

Abstract
The automatic recognition of gene names and their corresponding database identifiers in biomedical text is an important first step for many downstream text-mining applications. While current methods for tagging gene entities have been developed for biomedical literature, their performance on species other than human is sub-stantially lower due to the lack of annotation data. We therefore present the NLM-Gene corpus, a high-quality manually annotated corpus for genes developed at the US National Library of Medicine (NLM), covering ambiguous gene names, with an average of 29 gene mentions (10 unique identifiers) per document, and a broader representation of different species (including Homo sapiens, Mus musculus, Rattus norvegicus, Drosophila melanogaster, Arabidopsis thaliana, Danio rerio, etc.) when compared to previous gene annotation corpora. NLM-Gene consists of 550 PubMed abstracts from 156 biomedical journals, doubly annotated by six experienced NLM indexers, randomly paired for each document to control for bias. The annotators worked in three annotation rounds until they reached complete agreement. This gold-standard corpus can serve as a benchmark to develop & test new gene text mining algorithms. Using this new resource, we have developed a new gene finding algorithm based on deep learning which improved both on precision and recall from existing tools. The NLM-Gene annotated corpus is freely available at ftp://ftp.ncbi.nlm.nih.gov/pub/lu/NLMGene. We have also applied this tool to the entire PubMed/PMC with their results freely accessible through our web-based tool PubTator (www.ncbi.nlm.nih.gov/research/pubtator).
Keywords: Manual annotation, Gene entity recognition, Natural language processing, Biomedical Text Mining, Deep Learning
1. Introduction
Automated biomedical natural language processing (BioNLP) is increasingly important for today’s biomedical research [1–3]. PubMed® (pubmed.gov), built and maintained by the US National Library of Medicine (NLM), provides free access to more than 32 million biomedical literature articles, and PubMed Central® (PMC), the free full-text archive of biomedical and life sciences journal literature at the NLM currently comprises almost 7 million articles.
Text mining and BioNLP tools make it possible to automatically peruse this vast literature and extract key knowledge on specific biomedical topics, such as protein–protein/drug-drug interactions [4–9], protein functions [10,11], and genetic mutations and their associations with disease [12–15]. The first, crucial step in the pipeline of BioNLP tasks is the named entity recognition (NER) task: to automatically identify the names of biological entities (e.g., gene/protein) from unstructured texts [16].
Automatic identification of genes (note that we use gene and protein interchangeably in this paper) [17–19], in addition to diseases [20,21] and chemicals [22,23], has received much attention from the BioNLP research community [24–28] due to their central role in biomedical research. Their correct automatic recognition, however, remains challenging due to both language variation and ambiguity. For example, in the biomedical literature, the same gene can be referred to in multiple different ways by different authors in different articles. Such linguistic differences could include orthographical variations (e.g., “ESR1” and “ESR-1”), morphological variations (e.g., “GHF-1 transcriptional factor” and “GHF-1 transcription factor”), abbreviated terms (e.g., “estrogen receptor alpha (ERα)”), or composite mentions (e.g., “BRCA1/2” and “SMADs 1, 5, and 8”). In addition, a given gene mention in biomedical literature can also refer to different entities written by different authors in different articles. These types of ambiguity could include: multispecies (orthologous) ambiguity (e.g., erbb2 can be either a human gene or mouse gene name); same species, different gene ambiguity (e.g., “AP1” can refer to either “jun proto-oncogene”, Entrez Gene: 3725, or “FBJ murine osteosarcoma viral oncogene homolog”, Entrez Gene: 2353, both human); different biomedical entity ambiguity (e.g., “Dtd” can refer to “D-aminoacyl-tRNA deacylase (Drosophila melanogaster)”, Entrez Gene: 41371, or “Diastrophic dysplasia” disease MeSH ID: C536170); or English language ambiguity (e.g., FOX).
At the National Library of Medicine (NLM), our interest in developing NLM-Gene arose from the fact that, despite the previous work in BioNLP research to automatically identify genes in biomedical text, gene recognition tools have difficulty with: (1) articles that contain a large number of gene mentions, (2) articles that discuss genes in the context of multiple species, or (3) articles that contain mentions of other biomedical entities such as diseases, chemicals and mutations. Such articles are of great interest to biocurators because they discuss molecular interactions, and/or gene-disease relationships; however, they exceed the capabilities of current state-of-the-art automatic gene annotation tools.
In developing this work, we were inspired and encouraged by the significant progress made by the community-wide shared tasks on gene recognition [19,29–31]. In particular, our current success has a strong foundation on the previously developed gene entity annotated corpora that included both gene mentions and concept identifiers for the same set of articles [17,32].
Taken together, this work makes the following significant contributions:
First, we present a new high-quality, manually annotated corpus of genes from biomedical literature. The NLM-Gene corpus contains 550 PubMed abstracts, and it differs from previous corpora because it was selected to be rich in gene mentions, rich in other biomedical entities, and representative for multiple species. Also, these articles were published in many different journals to represent a wide range of language variation. These characteristics make this corpus invaluable for the advancement and improvement of text mining tools for accurate gene entity identification.
Second, we build a new end-to-end system that includes both name entity recognition and gene entity recognition modules, for improved performance. This system is the upgraded GNormPlus tool [17], which has been improved with a deep learning component for named entity recognition, and several other features to improve on accurate species identification, gene entity recognition, and false positive detection.
Finally, our new resource and upgraded tool are publicly available at ftp://ftp.ncbi.nlm.nih.gov/pub/lu/NLMGene and the gene finding results have been streamlined to process all PubMed and PMC articles (in daily updates via API: https://www.ncbi.nlm.nih.gov/research/bionlp/APIs/ and/or Pubtator: https://www.ncbi.nlm.nih.gov/research/pubtator/.
This manuscript is organized as follows: In Methods, we describe our corpus development, annotation process, annotation guidelines, annotation tool, and the details of the automatic gene recognition method. In Results, we detail the corpus characteristics, we compare it with previous gene annotation corpora, and detail the new advantages. Finally, our evaluation shows that the new corpus significantly improves the gene prediction results, not only on the NLM-Gene corpus when used as a benchmark, but also when validated on a previously unseen dataset.
2. Materials and methods
2.1. Corpus development
The development of the NLM-Gene corpus followed a systematic approach, which is schematically detailed in Fig. 1.
Fig. 1.
