

Abstract
The recent, unfortunate death of Alfred G. (“Al”) Gilman, M.D., Ph.D., represents a sad signpost for an era spanning over 40 years in molecular pharmacology. Gilman’s discoveries, influence, and persona were dominant forces in research and training in pharmacology. Here, we review the progression of ideas and knowledge that spawned early work by Gilman and collaborators (among them, one of the authors) and later efforts (including those of the other author) that have recently yielded a comprehensive and precise structural understanding of fundamental topics in pharmacology: the binding of ligands to G protein-coupled receptors (GPCRs) and the interaction of GPCRs with heterotrimeric G proteins and effector molecules. Those data provide new and important insights into the molecular basis that underlies affinity and efficacy, two of the most important features of drug action, which represent the latest chapter in the saga that Al Gilman’s work helped launch.
Alfred Goodman Gilman (who preferred to be called “Al” by all who met him) was a giant in science and especially molecular pharmacology. As he described himself, he was “born with a scientific/academic silver spoon in my mouth, or perhaps a pestle (but not a mortar)” (Gilman, 2012), verbiage that captures both the conditions of his birth and his incisive sense of humor. His father, Alfred Gilman, a professor of pharmacology at Yale at the time of Al’s birth, is best known for his coauthorship with Louis S. Goodman of the first edition (in 1941, the year of Al’s birth) and several subsequent editions of The Pharmacological Basis of Therapeutics, which became known as the “Bible of Pharmacology.” The close relationship of Drs. A. Gilman and L.S. Goodman led to Al being given Goodman as his middle name; perhaps in some magical way it was also his start on a path to be a molecular pharmacologist. Moreover, Al served as editor of several later editions of the “G & G” bible, thus keeping it in the family.
After graduating from Yale with a major in biochemistry, Al entered what is now Case-Western Reserve University in one of the first M.D./Ph.D. programs in the United States. He was encouraged by Earl Sutherland [the discoverer of cyclic adenosine monophosphate (cAMP)], for which he later received the Nobel Prize) to get a Ph.D. in pharmacology (“just biochemistry with a purpose,” Sutherland said) (Gilman, 2012). Al did his Ph.D. thesis with Theodore (Ted) Rall, Sutherland’s collaborator on discoveries related to cAMP. These included not only the identification of cAMP as a critical second messenger (for hormone, neurotransmitter, and drug agonist action and as the mediator of norepinephrine action at canine hepatic β-adrenergic receptors, β-ARs) (Rall and Sutherland, 1958) but also of the key enzymes (adenyl [now adenylyl] cyclase [AC] and cyclic nucleotide phosphodiesterase) that catalyze, respectively, cAMP formation and degradation (Sutherland and Rall, 1958; Sutherland et al., 1962). Sutherland and colleagues proposed that in addition to enzymatic activity AC was the receptor for norepinephrine (Fig. 1) (Butcher and Sutherland, 1962). Meanwhile, in his Ph.D. thesis work with Ted Rall, Al assessed cAMP formation and actions in the bovine thyroid gland (Robison et al., 1967; Gilman and Rall, 1968).
Fig. 1.

Sutherland and Rall’s proposed model from 1957, illustrating their concept that the hormone receptor/adenylyl cyclase is a single membrane-bound protein. (Taken with permission from Circulation, American Heart Association; Sutherland et al., 1969.)
Hormone Receptor and Adenylyl Cyclase Are Two Proteins
After postdoctoral training in the National Institutes of Health laboratory of Nobel laureate Marshall Nirenberg, during which time Al produced a single-authored paper describing an assay for cAMP that would became the standard for many investigators at the time (Gilman, 1970), in 1974 Al joined the Department of Pharmacology at the University of Virginia. There, he initiated studies to define the molecular basis of β-AR agonist action. His early work included efforts to improve upon β-AR radioligand-binding assays (Maguire et al., 1976b). The Gilman laboratory adopted the use of a radioiodinated β-AR antagonist, which set the stage for studies to test the proposal (Fig. 1) by Sutherland and coworkers that AC also possessed β-AR binding activity (Robison et al., 1967).
One way to examine this proposal was to employ a genetic approach: using a mutant S49 lymphoma cell that had been generated from wild-type (WT) S49 cells on the basis of resistance to killing by the β-AR agonist isoproterenol, which because of its absence of AC activity was named cyc−or AC−. The reasoning was that if membranes from cyc− cells possessed similar β-ARs as WT membranes, AC and β-ARs must be the products of separate genes and thus separate proteins. One of us (P.A.I.) worked for several weeks in 1975 (together with Mike Maguire) in the Gilman laboratory to test this idea, and we found that membranes from both WT and cyc− S49 cells did indeed have β-ARs with several similar properties. The results were published 40 years ago in Molecular Pharmacology (Insel et al., 1976).
Maguire conducted additional β-AR radioligand binding studies that yielded an unexpected and important result: binding of agonists, but not antagonists, to β-ARs was modulated by the addition of guanine nucleotides (Fig. 2); thus, β-AR “agonists and antagonists can be distinguished by means of binding alone; no assessment of ‘effect’ need be made,” as (Maguire et al. (1976a) reported in a Short Communication in Molecular Pharmacology that same year.
Fig. 2.

Guanine nucleotide sensitivity of agonist sensitivity to the β−AR. Inhibition of the binding of the antagonist [I125]iodohydroxybenzylpindolol ([I125]IHYP) by the agonist epinephrine in the absence (■) and presence (□) of the nonhydrolyzable GTP analog Gpp(NH)p. The inhibition of [I125]IHYP by epinephrine is multiphasic (high- and low-affinity binding) in comparison with the antagonist alprenolol. The addition of Gpp(NH)p affects only the high-affinity binding of epinephrine. (Taken from Maguire et al., 1976a,b.)
