) is inactive when added at 1 hour postinfection. Compounds 10, 40, and 41 have a dual effect by acting both upon virus entry and viral RNA synthesis. Data shown are the mean ± S.E.M. of two independent tests.
Discussion
The potential of influenza virus endonuclease inhibitors as an entirely novel concept in antiviral therapy was recognized about twenty years ago, when two prototype inhibitors, L-742,001 and flutimide were initially described (Hastings et al., 1996; Tomassini et al., 1996). The fact that these compounds were discovered in a screening approach combining enzymatic and cell-based methods probably explains why these molecules still serve as the two main lead PAIs today. Their presumed binding mode in the catalytic center of the enzyme was only recently revealed in cocrystallization experiments with PA-Nter (DuBois et al., 2012; Kowalinski et al., 2012). These studies further revealed several hydrophobic pockets surrounding the catalytic core of PA-Nter, and that the binding of flutimide, L-742,001, and its congener, DPBA, involves an induced-fit mechanism. On the basis of these structural insights, several groups embarked on in silico design of novel PAIs with unrelated chemical structures (Ishikawa and Fujii, 2011; Bauman et al., 2013; Chen et al., 2014).
We followed another strategy by evaluating, in complementary enzyme- and cell-based biological assays, available scaffolds of DKA derivatives, which were originally developed toward HIV integrase or other metal-dependent enzymes. By determining their inhibitory activity against PA-Nter, we aimed at identifying which structures (and their structural determinants) could serve as the basis for future design of PAIs. For this purpose, we created a three-dimensional pharmacophore model defining the optimal distances between the metal chelating parts of the ligands and the aromatic/hydrophobic moieties.
Efficient development of novel PAIs requires an enzymatic assay that is amenable to high-throughput screening. The plasmid-based electrophoretic assay used here for evaluating the DKA analogs is quite robust but too labor-intensive to be used in screening programs. In contrast, our real-time MB assay can be performed in miniaturized plate format and at low cost. Although similar fluorescent assays have been described by others (Kowalinski et al., 2012; Noble et al., 2012; Bauman et al., 2013; Datta et al., 2013; Chen et al., 2014), our MB assay is unique in having a superior signal-to-noise ratio. Besides this analytical aspect, we obtained some unanticipated findings that further broaden the relevance of the MB assay. First, the mere observation that a bent molecular beacon is a very efficient substrate for PA-Nter is reminiscent of what has been described for the type II endonuclease EcoRV, which requires bending of the nucleic acid substrate for catalysis to occur (Horton and Perona, 2004; Xiao et al., 2014). Second, PA-Nter proved to have low cleavage site specificity, since it was able to cleave molecular beacons with different DNA sequences, cutting not only the beacon itself but also subsequently formed smaller fragments. Third, whereas most enzymatic studies with PA-Nter have indicated that the enzyme is considerably more active in the presence of Mn2+ compared with Mg2+ (Dias et al., 2009; Crépin et al., 2010; Datta et al., 2013), our molecular beacon assay works equally well with Mg2+ as with Mn2+. We speculate that the intrinsic bending of this substrate may bring the scissile phosphodiester group in a prereactive conformation and drive catalysis; hence, cleavage occurs with the less reactive Mg2+. On the other hand, with more flexible substrates (such as the M13mp18 plasmid), the softer metal Mn2+ seems to be required for optimal enzymatic activity. Mg2+ has been proposed as the biologically relevant cofactor, given that its intracellular concentration is at least 1000-fold higher than that of Mn2+ (Zhao et al., 2009; Xiao et al., 2014). This may implicate that PA-Nter assays using Mg2+ are more stringent for evaluating potential PAIs. For most of the DKA inhibitors tested here, the inhibitory activity against PA-Nter was far less with Mg2+ than with Mn2+. A notable exception is L-742,001 (and its cyclohexyl analog 5), for which the activity was similar for both metal ions. It is tempting to speculate that this metal ion–independent behavior of L-742,001 (or, at least, its strong activity versus Mg2+) may contribute to its superior efficacy in cell culture. For comparison, compound 10 had a low IC50, similar to that of L-742,001, when evaluated against PA-Nter with Mn2+ but was 40-fold less active in the presence of Mg2+. In influenza virus–infected cells, 10 was only moderately active (despite the fact that this molecule is a strong inhibitor of human immunodeficiency virus in infected cells, indicating that it enters the cells quite efficiently) (Hazuda et al., 2000). The issue of cell penetration may also explain our observation that the anti–influenza virus activity in cell culture was higher for the DKA-indole derivatives carrying an N-benzyl (compared with ethyl or methyl).
