


Abstract
Ribosome-enhanced translational miscoding of the genetic code causes protein dysfunction and loss of cellular fitness. During evolution, open reading frame length increased, necessitating mechanisms for enhanced translation fidelity. Indeed, eukaryal ribosomes are more accurate than bacterial counterparts, despite their virtually identical, conserved active centers. During the evolution of eukaryotic organisms ribosome expansions at the rRNA and protein level occurred, which potentially increases the options for translation regulation and cotranslational events. Here we tested the hypothesis that ribosomal RNA expansions can modulate the core function of the ribosome, faithful protein synthesis. We demonstrate that a short expansion segment present in all eukaryotes’ small subunit, ES7S, is crucial for accurate protein synthesis as its presence adjusts codon-specific velocities and guarantees high levels of cognate tRNA selection. Deletion of ES7S in yeast enhances mistranslation and causes protein destabilization and aggregation, dramatically reducing cellular fitness. Removal of ES7S did not alter ribosome architecture but altered the structural dynamics of inter-subunit bridges thus affecting A-tRNA selection. Exchanging the yeast ES7S sequence with the human ES7S increases accuracy whereas shortening causes the opposite effect. Our study demonstrates that ES7S provided eukaryal ribosomes with higher accuracy without perturbing the structurally conserved decoding center.
Graphical Abstract
Graphical Abstract.

Introduction
Accurate translation of the genetic code into functional proteins is essential for cell viability. The machines performing this task, ribosomes, are highly conserved in the sequence and structure of their functional centers. Nevertheless, during evolution expansion occurred at the protein and the ribosomal RNA (rRNA) level adding further options for regulation and modulation of cotranslational processes to the ribosome (1–4). The addition of an amino acid to the nascent polypeptide chain, translation elongation, involves transfer RNA (tRNA) recognition at the ribosomal A-site, peptide bond formation and tRNA translocation; a cycle that is repeated until a stop codon is reached and protein synthesis is terminated. The individual sub-steps of the elongation cycle require ribosomal dynamics, especially within the small subunit (5–7). Cognate codon-anticodon interactions are recognized by base flipping of universally conserved 18S rRNA residues within the decoding center, A-site tRNA accommodation in the large ribosomal subunit triggers 40S subunit rolling along its long axis and rotation with respect to the 60S subunit. Translocation is achieved by back-rolling and rotation (6,7). The fidelity of each of these steps is essential for accurate protein synthesis and perturbances are rarely tolerated as they cause the accumulation of non-functional, possibly toxic proteins. During evolution, mRNA decoding became increasingly accurate at the cost of a decrease in translation rates (8). Bacterial ribosomes mis-incorporate amino acids at a frequency of 31 × 10−4 whereas Saccharomyces cerevisiae ribosomes perform ∼4.5 times better (6.9 × 10−4) (8,9). Simultaneously, the average protein length increased almost 1.5-fold from Escherichia coli to S. cerevisiae and almost 2-fold from E. coli to Homo sapiens, thus demanding reduced translation errors (10). To date, the mechanistic details of how this increase in accuracy was accomplished by the eukaryal translation machinery, despite the virtually identical core of the decoding center have remained enigmatic.
Apart from the crucial intrinsic mobility of ribosomal elements, expanded rRNA helices in eukaryal systems, expansion segments (ES), can be highly dynamic structures at the surface of the ribosome. While initial studies linked ES to ribosome biogenesis (4,11) more recent studies further elaborated their roles during translation. These mainly involve the recruitment of cotranslationally acting protein factors and the preferred association to individual transcripts (12–15). While the idea of a preferential association to mRNA transcripts has been challenged recently (16), further functions apart from partner recruitment during translation have not yet been described.
Compared to bacterial ribosomes only certain rRNA helices expanded further. The extent to which expansions emerged and increased in size during eukaryal evolution is highly ES-specific. The larger ES of the large ribosomal subunit (LSU) dramatically increased in size when comparing human and fungal ribosomes (e.g. ES7Lb: H. sapiens: 241 bases; S. cerevisiae 88 bases) whereas other ES maintained a similar size (e.g. ES7S: H. sapiens: 12 bases; S. cerevisiae 12 bases) but altered their sequence (17). Overall, ES within the small subunit (SSU) did not experience such dramatic expansion. Their potential to recruit cytosolic factors might thus be limited compared to the LSU possibly suggesting distinct roles during translation.