Discovery of G Proteins
Use of cyc−/AC− S49 cells proved to be well suited for a series of studies undertaken by Gilman, Elliott Ross, and their colleagues. In what are now considered classic experiments, they reconstituted AC activity in cyc−/AC− S49 cells through the addition of soluble extracts from WT S49 cells (Ross and Gilman, 1977). The reconstituted activity was dependent on both hormones and GTP. Further studies proved the existence of G proteins and their essential role along with a G protein-coupled receptor (GPCR) and AC as the three components that are sufficient for hormone/agonist-stimulated cAMP synthesis (Ross et al., 1977, 1978; Northup et al., 1980; Ross and Gilman, 1980; Sternweis et al., 1981; May et al., 1985).
These studies built on work conducted in the National Institute of Health laboratory of Martin (“Marty”) Rodbell, who first observed an obligatory role of guanine nucleotides in mediating the activation of AC by the hormone glucagon bound to its receptor. Rodbell et al. (1971) proposed that a “discriminator, which appears to be a separate entity from adenyl [sic] cyclase” mediates this action. Rodbell and Gilman shared the 1994 Nobel Prize in Physiology or Medicine for these discoveries.
In their efforts, Gilman and his colleagues used a reductionist approach: preparing purified plasma membranes then solubilization, isolation, and purification of the critical components required for receptor-stimulated AC activity and the proof of their roles in this signaling system through in vitro reconstitution of that activity (Ross and Gilman, 1977, 1980). Figure 3, taken from one of their studies, illustrates the capacity of soluble extracts from WT cells to restore hormone-stimulated AC activity in S49 cyc− cell membranes, an activity that is GTP-dependent and can bypass the receptor by the use of sodium fluoride. This landmark discovery required ingenuity, perspicacity, creativity, intense effort, and as Gilman has noted, “good luck” that needs to be recognized, embraced, and “whatever it takes to extract its full value” (Gilman, 2012).
Fig. 3.

Separation of a nucleotide-sensitive extract containing a heat-sensitive activator of adenylyl cyclase (AC). In this classic experiment, extracts from wild-type cells (donor membranes) were added to membrane fractions from S49 cyc− cells to restore hormone-stimulated AC activity. Concentration-dependent increases in AC activity in S49 cyc− membrane fractions were observed upon addition of detergent extracts from donor membranes (wild-type cells) in the presence of GTP with (□) or without agonist isoproterenol (▲), or with the addition of sodium fluoride (○). No such activity was observed using detergent extracts from S49 cyc− cells. (Reprinted from Ross and Gilman (1977), with the permission of Dr. Elliott Ross, University of Texas Southwestern.)
An example of these qualities was the demonstration—through a series of elegant purification and reconstitution studies spearheaded by postdoctoral fellows in the Gilman laboratory (John Northup, Emanuel Hanski, and Paul Sternweis)—that the Gα subunit was directly responsible for the GTP-dependent activation of AC and that its dissociation from the Gβ, also GTP-dependent, was critical to this activation (Northup et al., 1980; Hanski et al., 1981; Sternweis et al., 1981; Katada et al., 1984; May et al., 1985). Among these studies, Al3+ was identified as the counter ion for fluorine, which together as AlF4− mimics the γ-phosphate of GTP. It was later determined that GDP•Mg2+•AlF4− bound to Gα represented an approximation of the transition state (Coleman et al., 1994). It was through the detective work of Paul Sternweis in the Gilman laboratory that Al3+, at the time a contaminant of ATP preparations as well as a contaminant of detergents used to wash laboratory glassware, was discovered to be not only the culprit but also the companion in arms with the activator sodium fluoride (Sternweis and Gilman, 1982).
Through a series of in-depth and comprehensive studies, the Gilman laboratory (in work spearheaded by Gary Bokoch, Richard Kahn, Toshiaki Katada, and others in the early 1980s) also described the mechanism of action of two bacteria-derived toxins, cholera (CT) and pertussis toxin (PTX), as the ADP ribosylation of Gs and Gi proteins, respectively (Schleifer et al., 1982; Bokoch et al., 1983; Kahn and Gilman, 1984). This work, together with contributions from others, provided a sound understanding of the pathogenic mechanism for these toxins as well as helped to fortify understanding of receptor coupling (for PTX) and GTPase activity (for CT). In addition, their work yielded useful agents to assess the biologic roles of Gi and Gs.
Incidentally, Gβ was initially considered “that darn 35 kDa contaminant,” as Gilman referred to it, because it was not one of the proteins that could be ADP-ribosylated by CT or PTX, but usually copurified with Gα, depending on the quality of the preparations (i.e., the integrity of the heterotrimer). Gβ from Gα dissociation stimulated by AlFl4−, however, suggested that they were indeed a functional complex (Sternweis et al., 1981). Amusingly, although Gγ forms an obligate heterodimer with Gβ, it was initially missed because it ran off the sodium dodecyl sulfate polyacrylamide gel electrophoresis gels due its small size (#x223c;8 kDa) and did not stain well. In fact, the three components of the G protein (heterotrimer) were first identified in the visual G protein, transducin, by Fung and Stryer in 1980 (Fung et al., 1981), but a consensus on the parallels between the visual system and hormone regulation of AC were not agreed upon until contributions from the Bitensky laboratory (Bitensky et al., 1982) and Gilman laboratory (Manning and Gilman, 1983) compared them directly.
The functional relevance of Gβγ, other than being a cofactor for Gα, was not fully realized until work from David Clapham’s laboratory on an ion channel—later known as G protein inward rectifying K+ channels (GIRK channels), and not directly related to AC—was shown to be directly activated by Gβγ (Logothetis et al., 1987). This work stimulated a plethora of studies that suggested the direct interaction with Gα and Gβγ on various effector systems, ranging from AC (Tang and Gilman, 1991) to phospholipase C (PLC) (Blank et al., 1992).