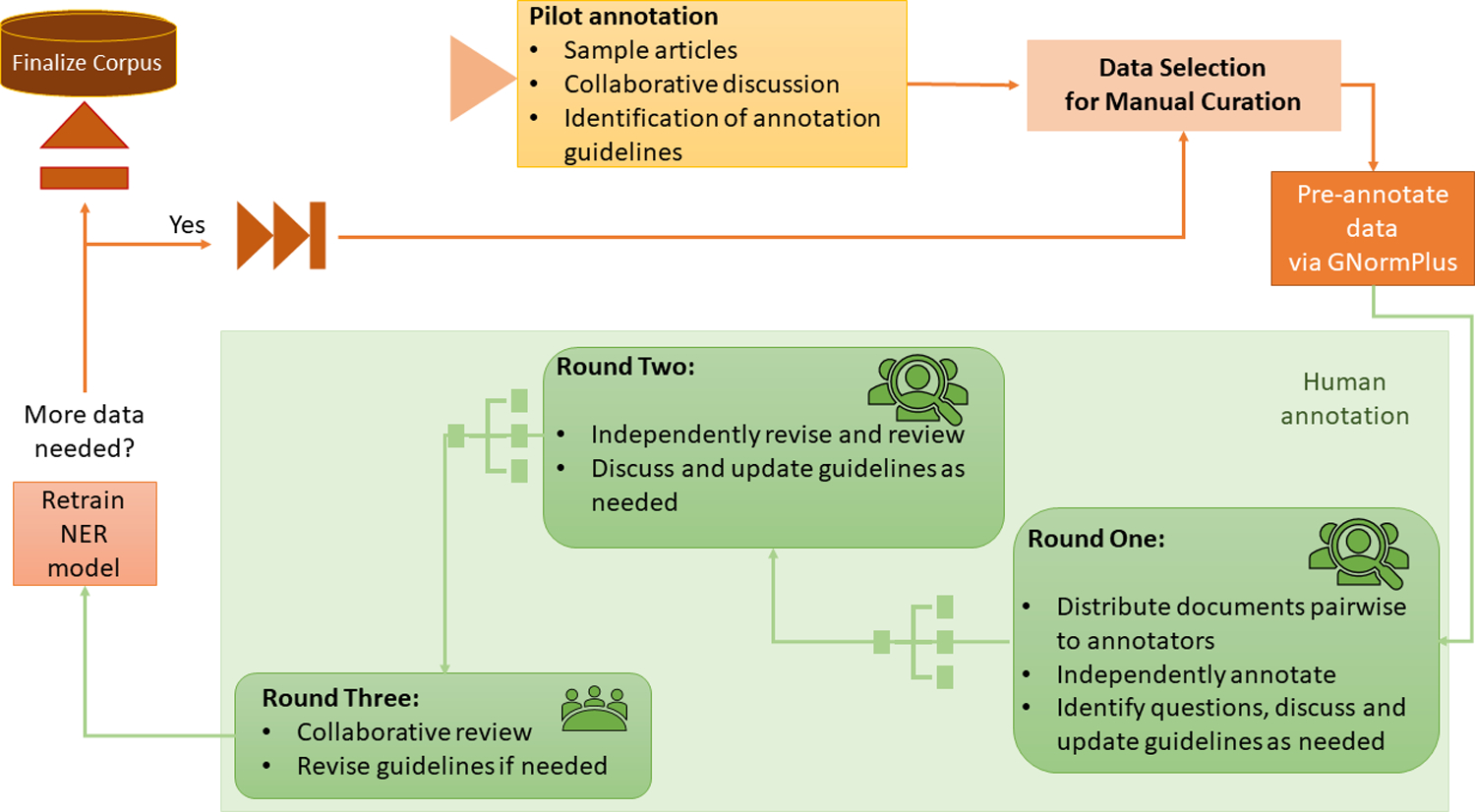
The NLM-Gene corpus annotation process.
Six National Library of Medicine (NLM) indexers, professional gene curators for the GENERIF project (https://www.ncbi.nlm.nih.gov/gene/about-generif/), with an average work experience of 20 years in biomedical data curation, participated in this project. GENERIF is a program at the NLM where the expert indexers create functional annotations for genes in the NCBI Gene database (https://www.ncbi.nlm.nih.gov/gene/), by linking PubMed articles describing a function or functions of that gene.
Based on our previous experience in corpora annotation [6,20,33], the NLM-Gene corpus annotation followed these steps, also detailed in Fig. 1:
Pilot phase: Initially 40 PubMed documents were randomly selected, following the criteria that they contained at least one predicted gene, and that the set contained genes from organisms other than human. The annotators reviewed the automatic gene identification in the sample articles, reviewed and corrected mention-level annotation, reviewed and corrected gene-ID normalization, and discussed how to annotate different cases, and categorize different possible cases. The initial draft of the annotation guidelines (supplementary file) was written.
Data Selection: Our goal was to identify documents where manual curation is useful for tool improvement, and automated tools do not produce accurate results. These articles have the following characteristics: they contain more gene mentions than average, they mention genes from a variety of organisms, and often more than one organism, they contain ambiguous gene mentions, and they discuss genes in relation to other biomedical topics such as diseases, chemicals, and mutations.
In order to optimize for the constraints listed above, we designed the following procedure:
We began with all articles in PubMed, with publicly available full text in the PubMed Central Open Access dataset and considered their titles and abstracts.
We ran our suite of biomedical named entity recognition tools (PubTator) and gave a higher weight to documents containing at least one non-gene biomedical entity in the abstract (either disease, chemical, mutation or species).
We gave a higher weight to documents containing genes from these organisms: human, mouse, rat, frog, zebrafish, thale cress, fruit fly, roundworm, yeast, fission yeast, and E. coli.
We gave a higher weight to documents containing multiple predicted genes.
We gave a higher weight to documents containing an ambiguous gene term.
We ran two different gene entity recognition models and gave a higher weight to documents with a high level of disagreement between the two different gene recognition tools.
The gene name entity recognition models were:
-
1
The gene annotations provided by PubTator, created using GNormPlus.
-
2
The gene annotations provided by the bluebert model trained using the GNormPlus corpus.
Fundamentally, our goal was to create a dataset that could train Gene NER algorithms to produce high-quality results in biomedical text, but also create a suitable dataset that could be used for other downstream biomedical text mining tasks.
-
3
Annotation process: Annotation was performed in four data batches. The first batch consisted of 100 PubMed documents, and the following three contained 150 each. Each batch was annotated in three annotation rounds using the newly developed annotation tool TeamTat (https://www.teamtat.org/) [34] (this corpus annotation project was instrumental in the development of TeamTat, because it gave real-time input on useful annotation features.)
Here is how we performed multi-user annotation for a batch of documents in the NLM-Gene corpus:
-
4
Round 0. Documents were pre-annotated with the GNormPlus [17] gene finding tool, and they were uploaded to TeamTat.
-
5
Round 1. Annotators were blindly paired per article, and they individually reviewed pre-annotations for each article in their set. Regular meetings brought up aspects of annotation that were not considered in the pilot, and annotation guidelines were updated accordingly.
Once all documents in the current batch were annotated, annotations and corrections from the individual annotators were merged into one copy of the document. TeamTat allows for visual cues to mark annotations where annotators disagree with each other. The project manager computed inter-annotator agreement (IAA) statistics. The IAA for round 1 of annotations in the NLM-Gene corpus, averaged over the four annotation batches, was 74%.
-
6
Round 2. Annotators worked individually, and still unaware of their annotation partner’s identities. They reviewed and revised all annotations. Because the tool facilitates the revision of disagreements, the work was naturally more focused, and therefore more efficient for the annotators.
Once all the documents in the batch were reviewed, all revised annotations were again merged to produce one copy, and the project leader produced the inter-annotator agreement. The IAA for round 2 of annotations in the NLM-Gene corpus, averaged over the four annotation batches, was 86%.
Regular annotator meetings provided opportunity for discussion of annotation issues discovered during annotation of new articles and annotation guidelines were updated accordingly.