We hypothesize that the less rigid coordination requirements of Mn2+ may create flexibility and allow binding of structurally diverse inhibitors (Bock et al., 1999). In contrast, Mg2+ may result in a tighter catalytic site that only accepts the best fitting compounds. Noble et al., (2012) showed a change in circular dichroism spectrum when comparing PA with Mn2+ to PA with Mg2+, which correlates with an increase in helical content. If the secondary structure of PA-Nter changes depending on the metal, it is possible that the metal ion also affects which hydrophobic pockets are available for binding of the inhibitor. Without crystallographic data of PA-Nter crystallized with inhibitors in the presence of only Mg2+ and/or more docking studies, this remains speculative. The finding that several DKA inhibitors prefer Mn2+ to exert their effect on PA-Nter is reminiscent of what was described for HIV IN (Marchand et al., 2003). In this case, both the aromatic and DKA part were shown to play a role in this metal selectivity, which was explained by a metal-dependent binding orientation of the inhibitor. Unfortunately, the understanding of how PAIs bind to PA-Nter is still quite limited. For a selection of PAIs (including L-742,001), the binding mode was recently revealed by cocrystallographic analyses (DuBois et al., 2012; Kowalinski et al., 2012). For L-742,001, our resistance studies in cells infected with a series of mutant influenza viruses established the role of several residues in the catalytic center of PA or surrounding hydrophobic pockets in positioning this inhibitor in the active site of PA-Nter (Stevaert et al., 2013).
Our enzyme inhibition data for PA-Nter convincingly demonstrate that the setup of the enzymatic assay (i.e., substrate, metal cofactor, and type of readout) should be carefully chosen when performing a compound screening for PAI development. We provide evidence that inhibitors targeting the catalytic site have similar activity whether assessed with PA-Nter or untruncated PA. Given the uncertainty on which metal ions (i.e., Mg2+ or Mn2+) are present in the active site, evaluation of potential PAIs against both metals (as possible with our MB assay) seems recommended. A number of recent reports have described novel PAIs with DKA-unrelated structures (Iwai et al., 2010; Baughman et al., 2012; Bauman et al., 2013; Chen et al., 2014). Only a few of these molecules were subsequently proven to have activity in influenza virus–infected cell cultures. The importance of proof-of-concept cell culture evaluation during early hit discovery of potential PAIs also appears from our cell culture studies using influenza virus vRNP reconstitution and replication assays. The fact that hit compounds with excellent activity against PA-Nter (i.e., comparable to that of L-742,001) may not survive proof-of-concept evaluation in cell culture can be related to different factors. As suggested above, an enzymatic PA-Nter assay using Mg2+ might be more stringent and have a better predictive value. Second, the hydrophilic/lipophilic balance for the chelating fragment and aromatic framework of DKA-based compounds may play an important role in explaining their antiviral potency. Besides, we previously demonstrated that the catechol compound epigallocatechin gallate, which is nicely active in an enzymatic PA-Nter assay, owes its excellent activity in virus-infected cells entirely to inhibition of virus entry (Stevaert et al., 2013). To avoid this confusion, we here evaluated the active DKA compounds in a basic time-of-addition experiment. Our data indicate that the pyrrole compound 10 and the indole compounds 40 and 41 do inhibit viral RNA synthesis, besides having a secondary effect on virus entry.