In this study, we asked if there are ES within the SSU that specifically modulate the core processes of translation. We hypothesized that larger, very mobile ES are likely to play a role in factor recruitment and thus focused on ES7S one of the smallest ES in yeast and H. sapiens which should have little possibility to recruit factors. Ablation of the ES from S. cerevisiae ribosomes severely decreased growth rate accompanied by perturbed protein homeostasis and reduced translation accuracy. Translational trajectories are altered in a codon-specific manner with an increased velocity at usually slowly translated codons. The increase in velocity is due to the frequent incorporation of non-cognate amino acids by error-prone ribosomes lacking ES7S. High-resolution structural analysis of the ES7S-truncated ribosomes indicates that the increased error rate does not result from biogenesis defects, nor from conformational rearrangements within the decoding center. Structural probing of translating ribosomes rather suggests prolonged dwelling in the ratcheted state and thus altered dynamics of translocation. Thus, for the first time, we report on an ES that directly modulates the velocity and accuracy of translation and consequently proteome integrity.
Materials and methods
Strain generation and maintenance
Strains were created as described earlier (14). Strain systems and plasmids were kindly provided by Prof. J Dinman. Briefly, strains were created by plasmid exchange in an SC-Uracil medium containing 300 μg/ml of hygromycin B. After selection, cells were grown and maintained at 30°C in YPD medium (1% (w/v) yeast extract, 2% (w/v) tryptone, 2% (w/v) glucose) containing 300 μg/ml hygromycin.
Growth curves and antibiotic treatment
Cells were streaked from cryo-stocks on YPD plates containing 300 μg/ml of hygromycin and later inoculated and expanded in YPD medium containing 300 μg/ml of hygromycin. 20 ml cultures were selected for at least 24 h at 30°C (220 rpm shaking). Cells were then inoculated to YPD without hygromycin B and maintained for 20 h. Cells were then diluted to OD600 = 0.1 and grown for 4 h (30°C, 220 rpm shaking). 300 μl cell suspension was transferred to a prewarmed 48-well plate and further incubated in a prewarmed Tecan plate reader. OD600 was measured in 15 min intervals for up to 72 h at 30 C (196 rpm shaking).
Cell growth during antibiotic treatment was measured identically in medium containing antibiotics with the following stock solutions: cycloheximide (Roth) 100 ng/ml; anisomycin (Lucerna Chem) 16.65 μg/ml; blasticidin S (Invivogen) 200 μg/ml; paromomycin (Abcam) 2 mg/ml; G418 (Invivogen) 16.67 μg/ml. The compound DTT (Roth) was used as a non-antibiotic compound and used as a stock of 5 mM. These stocks were serially diluted in YPD. Per strain and antibiotic eight measurements were performed comprising seven serial antibiotic dilutions and one culture in pure YPD. For each growth curve, the maximum doubling time was calculated from a semilogarithmic plot. From these growth curves IC50 was calculated using the Hill equation:
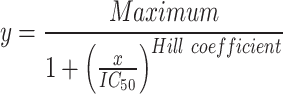 |
Each experiment was performed in biological replicates (n = 3–6).
Total RNA isolation and poisoned primer extension assays
Yeast total RNA was isolated as described earlier (14). Cells were grown to OD600 = 0.8 in YPD medium containing 300 μg/ml of hygromycin B. Cells were opened by repeated cycles of beat disruptions (FastPrep24, 20 s shaking per cycle). Cleared lysates were subjected to hot acid phenol extraction, RNA was precipitated in >2.5 volumes 100% ethanol and finally resuspended in water.