Later work by others showed that a purified GPCR (β2-AR), Gs protein, and biochemically resolved catalytic AC could reconstitute hormone-stimulated AC activity (Cerione et al., 1984). Efforts by numerous investigators—including Gilman’s laboratory, in part through work together with others, including Melvin Simon—revealed that heterotrimeric G proteins are composed of αβγ subunits and that the Gsα protein is part of a family of GTP-binding proteins, which includes Giα, Goα, Gtα (transducin), and others (Manning and Gilman, 1983; Hurley et al., 1984; Stryer and Bourne, 1986; Simon et al., 1991). Together, these signaling partners culminate in the mechanism for hormone-stimulated AC described in Fig. 4A, in which the G proteins function as timed molecular switches that are activated by agonist-occupied GPCRs, which promote the exchange of GTP for GDP bound to Gα proteins, thereby enhancing the ability of the G protein subunits to regulate effector molecules.
Fig. 4.

The G protein cycle: G proteins as timed molecular switches. (A) Cartoon of the G protein cycle. 1) Hormone binding to a cell surface GPCR activates the heterotrimeric G protein by promoting GDP release from the Gα-subunit. 2) High intracellular concentrations of GTP allow rapid binding to the Gα subunit, causing in a conformational change in Gα that affects its interaction with Gβγ, resulting in its functional dissociation. 3) The dissociated subunits can now interact with effectors, in this case allowing Gαs to bind to and activate AC. 4) Hydrolysis of GTP serves as a timing mechanism for effector regulation because the affinity of the GDP-bound form of Gα for the effector is lower. The hydrolysis can be accelerated by the effector itself or by GTPase accelerating proteins such as Regulators of G protein Signaling (RGS). 5) GDP-bound Gα prefers to rebind Gβγ rather than the effector, resulting in the reformation of the inactive heterotrimer. 6) Formation of a precoupled or preassociated complex between the inactive receptor and inactive heterotrimer. (B) High-resolution view of the G protein cycle. Complexes in A were replaced with models based on X-ray crystallographic data. Renderings for each step in the cycle were generated based on the following PDB files: 1) β2-AR•Gαsβγ (PDB: 3SN6), 2) Gβγ (model based on PDB:1TGB) and Gαs•GTPγS (PDB: 1AZT), 3) Gαs•GTPγS•AC (PDB:1AZS), 4) Gαs•GDP •AC (modeled on PDB:1AZS) and Gαi1•GDP•AlF4•RGS4 (1AGR), and 5) Gαiβγ (PDB: 1GP2) and β2-AR (2RH1).
Molecular Biology of G Proteins and Adenylyl Cyclase
The use of biochemical and pharmacologic approaches was followed by the application of molecular biologic techniques: after accrual of information from amino acid sequencing of purified Gα subunits, the synthesis of cDNA probes based on those sequences and screening of various cDNA libraries culminated in the molecular cloning of full-length G proteins, starting with Gαs (Harris et al., 1985). This effort, largely through the contributions in the Gilman laboratory of Bruce Harris and Janet Robishaw in the mid-1980s, led to the subsequent identification and molecular cloning of other Gα isoforms as well as the Gβ-and Gγ-subunits, in work conducted by several other laboratories in the field. The application of site-directed mutagenesis to ascertain the role of key residues of the proteins identified their functional domains and provided mechanistic insights regarding the function of GPCR, heterotrimeric G protein, and effector molecules (AC and others, including PLC and certain ion channels).
An unexpected outcome of such efforts was the discovery that each component was part of a family of structurally related members, some of which had unknown function. For Gα proteins, of which there are 23 family members, Gαs and Gαi were initially identified as stimulants and inhibitors, respectively, of AC. Gα proteins with other actions were subsequently discovered (e.g., Gαq/Gα11, which activate PLC-β); some Gα proteins initially had unclear functional activities (e.g., Gα15, Gα16). The focus shifted from Gα proteins being the exclusively mediators of functional effects to the recognition that Gβγ (the two proteins function as a dimer) has a role in signal transduction, not only by inhibiting the actions of Gα proteins (because the Gαβγ trimer is inactive); it also can in some settings promote or oppose Gα actions (e.g., to stimulate or inhibit certain isoforms of AC) or function alone (e.g., to regulate activity of certain ion channels and phosphoinositide 3-kinase, or to help “recruit” proteins, such as certain G protein–coupled receptor kinases, to the plasma membrane).
Through the use of biochemical and molecular biologic approaches, the Gilman laboratory played a major role in the initial cloning of a mammalian ACs, which revealed the unexpected: its deduced structure was akin to that of previously cloned channels and transporters, with two sets of 6-transmembrane spanning regions and two homologous cytosolic domains. The initial cloning of the Ca2+/CaM-sensitive type I AC in 1989 was largely through the heroic efforts of Jack Krupinski and others in the Gilman laboratory, together with the help of Randy Reed’s laboratory (Krupinski et al., 1989). Shortly thereafter, the Reed and Gilman laboratories isolated the cDNA for a new isoform, a Ca2+/CaM-insensitive type II AC (Feinstein et al., 1991).
The isolation of these cDNAs facilitated the biochemical characterization of these enzyme systems. Wei-Jen Tang and Ron Taussig in the Gilman laboratory used recombinant expression systems to express, purify, and characterize various AC isoforms and to reveal their complex and diverse regulation (Tang and Gilman, 1991; Taussig et al., 1993a,b). Work by the Gilman laboratory and others revealed the existence of seven more membrane-bound, G protein–regulated ACs (yet another example of the multiplicity among key signaling molecules); these AC isoforms also revealed large diversity in the way they are regulated (Sunahara et al., 1996).
