

Abstract
Wheat (Triticum aestivum) is one of the most important food crops with an urgent need for increase in its production to feed the growing world. Triticum timopheevii (2n = 4x = 28) is an allotetraploid wheat wild relative species containing the At and G genomes that has been exploited in many pre-breeding programmes for wheat improvement. In this study, we report the generation of a chromosome-scale reference genome assembly of T. timopheevii accession PI 94760 based on PacBio HiFi reads and chromosome conformation capture (Hi-C). The assembly comprised a total size of 9.35 Gb, featuring a contig N50 of 42.4 Mb and included the mitochondrial and plastid genome sequences. Genome annotation predicted 166,325 gene models including 70,365 genes with high confidence. DNA methylation analysis showed that the G genome had on average more methylated bases than the At genome. In summary, the T. timopheevii genome assembly provides a valuable resource for genome-informed discovery of agronomically important genes for food security.
Subject terms: Comparative genomics, Plant evolution, Genome informatics
Background & Summary
The Triticum genus comprises many wild and cultivated wheat species including diploid, tetraploid and hexaploid forms. The polyploid species originated after hybridisation between Triticum and the neighbouring Aegilops genus (goatgrass). The tetraploid species, Triticum turgidum (2n = 4x = 28, AABB), also known as emmer wheat, and Triticum timopheevii (2n = 4x = 28, AtAtGG) are polyphyletic. Triticum urartu Thum. ex Gandil (2n = 2x = 14, AA) is the A genome donor for both these species1 whereas, the B and G genomes are closely related to the S genome of Aegilops speltoides2. Both tetraploid species have wild and domesticated forms, i.e., T. turgidum L. ssp. dicoccoides (Körn. ex Asch. & Graebn.) Thell. and ssp. dicoccum (Schrank ex Schübl.) Thell., respectively, and T. timopheevii (Zhuk.) Zhuk. ssp. armeniacum (Jakubz.) Slageren and ssp. timopheevii, respectively. Additionally, tetraploid durum wheat T. turgidum L. ssp. durum (Desf.) Husn. (2n = 4x = 28, AABB), used for pasta production, and hexaploid bread wheat Triticum aestivum L. (2n = 6x = 42, BBAADD) evolved from domesticated emmer wheat with the latter originating through hybridisation with Aegilops tauschii (D genome donor) 6,000–7,000 years ago. Hexaploid Triticum zhukovskyi (AAGGAmAm) originated from hybridisation of cultivated T. timopheevii and cultivated einkorn Triticum monococcum3 (2n = 2x = 14, AmAm).
The G genome is only found in T. timopheevii and T. zhukovskyi and is virtually identical to the S genome on a molecular level4,5 but differs from it, and the B genome, due to a number of chromosomal rearrangements and translocations involving the At genome6. The most studied are the 6At/1 G/4 G and 4 G/4At/3At translocations in T. timopheevii7–10.
Triticum timopheevii ssp. timopheevii has been exploited in various studies for wheat improvement as it has been shown to be an abundant source for genetic variation for many traits such as resistance to leaf rust11–13, stem rust14–16, powdery mildew16–18, Fusarium head blight19,20 Hessian fly, Septoria blotch, wheat curl mite and tan spot21. It has also been shown to have tolerance to abiotic stresses such as salinity22,23 and be a good source for traits affecting grain quality such as milling yield and grain protein24 and grain mineral content25. During sequence analysis of reference quality assemblies (RQA) of 10 wheat cultivars, recent studies found two of them, cv. LongReach Lancer and cv. Julius, contained major introgressions on Chr2B (among others) potentially originating from T. timopheevii26,27. Introgressions from T. timopheevii have also been found in many other wheat accessions present in genebank collections28. Pre-breeding programmes involving the introgression of the whole genome of T. timopheevii, in small segments, into bread wheat10,29 with diagnostic KASP markers that can track these introgressions in wheat29,30 have provided promising new germplasm and tools to the wheat research community.
In this study, we report a chromosome-scale reference genome sequence assembly for T. timopheevii by integrating chromatin conformation capture (Hi-C) derived short-reads31 with PacBio HiFi long-reads32. The assembly was annotated for gene models and repeats. CpG methylation along the chromosomes was inferred from the PacBio CCS data. The high-quality T. timopheevii genome assembly obtained in this study provides a reference for the G genome of the Triticum genus. This new resource will form the basis to study chromosome rearrangements across different Triticeae species and will be explored to detect At and G genome introgressions in durum and bread wheat allowing future genome-informed gene discoveries for various agronomic traits.
Methods
Plant material, nucleic acid extraction and sequencing
High molecular weight (HMW) DNA was extracted from a young seedling (dark-treated for 48 hours) of T. timopheevii accession PI 94760 (United States National Plant Germplasm System, NPGS available at https://npgsweb.ars-grin.gov/gringlobal/search) using a modified Qiagen Genomic DNA extraction protocol (10.17504/protocols.io.bafmibk6)33. DNA was sheared to the appropriate size range (15–20 kb) and PacBio HiFi sequencing libraries were constructed by Novogene (UK) Company Limited. Sequencing was performed on 10 SMRT cells of the PacBio Sequel II system in CCS mode with kinetics option to generate ~267 Gb (~28-fold coverage) of long HiFi reads (Supplementary Table S1). Four Hi-C libraries were prepared using leaf samples (from the same plant used for HMW DNA extraction), at Phase Genomics (Seattle, USA) using the Proximo® Hi-C Kit for plant tissues according to the manufacturer’s protocol. The Hi-C libraries were sequenced on an Illumina NovaSeq 6000 S4 platform to generate ~2.8 billion of paired end 150 bp reads (~842 Gb raw data; ~89-fold coverage; Supplementary Table S2).
Total RNA was extracted from seedlings (3-leaf stage), seedlings at dusk, roots, flag leaves, spikes and grains. Flag leaf and whole spike were collected at 7 days post-anthesis and whole grains were collected at 15 days post-anthesis. In brief, 100 mg of ground powder from each tissue was used for RNA isolation using the RNeasy Plant Mini Kit (#74904, QIAGEN Ltd UK). The RNA samples were split into 2 aliquots, one for mRNA sequencing (RNA-Seq) and one for Iso-Seq34. Library construction for both types of sequencing was carried out by Novogene (UK) Company Limited. Illumina NovaSeq 6000 S4 platform was used for mRNA sequencing to generate on average 450 million reads (~67 Gb of 2 × 150 bp reads) for each sample (Supplementary Table S3). The second set of RNA aliquots from each of the six tissues were pooled into one sample and sequenced on the PacBio Sequel II system using the Iso-Seq pipeline to generate 4.47 Gb of Iso-Seq data (Supplementary Table S4a) which was analysed using PacBio Iso-Seq analysis pipeline (SMRT Link v12.0.0.177059).