-
7
Round 3. Annotators learned the identities of their annotation partners for each article, and they collaboratively revised and discussed the differences, reaching complete consensus in the articles that they annotated.
-
8
The 550 annotated PubMed documents constitute the NLM-Gene corpus.
This process may be generalized for generation of other gold-standard benchmark annotation corpora. We recommend the use of the TeamTat annotation tool, as it provides great flexibility on adapting to different annotation projects and provides an efficient and intuitive user interface.
2.2. TeamTat annotation tool
The development of the TeamTat annotation tool [34] was a byproduct of our NLM-Gene development work, aiming to ensure the high quality of the corpus and ease the burden of annotators, so that their time was used efficiently and productively. Collaborative text annotation is a complex process, and requires domain experts, project managers and a wide range of automatic pre-processing, user interface, and evaluation tools. Teamtat provided an interactive, intuitive user interface for project management and document annotation, supporting full text articles and figure display, highlighted pre-annotation to help achieve time and cost savings, corpus quality assessment, and the ability to organize annotation process until the desired corpus quality is achieved. A screenshot of the TeamTat annotation tool can be seen in Fig. 2.
Fig. 2.
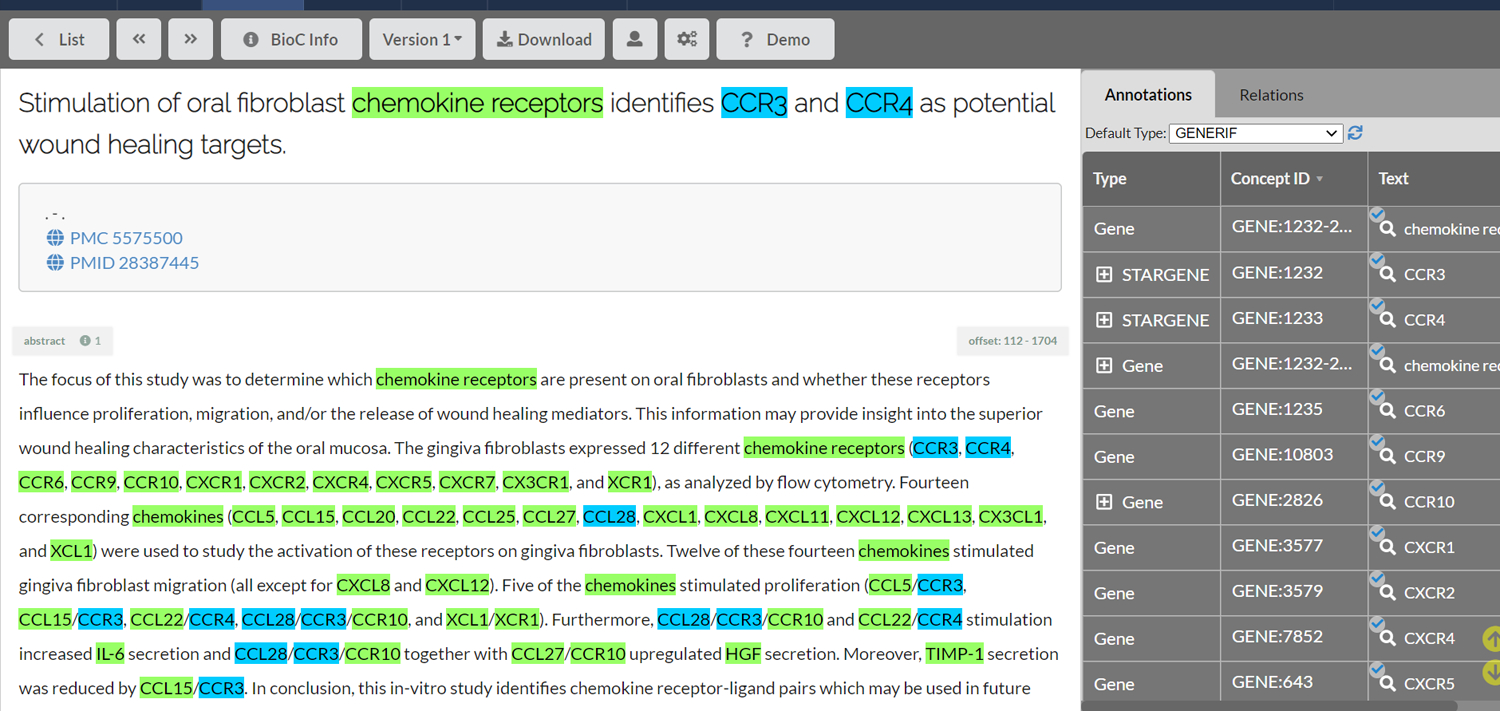
The Teamtat annotation tool. Shown, a document from the NLM-Gene annotation project. The interface allows easy access to the PubMed and PMC records, highlights all annotations, and distinguishes between different annotation types.
The TeamTat annotation tool automates all the steps listed in the Corpus Development section above, allows the user to define a benchmark corpus annotation project with ease, and coordinate a team of human experts to label the data with efficiency.
2.3. Annotation guidelines
This manuscript is accompanied with a detailed description of the NLM-Gene corpus annotation guidelines, given in a supplementary file. The following is a summary of the most salient points:
The NLM-Gene annotators annotated the title and abstract of a journal article, however they had access to the full text, and referred to the full text as needed to fully annotate each entity mentioned in the title/abstract.
- The NLM-Gene annotators distinguished between genes being the focus of an article, and other gene mentions, and marked them accordingly:
- GENERIF – the annotated gene meets the criteria for creating a GeneRIF - the basic biology or clinical significance of a gene/gene product is the primary point of the article.
- STARGENE – the annotated gene is a main point of the article but does not meet the criteria for creating a GeneRIF. (It is implied that a gene mention tagged as GENERIF is automatically the STARGENE of the article)
- GENE – the annotated gene is mentioned, but is not a main point.
- DOMAIN – the word denotes a protein domain
- OTHER – the gene mentioned is either a gene product used as therapeutic or pharmacological agent, or a gene used as a tool (e. g. marker gene, gene used in techniques, etc.)
A composite gene mention, e.g., Smad 1, 2, and 8, was annotated as one string, linked to three identifiers, that correspond to the order of appearance for Smad 1, Smad 2 and Smad 8. These identifiers are separated by a semi-colon.
A gene mention that refers to two genes (e.g., gene mention referring to both human and mouse organisms) is linked to both corresponding gene identifiers, separated by a comma. The annotation tool automatically sorts the identifiers in numerical order.
The NLM-Gene annotators distinguished between gene mentions discussing a general characteristic of a gene, versus a specific gene of a specific organism which is linked to experimental evidence.
The NLM-Gene annotators distinguished between individual gene mentions and family (class, group, complex) gene mentions. Since there exists no standardized vocabulary for gene families, gene families in the NLM-Gene corpus were annotated with all gene identifiers of their respective members mentioned in the title and abstract. For specific details and examples, see annotation guidelines.
NLM-Gene corpus does not contain annotations for non-standard gene references within articles. Gene references that do not use a standard name, synonym, or abbreviation (e.g. gene a, compound x) are not annotated.
The NLM-Gene corpus annotates the genes for the organism that is the source of the gene, in the articles describing experimental organisms (e.g., mouse gene transfected into human cells is annotated to record for mouse gene).