In conclusion, we demonstrated that diverse DKA scaffolds can lead to potent inhibition of PA-Nter in enzymatic assays, which agrees with the notion that the catalytic center of PA-Nter is quite spacious and flexible, and subject to induced-fit inhibitor binding (DuBois et al., 2012; Kowalinski et al., 2012). Our pharmacophore model defines strict structural requirements along with chemical features, and serves as a basis for design of novel PAIs with DKA-based structures or bioisosteres thereof. For high-throughput compound screening, the novel molecular beacon assay appears well suited. This method enables determination of the activity of potential PAIs versus either Mn2+ or Mg2+, the latter probably being the biologically relevant cofactor. Any hit-to-lead process on novel PAIs requires rapid progression of potential hit compounds to relevant cell culture assays, such as the vRNP reconstitution, virus yield, and time-of-addition methods, which we used to identify some relevant scaffolds for further design of PAIs. Successful development of this new class of anti–influenza virus therapeutics will require an integrated biologic approach such as the one described here.
Acknowledgments
The authors thank W. van Dam and S. Stevens for their dedicated technical assistance and M. Kim, W. Barclay, J. McCullers, and K. Nagata for the generous donation of plasmids. M.S. thanks Dr. Andrea Brancale for the use of software for molecular modeling calculation.
Abbreviations
- 1
(Z)-4-(1-benzyl-4-(4-chlorobenzyl)piperidin-4-yl)-2-hydroxy-4-oxobut-2-enoic acid
- 2
(Z)-methyl 4-(1-benzyl-4-(4-chlorobenzyl)piperidin-4-yl)-2-hydroxy-4-oxobut-2-enoate
- 3
(Z)-4-(4-(4-chlorobenzyl)-1-(4-fluorobenzyl)piperidin-4-yl)-2-hydroxy-4-oxobut-2-enoic acid
- 4
(Z)-methyl 4-(4-(4-chlorobenzyl)-1-(4-fluorobenzyl)piperidin-4-yl)-2-hydroxy-4-oxobut-2-enoate
- 5
(Z)-4-(4-(4-chlorobenzyl)-1-(cyclohexylmethyl)piperidin-4-yl)-2-hydroxy-4-oxobut-2-enoic acid
- 6
(Z)-methyl 4-(4-(4-chlorobenzyl)-1-(cyclohexylmethyl)piperidin-4-yl)-2-hydroxy-4-oxobut-2-enoate
- 7
2,3-dihydroxyfumaric acid
- 8
(Z)-3-hydroxy-1-phenyl-3-(1H-1,2,4-triazol-5-yl)prop-2-en-1-one
- 9
(Z)-3-hydroxy-1-(2-hydroxyphenyl)-3-phenylprop-2-en-1-one
- 10
(Z)-4-(1-(4-fluorobenzyl)-1H-pyrrol-2-yl)-2-hydroxy-4-oxobut-2-enoic acid
- 11
(Z)-methyl 4-(1-(4-fluorobenzyl)-1H-pyrrol-2-yl)-2-hydroxy-4-oxobut-2-enoate
- 12
(Z)-2-hydroxy-4-oxo-4-phenylbut-2-enoic acid
- 13
(Z)-methyl 2-hydroxy-4-oxo-4-phenylbut-2-enoate
- 14
(Z)-2-hydroxy-4-(2-methoxyphenyl)-4-oxobut-2-enoic acid
- 15
(Z)-methyl 2-hydroxy-4-(2-methoxyphenyl)-4-oxobut-2-enoate
- 16
(Z)-2-hydroxy-4-(4-methoxyphenyl)-4-oxobut-2-enoic acid
- 17
(Z)-4-(4-chlorophenyl)-2-hydroxy-4-oxobut-2-enoic acid
- 18
(Z)-4-(2-chlorophenyl)-2-hydroxy-4-oxobut-2-enoic acid
- 19
(Z)-methyl 4-(2-chlorophenyl)-2-hydroxy-4-oxobut-2-enoate