Plants were grown in a glasshouse in 2 L pots containing John Innes No. 2 soil and maintained at 18–25 °C under 16 h light and 8 h dark conditions. All sequencing was carried out by Novogene (UK) Company Limited.
Cleaning of sequencing data
The HiFi sequencing read files in BAM format were converted and combined into one fastq file using bam2fastq v1.3.1 (available at https://github.com/jts/bam2fastq). Reads with PacBio adapters were removed using cutadapt v4.135 with parameters:–error-rate = 0.1–times = 3–overlap = 35–action = trim–revcomp–discard-trimmed. Hi-C reads were trimmed to remove Illumina adapters using Trimmomatic v0.3936 with parameters ILLUMINACLIP:TruSeq 3-PE-2.fa:2:30:10:2:keepBothReads SLIDINGWINDOW:4:20 MINLEN:40 CROP:150.
Genome size estimation
The size of the T. timopheevii genome was estimated by using k-mer (k = 32) distribution analysis with Jellyfish v2.2.1037 on the cleaned HiFi reads38. A k-mer count histogram was generated and the size of the T. timopheevii genome was estimated as ~9.46 Gb with heterozygosity of 0.001% (Fig. 1), using GenomeScope v2.039 (available at http://qb.cshl.edu/genomescope/genomescope2.0/) with parameters: ploidy = 2, k-mer length = 32, max k-mer coverage = 1000000 and average k-mer coverage = 10.
Fig. 1.
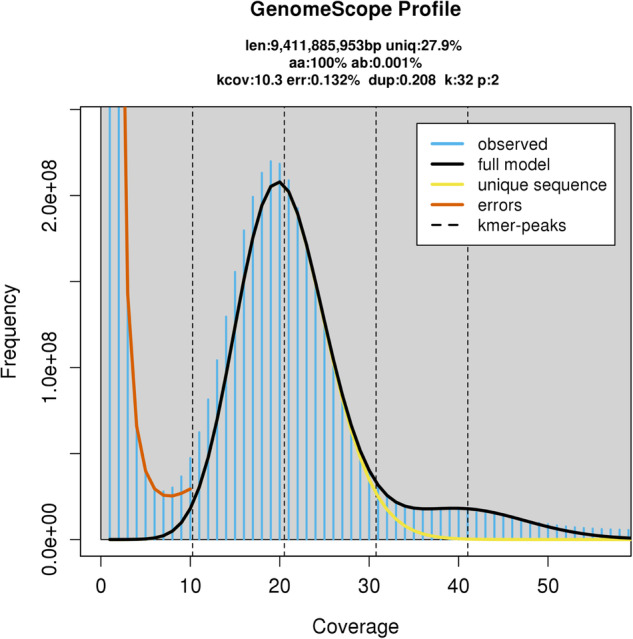
Genomescope profile for 32-mers based on HiFi reads.
Chromosome-scale genome assembly
The cleaned HiFi reads were assembled into the initial set of contigs using hifiasm v.0.16.140 with default parameters and the dataset was assessed using gfastats v1.3.141. The contig assembly had a total size of ~9.41 Gb, with a contig N50 value of 43.12 Mb. Genome completeness was assessed for the contig assembly using the Benchmarking Universal Single-Copy Orthologs (BUSCO v5.3.2)42 program with the embryophyta_odb10 database which yielded 99% of the complete BUSCO genes. Contaminants (contigs other than those categorised as Streptophyta or no hit) were identified using BlobTools v1.1.143 and removed.
To achieve chromosome-level assembly, the trimmed Hi-C data44 was mapped onto the decontaminated contig assembly using the Arima Genomics® mapping pipeline (available at https://github.com/ArimaGenomics/mapping_pipeline) and chromosome construction was conducted using the Salsa245 pipeline (available at https://github.com/marbl/SALSA) with default parameters and GATC as the cutting site for the restriction enzyme (DpnII). The Hi-C contact map for the scaffold assembly was constructed using PretextMap v0.1.9 and the chromatin contact matrix was manually corrected using PretextView v0.2.5 by following the Rapid Curation pipeline46 (https://gitlab.com/wtsi-grit/rapid-curation). The curated assembly was assessed using gfastats to consist of 14 pseudomolecules and 1656 unplaced scaffolds with a total length of 9,350,839,849 bp (including gaps) and a contig N50 of 42.4 Mb (Table 1). The orientation and the chromosome name of each pseudomolecule were determined based on homology with the wheat cv. Chinese Spring assembly RefSeq 2.147 A and B subgenomes, using dotplot comparison of sequence alignments produced by MUMmer’s (v3.2348) nucmer aligner and viewed on Dot (available at https://github.com/marianattestad/dot). The pseudomolecules were thus, renamed into the 14 T. timopheevii chromosomes, seven At genome chromosomes with a total length of ~4.85 Gb and consisting of 119 contigs and seven G genome chromosomes with a total length of ~4.40 Gb and consisting of 529 contigs (Table 2).
Table 1.
Summary statistics for genome assembly of Triticum timopheevii.
| Assembly characteristics | Value |
|---|---|
| Number of scaffolds | 1,670 |
| Total scaffold length (bp) | 9,350,839,849 |
| Scaffold N50 (bp) | 671,191,297 |
| Largest scaffold (bp) | 771,176,557 |
| No. of contigs | 2,304 |
| Total contig length (bp) | 9,350,587,949 |
| Average contig length (bp) | 4,058,415 |
| Contig N50 (bp) | 42,410,373 |
| Largest contig (bp) | 311,469,246 |
| GC content (%) | 46 |
| BUSCO evaluation (% of complete BUSCO genes) | 99.1 |
Table 2.