Regulators of G Protein Signaling (RGS Molecules)
A major subsequent discovery made by several groups and related to heterotrimeric G protein signaling was that a family of proteins, termed the regulators of G protein signaling (RGS), modulate the activity of the proteins. Gilman and his colleagues advanced our understanding of RGS proteins by demonstrating their capacity to serve as GTPase-activating proteins (GAPs) for certain Gα subunits. This discovery revealed key functional similarities between low-molecular-weight (#x223c;20 kDa) G proteins (such as Ras, Rho, and Rac) and Gα subunits (#x223c;40–50 kDa) proteins in terms of GTP hydrolysis. Heterotrimeric G proteins use GPCRs as GTP exchange factors (GEFs) to catalyze the release of prebound GDP and the binding of GTP. Owing to the high intracellular GTP concentration, the rapid GTP binding event activates the G protein. Agonist occupancy of GPCRs promotes this GEF function for Gα protein activation. Low-molecular-weight G proteins (ras-like), on the other hand, use cytosolic proteins as GEFs. To terminate signaling through hydrolysis of GTP to GDP, ras-like G proteins have separate GAPs whereas heterotrimeric G proteins can use RGS proteins to help foster GTP hydrolysis. In an interesting integration between ras-like and heterotrimeric G proteins are certain members of the RhoGEF family (P115, PDZ-RhoGEF, LARG, p63RhoGEF, and Trio). The Gilman and Sternweis laboratories determined that these multidomain proteins contain both an RGS domain that interacts and GAPs the Gα12/13 isoforms and a GEF motif that facilitates exchange on the rhoA family of low-molecular-weight G proteins (Hart et al., 1998; Kozasa et al., 1998).
Structural Biology of G Proteins
Through a strong and lasting collaboration with Stephen Sprang’s laboratory, the Gilman laboratory brought together biochemistry and molecular biology to investigate the structural biology of the G protein signaling pathway. These efforts in the mid-1990s, along with independent contributions by Paul Sigler’s and Heidi Hamm’s laboratories, replaced many of the characters illustrated in Fig. 4A with high-resolution structures (Fig. 4B). Through a series of seminal studies by the Sprang and Gilman laboratories, largely through the heroic efforts of a young graduate student, Ethan Lee, and postdoctoral fellow in the Sprang laboratory, David Coleman, each of these species was isolated and trapped, their X-ray crystal structures were determined, and their mechanisms of actions were elucidated. Gilman’s early studies identifying the γ-phosphate of GTP as critical for the functional dissociation of the Gβγ subunits as well as for enhancing the capacity of Gα subunits to regulate AC activity proved to be critical for the structural studies. Indeed, the structures reveal that the γ-phosphate of GTP helps to stabilize three regions, termed switch regions (swI, swII, and swIII), one of which, swII, interacts directly with Gβ but also with the effectors such as AC (Fig. 5) (Coleman et al., 1994; Mixon et al., 1995). GTP appears to stabilize an unfavorable swII conformation for binding Gβ, but it also is well suited to bind and activate AC (Fig. 5, B and C). In contrast, the GDP-bound form has much higher affinity for Gβγ but only modestly lower affinity for AC (10-fold), suggesting that the signal-terminating role of GTP hydrolysis enhances the interaction with Gβγ rather than lowering the affinity for the effector (Sunahara et al., 1997).
Fig. 5.
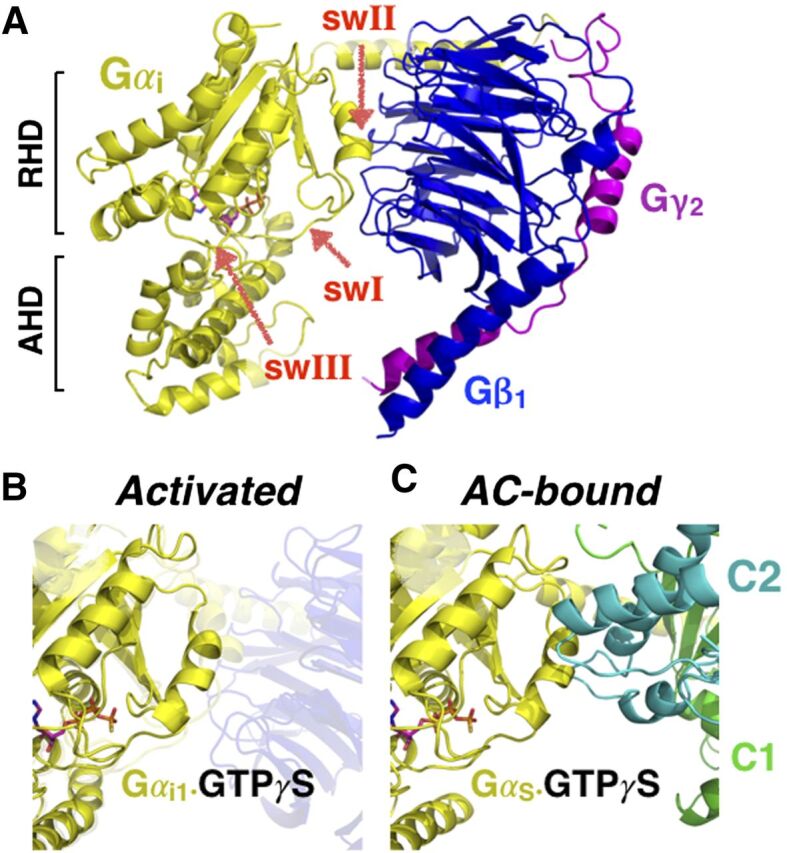
Structure of the G protein heterotrimer. (A) The GDP-bound Gα subunit (yellow) bound to Gβ1 (blue) and Gγ2 (magenta) through the switch II (swII) and the Gα-amino terminus. Gα is composed of the ras homology domain (RHD), named for its structural similarity to the ras family of small-molecular-weight G proteins, and the α-helical domain (AHD). (B) Activation of the Gα through GTP-binding restructures the swII region decreasing its affinity for Gβγ but increasing its affinity for the effector (C), in this Gsα•GTPγS bound to AC.