- 20
(Z)-4-(3,5-bis(benzyloxy)phenyl)-2-hydroxy-4-oxobut-2-enoic acid
- 21
(2Z,5Z)-2,6-dihydroxy-4-oxo-6-phenylhexa-2,5-dienoic acid
- 22
(2Z,5Z)-methyl 2,6-dihydroxy-4-oxo-6-phenylhexa-2,5-dienoate
- 23
(2Z,5Z)-2,6-dihydroxy-6-(2-methoxyphenyl)-4-oxohexa-2,5-dienoic acid
- 24
(2Z,5Z)-6-(2-chlorophenyl)-2,6-dihydroxy-4-oxohexa-2,5-dienoic acid
- 25
6-benzyl-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
- 26
6-benzyl-1-(2-hydroxyethyl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
- 27
1,6-dibenzyl-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
- 28
ethyl 1,6-dibenzyl-4-oxo-1,4-dihydroquinoline-3-carboxylate
- 29
N-(4-fluorobenzyl)-5-hydroxy-2-isopropyl-1-methyl-6-oxo-1,6-dihydropyrimidine-4-carboxamide
- 30
(Z)-2-hydroxy-4-(1-methyl-1H-indol-2-yl)-4-oxobut-2-enoic acid
- 31
(Z)-4-(1-benzyl-1H-indol-2-yl)-2-hydroxy-4-oxobut-2-enoic acid
- 32
(Z)-methyl 2-hydroxy-4-(1-methyl-1H-indol-2-yl)-4-oxobut-2-enoate
- 33
(Z)-methyl 4-(1-ethyl-1H-indol-2-yl)-2-hydroxy-4-oxobut-2-enoate
- 34
(Z)-2-hydroxy-4-(5-methyl-5H-[1,3]dioxolo[4,5-f]indol-6-yl)-4-oxobut-2-enoic acid
- 35
(Z)-4-(5-ethyl-5H-[1,3]dioxolo[4,5-f]indol-6-yl)-2-hydroxy-4-oxobut-2-enoic acid
- 36
(Z)-4-(5-benzyl-5H-[1,3]dioxolo[4,5-f]indol-6-yl)-2-hydroxy-4-oxobut-2-enoic acid
- 37
(Z)-methyl 4-(5-benzyl-5H-[1,3]dioxolo[4,5-f]indol-6-yl)-2-hydroxy-4-oxobut-2-enoate
- 38
(Z)-methyl 2-hydroxy-4-(5-methyl-5H-[1,3]dioxolo[4,5-f]indol-6-yl)-4-oxobut-2-enoate
- 39
(Z)-2-hydroxy-4-(1-methyl-1H-indol-3-yl)-4-oxobut-2-enoic acid
- 40
(Z)-4-(1-ethyl-1H-indol-3-yl)-2-hydroxy-4-oxobut-2-enoic acid
- 41
(Z)-4-(1-benzyl-1H-indol-3-yl)-2-hydroxy-4-oxobut-2-enoic acid
- 42
(Z)-2-hydroxy-4-(5-methyl-5H-[1,3]dioxolo[4,5-f]indol-7-yl)-4-oxobut-2-enoic acid
- 43
(Z)-methyl 2-hydroxy-4-(5-methyl-5H-[1,3]dioxolo[4,5-f]indol-7-yl)-4-oxobut-2-enoate
- 44
3-(1-ethyl-1H-indol-3-yl)-4-hydroxy-1H-pyrrole-2,5-dione
- 45
(E)-2-hydroxy-2-(2-oxoindolin-3-ylidene)acetic acid
- 46
(E)-methyl 2-hydroxy-2-(2-oxoindolin-3-ylidene)acetate
- 47
(E)-methyl 2-hydroxy-2-(2-oxo-1-phenylindolin-3-ylidene)acetate
- DFO
Dragonfly Orange dye
- DKA
β-diketo acid
- DPBA
2,4-dioxo-4-phenylbutanoic acid
- fluc
firefly luciferase
- HIV IN
human immunodeficiency virus integrase
- MB
molecular beacon
- MDCK
Madin-Darby canine kidney
- ML
metal ligator
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- PAI
influenza virus PA inhibitor
- PA-Nter
N-terminal part of influenza virus PA
- PSA
polar surface area
- RT-PCR
reverse transcription–polymerase chain reaction
- vRNA
viral RNA
- vRNP
viral ribonucleoprotein
Authorship Contributions
Participated in research design: Stevaert, Naesens, Sechi, Rogolino, Kim.
Conducted experiments: Stevaert, Nurra, Pala, Carcelli, Shepard, Domaoal.
Contributed new reagents or analytic tools: Marras.