Statistics of the Triticum timopheevii pseudomolecules.
| Chromosome | Length (bp) | Number of contigs | Number of gene models |
|---|---|---|---|
| Chr1At | 614,431,332 | 14 | 9,982 |
| Chr1G | 495,016,746 | 50 | 8,777 |
| Chr2At | 767,071,137 | 10 | 12,729 |
| Chr2G | 671,256,291 | 72 | 13,941 |
| Chr3At | 670,741,101 | 10 | 9,489 |
| Chr3G | 671,191,297 | 75 | 13,452 |
| Chr4At | 771,176,557 | 23 | 12,878 |
| Chr4G | 643,128,204 | 68 | 9,936 |
| Chr5At | 694,350,238 | 12 | 11,821 |
| Chr5G | 641,290,954 | 78 | 13,079 |
| Chr6At | 585,824,631 | 33 | 9,011 |
| Chr6G | 589,079,669 | 87 | 11,406 |
| Chr7At | 745,638,687 | 17 | 12,863 |
| Chr7G | 692,654,486 | 99 | 14,851 |
| Unplaced scaffolds | 97,988,519 | 1656 | 2,110 |
| Total | 9,350,839,849 | 2,304 | 166,325 |
Organellar genome assembly
De novo assembly of the organelle genomes was carried out using the Oatk pipeline (available at https://github.com/c-zhou/oatk) with the cleaned HiFi reads. The circular chloroplast and mitochondrial contigs were assembled with a total size of 136,158 bp and 443,464 bp, respectively. Any unanchored contigs that aligned to these extranuclear genomes were removed from the final assembly.
Genome annotation
Gene models were generated from the T. timopheevii assembly using REAT - Robust and Extendable eukaryotic Annotation Toolkit (https://github.com/EI- CoreBioinformatics/reat) and Minos (https://github.com/EI-CoreBioinformatics/minos) which make use of Mikado49 (https://github.com/EI-CoreBioinformatics/mikado), Portcullis (https://github.com/EI-CoreBioinformatics/portcullis) and many third-party tools (listed in the above repositories). A consistent gene naming standard50 was used to make the gene models uniquely identifiable.
Repeat identification
Repeat annotation was performed using EI-Repeat version 1.3.4 pipeline (https://github.com/EI- CoreBioinformatics/eirepeat) which uses third party tools for repeat calling. In the pipeline, RepeatModeler (v1.0.11 - http://www.repeatmasker.org/RepeatModeler/) was used for de novo identification of repetitive elements from the assembled T. timopheevii genome. High copy protein coding genes potentially included in the RepeatModeler library were identified and effectively removed by running RepeatMasker v4.0.7 using a curated set of high confidence T. aestivum coding genes to hard mask the RepeatModeler library; transposable element genes were first excluded from the T. aestivum coding gene set by running TransposonPSI (r08222010). Unclassified repeats were searched in a custom BLAST database of organellar genomes (mitochondrial and chloroplast sequences from Triticum in the NCBI nucleotide division). Any repeat families matching organellar DNA were also hard-masked. Repeat identification was completed by running RepeatMasker v4.0.72 with a RepBase embryophyte library and with the customized RepeatModeler library (i.e. after masking out protein coding genes), both using the -nolow setting. Overall, 78.43% of the assembly was classified as repetitive sequences (Table 3). The consolidated set of repeat features (i.e. RepeatMasker outputs from the embryophyte and customized RepeatModeler libraries) were given as input to the evidence guided gene prediction (REAT prediction) and gene model consolidation (Minos) steps. All other annotation steps utilised the unmasked genome.
Table 3.
Classification of repeat annotation in Triticum timopheevii.
| Class | Number of elements | Length occupied (bp) | Percentage of sequence | |
|---|---|---|---|---|
| Retrotransposons | SINEs | 20,589 | 1,759,774 | 0.02 |
| LINEs | 150,497 | 116,697,520 | 1.25 | |
| LTRs: Copia | 535,455 | 1,620,870,187 | 17.33 | |
| LTRs: Gypsy | 1,690,034 | 3,873,777,180 | 41.43 | |
| LTRs: Unknown | 1,501,064 | 139,512,022 | 1.49 | |
| DNA transposons | hobo-Activator | 20,177 | 6,117,182 | 0.07 |
| Tc1-IS630-Pogo | 118,082 | 16,100,160 | 0.17 | |
| Tourist/Harbinger | 54,914 | 16,353,000 | 0.17 | |
| Other | 1,582,537 | 948,759,196 | 10.15 | |
| Unclassified | — | 1,164,179 | 593,808,513 | 6.35 |
| Total | 6,837,528 | 7,333,754,734 | 78.43 |
-
2.
Reference guided transcriptome reconstruction
Gene models were derived from the RNA-Seq reads, Iso-Seq transcripts (122,253 high quality and 82 low quality isoforms; Supplementary Table S4b) and Full-Length Non- Concatamer Reads (FLNC) using the REAT transcriptome workflow. HISAT2 v2.2.151 was selected as the short read aligner with Iso-Seq transcripts aligned with minimap2 v2.18-r101552, maximum intron length was set as 50,000 bp and minimum intron length to 20 bp. Iso-Seq alignments were required to meet 95% coverage and 90% identity. High-confidence splice junctions were identified by Portcullis v 1.2.453. RNA-Seq Illumina reads were assembled for each tissue with StringTie2 v2.1.554 and Scallop v0.10.555, while FLNC reads were assembled using StringTie2 (Supplementary Table S5). Gene models were derived from the RNA-Seq assemblies and Iso-Seq and FLNC alignments with Mikado. Mikado was run with all Scallop, StringTie2, Iso-Seq and FLNC alignments and a second run with only Iso-Seq and FLNC alignments (Supplementary Table S6).
-
3.
Cross-species protein alignment
Protein sequences from 10 Poaceae species (Supplementary Table S7) were aligned to the T. timopheevii assembly using the REAT Homology workflow with options–annotation_filters aa_len–alignment_species Angiosp–filter_max_intron 20000–filter_min_exon 10–alignment_filters aa_len internal_stop intron_len exon_len splicing–alignment_min_coverage 90–junction_f1_filter 40–post_alignment_clip clip_term_intron-exon–term5i_len 5000–term3i_len 5000–term5c_len 36–term3c_len 36. The REAT Homology workflow aligns proteins with spaln v2.4.756 and filters and generates metrics to remove misaligned proteins. Simultaneously, the same protein set were also aligned using miniprot v0.357 and similarly filtered as in the REAT homology workflow. The aligned proteins from both methods were clustered into loci and a consolidated set of gene models were derived via Mikado.
-
4.