To assess the purity of mutant rRNA, Morgan analysis was carried out as previously described (18) with the following modifications: primer 5′-TTTCTCGTAAGGTGCCGA-3′ was 5′-labeled with 32P, added along 0.5 μg of total RNA in a reverse transcription reaction containing 66.7 μM of ddATP and 833 μM of the other three nucleotides as dNTPs (dTTP, dGTP and dCTP), using AMV reverse transcriptase (Promega). The reaction was resolved on a 15% polyacrylamide-urea gel and visualized using a Typhoon FLA1000 phosphoimager to detect the wild-type and mutant cDNA products.
rRNA northern blots
20 μg total RNA was mixed with 2× RNA loading dye containing ethidium bromide, denatured (3 min, 80°C), loaded on a 1% denaturing agarose gel and electrophoresed for ∼3.5 h at 100 V. Total RNA integrity was assessed by rRNA signal before passive transfer to an H+ HYBOND nylon membrane in 20× SSC buffer (6 M NaCl,690 mM Tri-Natrium-Citrate, pH 7). Membranes were crosslinked and hybridized using described rRNA precursor-specific 32P-labelled probes for ITS1 (FL185: GGCCAGCAATTTCAAGTTA), 3′ ETS (TCCTGCCAGTACCCACTT) and ITS1 upstream of the A2 cut (CGGTTTTAATTGTCCTA). Membranes were washed thrice, and an autoradiogram was developed using Typhoon FLA1000 phosphoimager.
Pulse-chase experiments
Five OD600 units of cells in exponential phase were inoculated to 5 ml SC–Met medium containing Glucose and 15 μCi/ml 35S l-methionine. Cells were pulsed for 10 min at 30°C at 220 rpm shaking. After the pulse, a zero-minute sample was taken and cells were centrifuged (4000 × g, 3 min, room temperature (RT)). The radioactive medium was removed and cells were resuspended in 4 ml SC–Met medium containing 2% glucose and 1 mg/ml methionine. Cells were incubated further (30°C, 220 rpm) and 500 μl samples were taken at indicated time points. Cells were centrifuged and resuspended in 500 μl ice-cold 20% TCA, stored on ice for 20 min, precipitated and acetone washed. Samples were electrophoresed and radioactive signals were scanned using the TyphoonFLA1000 Phosphoimager. Quantification was performed using ImageJ software.
In vitro translation
400 ml of yeast culture was grown to OD = 0.8. Cells were harvested by centrifugation (5000 × g, 4°C, 10 min), pellets were resuspended in ice-cold Ribosome buffer (30 mM HEPES pH 7.5, 100 mM KOAc, 2 mM Mg(OAc)2, 2 mM DTT, 0.1 mM PMSF, 8.5% manitol) and centrifuged again (4000 × g, 4°C, 3 min). The pellet was resuspended in Ribobuffer and cells were opened by glass bead homogenization (5 cycles of 20 s with intermittent cooling). Lysates were centrifuged (5000 × g, 30 s, 4°C) and the supernatant was transferred to an ultracentrifuge tube and centrifuged (18 000 rpm, 8 min, 4°C). The clear layer was transferred to a new tube and centrifugation was repeated. Lysates were mixed with 35S-methionine and global translation was performed at 23°C for 0–20 min. Samples were mixed with Laemmli buffer, denatured (5 min, 95°C) and separated on a 12% SDS-PAGE gel. Gels were stained, destained, and dried prior to exposure and detection of radioactive signal in a TyphoonFLA1000 Phosphoimager.
Aggregation assays
50 ml cultures were grown in YPD to OD600 = 0.8. In case of heat shock, 50 ml cultures at OD600 = 0.8 were pelleted (4000 × g, 2 min, RT), resuspended in prewarmed (37°C) YPD and incubated in 50 ml for 30 min at 37°C before harvesting. Cells were quickly mixed with 250 μl 3 M Na-azide and centrifuged (4000 × g, 2 min, 4°C). Pellets were resuspended in 15 mM Na-azide and centrifuged again (14 000 × g, 1 min, 4°C). The supernatant was removed and the pellets were snap-frozen. Pellets were resuspended in lysis buffer (20 mM KH2PO4 pH = 6.8, 10 mM DTT, 1 mM EDTA, pH 8.0, 0.1% Tween, 2× Easy cOmplete™ protease inhibitor (Roche), 2 mM PMSF) and cells were opened by sonication (Bioruptor® ultrasonicator; 4 × of 20 s ON/20 s OFF cycles, level setting: H. Lysates were passed through a 23 G needle before centrifugation (500 × g, 5 min, 4°C). Concentrations were adjusted to 6 mg/ml in lysis buffer and adjusted to a final volume of 770 μl. 20 μl lysates were used for total protein comparisons. The remaining lysates were centrifuged (16 000 × g, 20 min, 4°C) and pellets were washed in wash buffer I (20 mM KH2PO4 pH 6.8, 2% NP40 (Sigma), 1× Easy cOmplete™ protease inhibitor (Roche), 2 mM PMSF) followed by another round of sonification (4 × 15 s ON/15 s OFF, level setting: H) and centrifugation. Again, pellets were resuspended in wash buffer I, sonicated (3 × 15 s ON/15 s OFF, level setting: M) and pelleted again as before. Pellets were then washed in wash buffer II (20 mM KH2PO4 pH 6.8, 1 × Easy cOmplete™ protease inhibitor (Roche), 2 mM PMSF), sonicated (3 × 10 s ON/5 s OFF, level setting: L) and centrifuged as before. The resulting pellets were resuspended in Laemmli buffer and electrophoresed (10% PAA SDS-PAGE). Proteins were stained using a colloidal Coomassie staining solution (19).