Hydrolysis of GTP
The structures also reveal that swII and swI contain residues critical in supporting the hydrolysis of GTP to GDP•Pi. To approximate the transition state, Gαi was crystallized bound to GDP•AlF4, in which AlF4− mimics the leaving γ-phosphate (Fig. 6A) (Coleman et al., 1994). In the structure, an attacking water molecule was identified as well as the residues responsible for coordinating the water (e.g., Gln204 in Gαi1, Gln227 in Gαs, or Gln209 in Gαq) and residues previously identified by mutagenesis to be critical for hydrolysis of GTP. Indeed, several somatic mutations of Gln227 (to leucine) in Gαs as well as similar Gln substitutions in Gαq (Gln205Leu) have been found in several human tumors (O’Hayre et al., 2013). The interaction of RGS molecules with Gα-subunits (all isoforms except Gαs) likewise helps to orient swII (and swI) in a more optimal manner and position the attacking water molecule (Fig. 6) (Tesmer et al., 1997a).
Fig. 6.

Hydrolysis of GTP by Gα. (A) Transition state of the Gα based on the Gαi1•GDP•AlF4 structure superimposed onto the structure of Gαi1•GTP. The highly conserved Gln204 in the swII region coordinates an attacking water molecule, leading to the nucleophilic attack of the γ-phosphate of GTP (PDB: 1GFI). (B) Switch II can also be further stabilized to promote better geometry for the Gln204 through an interaction with RGS4, a GTPase accelerating protein (PDB: 1AGR). (C) Hydrolysis results in the subtle relaxation of the swII region (PDB: 1GIT). (D) Pi release results in the complete disordering of swII (PDB: 1GDD).
The other catalytic residue accelerating the GTPase activity is an arginine (Arg201 in Gαs, Arg178 in Gαi1, or Arg193 in Gαq), located in the α-helical domain (AHD in Fig. 5, A and B). As with Gln227 in Gαs, substitution of Arg201 (to His or Cys) severely impairs GTPase activity and, of note, has appeared in several tumors (O’Hayre et al., 2013). Interestingly, the catalytic arginine in Gs (Arg201) and Gt (Arg174) are the substrates for ADP ribosylation by CT (Angus et al., 1986), suggesting that modification of the arginine side chain impairs its capacity to support GTP hydrolysis.
Adenylyl Cyclase
Although the structural analysis of full length-AC with its 12 transmembrane domains had remained elusive, Wei-Jen Tang, Richard Wisnant, Carmen Dessauer, and one of us (R.K.S.) were able to isolate the highly conserved cytoplasmic domains and demonstrate full catalytic activity in response to Gαs and the diterpene forskolin. In collaboration with John Tesmer (Sprang laboratory), we were able to purify a complex and eventually delineate the crystal structure of the catalytic core of AC bound to Gαs•GTPγS and forskolin.
As illustrated in Fig. 7 the catalytic core of AC is composed of the first cytoplasmic loop (C1, mauve) and the closely sequence-related second cytoplasmic loop (C2, cyan), arranged with 2-fold pseudo-symmetry (Tesmer et al., 1997b). The location of the forskolin-binding site came as a surprise: first, because the relatively hydrophobic polycyclic compound was thought to bind to the transmembrane domain; second, because it bound the pseudo-symmetrically related site to the ATP or catalytic site (Fig. 7B). Both activators (Gs, forskolin) and substrate ATP make contacts with the C1 and C2 domains, suggesting that AC activation involves stabilization of the C1•C2 dimer and formation of the active site. Although the stimulatory Gαs (yellow) engages both domains, its binding site is proximal to forskolin such that the pseudo-symmetry of the catalytic core would allow the binding of the inhibitory G protein Gαi (Fig. 7A, modeled in transparent orange) (Dessauer et al., 1998).
Fig. 7.

Structure of Gαs-bound AC. (A) The structure of Gαs•GTPγS (yellow) bound to the C1 (mauve) and C2 (cyan) domains of the catalytic core of AC. Also illustrated is a model of the Gαi-bound form (B) Forskolin and ATP bind to the interface of the pseudo-symmetrically arranged C1 and C2 domains. (C) Structure of Gαs•GTPγS•AC with 2′,5′-dideoxy-3′ATP•Mg2+, as a mimic of substrate ATP (PDB: 1CJU). Note the two bound Mg2+ ions important for catalysis. (D) Model of structure of Gαs•GTPγS•AC bound to products pyrophosphate (PPi) and cAMP. Cyclic AMP was modeled based on the structure of 2′-deoxy-3′AMP• PPi (PDB: 1CS4).
Crystal soaks with adenosine phosphate analogs, 2′-deoxy 3′-AMP (with pyrophosphate, PPi, Fig. 8C) and 2′,5′ dideoxy-3ʹ-ATP (Fig. 8D), provided suitable models for the product cAMP• PPi and substrate ATP, respectively (Tesmer et al., 1999, 2000). Remarkably, two divalent cations (Mg2+) were found to be bound between the 2′,5′ dideoxy-3′-ATP and two highly conserved aspartate residues in the C1 domain (Fig. 8C). This geometry and organization of the active site are reminiscent of that found in DNA and RNA polymerases. Thus, like nucleotide polymerases, one of these Mg2+ ions is catalytic and aids in the deprotonation of the 3′-OH− on the ribose ring so that it may attack the α-phosphate of ATP. In AC the α-phosphate is on the same ATP molecule and yields cAMP and the leaving group pyrophosphate, whereas in polymerases the deprotonated 3′-O− on the ribose attacks the α-phosphate of an incoming nucleotide to yield single stranded DNA or RNA and the leaving group pyrophosphate (PPi). Indeed, the structure and chemistry of cAMP formation served as good templates for the guanylyl cyclases and soluble ACs, as was later demonstrated by X-ray crystallography (Steegborn et al., 2005; Winger et al., 2008).