Evidence guided gene prediction
The evidence guided annotation of protein coding genes based on repeats, RNA-Seq mappings, transcript assembly and alignment of protein sequences was created using the REAT prediction workflow. The pipeline has four main steps: (1) the REAT transcriptome and homology Mikado models are categorised based on alignments to UniProt proteins to identify models with likely full-length CDS and which meet basic structural checks i.e., having complete but not excessively long UTRs and not exceeding a minimum CDS/cDNA ratio. A subset of gene models is then selected from the classified models and used to train the AUGUSTUS gene predictor58; (2) Augustus is run in both ab initio mode and with extrinsic evidence generated in the REAT transcriptome and homology runs (repeats, protein alignments, RNA-Seq alignments, splice junctions, categorised Mikado models). Three evidence guided AUGUSTUS predictions are created using alternative bonus scores and priority based on evidence type; (3) AUGUSTUS models, REAT transcriptome/homology models, protein and transcriptome alignments are provided to EVidenceModeler59 (EVM) to generate consensus gene structures; (4) EVM models are processed through Mikado to add UTR features and splice variants.
-
5.
Projection of gene models from Triticum aestivum
A reference set of hexaploid wheat gene models was derived from public gene sets (IWGSC60 and 10+ wheat26) projected onto the IWGSC RefSeq v1.0 assembly60; a filtered and consolidated set of models was derived with Minos, with a primary model defined for each gene. Models were scored on a combination of intrinsic gene structure characteristics, evidence support (protein and transcriptome data) and consistency in gene structure across the input gene models. The Minos primary models were classified as full-length or partial based on alignment to a filtered magnoliopsida Swiss-Prot TrEMBL database. This assignment, together with criteria for gene structure characteristics and the original confidence classification, was used to classify models into 6 categories (Platinum, Gold, Silver, Bronze, Stone and Paper), with Platinum being the highest confidence category for models assessed as full-length, with an original confidence classification of “high”, meeting structural checks for number of UTR and CDS/cDNA ratio and which were assessed as consistently annotated across the input gene sets. Reclassification resulted in 55,319 Platinum, 24,789 Gold, 11,968 Silver, 61,845 Bronze, 110,518 Stone and 115,336 Paper genes. The four highest confidence categories Platinum, Gold, Silver and Bronze were projected onto the T. timopheevii assembly with Liftoff v1.5.161, only those models transferred fully with no loss of bases and identical exon/intron structure were retained (https://github.com/lucventurini/ei-liftover). Similarly, high confidence genes annotated in the hexaploid wheat cv. Chinese Spring RefSeq v2.1 assembly47 were projected onto the T. timopheevii genome assembly with Liftoff, and only those models transferred fully with no loss of bases and identical exon/intron structure were retained. Among these, gene models with the attribute “manually_curated” in the original Refseq v2.1 assembly were extracted as a set.
-
6.
Gene model consolidation
The final set of gene models was selected using Minos (Table 4). Minos is a pipeline that generates and utilises metrics derived from protein, transcript, and expression data sets to create a consolidated set of gene models. In this annotation, the following gene models were filtered and consolidated into a single set of gene models using Minos:
The three alternative evidence guided Augustus gene builds described earlier.
The gene models derived from the REAT transcriptome runs described earlier.
The gene models derived from the REAT homology runs described earlier.
The gene models derived from the REAT prediction run (AUGUSTUS and EVM-Mikado) described earlier.
The gene models derived from projecting public and curated T. aestivum gene models of varying confidence levels onto the T. timopheevii genome as described earlier.
IWGSC Refseq v2.1 models identified as “manually_curated” projected onto the T. timopheevii genome as described earlier.
Table 4.
Summary statistics for the final structural annotation of the T. timopheevii genome.
| Stat | Value |
|---|---|
| Number of genes | 166,325 |
| Number of Transcripts | 218,100 |
| Transcripts per gene | 1.31 |
| Number of monoexonic genes | 51,702 |
| Monoexonic transcripts | 53,192 |
| Transcript mean size cDNA (bp) | 1,658.27 |
| Transcript median size cDNA (bp) | 1412 |
| Min cDNA | 96 |
| Max cDNA | 20,589 |
| Total exons | 997,779 |
| Exons per transcript | 4.57 |
| Exon mean size (bp) | 362.47 |
| CDS mean size (bp) | 277.55 |
| Transcript mean size CDS (bp) | 1,171.61 |
| Transcript median size CDS (bp) | 957 |
| Min CDS | 0 |
| Max CDS | 20,283 |
| Intron mean size (bp) | 628.4 |
| 5’UTR mean size (bp) | 182.93 |
| 3’UTR mean size (bp) | 294.58 |
Gene models were classified as biotypes protein_coding_gene, predicted_gene and transposable_element_gene, and assigned as high or low confidence (Table 5) based on the criteria below:
High confidence (HC) protein_coding_gene: Any protein coding gene where any of its associated gene models have a BUSCO v5.4.762 protein status of Complete/Duplicated OR have diamond v0.9.36 coverage (average across query and target coverage) >= 90% against the listed Poaceae protein datasets (Supplementary Table S7) or UniProt magnoliopsida proteins. Or alternatively have average blastp coverage (across query and target coverage) >= 80% against the listed protein datasets/UniProt magnoliopsida AND have transcript alignment F1 score (average across nucleotide, exon and junction F1 scores based on RNA-Seq transcript assemblies) >= 60%.
Low confidence (LC) protein_coding_gene: Any protein coding gene where all its associated transcript models do not meet the criteria to be considered as high confidence protein coding transcripts.
HC transposable_element_gene: Any protein coding gene where any of its associated gene models have coverage >= 40% against the combined interspersed repeats (see section 1).
LC transposable_element_gene: Any protein coding gene where all its associated transcript models do not meet the criteria to be considered as high confidence and assigned as a transposable_element_gene (see c).
LC predicted_gene: Any protein coding gene where all the associated transcript models do not meet the criteria to be considered as high confidence protein coding transcripts. In addition, where any of the associated gene models have average blastp coverage (across query and target coverage) <30% against the listed protein datasets AND having a protein-coding potential score <0.25 calculated using CPC2 0.163.
LC ncRNA gene: Any gene model with no CDS features AND a protein-coding potential score <0.3 calculated using CPC2 0.1.