The NLM-Gene corpus does not annotate gene products used as therapeutic or pharmacological agents, or genes used as tools (e.g. marker genes, genes used in techniques). If identified, these mentions are given the annotation type “Other”.
2.4. Gene recognition method overview
Gene name entity recognition is the process in natural language processing that helps identify which words or phrases mentioned in the text are gene names. Gene name normalization, on the other hand, requires that the entity of interest is mapped from the word or phrase in the text to a corresponding known entity catalogued in a target knowledge base, in our case, the NCBI GENE database (https://www.ncbi.nlm.nih.gov/gene/).
We use GNormPlus [17] as our gene NER tool. GNormPlus is an end-to-end system that handles both gene/protein name and identifier detection in biomedical literature, including gene/protein mentions, family names and domain names. GNormPlus has compared favorably with previously reported gene finding systems and is the system of choice for PubTator [35]. The GNormPlus system consists of two major components: the Name Entity Recognition component, which recognizes the gene mentions in the text, and the Gene Entity Normalization component, which recognizes which gene is mentioned in the text and pairs the mention with a database identifier.
GNormPlus uses a Conditional Random Fields (CRF) [36] model to recognize the boundary of the gene names and combines SR4GN [37] with a lexicon of gene/protein family names and the domain information to disambiguate the corresponding species and normalize the gene identifiers. GNormPlus further integrates Ab3P [38], an abbreviation resolution and composite mention simplification tool, and SimConcept [39], a composite name entity resolution tool (BRCA1/2 entity refers to two separate concepts) to optimize the performance.
We first used the annotated NLM-Gene corpus batches to enrich the GNormPlus corpus and obtain a larger training corpus for gene name recognition. Next, upon analyzing the testing errors, we decided to incorporate several improvements and upgrades to the GNormPlus system to better improve its accuracy. These upgrades are shown in Fig. 3 and detailed below.
Fig. 3.
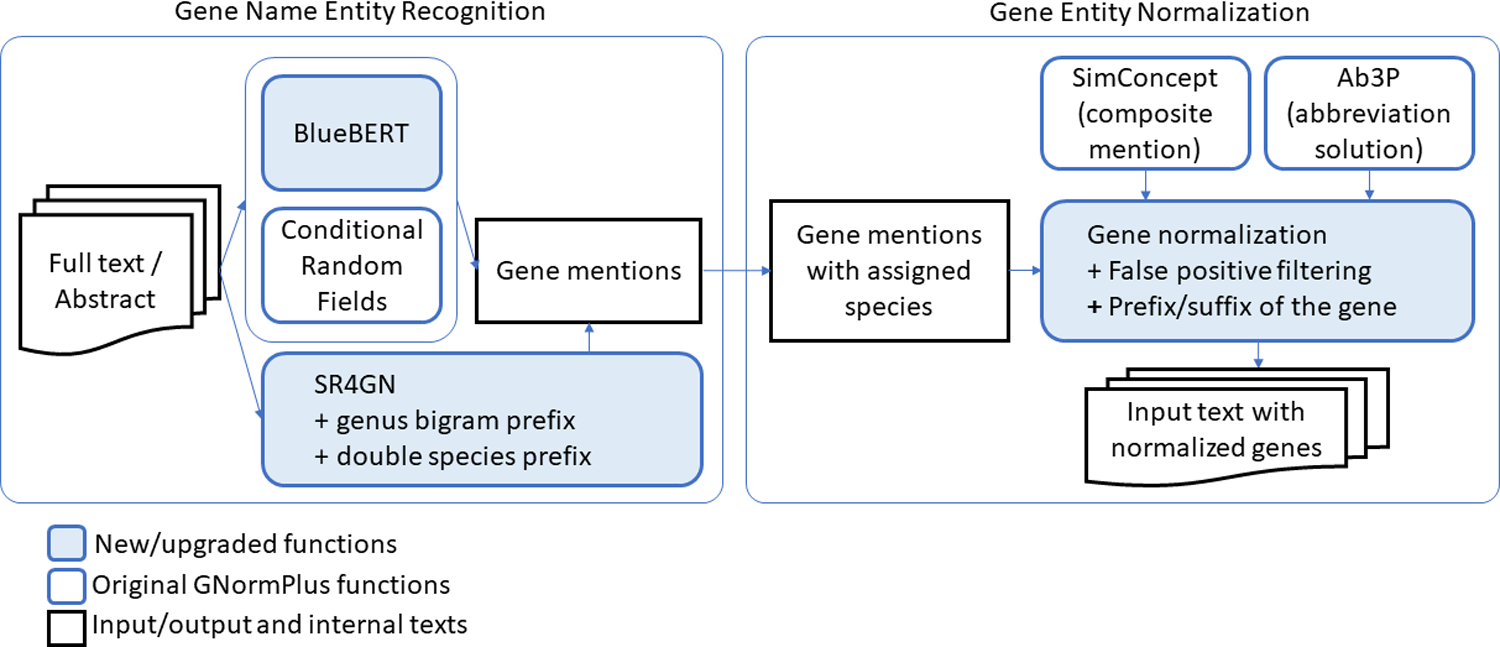
Upgraded functions of GNormPlus, to improve gene name recognition in biomedical literature.
Our analysis showed that there is room to improve both the Name Entity Recognition, as well as the Gene Entity Normalization. Regarding the Name Entity Recognition component, we made two important updates in the GNormPlus system:
BlueBERT [40]: This component allows the GNormPlus system to switch between a conditional random field named entity recognition training model and the BlueBERT deep learning model. The tradeoff is the required time available for training.
SR4GN: This GNormPlus component identifies a species for each gene mention in the text given the clues in the surrounding text. This component has been upgraded to account for a new feature that recognizes a prefix term in a species mention (i.e. Ae. aegypti stands for Aedes aegypti) and a species prefix term in a gene mention (i.e. NtHAK1 stands for Nicotiana tabacum HAK1, and TgCPL stands for Toxoplazma gondii cathepsin L). Better species recognition improves gene entity normalization. Our analysis of PubMed/PMC revealed that there were more than 300 thousand articles containing mentions of species expressed in this form, and more that 27 thousand articles containing mentions of genes with a species prefix of this form.
For the Gene Entity Normalization component, we added these refinement steps that improved accuracy:
Recognition of abbreviations defined in the article to filter false positives. For example, in an article where we find the text “… the associations between public stigma, desire for social distance, familiarity with mental illness and CSE in community members. The CSE of those with MHCs correlated positively with their personal recovery…”, given that CSE is the official symbol for the human gene 1433, coding for episodic choreoathetosis/spasticity, the gene finding program will confidently label it as such. However, upon further inspection, the abbreviation finding algorithm identifies that the term CSE is else-where in the article defined as short term notation for creative self-efficacy. This information allows the Gene Entity Normalization component to review its decision and remove the labelling. Our analysis of PubMed/PMC revealed that there were more than 5 million articles containing such ambiguous abbreviated term mentions, and the NLM-Gene corpus contains 47 documents with such examples.