Performed data analysis: Stevaert, Naesens, Sechi, Rogolino, Kim, Alfonso-Prieto.
Wrote or contributed to the writing of the manuscript: Stevaert, Naesens, Sechi, Alfonso-Prieto.
Footnotes
A. Stevaert is holder of a PhD grant from the Flemish Agency for Innovation by Science and Technology (IWT). The authors acknowledge financial support by the Geconcerteerde Onderzoeksacties [GOA/15/019/TBA] from the KU Leuven; the Emory Pediatric Center for Drug Discovery fund; the Fondazione Banco di Sardegna; and the Italian Ministero dell’Istruzione, dell’Università e della Ricerca [PRIN 2010, 2010W2KM5L_003]. S.A.E. Marras is among a group of inventors who earn royalties for molecular beacon usage.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Agrawal A, DeSoto J, Fullagar JL, Maddali K, Rostami S, Richman DD, Pommier Y, Cohen SM. (2012) Probing chelation motifs in HIV integrase inhibitors. Proc Natl Acad Sci USA 109:2251–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacchi A, Biemmi M, Carcelli M, Carta F, Compari C, Fisicaro E, Rogolino D, Sechi M, Sippel M, Sotriffer CA, et al. (2008) From ligand to complexes. Part 2. Remarks on human immunodeficiency virus type 1 integrase inhibition by beta-diketo acid metal complexes. J Med Chem 51:7253–7264. [DOI] [PubMed] [Google Scholar]
- Bacchi A, Carcelli M, Compari C, Fisicaro E, Pala N, Rispoli G, Rogolino D, Sanchez TW, Sechi M, Sinisi V, et al. (2011) Investigating the role of metal chelation in HIV-1 integrase strand transfer inhibitors. J Med Chem 54:8407–8420. [DOI] [PubMed] [Google Scholar]
- Baughman BM, Jake Slavish P, DuBois RM, Boyd VA, White SW, Webb TR. (2012) Identification of influenza endonuclease inhibitors using a novel fluorescence polarization assay. ACS Chem Biol 7:526–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman JD, Patel D, Baker SF, Vijayan RS, Xiang A, Parhi AK, Martínez-Sobrido L, LaVoie EJ, Das K, Arnold E. (2013) Crystallographic fragment screening and structure-based optimization yields a new class of influenza endonuclease inhibitors. ACS Chem Biol 8:2501–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock CW, Katz AK, Markham GD, Glusker JP. (1999) Manganese as a replacement for magnesium and zinc: functional comparison of the divalent ions. J Am Chem Soc 121:7360–7372. [Google Scholar]
- Bouloy M, Plotch SJ, Krug RM. (1978) Globin mRNAs are primers for the transcription of influenza viral RNA in vitro. Proc Natl Acad Sci USA 75:4886–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carcelli M, Rogolino D, Bacchi A, Rispoli G, Fisicaro E, Compari C, Sechi M, Stevaert A, Naesens L. (2014) Metal-chelating 2-hydroxyphenyl amide pharmacophore for inhibition of influenza virus endonuclease. Mol Pharm 11:304–316. [DOI] [PubMed] [Google Scholar]
- Chen E, Swift RV, Alderson N, Feher VA, Feng GS, Amaro RE. (2014) Computation-guided discovery of influenza endonuclease inhibitors. ACS Med Chem Lett 5:61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianci C, Chung TDY, Meanwell N, Putz H, Hagen M, Colonno RJ, Krystal M. (1996) Identification of N-hydroxamic acid and N-hydroxy-imide compounds that inhibit the influenza virus polymerase. Antivir Chem Chemother 7:353–360. [Google Scholar]
- Crépin T, Dias A, Palencia A, Swale C, Cusack S, Ruigrok RW. (2010) Mutational and metal binding analysis of the endonuclease domain of the influenza virus polymerase PA subunit. J Virol 84:9096–9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta K, Wolkerstorfer A, Szolar OH, Cusack S, Klumpp K. (2013) Characterization of PA-N terminal domain of Influenza A polymerase reveals sequence specific RNA cleavage. Nucleic Acids Res 41:8289–8299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias A, Bouvier D, Crépin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, Ruigrok RW. (2009) The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458:914–918. [DOI] [PubMed] [Google Scholar]