Discarded models: Any models having no BUSCO protein hit AND a protein alignment score (average across nucleotide, exon and junction F1 scores based on protein alignments) <0.2 AND a transcript alignment F1 score (average across nucleotide, exon and junction F1 scores based on RNA-Seq transcript assemblies) <0.2 AND a diamond coverage (target coverage) <0.3 AND Kallisto v0.4464 expression score <0.2 from across RNA-Seq reads OR having short CDS <30 bps. Any ncRNA genes (no CDS features) not meeting the ncRNA gene requirements (f) were also excluded.
Table 5.
Minos classified gene models.
| Biotype | Confidence | Gene | Transcript |
|---|---|---|---|
| protein_coding_gene | Low | 73,844 | 79,329 |
| protein_coding_gene | High | 67,107 | 112,338 |
| transposable_element_gene | Low | 15,871 | 16,231 |
| predicted_gene | Low | 4,974 | 5,033 |
| transposable_element_gene | High | 3,258 | 3,410 |
| ncRNA_gene | Low | 1,271 | 1,759 |
| Total | 166,325 | 218,100 |
Gene model distribution across the pseudomolecules and unplaced scaffolds is shown in Table 2 and gene density of 164,617 protein coding genes across the T. timopheevii genome was calculated using deepStats v0.465 in 10 Mb bins (Fig. 2b).
-
7.
Functional annotation
Fig. 2.

Circos plot84 of features of the chromosome-scale assembly of T. timopheevii showing (a) major translocations with the T. timopheevii genome as observed through collinearity analysis against T. turgidum, (b) gene density (of all gene models), (c) NLR density (max count 87), (d) DNA methylation (5mC modification) density, (e) distribution of KASP markers based on SNPs with bread wheat cv. Chinese Spring29 and (f) GC content (in %). Tt in chromosome name represents T. timopheevii. Y-axis for tracks c and f have an interval of 18 and 20 units, respectively.
All proteins were annotated using AHRD v.3.3.3 (available at https://github.com/groupschoof/AHRD/blob/master/README.textile). Sequences were compared against the reference proteins (Arabidopsis thaliana TAIR10, TAIR10_pep_20101214_updated.fasta.gz - https://www.araport.org) and the UniProt viridiplantae sequences66 (data download 06-May-2023), both Swiss-Prot and TrEMBL datasets using blastp v2.6.0 with an e-value of 1e- 5. InterproScan v5.22.6167 results were also provided to AHRD. The standard AHRD example configuration file path test/resources/ahrd_example_input_go_prediction.yml, distributed with the AHRD tool, was adapted apart from the location of input and output files. The GOA mapping from UniProt (ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/UNIPROT/goa_uniprot_all.gaf.gz) was included as parameter ‘gene_ontology_result’. The interpro database (ftp://ftp.ebi.ac.uk/pub/databases/interpro/61.0/interpro.xml.gz) was included as parameter ‘interpro_database’. The parameter ‘prefer_reference_with_go_annos’ was changed to ‘false’ and the blast database specific weights used were:
blast_dbs:
swissprot:
weight: 100
description_score_bit_score_weight: 0.2
trembl:
weight: 50
description_score_bit_score_weight: 0.4
tair:
weight: 50
description_score_bit_score_weight: 0.4
Since T. timopheevii is known as an important source for genetic variation for resistance against major diseases of wheat as described above and as the majority of cloned disease-resistance genes encode nucleotide-binding leucine-rich repeats (NLRs)68,69, we validated the annotation of all gene models annotated as NB-ARC domain-containing/disease resistance proteins in the genome assembly (2399 gene models) using NLR-Annotator v270 and found an additional 166 NLRs (total 2565). We plotted the genomic distribution of the larger set (Fig. 2c), by calculating the density in 10 Mb bins using deepStats v0.4, which shows concentration of these NLRs at mostly distal ends of the chromosomes of Triticum timopheevii.
Generation of PacBio DNA methylation profile
Methylation in CpG context was inferred with ccsmeth v0.3.271, using the kinetics data from PacBio CCS subreads obtained during HMW DNA sequencing. The methylation prediction for CCS reads were called using the model “model_ccsmeth_5mCpG_call_mods_attbigru2s_b21.v2.ckpt”. The reads with the MM + ML tags were aligned to the pseudomolecules in the T. timopheevii assembly using BWA v0.7.1772. The methylation frequency was calculated at genome level with the modbam files and the aggregate mode of ccsmeth with the model “model_ccsmeth_5mCpG_aggregate_attbigru_b11.v2p.ckpt”. The genomic distribution of 5mC modifications across T. timopheevii (Fig. 2d) shows that G genome chromosomes have more methylation with an average of ~401.8 Kbp methylated bases per 10 Mb bin as compared to the At genome chromosomes with an average of ~385.5 Kbp per 10 Mb bin (calculated using deepStats v0.4).
Data Records
The raw sequence files for the HiFi, Hi-C, RNA-Seq and IsoSeq reads were deposited in the European Nucleotide Archive (ENA) under accession number PRJEB7166073. The final chromosome-scale assembly consisting of the nuclear and organelle genomes was deposited at ENA under the accession number GCA_963921465.174.
The genome assemblies, gene model and repeat annotations, methylation profile and Hi-C contact map are also available at on DRYAD Digital Repository75.
Technical Validation
Assessment of genome assembly and annotation
The final curated assembly was assessed by mapping the trimmed Hi-C reads to the post-curated assembly (as described above for scaffolding) and generating a final Hi-C contact map using PretextMap v0.1.9 and viewed using PretextView v0.2.5. It showed a dense dark blue pattern along the diagonal revealing no potential mis-assemblies (Fig. 3). The anti-diagonals in the Hi-C contact matrix were expected and have been reported for other relatively large plant genomes such as those from the Triticeae tribe76,77 as they correspond to the typical Rabl configuration of Triticeae chromosomes78,79.
Fig. 3.