Endogenous tagging of proteins
We employed the pFA6a-3HA-KanMX C-terminal tagging system. The triple HA-tag and the adjacent kanamycin cassette were PCR amplified with primers containing overhangs homologous to the region of interest. PCR products were gel purified and 1 μg of product was transformed. Cells were grown on G418-containing plates and clones were tested for the correct insertion via PCR and western blotting detecting the HA-tagged protein.
Sucrose density centrifugation and mass spectrometric analysis
Cells were grown to OD600 = 0.8 in YPD and harvested by filtration on a Nitrocellulose filter. Cell pellets were snap frozen and mixed with 500 μl polysome buffer (20 mM Tris–HCl, pH 7.5, 100 mM NaCl, 10 mM MgCl2, 1% Triton X100, 100 μg/ml cycloheximide, 1 mM DTT) before grinding in a freezer mill at liquid nitrogen temperature. Powders were stored at −80°C. Lysates were prepared by adding 500 μl polysome buffer to powders and thawing the material for a maximum of 2 min in a 30°C water bath. Lysates were transferred to precooled tubes and centrifuged (5000 × g, 3 min, 4°C). Supernatants were transferred to new tubes and centrifuged again (14 000 × g, 5 min, 4°C). Cleared lysates were used for ultracentrifugation in sucrose gradients (10–40%) supplemented with 100 μg/ml cycloheximide. Samples were centrifuged (3 h 15 min, 4°C, 39 000 rpm) in an SW-41Ti rotor (Beckman Coulter) and fractionated. For mass spectrometric analysis, fractions corresponding to the monosome peak or heavy polysomes (4 or more ribosomes) were pooled and centrifuged again (16 h, 4°C, 39 000 rpm) in an SW-41Ti rotor. Pellets were snap-frozen and used as input material for shotgun mass spectrometric protein identification/quantification alongside a whole cell lysate sample precipitated in 10% TCA. Trypsinized peptides were detected by LC–MS using a Fusion Lumos ETD connected to a nano-UPLC column. Peptide intensities were quantified by the MaxQuant LFQ algorithm (20). For protein detection in western blots, fractions were mixed with TCA (f.c. 10% (w/v)) and precipitated.
Western blots
Cells were grown in YPD to OD600 = 0.8 and harvested by filtration. Snap-frozen samples were mixed with 300 μl MNT buffer (20 mM MES, 100 mM NaCl, 30 mM Tris–HCl, pH 7.5, 1% Triton X-100, 1× complete protease inhibitor (Roche)). Pellets were ground as described above. Lysates were thawed in a 30°C water bath for 1 min and cleared via centrifugation (14 000 × g, 4°C, 10 min). Protein content was measured using a NanoDrop spectrophotometer and 200 mg were diluted in a final volume of 1× SDS loading buffer. Samples were electrophoresed in 8–12% SDS gels. Proteins were transferred to nitrocellulose membranes in a semi-dry setup (0.8 mA/cm2, 2 h). Membranes were blocked in 5% Milk in TBST for 1 h at RT and incubated with anti-HA antibody (12CA5, Merck) overnight at 4°C according to the manufacturer's instructions. Membranes were then washed in TBST and incubated with respective secondary antibodies in 1% milk in TBST. Finally, membranes were washed in TBST and dried for 1 h at RT before signal detection on a LI-COR device. The Signal was quantified using ImageJ software.