- DuBois RM, Slavish PJ, Baughman BM, Yun MK, Bao J, Webby RJ, Webb TR, White SW. (2012) Structural and biochemical basis for development of influenza virus inhibitors targeting the PA endonuclease. PLoS Pathog 8:e1002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilligay D, Kadlec J, Crépin T, Lunardi T, Bouvier D, Kochs G, Ruigrok RW, Cusack S. (2014) Comparative structural and functional analysis of orthomyxovirus polymerase cap-snatching domains. PLoS ONE 9:e84973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Schmidt FI, Crow M, Brownlee GG. (2006) Amino acid residues in the N-terminal region of the PA subunit of influenza A virus RNA polymerase play a critical role in protein stability, endonuclease activity, cap binding, and virion RNA promoter binding. J Virol 80:7789–7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings JC, Selnick H, Wolanski B, Tomassini JE. (1996) Anti-influenza virus activities of 4-substituted 2,4-dioxobutanoic acid inhibitors. Antimicrob Agents Chemother 40:1304–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, et al. (2000) Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 287:646–650. [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Mahmood K, Yang CF, Webster RG, Greenberg HB, Kemble G. (2002) Rescue of influenza B virus from eight plasmids. Proc Natl Acad Sci USA 99:11411–11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton NC, Perona JJ. (2004) DNA cleavage by EcoRV endonuclease: two metal ions in three metal ion binding sites. Biochemistry 43:6841–6857. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Fujii S. (2011) Binding mode prediction and inhibitor design of anti-influenza virus diketo acids targeting metalloenzyme RNA polymerase by molecular docking. Bioinformation 6:221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai Y, Takahashi H, Hatakeyama D, Motoshima K, Ishikawa M, Sugita K, Hashimoto Y, Harada Y, Itamura S, Odagiri T, et al. (2010) Anti-influenza activity of phenethylphenylphthalimide analogs derived from thalidomide. Bioorg Med Chem 18:5379–5390. [DOI] [PubMed] [Google Scholar]
- Kim M, Kim SY, Lee HW, Shin JS, Kim P, Jung YS, Jeong HS, Hyun JK, Lee CK. (2013) Inhibition of influenza virus internalization by (-)-epigallocatechin-3-gallate. Antiviral Res 100:460–472. [DOI] [PubMed] [Google Scholar]
- Kowalinski E, Zubieta C, Wolkerstorfer A, Szolar OH, Ruigrok RW, Cusack S. (2012) Structural analysis of specific metal chelating inhibitor binding to the endonuclease domain of influenza pH1N1 (2009) polymerase. PLoS Pathog 8:e1002831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzuhara T, Iwai Y, Takahashi H, Hatakeyama D, Echigo N. (2009) Green tea catechins inhibit the endonuclease activity of influenza A virus RNA polymerase. PLoS Curr 1:RRN1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26. [DOI] [PubMed] [Google Scholar]
- Marchand C, Johnson AA, Karki RG, Pais GC, Zhang X, Cowansage K, Patel TA, Nicklaus MC, Burke TR, Jr, Pommier Y. (2003) Metal-dependent inhibition of HIV-1 integrase by beta-diketo acids and resistance of the soluble double-mutant (F185K/C280S). Mol Pharmacol 64:600–609. [DOI] [PubMed] [Google Scholar]
- Maret W. (2010) Metalloproteomics, metalloproteomes, and the annotation of metalloproteins. Metallomics 2:117–125. [DOI] [PubMed] [Google Scholar]
- Marras SAE, Tyagi S, Kramer FR. (2006) Real-time assays with molecular beacons and other fluorescent nucleic acid hybridization probes. Clin Chim Acta 363:48–60. [DOI] [PubMed] [Google Scholar]
- Maurin C, Bailly F, Cotelle P. (2004) Improved preparation and structural investigation of 4-aryl-4-oxo-2-hydroxy-2-butenoic acids and methyl esters. Tetrahedron 60:6479–6486. [Google Scholar]