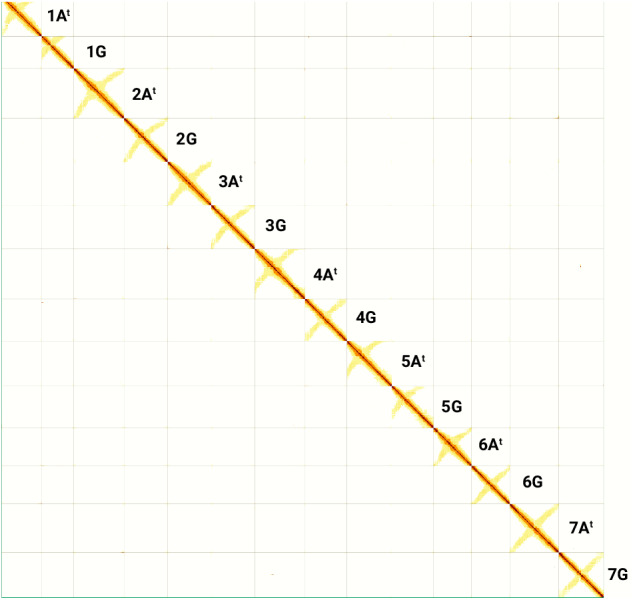
Contact map after the integration of the Hi-C data and manual correction using PretextView.
The BUSCO v5.3.242 (-l poales_odb10) score of 99.1% (0.1% fragmented and 0.8% missing BUSCOs; Supplementary Table S8a) at the genome level indicates a high completeness of the T. timopheevii assembly. The quality of the T. timopheevii assembly was assessed with Merqury80 based on the PacBio HiFi reads using 31-mers. The QV (consensus quality value) and k-mer completeness scores were 65.5 and 97.8%, respectively. The quality of the assembly was further assessed by determining the LTR Assembly Index (LAI) and attainment of a value of 13.62 suggests that the assembly meets the criteria for a reference quality genome81 indicating a high level of accuracy and completeness in capturing genomic features, particularly those related to LTR retrotransposons.
Completeness of the gene model prediction was also evaluated using BUSCO (-l poales_odb10) and produced a score of 99.9% (0.0% fragmented and 0.1% missing BUSCOs; Supplementary Table S8b). The number of HC gene models (70,365) is in the range of a tetraploid Triticeae species (34,000–43,000 high-confidence gene models per haploid genome)82.
Of the total 14 chromosomes, we found telomeric repeats on both ends for 5 chromosomes (1At, 2 G, 3At, 6At, and 7 G) and on one end for 7 chromosomes (1GL, 2AtS, 3GL, 4GS, 5GL, 6GL and 7AtL).
Usage Notes
A genome browser for the assembly of T. timopheevii generated in this study is currently being hosted at GrainGenes83 (https://wheat.pw.usda.gov/jb?data=/ggds/whe-timopheevii) with tracks for annotated gene models and repeats and BLAST functionality available at https://wheat.pw.usda.gov/blast/.
Supplementary information
Acknowledgements
This work was supported by the Biotechnology and Biological Sciences Research Council [grant number BB/P016855/1] as part of the Developing Future Wheat (DFW) programme. We are grateful for access to the University of Nottingham’s Augusta HPC service. Part of this work was also delivered via Transformative Genomics the BBSRC funded National Bioscience Research Infrastructure (BBS/E/ER/23NB0006) at Earlham Institute by members of the Genomics Pipelines and Core Bioinformatics Groups. E.Y. and T.S. were supported by the US. Department of Agriculture, Agricultural Research Service, Project No. 2030–21000-056-00D.
Author contributions
Su.G., J.K. and I.K. designed the study and obtained funding for it. C.Y., Du.S. and S.A. carried out plant maintenance and nucleic acid extraction. Su.G., M.W. and L.Y. generated the genome assembly. Su.G and J.C. carried out manual curation of the assembly. Sr.G. and Da.S. carried out the genome annotation. E.Y. and T.S. generated the genome browser. Su.G. wrote the initial manuscript. All authors have read and approved the final manuscript.
Code availability
All software and pipelines were executed according to the manual and protocol of published tools. No custom code was generated for these analyses.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41597-024-03260-w.
References
- 1.Dvořák J, Terlizzi PD, Zhang H-B, Resta P. The evolution of polyploid wheats: identification of the A genome donor species. Genome. 1993;36:21–31. doi: 10.1139/g93-004. [DOI] [PubMed] [Google Scholar]
- 2.Dvorak J, Zhang H-B. Variation in repeated nucleotide sequences sheds light on the phylogeny of the wheat B and G genomes. Proceedings of the National Academy of Sciences. 1990;87:9640–9644. doi: 10.1073/pnas.87.24.9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed HI, et al. Einkorn genomics sheds light on history of the oldest domesticated wheat. Nature. 2023;620:830–838. doi: 10.1038/s41586-023-06389-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodriguez S, Maestra B, Perera E, Diez M, Naranjo T. Pairing affinities of the B-and G-genome chromosomes of polyploid wheats with those of Aegilops speltoides. Genome. 2000;43:814–819. doi: 10.1139/g00-055. [DOI] [PubMed] [Google Scholar]
- 5.Li LF, et al. Genome sequences of five Sitopsis species of Aegilops and the origin of polyploid wheat B subgenome. Molecular plant. 2022;15:488–503. doi: 10.1016/j.molp.2021.12.019. [DOI] [PubMed] [Google Scholar]
- 6.Dvořák, J. Triticum Species (Wheat). Encyclopedia of Genetics, 2060–2068 (2001).