Gene mention needs to be modified based on a prefix/suffix. For example, the term HSD1KO is used as a notation for HSD1 knockout gene. Being able to recognize what the -KO suffix stands for knockout, allows the program to re-adjust the boundaries of the gene mention and map it to the correct gene identifier. Our analysis of PubMed/PMC revealed that there were more than 60 thousand articles containing such prefix/suffix terms embedded in the gene mentions, and the NLM-Gene corpus contains 25 documents with such examples.
3. Results
3.1. NLM-Gene corpus characteristics
The NLM-Gene corpus is currently the most refined gene annotation corpus (Table 1), covering a variety of gene names, with an average of 29 gene mentions (10 unique identifiers) per document, and a broader representation of different species (28 different species, including human, mouse, rat, fruit fly, thale cress, zebrafish, worm, frog, yeast, etc.). Annotators annotated the title and abstract of a PubMed document. These articles were published in 156 different journals.
Table 1.
NLM-Gene corpus characteristics, compared with GNormPlus corpus.
| Corpus | GNormPlus | NLM-Gene |
|---|---|---|
| Number of PubMed abstracts | 694 | 550 |
| Number of journals | 117 | 156 |
| Number of total gene mentions (unique) | 9,986 (3,861) | 15,553 (5,298) |
| Number of unique PMID – GENE ids | 2,025 | 6,366 |
| Number of species | 30 | 28 |
| Min gene mentions (ID) per document | 1 (0) | 2(1) |
| Max gene mentions (ID) per document | 56 (11) | 86 (40) |
| Ave gene mentions (ID) per document | 14.4 (5.6) | 28.3 (9.6) |
| Median gene mentions (ID) per document | 9(2) | 26 (10) |
NLM-Gene was annotated so that it could work in complement with other previously annotated gene corpora, such as GNormPlus. In addition to the GNormPlus corpus, NLM-gene contributes valuable annotations increasing both the diversity of the gene mentions and identifiers, as well as the number of species. Table 1 compares the number of articles, number of journals, number of unique gene mentions and identifiers in both corpora, and clearly displays that NLM-Gene documents are significantly denser in gene mention/entity content. When comparing the gene annotations per document by their respective species, we notice that a typical document in the NLM-Gene corpus contains gene annotations from a number of species ranging from 1 to 4, with an average of 1.38. In comparison, the GNormPlus corpus contains a maximum of two species, and the average number is 0.96. Fig. 4 shows the rate of common occurrences for the gene mentions and gene identifiers that appear in both corpora. The NLM-Gene corpus brings a 247% increase in new gene identifiers, and a significantly broader representation of different species.
Fig. 4.
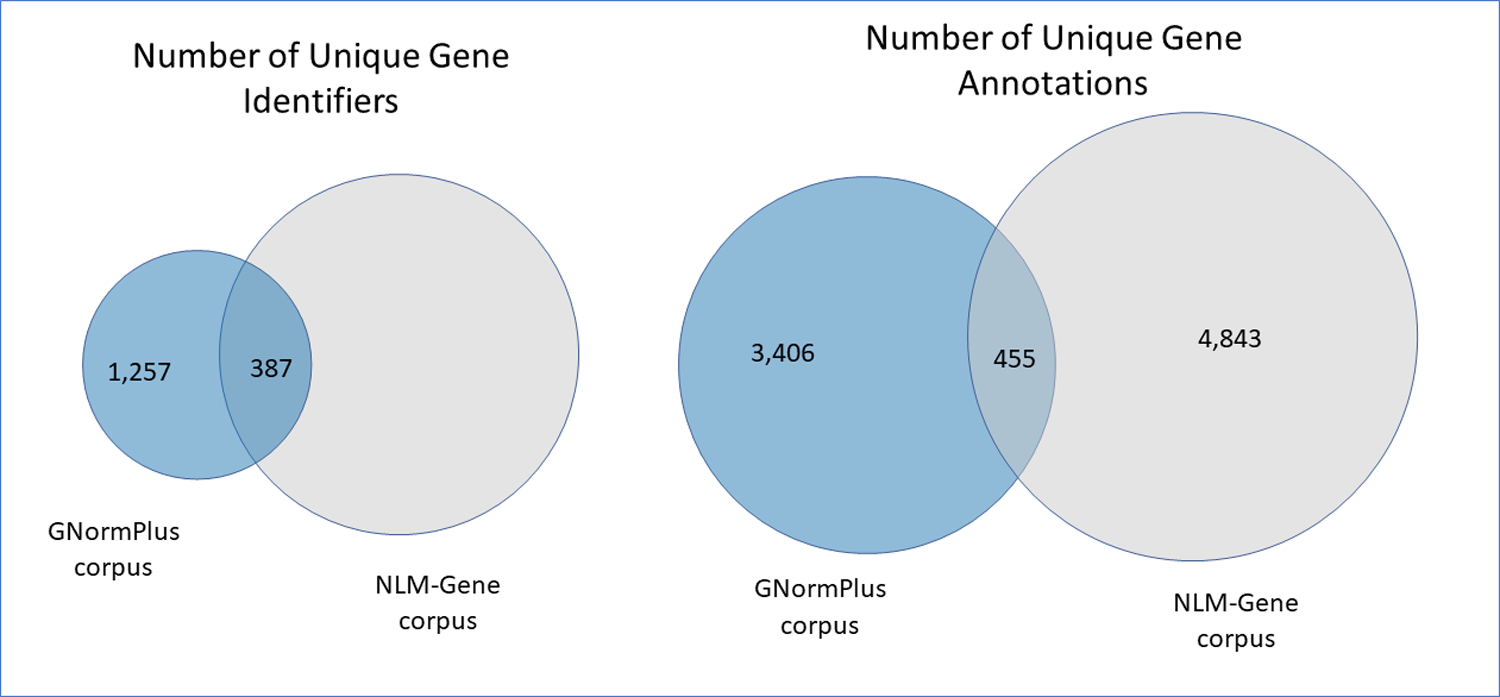
Comparison of the gene names, and gene identifiers between the GNormPlus and NLM-Gene corpora.
Table 2 shows the top ten species in the NLM-Gene corpus, compared with the corresponding distribution in the GNormPlus corpus. Here we count the number of (PMID-Gene ID) pairs for each species for both corpora, which means that for each normalized gene, we count its occurrence once for each document it appears in. While both corpora have a similar number of species, the NLM-Gene has more than triple the number of PMID-Gene ID pairs. GNormPlus is mostly concentrated on human genes, with only 6 species having more than ten genes in the corpus. NLM-Gene, on the other hand, has balanced representation between human and mouse, and contains sufficient examples for many more organisms.
Table 2.