Fig. 8.

Structure of the Gsαβγ•β2-AR complex. Structure of the nucleotide-free Gsαβγ bound to agonist (Bi 1670107) in a complex with β2-AR (PDB: 3SN6). Structure of the GDP-bound heterotrimer of Gi (left, PDB: 1GP2) compared with the nucleotide-free form of Gs heterotrimer (far right panel, receptor excluded) reveals the conformational flexibility of the Gα subunit. Loss of GDP results in a large, rigid body translation (indicated by the blue arrow) of the α-helical domain (AHD) away from the ras homology domain (RHD) (right four panels).
Although Al Gilman himself was keenly interested in the contributions of the transmembrane domains, delineation of the structure and mechanism by which AC catalyzed the conversion of ATP into cAMP was one of his longtime dreams. That being said, he felt a great degree of satisfaction in being able to replace hypothesized components in Fig. 1 (by Sutherland) with a more complex signaling pathway (Fig. 4A), and then replacing these cartoons with high-resolution atomic representations (Fig. 4B). The only remaining structure, just out of his reach, was the hormone-bound GPCR in complex with the G protein. One of our laboratories (R.K.S.) and that of Brian Kobilka succeeded in filling this missing link (Fig. 8). The structure of the nucleotide-free Gsαβγ heterotrimer bound to the β2-AR reveals not only the interaction site of Gs and β2-AR but dramatic conformational change in Gαs. These data have helped generate models for receptor-mediated G protein activation (Rasmussen et al., 2011).
Understanding of the molecular basis of drug action has advanced dramatically since Al Gilman’s entry into pharmacology—as a graduate student, at approximately the time (1965) Molecular Pharmacology was launched (Brown et al., 2015). His use of reductionist approaches to study mechanisms inspired many who followed him (including the authors of this article). In addition, his mentorship of trainees in these efforts helped many launch independent careers and become scientific leaders in their own right.
The immense scientific contributions of Al Gilman in transforming the original 1957 Sutherland model and concept of receptor-AC (Fig. 9) into an atomic resolution description is remarkable (Fig. 9, inset). The importance of GPCR signaling in physiology and medicine is undisputable, and Al Gilman’s shaping of the field is incontrovertible. In addition to training many outstanding scientists who have become leaders, predominantly in the G protein area, his influence in the molecular pharmacology of signaling is particularly outstanding. Of note, though, in his later efforts Gilman sought to place signal transduction components and their mechanisms of action into a cellular, tissue, and disease context, as reflected in his creation and leadership of the Alliance for Cellular Signaling and his subsequent scientific leadership of the Cancer Prevention and Research Institute of Texas.
Fig. 9.

Hormone regulation adenylyl cyclase activity then and now. When Al Gilman entered the field, the state of the art representation of hormone regulation of cAMP production suggested a single-membrane protein AC also regulated hormone binding. (Inset) Current model of the receptor•G protein•AC complex generated from crystallography and modeling studies, based largely on Al Gilman’s contributions. Illustrated is Gαs (yellow) activating AC (green and cyan) while still bound to the β2-AR (blue metal). The membrane-bound for of AC was modeled after the structures of Gαs•AC(C1C2) whereas the transmembrane domains were modeled after the structure of glucose transporter (PDB: 4PYP).
In one sense Gilman’s passing represents the end of an era, one characterized by fundamental discoveries that addressed major questions regarding drug and hormone action. The application of new techniques played a major part in this progress. However, we believe important questions remain, and new techniques will likely be needed to address them.
Sadly, what has ended is the guidance that Gilman has given to us and others: he led by example, using rigorous thinking, creativity, hard work, and humor to discover, inspire, and motivate. Al Gilman may have been born with an academic/scientific silver spoon (Gilman, 2012), but he used it to ingest, digest, and reshape the scientific world that he entered. We and molecular pharmacology are better for his discoveries but perhaps even more for the mentorship, friendship, vision, and values that he lived by—all of which continue to inspire us and the many others with whom he interacted.
Abbreviations
- AC
adenylyl cyclase
- β-AR
β-adrenergic receptor
- cAMP
cyclic adenosine monophosphate
- CT
cholera
- GPCR
G protein-coupled receptors
- PPi
pyrophosphate
- PTX
pertussis toxin
- WT
wild-type
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Sunahara, Insel.