- 7.Jiang J, Gill BS. Different species-specific chromosome translocations inTriticum timopheevii and T. turgidum support the diphyletic origin of polyploid wheats. Chromosome Research. 1994;2:59–64. doi: 10.1007/BF01539455. [DOI] [PubMed] [Google Scholar]
- 8.Maestra B, Naranjo T. Structural chromosome differentiation between Triticum timopheevii and T. turgidum and T. aestivum. Theoretical and Applied Genetics. 1999;98:744–750. doi: 10.1007/s001220051130. [DOI] [Google Scholar]
- 9.Rodriguez S, Perera E, Maestra B, Díez M, Naranjo T. Chromosome structure of Triticum timopheevii relative to T. turgidum. Genome. 2000;43:923–930. doi: 10.1139/g00-062. [DOI] [PubMed] [Google Scholar]
- 10.Devi U, et al. Development and characterisation of interspecific hybrid lines with genome-wide introgressions from Triticum timopheevii in a hexaploid wheat background. BMC Plant Biol. 2019;19:183. doi: 10.1186/s12870-019-1785-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown-Guedira GL, Singh S, Fritz AK. Performance and Mapping of Leaf Rust Resistance Transferred to Wheat from Triticum timopheevii subsp. armeniacum. Phytopathology. 2003;93:784–789. doi: 10.1094/PHYTO.2003.93.7.784. [DOI] [PubMed] [Google Scholar]
- 12.Singh AK, et al. Genetics and mapping of a new leaf rust resistance gene in Triticum aestivum L. × Triticum timopheevii Zhuk. derivative ‘Selection G12. J Genet. 2017;96:291–297. doi: 10.1007/s12041-017-0760-4. [DOI] [PubMed] [Google Scholar]
- 13.Leonova IN, et al. Microsatellite mapping of a leaf rust resistance gene transferred to common wheat from Triticum timopheevii. Cereal Research Communications. 2010;38:211–219. doi: 10.1556/CRC.38.2010.2.7. [DOI] [Google Scholar]
- 14.McIntosh R, Gyarfas J. Triticum timopheevii as a source of resistance to wheat stem rust. Zeitschrift fur Pflanzenzuchtung. 1971;66:240–248. [Google Scholar]
- 15.Wu S, Pumphrey M, Bai G. Molecular Mapping of Stem-Rust-Resistance Gene Sr40 in Wheat. Crop Science. 2009;49:1681–1686. doi: 10.2135/cropsci2008.11.0666. [DOI] [Google Scholar]
- 16.Allard R, Shands R. Inheritance of resistance to stem rust and powdery mildew in cytologically stable spring wheats derived from Triticum timopheevii. Phytopathology. 1954;44:266–274. [Google Scholar]
- 17.Perugini LD, Murphy JP, Marshall D, Brown-Guedira G. Pm37, a new broadly effective powdery mildew resistance gene from Triticum timopheevii. Theoretical and Applied Genetics. 2008;116:417–425. doi: 10.1007/s00122-007-0679-x. [DOI] [PubMed] [Google Scholar]
- 18.Qin B, et al. Collinearity-based marker mining for the fine mapping of Pm6, a powdery mildew resistance gene in wheat. Theoretical and Applied Genetics. 2011;123:207–218. doi: 10.1007/s00122-011-1577-9. [DOI] [PubMed] [Google Scholar]
- 19.Steed, A. et al. Identification of Fusarium Head Blight Resistance in Triticum timopheevii Accessions and Characterization of Wheat-T. timopheevii Introgression Lines for Enhanced Resistance. Frontiers in Plant Science13 (2022). [DOI] [PMC free article] [PubMed]
- 20.Malihipour A, Gilbert J, Fedak G, Brûlé-Babel A, Cao W. Characterization of agronomic traits in a population of wheat derived from Triticum timopheevii and their association with Fusarium head blight. European Journal of Plant Pathology. 2016;144:31–43. doi: 10.1007/s10658-015-0744-2. [DOI] [Google Scholar]
- 21.Brown-Guedira G, et al. Evaluation of a collection of wild timopheevi wheat for resistance to disease and arthropod pests. Plant disease. 1996;80:928–933. doi: 10.1094/PD-80-0928. [DOI] [Google Scholar]
- 22.Badridze G, Weidner A, Asch F, Börner A. Variation in salt tolerance within a Georgian wheat germplasm collection. Genetic resources and crop evolution. 2009;56:1125–1130. doi: 10.1007/s10722-009-9436-0. [DOI] [Google Scholar]
- 23.Yudina R, Leonova I, Salina E, Khlestkina E. Change in salt tolerance of bread wheat as a result of the introgression of the genetic material of Aegilops speltoides and Triticum timopheevii. Russian Journal of Genetics: Applied Research. 2016;6:244–248. [Google Scholar]
- 24.Lehmensiek, A., Bovill, W., Banks, P., Sutherland, M. Molecular characterization of a Triticum timopheevii introgression in a Wentworth/Lang population. (2008).
- 25.Hu X, et al. Zn and Fe concentration variations of grain and flag leaf and the relationship with NAM-G1 gene in Triticum timopheevii (Zhuk.) Zhuk. ssp. timopheevii. Cereal Research Communications. 2017;45:421–431. doi: 10.1556/0806.45.2017.022. [DOI] [Google Scholar]
- 26.Walkowiak S, et al. Multiple wheat genomes reveal global variation in modern breeding. Nature. 2020;588:277–283. doi: 10.1038/s41586-020-2961-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keilwagen J, et al. Detecting major introgressions in wheat and their putative origins using coverage analysis. Scientific Reports. 2022;12:1908. doi: 10.1038/s41598-022-05865-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keilwagen, J. et al. Finding needles in a haystack: identification of inter-specific introgressions in wheat genebank collections using low-coverage sequencing data. Frontiers in Plant Science14 (2023). [DOI] [PMC free article] [PubMed]
- 29.King, J. et al. Introgression of the Triticum timopheevii Genome Into Wheat Detected by Chromosome-Specific Kompetitive Allele Specific PCR Markers. Frontiers in Plant Science13 (2022). [DOI] [PMC free article] [PubMed]
- 30.Grewal S, et al. Rapid identification of homozygosity and site of wild relative introgressions in wheat through chromosome-specific KASP genotyping assays. Plant Biotechnol J. 2020;18:743–755. doi: 10.1111/pbi.13241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belton JM, et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods. 2012;58:268–276. doi: 10.1016/j.ymeth.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wenger AM, et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nature Biotechnology. 2019;37:1155–1162. doi: 10.1038/s41587-019-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Driguez P, et al. LeafGo: Leaf to Genome, a quick workflow to produce high-quality de novo plant genomes using long-read sequencing technology. Genome biology. 2021;22:256. doi: 10.1186/s13059-021-02475-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong L, et al. Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genomics. 2015;16:1039. doi: 10.1186/s12864-015-2257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17, 3 (2011).
- 36.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang, H. et al. Estimation of genome size using k-mer frequencies from corrected long reads. arXiv:200311817 [q-bioGN] (2020).
- 39.Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature Communications. 2020;11:1432. doi: 10.1038/s41467-020-14998-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng H, Concepcion GT, Feng X, Zhang H, Li H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods. 2021;18:170–175. doi: 10.1038/s41592-020-01056-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Formenti G, et al. Gfastats: conversion, evaluation and manipulation of genome sequences using assembly graphs. Bioinformatics. 2022;38:4214–4216. doi: 10.1093/bioinformatics/btac460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 43.Laetsch, D., Blaxter, M. BlobTools: Interrogation of genome assemblies. F1000Research6 (2017).