Ribosome profiling
Ribosome profiling experiments were performed as described before (21) with minor adjustments. Briefly, 10 OD260nm units of whole cell lysates in a total volume of 100 μl were digested (35 min, 23°C, 400 rpm) using 400 units of RNase I (Ambion). Reactions were stopped by adding 12 μl SUPERase•In™ RNase Inhibitor (Thermo Scientific) and 100 μl ice-cold polysome buffer. Samples were ultracentrifuged, monosomes were isolated and RNA extracted by hot acid phenol extraction. 20 μg digested RNA was diluted in a final volume of 20 μl water. In parallel, total RNA from lysates was extracted and DNase 1 treated (30 min, 37°C, supplemented with Superase Inhibitor). 100 μg DNase 1 treated total RNA was subjected to polyA-enrichment using the (Lexogen, Poly(A) RNA Selection Kit) according to the manufacturer's instructions. PolyA-enriched RNA was immediately fragmented in alkaline fragmentation buffer (0.5 Vol 0.5 M EDTA, 15 Vol 100 mM Na2CO3, 110 Vol 100 mM NaHCO3, 95°C, 45 min, with spinning down every 10 min). Fragmentation was stopped by adding ice-cold stop-solution (60 μl 3 M Na-acetate (pH 5.5), 8 μl Glycoblue, 500 μl RNase-free H2O). RNA was precipitated and resuspended in 20 μl water.
Ribosome-protected fragments and fragmented RNA were size-excluded in 15% denaturing TBE gels and fragments ranging from 28–32 nt were cut and eluted. Fragments were dephosphorylated before adapter ligation. Ligated RNA was reverse transcribed (Superscript IV) and fragments corresponding to the ligated product were isolated after denaturing gel electrophoresis. cDNA was circularized and libraries were PCR amplified for sequencing. Libraries were sequenced on a (device) and libraries were obtained. Reads were mapped to (GENOME) allowing 1 mismatch per read. Reads per feature were counted and differential expression analysis was performed using the DESeq2 software (22).
Ribosome A-site occupancies were calculated as described earlier (23). Differences in velocity were calculated based on each individual transcripts sequence. A-site-occupancies were summed for each codon and the differences normalized to the length of the open reading frame. Translation profiles of individual transcripts were calculated based on their sequence using A-site occupancies from separate biological replicates (n = 3). The values were smoothened using a 20-codon window.
CGA stalling assay
Wild-type and ΔES7S cells were transformed with a plasmid containing the stalling reporter and a Leu2 selection marker and streaked to -Leucine plates. Individual colonies were grown in -Leucine medium and tested initially for fluorescence. Positive colonies were stored as cryostocks. Cells were plated on -Leucine plates, colonies inoculated to 5 ml -Leu medium and incubated for 24 h at 30°C. Cells were then diluted to OD600 = 0.03 and OD600 = 0.08 respectively in 20 ml fresh -Leucine medium. At OD600 = 0.8, cells were harvested by centrifugation (4000 x g, 2 min, 4°C), pellets were washed in 500 μl water and pelleted again (6000 × g, 30 s, 4°C) before snap freezing. Pellets were mixed with 100 μl acid-washed glass beads and 500 μl MNT buffer (20 mM MES, 100 mM NaCl, 30 mM Tris–HCl, pH 7.5, 1% Triton X-100, 1× complete protease inhibitor (Roche)) and opened by bead disruption (FastPrep24, 20 s shaking per cycle, 4 cycles). Lysates were cleared by centrifugation (14 000 × g, 10 min, 4°C) and fluorescence was measured in a Tecan plate reader.