Footnotes
References
- Angus CW, Van Meurs KP, Tsai SC, Adamik R, Miedel MC, Pan YC, Kung HF, Moss J, Vaughan M. (1986) Identification of the probable site of choleragen-catalyzed ADP-ribosylation in a Go alpha-like protein based on cDNA sequence. Proc Natl Acad Sci USA 83:5813–5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitensky MW, Wheeler MA, Rasenick MM, Yamazaki A, Stein PJ, Halliday KR, Wheeler GL. (1982) Functional exchange of components between light-activated photoreceptor phosphodiesterase and hormone-activated adenylate cyclase systems. Proc Natl Acad Sci USA 79:3408–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank JL, Brattain KA, Exton JH. (1992) Activation of cytosolic phosphoinositide phospholipase C by G-protein beta gamma subunits. J Biol Chem 267:23069–23075. [PubMed] [Google Scholar]
- Bokoch GM, Katada T, Northup JK, Hewlett EL, Gilman AG. (1983) Identification of the predominant substrate for ADP-ribosylation by islet activating protein. J Biol Chem 258:2072–2075. [PubMed] [Google Scholar]
- Brown JH, Catterall WA, Conn PJ, Cull-Candy SG, Dingledine R, Harden TK, Insel PA, Milligan G, Traynelis SF. (2015) The first 50 years of molecular pharmacology. Mol Pharmacol 88:139–140. [DOI] [PubMed] [Google Scholar]
- Butcher RW, Sutherland EW. (1962) Adenosine 3′,5′-phosphate in biological materials. I. Purification and properties of cyclic 3′,5′-nucleotide phosphodiesterase and use of this enzyme to characterize adenosine 3′,5′-phosphate in human urine. J Biol Chem 237:1244–1250. [PubMed] [Google Scholar]
- Cerione RA, Sibley DR, Codina J, Benovic JL, Winslow J, Neer EJ, Birnbaumer L, Caron MG, Lefkowitz RJ. (1984) Reconstitution of a hormone-sensitive adenylate cyclase system. The pure beta-adrenergic receptor and guanine nucleotide regulatory protein confer hormone responsiveness on the resolved catalytic unit. J Biol Chem 259:9979–9982. [PubMed] [Google Scholar]
- Coleman DE, Berghuis AM, Lee E, Linder ME, Gilman AG, Sprang SR. (1994) Structures of active conformations of Gi alpha 1 and the mechanism of GTP hydrolysis. Science 265:1405–1412. [DOI] [PubMed] [Google Scholar]
- Dessauer CW, Tesmer JJ, Sprang SR, Gilman AG. (1998) Identification of a Giα binding site on type V adenylyl cyclase. J Biol Chem 273:25831–25839. [DOI] [PubMed] [Google Scholar]
- Feinstein PG, Schrader KA, Bakalyar HA, Tang WJ, Krupinski J, Gilman AG, Reed RR. (1991) Molecular cloning and characterization of a Ca2+/calmodulin-insensitive adenylyl cyclase from rat brain. Proc Natl Acad Sci USA 88:10173–10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung BK, Hurley JB, Stryer L. (1981) Flow of information in the light-triggered cyclic nucleotide cascade of vision. Proc Natl Acad Sci USA 78:152–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman AG. (1970) A protein binding assay for adenosine 3′:5′-cyclic monophosphate. Proc Natl Acad Sci USA 67:305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman AG. (2012) Silver spoons and other personal reflections. Annu Rev Pharmacol Toxicol 52:1–19. [DOI] [PubMed] [Google Scholar]
- Gilman AG, Rall TW. (1968) The role of adenosine 3′,5′-phosphate in mediating effects of thyroid-stimulating hormone on carbohydrate metabolism of bovine thyroid slices. J Biol Chem 243:5872–5881. [PubMed] [Google Scholar]
- Hanski E, Sternweis PC, Northup JK, Dromerick AW, Gilman AG. (1981) The regulatory component of adenylate cyclase. Purification and properties of the turkey erythrocyte protein. J Biol Chem 256:12911–12919. [PubMed] [Google Scholar]
- Harris BA, Robishaw JD, Mumby SM, Gilman AG. (1985) Molecular cloning of complementary DNA for the alpha subunit of the G protein that stimulates adenylate cyclase. Science 229:1274–1277. [DOI] [PubMed] [Google Scholar]
- Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G. (1998) Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Galpha13. Science 280:2112–2114. [DOI] [PubMed] [Google Scholar]
- Hurley JB, Simon MI, Teplow DB, Robishaw JD, Gilman AG. (1984) Homologies between signal transducing G proteins and ras gene products. Science 226:860–862. [DOI] [PubMed] [Google Scholar]
- Insel PA, Maguire ME, Gilman AG, Bourne HR, Coffino P, Melmon KL. (1976) Beta adrenergic receptors and adenylate cyclase: products of separate genes? Mol Pharmacol 12:1062–1069. [PubMed] [Google Scholar]
- Kahn RA, Gilman AG. (1984) ADP-ribosylation of Gs promotes the dissociation of its alpha and beta subunits. J Biol Chem 259:6235–6240. [PubMed] [Google Scholar]
- Katada T, Bokoch GM, Smigel MD, Ui M, Gilman AG. (1984) The inhibitory guanine nucleotide-binding regulatory component of adenylate cyclase. Subunit dissociation and the inhibition of adenylate cyclase in S49 lymphoma cyc- and wild type membranes. J Biol Chem 259:3586–3595. [PubMed] [Google Scholar]
- Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC. (1998) p115 RhoGEF, a GTPase activating protein for Gα12 and Gα13. Science 280:2109–2111. [DOI] [PubMed] [Google Scholar]
- Krupinski J, Coussen F, Bakalyar HA, Tang WJ, Feinstein PG, Orth K, Slaughter C, Reed RR, Gilman AG. (1989) Adenylyl cyclase amino acid sequence: possible channel- or transporter-like structure. Science 244:1558–1564. [DOI] [PubMed] [Google Scholar]
- Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE. (1987) The beta gamma subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature 325:321–326. [DOI] [PubMed] [Google Scholar]
- Maguire ME, Van Arsdale PM, Gilman AG. (1976a) An agonist-specific effect of guanine nucleotides on binding to the beta adrenergic receptor. Mol Pharmacol 12:335–339. [PubMed] [Google Scholar]
- Maguire ME, Wiklund RA, Anderson HJ, Gilman AG. (1976b) Binding of (125I)iodohydroxybenzylpindolol to putative beta-adrenergic receptors of rat glioma cells and other cell clones. J Biol Chem 251:1221–1231. [PubMed] [Google Scholar]