Distribution of annotations for genes of different species in GNormPlus and NLM-Gene. First column lists the Species name and ID from the Taxonomy database (https://www.ncbi.nlm.nih.gov/taxonomy), second and third column show the percentage and raw numbers of genes for each species in the GNormPlus and NLM-Gene corpora.
| TaxID – Taxonomy name | GNormPLUS | NLM-GENE |
|---|---|---|
| 9606 (H. sapiens – human) | 85.35% (1,724) | 47.84% (3,141) |
| 10090 (M. musculus – mouse) | 5.10% (1033) | 31.08% (2,041) |
| 10116 (R. norvegicus – rat) | 2.72% (55) | 8.35% (548) |
| 559292 (S. cerevisiae – yeast) | 2.37% (48) | 1.80% (118) |
| 7227 (D. melanogaster – fly) | 1.04% (21) | 1.52% (100) |
| 3702 (A. thaliana – thale cress) | 0.05% (1) | 1.20% (79) |
| 6239 (C. elegans – worm) | 0.40% (8) | 0.99% (65) |
| 7955 (D. rerio – zebrafish) | 0.10% (2) | 0.84% (55) |
| 8355 (X. laevis – frog) | 0.54% (11) | 0.82% (54) |
| 9940 (Ovis aries – sheep) | 0% (0) | 0.44% (29) |
3.2. Improved performance of automatic gene identification
An important step in corpus development is identifying a good split between the set of documents to be designated as the train set and test set. An optimal split is useful for the development of new algorithms, meaning that the data needs to be similar and not have unexpected biases. For the NLM-Gene corpus, we prepared a split of 450/100 articles.
To ensure a similar distribution of documents in all the sets, we sampled proportionally from each annotation batch, selecting the articles so that the gene mention and gene ID distribution approximated those of the full corpus. This step ensured that we did not inadvertently split the dataset into defined clusters.
Table 3 summarizes our evaluations for automatic gene recognition. The first evaluation shows the performance of our previous system GNormPlus, for joint named entity recognition and normalization of genes, when trained on the original corpus (the GNormPlus corpus) and evaluated on the NLM-Gene test dataset. The second evaluation shows the performance of the GNormPlus system trained on the GNormPlus corpus and the NLM-Gene train dataset and evaluated on the NLM-Gene test dataset. The difference between these two evaluations clearly shows an improvement in performance, due to the much richer training data including many more organisms contributed by the NLM-Gene corpus.
Table 3.
Text mining evaluation, showing performance results on the NLM-Gene test set, when the training is performed on the listed combinations of training sets. The results show the effect on performance improvement due to the NLM-Gene corpus, and specific system upgrades.
| Model | Gene name entity recognition | Gene entity normalization | |||||
|---|---|---|---|---|---|---|---|
| Train | Precision | Recall | f-measure | Precision | Recall | f-measure | |
| GNormPlus (original) | GN | 0.919 | 0.762 | 0.833 | 0.714 | 0.619 | 0.663 |
| GNormPlus (original) | GN + NLM | 0.922 | 0.825 | 0.871 | 0.724 | 0.697 | 0.710 |
| GNormPlus (original) + Deep Learning | GN + NLM | 0.929 | 0.821 | 0.872 | 0.726 | 0.697 | 0.711 |
| GNormPlus (upgraded) | GN + NLM | 0.933 | 0.834 | 0.881 | 0.748 | 0.707 | 0.727 |
| GNormPlus (upgraded) + (species insensitive) | GN + NLM | 0.933 | 0.834 | 0.881 | 0.879 | 0.840 | 0.859 |
The third evaluation shows the BlueBERT model upgrade of the GNormPlus system, when trained on PubMed and fine-tuned on the new training data, tested on the NLM-Gene test dataset. For this experiment we use all other components of original GNormPlus but substitute the Conditional Random Fields component for BlueBERT. We see a further improvement in the precision of gene name entity recognition, which translates into an improvement in the precision of correctly identifying genes. This evaluation shows the contribution of the deep learning component to the performance improvement.
The fourth row shows the performance when all upgrades of the GNormPlus are included in addition to the deep learning component. It is noticeable that we gain both in name entity recognition, as well as in the gene entity recognition. Note that the inter-annotator agreement (after the first round) for the gene annotation task was 74%, so this evaluation result is on par with human assessment.
The last row shows a relaxed normalization evaluation, that accepts an incorrect species assignment for a given gene, so long as the prediction is a known homologue of the correct gene. Most information retrieval tasks as well as database curation tasks are likely to prefer this mode of operation.
3.3. Improved performance of automatic gene recognition for different species
We applied the new gene finding model trained on the NLM-Gene/GNormPlus corpus to a previously unseen set of 30,000 articles from the GENERIF curated articles. The GENERIF curated articles, constitute a gold-standard dataset of more than 800,000 articles that have been manually annotated over the years. The articles we used for this experiment were randomly selected from this larger set.
Table 4 shows the top 15 organisms (from 618) comprising nearly 95% of all curated gene links (in the 30,000 articles) and the number of PMID-Gene ID pairs for each organism in this dataset. In Table 4, we also list the recall accuracy score that our new and improved gene recognition tool achieves for each species. This score is computed by giving full credit to the algorithm when the manually annotated gene ID is correctly predicted, and a weighted credit when an ortholog gene is reported instead (For this experiment, the algorithm reported an ortholog when not able to identify the correct species in 6% of the cases). The highlighted values note the organisms for which the improvement in score compared to the original GNormPlus is statistically significant. Furthermore, if we considered the prediction scores for all the genes from all 618 species, the majority of which was not seen in the training data, the improvement is statistically significant with p-value = 3.44E-05. The algorithm has high confidence for a variety of organisms with the exception of bacteria, which are poorly represented in the NLM-Gene corpus. We recommend that future manual gene annotation efforts be applied on the bacterial genomes to further improve automatic gene recognition.
Table 4.
Gene recognition recall of the GENERIF genes, on a randomly selected set of 30,000 articles, with a total of 49,221 manually curated links between the GENE and PubMed databases. We list the recall for different species, ranked by their number of curated genes, and mark as bold the cases when our results are statistically significantly better than what we could achieve before the improvements listed in this paper.
| Species | Taxonomy ID | Number of Genes | Ratio | Recall |
|---|---|---|---|---|
| Homo sapiens (human) | 9606 | 30,760 | 0.625 | 0.764 |
| Mus musculus (mouse) | 10090 | 9,319 | 0.189 | 0.835 |
| Rattus norvegicus (rat) | 10116 | 2,962 | 0.060 | 0.760 |
| Saccharomyces cerevisiae (budding yeast) | 559292 | 593 | 0.012 | 0.762 |
| Arabidopsis thaliana (thale cress) | 3702 | 682 | 0.014 | 0.774 |
| Drosophila melanogaster (fruit fly) | 7227 | 643 | 0.013 | 0.745 |
| Danio rerio (zebrafish) | 7955 | 309 | 0.006 | 0.790 |
| Caenorhabditis elegans (roundworm) | 6239 | 258 | 0.005 | 0.713 |
| Bos taurus (cattle) | 9913 | 242 | 0.005 | 0.727 |
| Gallus gallus (chicken) | 9031 | 199 | 0.004 | 0.653 |
| Sus scrofa (pig) | 9823 | 176 | 0.004 | 0.358 |
| Xenopus laevis (African clawed frog) | 8355 | 114 | 0.002 | 0.465 |
| Escherichia coli str. K-12 substr. MG1655 | 511,145 | 248 | 0.005 | 0.016 |
| Canis lupus familiaris (dog) | 9615 | 99 | 0.002 | 0.798 |
| Human immunodeficiency virus 1 | 11,676 | 92 | 0.002 | 0.489 |
3.4. The practical use of automatic gene prediction in biomedical literature
To measure the utility of the gene tool for a real-life application we tested the actual gene recognition in PubMed. We ran our upgraded GNormPlus tool over all the articles with PubMed publication dates between Feb 11 and 25, 2020. Since this set consisted of more than 200 thousand PubMed abstracts, 200 documents were randomly selected, and the gene prediction results were given to six NLM indexers to review. This set contained a random distribution of organisms, including some not seen in the training set. The indexers reported: (1) ease of indexing for gene terms, due to the highlighting of gene terms and accurate identification of gene identifiers, and (2) significant drop in the review time, due to the automatic annotations. In response, our team has incorporated the new and upgraded GNormPlus to the PubTator system, and all articles received in PubMed daily are tagged for genes, species and other bio-entities. The data are available to the public the next day.