- Meneghesso S, Vanderlinden E, Stevaert A, McGuigan C, Balzarini J, Naesens L. (2012) Synthesis and biological evaluation of pyrimidine nucleoside monophosphate prodrugs targeted against influenza virus. Antiviral Res 94:35–43. [DOI] [PubMed] [Google Scholar]
- Moncorgé O, Mura M, Barclay WS. (2010) Evidence for avian and human host cell factors that affect the activity of influenza virus polymerase. J Virol 84:9978–9986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin B, Coutard B, Lelke M, Ferron F, Kerber R, Jamal S, Frangeul A, Baronti C, Charrel R, de Lamballerie X, et al. (2010) The N-terminal domain of the arenavirus L protein is an RNA endonuclease essential in mRNA transcription. PLoS Pathog 6:e1001038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa M, Kadowaki SE, Watanabe I, Kadowaki Y, Takei M, Fukuda H. (2008) PA subunit of RNA polymerase as a promising target for anti-influenza virus agents. Antiviral Res 78:194–201. [DOI] [PubMed] [Google Scholar]
- Noble E, Cox A, Deval J, Kim B. (2012) Endonuclease substrate selectivity characterized with full-length PA of influenza A virus polymerase. Virology 433:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parhi AK, Xiang A, Bauman JD, Patel D, Vijayan RS, Das K, Arnold E, Lavoie EJ. (2013) Phenyl substituted 3-hydroxypyridin-2(1H)-ones: inhibitors of influenza A endonuclease. Bioorg Med Chem 21:6435–6446. [DOI] [PubMed] [Google Scholar]
- Parkes KE, Ermert P, Fässler J, Ives J, Martin JA, Merrett JH, Obrecht D, Williams G, Klumpp K. (2003) Use of a pharmacophore model to discover a new class of influenza endonuclease inhibitors. J Med Chem 46:1153–1164. [DOI] [PubMed] [Google Scholar]
- Plotch SJ, Bouloy M, Ulmanen I, Krug RM. (1981) A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 23:847–858. [DOI] [PubMed] [Google Scholar]
- Reddy TR, Li C, Guo X, Myrvang HK, Fischer PM, Dekker LV. (2011) Design, synthesis, and structure-activity relationship exploration of 1-substituted 4-aroyl-3-hydroxy-5-phenyl-1H-pyrrol-2(5H)-one analogues as inhibitors of the annexin A2-S100A10 protein interaction. J Med Chem 54:2080–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reguera J, Weber F, Cusack S. (2010) Bunyaviridae RNA polymerases (L-protein) have an N-terminal, influenza-like endonuclease domain, essential for viral cap-dependent transcription. PLoS Pathog 6:e1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogolino D, Carcelli M, Sechi M, Neamati N. (2012) Viral enzymes containing magnesium: metal binding as a Successful Strategy in Drug Design. Coord Chem Rev 256:3063–3086. [Google Scholar]
- Ruigrok RW, Crépin T, Hart DJ, Cusack S. (2010) Towards an atomic resolution understanding of the influenza virus replication machinery. Curr Opin Struct Biol 20:104–113. [DOI] [PubMed] [Google Scholar]
- Sagong HY, Parhi A, Bauman JD, Patel D, Vijayan RSK, Das K, Arnold E, LaVoie EJ. (2013) 3-Hydroxyquinolin-2(1H)-ones as inhibitors of influenza A endonuclease. ACS Med Chem Lett 4:547–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechi M, Derudas M, Dallocchio R, Dessì A, Bacchi A, Sannia L, Carta F, Palomba M, Ragab O, Chan C, et al. (2004) Design and synthesis of novel indole beta-diketo acid derivatives as HIV-1 integrase inhibitors. J Med Chem 47:5298–5310. [DOI] [PubMed] [Google Scholar]
- Sechi M, Sannia L, Carta F, Palomba M, Dallocchio R, Dessì A, Derudas M, Zawahir Z, Neamati N. (2005) Design of novel bioisosteres of beta-diketo acid inhibitors of HIV-1 integrase. Antivir Chem Chemother 16:41–61. [DOI] [PubMed] [Google Scholar]
- Sechi M, Bacchi A, Carcelli M, Compari C, Duce E, Fisicaro E, Rogolino D, Gates P, Derudas M, Al-Mawsawi LQ, et al. (2006) From ligand to complexes: inhibition of human immunodeficiency virus type 1 integrase by beta-diketo acid metal complexes. J Med Chem 49:4248–4260. [DOI] [PubMed] [Google Scholar]