- Manning DR, Gilman AG. (1983) The regulatory components of adenylate cyclase and transducin. A family of structurally homologous guanine nucleotide-binding proteins. J Biol Chem 258:7059–7063. [PubMed] [Google Scholar]
- May DC, Ross EM, Gilman AG, Smigel MD. (1985) Reconstitution of catecholamine-stimulated adenylate cyclase activity using three purified proteins. J Biol Chem 260:15829–15833. [PubMed] [Google Scholar]
- Mixon MB, Lee E, Coleman DE, Berghuis AM, Gilman AG, Sprang SR. (1995) Tertiary and quaternary structural changes in Gi alpha 1 induced by GTP hydrolysis. Science 270:954–960. [DOI] [PubMed] [Google Scholar]
- Northup JK, Sternweis PC, Smigel MD, Schleifer LS, Ross EM, Gilman AG. (1980) Purification of the regulatory component of adenylate cyclase. Proc Natl Acad Sci USA 77:6516–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hayre M, Vázquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, Gutkind JS. (2013) The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer 13:412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rall TW, Sutherland EW. (1958) Formation of a cyclic adenine ribonucleotide by tissue particles. J Biol Chem 232:1065–1076. [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477:549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison GA, Butcher RW, Sutherland EW. (1967) Adenyl cyclase as an adrenergic receptor. Ann N Y Acad Sci 139:703–723. [DOI] [PubMed] [Google Scholar]
- Rodbell M, Birnbaumer L, Pohl SL, Krans HM. (1971) The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. V. An obligatory role of guanylnucleotides in glucagon action. J Biol Chem 246:1877–1882. [PubMed] [Google Scholar]
- Ross EM, Gilman AG. (1980) Biochemical properties of hormone-sensitive adenylate cyclase. Annu Rev Biochem 49:533–564. [DOI] [PubMed] [Google Scholar]
- Ross EM, Gilman AG. (1977) Reconstitution of catecholamine-sensitive adenylate cyclase activity: interactions of solubilized components with receptor-replete membranes. Proc Natl Acad Sci USA 74:3715–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross EM, Howlett AC, Ferguson KM, Gilman AG. (1978) Reconstitution of hormone-sensitive adenylate cyclase activity with resolved components of the enzyme. J Biol Chem 253:6401–6412. [PubMed] [Google Scholar]
- Ross EM, Maguire ME, Sturgill TW, Biltonen RL, Gilman AG. (1977) Relationship between the beta-adrenergic receptor and adenylate cyclase. J Biol Chem 252:5761–5775. [PubMed] [Google Scholar]
- Schleifer LS, Kahn RA, Hanski E, Northup JK, Sternweis PC, Gilman AG. (1982) Requirements for cholera toxin-dependent ADP-ribosylation of the purified regulatory component of adenylate cyclase. J Biol Chem 257:20–23. [PubMed] [Google Scholar]
- Simon MI, Strathmann MP, Gautam N. (1991) Diversity of G proteins in signal transduction. Science 252:802–808. [DOI] [PubMed] [Google Scholar]
- Steegborn C, Litvin TN, Levin LR, Buck J, Wu H. (2005) Bicarbonate activation of adenylyl cyclase via promotion of catalytic active site closure and metal recruitment. Nat Struct Mol Biol 12:32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternweis PC, Gilman AG. (1982) Aluminum: a requirement for activation of the regulatory component of adenylate cyclase by fluoride. Proc Natl Acad Sci USA 79:4888–4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternweis PC, Northup JK, Smigel MD, Gilman AG. (1981) The regulatory component of adenylate cyclase. Purification and properties. J Biol Chem 256:11517–11526. [PubMed] [Google Scholar]
- Stryer L, Bourne HR. (1986) G proteins: a family of signal transducers. Annu Rev Cell Biol 2:391–419. [DOI] [PubMed] [Google Scholar]
- Sunahara RK, Dessauer CW, Gilman AG. (1996) Complexity and diversity of mammalian adenylyl cyclases. Annu Rev Pharmacol Toxicol 36:461–480. [DOI] [PubMed] [Google Scholar]
- Sunahara RK, Dessauer CW, Whisnant RE, Kleuss C, Gilman AG. (1997) Interaction of Gsalpha with the cytosolic domains of mammalian adenylyl cyclase. J Biol Chem 272:22265–22271. [DOI] [PubMed] [Google Scholar]
- Sutherland EW, Rall TW. (1958) Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J Biol Chem 232:1077–1091. [PubMed] [Google Scholar]
- Sutherland EW, Rall TW, Menon T. (1962) Adenyl cylase. I. Distribution, preparation, and properties. J Biol Chem 237:1220–1227. [PubMed] [Google Scholar]
- Sutherland EW, Robison GA, Butcher RW. (1969) Some aspects of the biological role of adenosine 3′,5′-monophosphate (cyclic AMP). Circulation 37:279–306. [Google Scholar]
- Tang W-J, Gilman AG. (1991) Type-specific regulation of adenylyl cyclase by G protein beta gamma subunits. Science 254:1500–1503. [DOI] [PubMed] [Google Scholar]
- Taussig R, Iñiguez-Lluhi JA, Gilman AG. (1993a) Inhibition of adenylyl cyclase by Gi alpha. Science 261:218–221. [DOI] [PubMed] [Google Scholar]
- Taussig R, Quarmby LM, Gilman AG. (1993b) Regulation of purified type I and type II adenylylcyclases by G protein beta gamma subunits. J Biol Chem 268:9–12. [PubMed] [Google Scholar]
- Tesmer JJ, Berman DM, Gilman AG, Sprang SR. (1997a) Structure of RGS4 bound to AlF4--activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell 89:251–261. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Dessauer CW, Sunahara RK, Murray LD, Johnson RA, Gilman AG, Sprang SR. (2000) Molecular basis for P-site inhibition of adenylyl cyclase. Biochemistry 39:14464–14471. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Sunahara RK, Gilman AG, Sprang SR. (1997b) Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gα.GTPγS. Science 278:1907–1916. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Sunahara RK, Johnson RA, Gosselin G, Gilman AG, Sprang SR. (1999) Two-metal-Ion catalysis in adenylyl cyclase. Science 285:756–760. [DOI] [PubMed] [Google Scholar]
- Winger JA, Derbyshire ER, Lamers MH, Marletta MA, Kuriyan J. (2008) The crystal structure of the catalytic domain of a eukaryotic guanylate cyclase. BMC Struct Biol 8:42. [DOI] [PMC free article] [PubMed] [Google Scholar]