4. Discussion
In this manuscript we presented NLM-Gene, a new benchmark resource for gene entity recognition in biomedical literature. NLM-Gene is a high-quality corpus, doubly annotated by six NLM indexers, in three rounds of annotation, and all annotator disagreements have been resolved. NLM-Gene consists of 550 PubMed documents, from 156 journals, and contains more than 15 thousand unique gene names, corresponding to more than five thousand gene identifiers (NCBI Gene taxonomy). This corpus contains gene annotation data from 28 organisms.
The annotated documents were selected so that they were rich in gene mentions and other biomedical entities, such as chemicals, diseases and/or mutations, and as such the collection contains on average 29 gene names, and 10 gene identifiers per document. These characteristics demonstrate that this dataset is an important benchmark to test the accuracy of gene recognition algorithms both on multi-species and ambiguous data. We believe the NLM-Gene corpus will be invaluable for advancing text-mining techniques for gene identification tasks in biomedical text.
In order to achieve a robust result of gene entity recognition that could translate to real life applications, we upgraded the GNormPlus system with deep learning for the name entity recognition component and several features that ensured better accuracy for species recognition, and false positive prediction detection. The new results are superior and identify genes in the NLM-Gene test dataset close to the performance of human inter-annotator agreement.
Because the goal of our NLM research is to provide practical benefits, NLM-Gene is available at Dryad [41] and at ftp://ftp.ncbi.nlm.nih.gov/pub/lu/NLMGene, the gene entity recognition results have been streamlined to process all PubMed articles in daily updates: https://www.ncbi.nlm.nih.gov/research/bionlp/APIs/, and the corpus development process can be adapted to create other gold-standard annotation corpora via our team annotation tool TeamTat (www.teamtat.org).
Supplementary Material
Acknowledgements
This RESEARCH was supported by the NIH Intramural Research Program, National Library of Medicine.
Footnotes
CRediT authorship contribution statement
Rezarta Islamaj: Methodology, Software, Validation, Formal analysis, Writing - review & editing, Visualization, Project administration. Chih-Hsuan Wei: Software, Methodology, Visualization. David Cissel: Data curation. Nicholas Miliaras: Data curation, Writing - review & editing. Olga Printseva: Data curation. Oleg Rodionov: Data curation. Keiko Sekiya: Data curation. Janice Ward: Data curation, Writing - review & editing. Zhiyong Lu: Conceptualization, Methodology, Supervision, Writing - review & editing, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jbi.2021.103779.
References
- [1].Khare R, Leaman R, Lu Z, Accessing biomedical literature in the current information landscape, Methods Mol. Biol 1159 (2014) 11–13. Epub 2014/05/03. doi: 10.1007/978-1-4939-0709-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rindflesch TC, Blake CL, Fiszman M, Kilicoglu H, Rosemblat G, Schneider J, et al. , Informatics support for basic research in biomedicine, ILAR J. 58 (1) (2017) 80–89. Epub 2017/08/26. doi: 10.1093/ilar/ilx004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Singhal A, Leaman R, Catlett N, Lemberger T, McEntyre J, Polson S, et al. , Pressing needs of biomedical text mining in biocuration and beyond: opportunities and challenges, Database (Oxford) 2016 (2016). Epub 2016/12/28. doi: 10.1093/database/baw161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Papanikolaou N, Pavlopoulos GA, Theodosiou T, Iliopoulos I, Protein-protein interaction predictions using text mining methods, Methods 74 (2015) 47–53. Epub 2014/12/03. doi: 10.1016/j.ymeth.2014.10.026. [DOI] [PubMed] [Google Scholar]
- [5].Oughtred R, Stark C, Breitkreutz BJ, Rust J, Boucher L, Chang C, et al. , The BioGRID interaction database: 2019 update, Nucleic Acids Res. 47 (D1) (2019) D529–D541. Epub 2018/11/27. doi: 10.1093/nar/gky1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Islamaj Dogan R, Kim S, Chatr-Aryamontri A, Chang CS, Oughtred R, Rust J, et al. , The BioC-BioGRID corpus: full text articles annotated for curation of protein-protein and genetic interactions, Database (Oxford) 2017 (2017). Epub 2017/01/13. doi: 10.1093/database/baw147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Thompson P, Daikou S, Ueno K, Batista-Navarro R, Tsujii J, Ananiadou S, Annotation and detection of drug effects in text for pharmacovigilance, J. Cheminform 10 (1) (2018) 3. Epub 2018/08/15. doi: 10.1186/s13321-018-0290-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Levy RH, Ragueneau-Majlessi I, Past, present, and future of drug-drug interactions, Clin. Pharmacol. Ther 105 (6) (2019) 1286. Epub 2019/02/19. doi: 10.1002/cpt.1349. [DOI] [PubMed] [Google Scholar]
- [9].Ben Abacha A, Chowdhury MFM, Karanasiou A, Mrabet Y, Lavelli A, Zweigenbaum P, Text mining for pharmacovigilance: Using machine learning for drug name recognition and drug-drug interaction extraction and classification, J. Biomed. Inf 58 (2015) 122–132. Epub 2015/10/04. doi: 10.1016/j.jbi.2015.09.015. [DOI] [PubMed] [Google Scholar]
- [10].Ruch P, Text mining to support gene ontology curation and vice versa, Methods Mol. Biol 1446 (2017) 69–84. Epub 2016/11/05. doi: 10.1007/978-1-4939-3743-1_6. [DOI] [PubMed] [Google Scholar]
- [11].Wang Q, Ross KE, Huang H, Ren J, Li G, Vijay-Shanker K, et al. , Analysis of protein phosphorylation and its functional impact on protein-protein interactions via text mining of the scientific literature, Methods Mol. Biol 1558 (2017) 213–232. Epub 2017/02/06. doi: 10.1007/978-1-4939-6783-4_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wei CH, Harris BR, Kao HY, Lu Z, tmVar: a text mining approach for extracting sequence variants in biomedical literature, Bioinformatics 29 (11) (2013) 1433–1439. Epub 2013/04/09. doi: 10.1093/bioinformatics/btt156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wei CH, Phan L, Feltz J, Maiti R, Hefferon T, Lu Z, tmVar 2.0: integrating genomic variant information from literature with dbSNP and ClinVar for precision medicine, Bioinformatics 34 (1) (2018) 80–87. Epub 2017/10/03. doi: 10.1093/bioinformatics/btx541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Allot A, Peng Y, Wei CH, Lee K, Phan L, Lu Z, LitVar: a semantic search engine for linking genomic variant data in PubMed and PMC, Nucleic Acids Res. 46 (W1) (2018) W530–W536. Epub 2018/05/16. doi: 10.1093/nar/gky355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Islamaj Dogan R, Kim S, Chatr-Aryamontri A, Wei CH, Comeau DC, Antunes R, et al. , Overview of the BioCreative VI Precision Medicine Track: mining protein interactions and mutations for precision medicine, Database (Oxford) 2019 (2019). Epub 2019/01/29. doi: 10.1093/database/bay147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sekine S, Ranchhord E, Named Entities: Recognition, Classification and Use, John Benjamins, 2009. [Google Scholar]