- Singh SB, Tomassini JE. (2001) Synthesis of natural flutimide and analogous fully substituted pyrazine-2,6-diones, endonuclease inhibitors of influenza virus. J Org Chem 66:5504–5516. [DOI] [PubMed] [Google Scholar]
- Stevaert A, Dallocchio R, Dessì A, Pala N, Rogolino D, Sechi M, Naesens L. (2013) Mutational analysis of the binding pockets of the diketo acid inhibitor L-742,001 in the influenza virus PA endonuclease. J Virol 87:10524–10538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomassini J, Selnick H, Davies ME, Armstrong ME, Baldwin J, Bourgeois M, Hastings J, Hazuda D, Lewis J, McClements W, et al. (1994) Inhibition of cap (m7GpppXm)-dependent endonuclease of influenza virus by 4-substituted 2,4-dioxobutanoic acid compounds. Antimicrob Agents Chemother 38:2827–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomassini JE, Davies ME, Hastings JC, Lingham R, Mojena M, Raghoobar SL, Singh SB, Tkacz JS, Goetz MA. (1996) A novel antiviral agent which inhibits the endonuclease of influenza viruses. Antimicrob Agents Chemother 40:1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treanor JJ, Talbot HK, Ohmit SE, Coleman LA, Thompson MG, Cheng PY, Petrie JG, Lofthus G, Meece JK, Williams JV, et al. US Flu-VE Network (2012) Effectiveness of seasonal influenza vaccines in the United States during a season with circulation of all three vaccine strains. Clin Infect Dis 55:951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderlinden E, Göktas F, Cesur Z, Froeyen M, Reed ML, Russell CJ, Cesur N, Naesens L. (2010) Novel inhibitors of influenza virus fusion: structure-activity relationship and interaction with the viral hemagglutinin. J Virol 84:4277–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderlinden E, Vanstreels E, Boons E, ter Veer W, Huckriede A, Daelemans D, Van Lommel A, Rőth E, Sztaricskai F, Herczegh P, et al. (2012) Intracytoplasmic trapping of influenza virus by a lipophilic derivative of aglycoristocetin. J Virol 86:9416–9431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderlinden E, Naesens L. (2014) Emerging antiviral strategies to interfere with influenza virus entry. Med Res Rev 34:301–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakai C, Iwama M, Mizumoto K, Nagata K. (2011) Recognition of cap structure by influenza B virus RNA polymerase is less dependent on the methyl residue than recognition by influenza A virus polymerase. J Virol 85:7504–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Klein ML, LeBard DN, Levine BG, Liang H, MacDermaid CM, Alfonso-Prieto M. (2014) Magnesium-dependent RNA binding to the PA endonuclease domain of the avian influenza polymerase. J Phys Chem B 118:873–889. [DOI] [PubMed] [Google Scholar]
- Yang W, Lee JY, Nowotny M. (2006) Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol Cell 22:5–13. [DOI] [PubMed] [Google Scholar]
- Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, He X, Lv Z, Ge R, Li X, Deng T, et al. (2009) Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 458:909–913. [DOI] [PubMed] [Google Scholar]
- Zeng LF, Jiang XH, Sanchez T, Zhang HS, Dayam R, Neamati N, Long YQ. (2008) Novel dimeric aryldiketo containing inhibitors of HIV-1 integrase: effects of the phenyl substituent and the linker orientation. Bioorg Med Chem 16:7777–7787. [DOI] [PubMed] [Google Scholar]
- Zhao C, Lou Z, Guo Y, Ma M, Chen Y, Liang S, Zhang L, Chen S, Li X, Liu Y, et al. (2009) Nucleoside monophosphate complex structures of the endonuclease domain from the influenza virus polymerase PA subunit reveal the substrate binding site inside the catalytic center. J Virol 83:9024–9030. [DOI] [PMC free article] [PubMed] [Google Scholar]