- 44.Korbel JO, Lee C. Genome assembly and haplotyping with Hi-C. Nature Biotechnology. 2013;31:1099–1101. doi: 10.1038/nbt.2764. [DOI] [PubMed] [Google Scholar]
- 45.Ghurye J, et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLOS Computational Biology. 2019;15:e1007273. doi: 10.1371/journal.pcbi.1007273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Howe, K. et al. Significantly improving the quality of genome assemblies through curation. GigaScience10 (2021). [DOI] [PMC free article] [PubMed]
- 47.Zhu T, et al. Optical maps refine the bread wheat Triticum aestivum cv. Chinese Spring genome assembly. The Plant Journal. 2021;107:303–314. doi: 10.1111/tpj.15289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome biology. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Venturini, L., Caim, S., Kaithakottil, G. G., Mapleson, D. L., Swarbreck, D. Leveraging multiple transcriptome assembly methods for improved gene structure annotation. GigaScience7 (2018). [DOI] [PMC free article] [PubMed]
- 50.Boden SA, et al. Updated guidelines for gene nomenclature in wheat. Theoretical and Applied Genetics. 2023;136:72. doi: 10.1007/s00122-023-04253-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mapleson, D., Venturini, L., Kaithakottil, G., Swarbreck, D. Efficient and accurate detection of splice junctions from RNA-seq with Portcullis. GigaScience7 (2018). [DOI] [PMC free article] [PubMed]
- 54.Kovaka S, et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology. 2019;20:278. doi: 10.1186/s13059-019-1910-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shao M, Kingsford C. Accurate assembly of transcripts through phase-preserving graph decomposition. Nature Biotechnology. 2017;35:1167–1169. doi: 10.1038/nbt.4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gotoh O. A space-efficient and accurate method for mapping and aligning cDNA sequences onto genomic sequence. Nucleic Acids Research. 2008;36:2630–2638. doi: 10.1093/nar/gkn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li, H. Protein-to-genome alignment with miniprot. Bioinformatics39 (2023). [DOI] [PMC free article] [PubMed]
- 58.Stanke M, Morgenstern B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Research. 2005;33:W465–W467. doi: 10.1093/nar/gki458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haas BJ, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome biology. 2008;9:R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.IWGSC et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science361 (2018). [DOI] [PubMed]
- 61.Shumate A, Salzberg SL. Liftoff: accurate mapping of gene annotations. Bioinformatics. 2021;37:1639–1643. doi: 10.1093/bioinformatics/btaa1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seppey, M., Manni, M., Zdobnov, E. M. in Gene Prediction: Methods and Protocols (ed. Kollmar M.) BUSCO: Assessing Genome Assembly and Annotation Completeness (Springer New York, 2019). [DOI] [PubMed]
- 63.Kong L, et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Research. 2007;35:W345–W349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 65.Gautier R. 2020. gtrichard/deepStats: New tools and much needed fixes. Zenodo. [DOI]
- 66.Consortium U. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jones P, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kourelis J, Van Der Hoorn RA. Defended to the nines: 25 years of resistance gene cloning identifies nine mechanisms for R protein function. The Plant cell. 2018;30:285–299. doi: 10.1105/tpc.17.00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen R, Gajendiran K, Wulff BBH. R we there yet? Advances in cloning resistance genes for engineering immunity in crop plants. Current opinion in plant biology. 2024;77:102489. doi: 10.1016/j.pbi.2023.102489. [DOI] [PubMed] [Google Scholar]
- 70.Steuernagel B, et al. The NLR-Annotator Tool Enables Annotation of the Intracellular Immune Receptor Repertoire1 [OPEN] Plant Physiology. 2020;183:468–482. doi: 10.1104/pp.19.01273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ni P, et al. DNA 5-methylcytosine detection and methylation phasing using PacBio circular consensus sequencing. Nature communications. 2023;14:4054. doi: 10.1038/s41467-023-39784-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.2024. NCBI Sequence Read Archive. ERP156445
- 74.2024. NCBI GenBank. GCA_963921465.1
- 75.Grewal S, 2024. Data from: Chromosome-scale genome assembly of bread wheat’s wild relative Triticum timopheevii [Dataset] Dryad. [DOI] [PMC free article] [PubMed]
- 76.Dong P, et al. 3D chromatin architecture of large plant genomes determined by local A/B compartments. Molecular plant. 2017;10:1497–1509. doi: 10.1016/j.molp.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 77.Mascher M, et al. A chromosome conformation capture ordered sequence of the barley genome. Nature. 2017;544:427–433. doi: 10.1038/nature22043. [DOI] [PubMed] [Google Scholar]
- 78.Anamthawat-Jónsson K, Heslop-Harrison J. Centromeres, telomeres and chromatin in the interphase nucleus of cereals. Caryologia. 1990;43:205–213. doi: 10.1080/00087114.1990.10796999. [DOI] [Google Scholar]
- 79.Cowan CR, Carlton PM, Cande WZ. The polar arrangement of telomeres in interphase and meiosis. Rabl organization and the bouquet. Plant Physiology. 2001;125:532–538. doi: 10.1104/pp.125.2.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rhie A, Walenz BP, Koren S, Phillippy AM. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome biology. 2020;21:1–27. doi: 10.1186/s13059-020-02134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ou S, Chen J, Jiang N. Assessing genome assembly quality using the LTR Assembly Index (LAI) Nucleic acids research. 2018;46:e126–e126. doi: 10.1093/nar/gky730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Poretti M, Praz CR, Sotiropoulos AG, Wicker T. A survey of lineage‐specific genes in Triticeae reveals de novo gene evolution from genomic raw material. Plant Direct. 2023;7:e484. doi: 10.1002/pld3.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yao, E. et al. GrainGenes: a data-rich repository for small grains genetics and genomics. Database2022 (2022). [DOI] [PMC free article] [PubMed]
- 84.Krzywinski M, et al. Circos: An information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Gautier R. 2020. gtrichard/deepStats: New tools and much needed fixes. Zenodo. [DOI]
- 2024. NCBI Sequence Read Archive. ERP156445
- 2024. NCBI GenBank. GCA_963921465.1
- Grewal S, 2024. Data from: Chromosome-scale genome assembly of bread wheat’s wild relative Triticum timopheevii [Dataset] Dryad. [DOI] [PMC free article] [PubMed]
Supplementary Materials
Data Availability Statement
All software and pipelines were executed according to the manual and protocol of published tools. No custom code was generated for these analyses.