- [17].Wei CH, Kao HY, Lu Z, GNormPlus: An integrative approach for tagging genes, gene families, and protein domains, Biomed. Res. Int 2015 (2015). Epub 2015/09/18. doi: 10.1155/2015/918710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Krallinger M, Vazquez M, Leitner F, Salgado D, Chatr-Aryamontri A, Winter A, et al. , The Protein-Protein Interaction tasks of BioCreative III: classification/ranking of articles and linking bio-ontology concepts to full text, BMC Bioinformatics. 12 (Suppl 8) (2011) S3. Epub 2011/12/22. doi: 10.1186/1471-2105-12-S8-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Morgan AA, Lu Z, Wang X, Cohen AM, Fluck J, Ruch P, et al. , Overview of BioCreative II gene normalization, Genome Biol. 9 (Suppl 2) (2008) S3. Epub 2008/10/18. doi: 10.1186/gb-2008-9-s2-s3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dogan RI, Leaman R, Lu Z, NCBI disease corpus: a resource for disease name recognition and concept normalization, J. Biomed. Inf 47 (2014) 1–10. Epub 2014/01/08. doi: 10.1016/j.jbi.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Leaman R, Islamaj Dogan R, Lu Z, DNorm: disease name normalization with pairwise learning to rank, Bioinformatics 29 (22) (2013) 2909–2917. Epub 2013/08/24. doi: 10.1093/bioinformatics/btt474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Krallinger M, Rabal O, Leitner F, Vazquez M, Salgado D, Lu Z, et al. , The CHEMDNER corpus of chemicals and drugs and its annotation principles, J Cheminform. 7 (Suppl 1 Text mining for chemistry and the CHEMDNER track) (2015) S2. Epub 2015/03/27. doi: 10.1186/1758-2946-7-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Leaman R, Wei CH, Lu Z, tmChem: a high performance approach for chemical named entity recognition and normalization, J. Cheminf 7 (Suppl 1 Text mining for chemistry and the CHEMDNER track) (2015) S3. Epub 2015/03/27. doi: 10.1186/1758-2946-7-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen L, Liu H, Friedman C, Gene name ambiguity of eukaryotic nomenclatures, Bioinformatics 21 (2) (2005) 248–256. Epub 2004/08/31. doi: 10.1093/bioinformatics/bth496. [DOI] [PubMed] [Google Scholar]
- [25].Hakenberg J, Gerner M, Haeussler M, Solt I, Plake C, Schroeder M, et al. , The GNAT library for local and remote gene mention normalization, Bioinformatics 27 (19) (2011) 2769–2771. Epub 2011/08/05. doi: 10.1093/bioinformatics/btr455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Huang M, Liu J, Zhu X, GeneTUKit: a software for document-level gene normalization, Bioinformatics 27 (7) (2011) 1032–1033. Epub 2011/02/10. doi: 10.1093/bioinformatics/btr042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tsai RT, Lai PT, Multi-stage gene normalization for full-text articles with context-based species filtering for dynamic dictionary entry selection, BMC Bioinf. 12 (Suppl 8) (2011) S7. Epub 2011/12/22. doi: 10.1186/1471-2105-12-S8-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wei CH, Kao HY, Cross-species gene normalization by species inference, BMC Bioinf. 12 (Suppl 8) (2011) S5. Epub 2011/12/22. doi: 10.1186/1471-2105-12-S8-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hirschman L, Colosimo M, Morgan A, Yeh A, Overview of BioCreAtIvE task 1B: normalized gene lists, BMC Bioinf. 6 (Suppl 1) (2005) S11. Epub 2005/06/18. doi: 10.1186/1471-2105-6-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lu Z, Kao HY, Wei CH, Huang M, Liu J, Kuo CJ, et al. , The gene normalization task in BioCreative III, BMC Bioinf. 12 (Suppl 8) (2011) S2. Epub 2011/12/22. doi: 10.1186/1471-2105-12-S8-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Huang CC, Lu Z, Community challenges in biomedical text mining over 10 years: success, failure and the future, Brief Bioinf. 17 (1) (2016) 132–144. Epub 2015/05/04. doi: 10.1093/bib/bbv024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dai HJ, Wu JC, Tsai RT, Collective instance-level gene normalization on the IGN corpus, PLoS One 8 (11) (2013). Epub 2013/11/28. doi: 10.1371/journal.pone.0079517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Islamaj R, Wilbur WJ, Xie N, Gonzales NR, Thanki N, Yamashita R, et al. , PubMed text similarity model and its application to curation efforts in the conserved domain database, Database (Oxford) 2019 (2019). Epub 2019/07/04. doi: 10.1093/database/baz064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Islamaj R, Kwon D, Kim S, Lu Z, TeamTat: a collaborative text annotation tool, Nucleic Acids Res. 48 (W1) (2020) W5–W11. Epub 2020/05/10. doi: 10.1093/nar/gkaa333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wei CH, Allot A, Leaman R, Lu Z, PubTator central: automated concept annotation for biomedical full text articles, Nucleic Acids Res. 47 (W1) (2019) W587–W593. Epub 2019/05/23. doi: 10.1093/nar/gkz389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lafferty JD, McCallum A, Pereira FCN, Conditional random fields: Probabilistic models for segmenting and labeling sequence data, in: Proceedings of the Eighteenth International Conference on Machine Learning, Morgan Kaufmann Publishers Inc., 2001, pp. 282–289. [Google Scholar]
- [37].Wei CH, Kao HY, Lu Z, SR4GN: a species recognition software tool for gene normalization, PLoS One 7 (6) (2012). Epub 2012/06/09. doi: 10.1371/journal.pone.0038460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sohn S, Comeau DC, Kim W, Wilbur WJ, Abbreviation definition identification based on automatic precision estimates, BMC Bioinf. 9 (2008) 402. Epub 2008/09/27. doi: 10.1186/1471-2105-9-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wei CH, Leaman R, Lu Z, SimConcept: a hybrid approach for simplifying composite named entities in biomedical text, IEEE J. Biomed. Health Inf 19 (4) (2015) 1385–1391. Epub 2015/04/17. doi: 10.1109/JBHI.2015.2422651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Peng Y, Yan S, Lu Z (Eds.), Transfer Learning in Biomedical Natural Language Processing: An Evaluation of BERT and ELMo on Ten Benchmarking Datasets. 18th BioNLP Workshop and Shared Task, Association for Computational Linguistics, Florence, Italy, 2019. [Google Scholar]
- [41].Islamaj R, Lu Z, NLM-Gene, a richly annotated gold standard dataset for gene entities that addresses ambiguity and multi-species gene recognition, Dryad (2020), 10.5061/dryad.dv41ns1wt. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.