Double luciferase assays
A series of plasmids containing a Firefly-Renilla double Luciferase reporter was prepared, which carried a mutation at Firefly luciferase position 529 or 245 altering the active site lysine or histidine to different amino acids or introducing stop codons. Plasmids further carried a Leu2 selection marker. Codon identity after mutagenesis was confirmed via Sanger sequencing. Cells were transformed and plated on -Leucine plates. Cells were inoculated in 5 ml -Leucine medium and incubated for 24 h at 30°C. Cells were then diluted to OD600 = 0.03 and OD600 = 0.08 respectively in 20 ml fresh -Leu medium. At OD600 = 0.8, 8 ml of the culture were harvested by centrifugation (4000 × g, 2 min, 4°C), pellets were washed in 500 μl water and pelleted again (6000 × g, 30 s, 4°C) before snap freezing. Pellets were mixed with 100 μl acid-washed glass beads and 500 μl LAR solution and opened by bead disruption (FastPrep24, 20 s shaking per cycle, 4 cycles). Lysates were cleared by centrifugation (14 000 × g, 10 min, 4°C) and luminescence was measured in a Tecan plate reader using the Dual-Luciferase® Reporter Assay System according to the manufacturer's instructions.
Cryo-EM data collection, processing and model building
Yeast cells were selected for 24 h in YPD containing hygromycin B. Cultures were expanded to 200 ml and cells were grown to OD600 = 0.8. Cells were harvested by filtration and snap-frozen. Pellets were mixed with 500 μl EM-buffer (10 mM Hepes-KOH, pH 7.5, 50 mM KOAc, 10 mM NH4OAc, 2 mM DTT, 5 mM Mg(OAc)2) and homogenized in a cryomill. Powders were thawed and lysates cleared by centrifugation (14 000 × g, 4°C, 10 min). Lysates were loaded on a 10–35% sucrose gradient in EM buffer and centrifuged (2 h, 40 000 rpm, SW-41Ti rotor, 4°C). 80S peaks were collected, and ribosomes enriched in a centrifugational filter unit (100 kDa cut off, Thermo Fisher, 30 min, 4°C, 4000 × g). Ribosomes were snap-frozen and stored at −80°C. Quantifoil R3/3 Cu300 grids with 3 nm holey carbon were glow discharged using the GloQube glow discharge system (negative charge, 25 mA, 90 s). 4 μl of ΔES7S-80S ribosomes with 10 OD260/ml concentration was applied to the grids, using a Vitrobot Mark IV (FEI) and flash frozen in an ethane/propane mixture. The data was collected using a 200 kV Talos Arctica (FEI) with a K2 (Gatan) direct detection camera, at a pixel size of 0.783. Processing was performed using Relion 4.0 (24). Movie frames were aligned with MotionCor2 using 5 × 5 patches (25) as implemented within RELION 4.0. The CTF of the resulting micrographs was estimated using CTFFind 4 (Rohou and Grigorieff, 2015). Only micrographs with an estimated resolution of 15 Å or better were retained (4578 out of 4941 micrographs). 254202 particles were picked using crYOLO with the default model (‘gmodel_phosnet_202005_N63_c17.h5’) (26). The particles were then 2D classified and 251 636 particles were selected for initial 3D refinement. The initial 3D refinement was performed using a yeast 80S structure (PDB ID: 4U5O) (27), lowpass-filtered to 60 Å. From these aligned particles the following steps of 3D classification were performed without further angular sampling. The initial 3D classification yielded 3 classes containing 80S particles containing 14.7% (class 1), 53.3% (class 2) and 32% (class 3) of the particles. Class 1 was further sorted into two subclasses, one containing 80S ribosomes (11.1% particles) and one containing 60S subunits (3.6% particles). The 80S subclass (subclass 1) was then further sub-sorted using a mask encompassing the A, P and E sites. This subclass divided into three further subclasses, one containing ABCE/RF1 (2.2%) and two containing the Lso1 hibernation factor in two different states (4.3% and 4.6% particles). The class with the majority of particles (class 2) from the initial 3D classification was further classified using a mask encompassing the P- and E-sites. This yielded three subclasses with 2.5%, 29% and 21.8% of the particles. The smallest class contained density for A/P- and P/E-tRNAs while the other two classes were nearly identical and contained density for eIF5A. For further analysis, the two eIF5A classes were then merged. The initial 3D class, which contained 32% of the particles was lower resolution and did not split further. After 3D classification, the combined eIF5A classes were 3D refined, postprocessed, 3D refined, Bayesian polished, CTF refined and postprocessed to yield a final reconstruction with an overall resolution of 2.4 Å. To build the model of the 80S-eIF5A containing ribosomes, a structure of the 80S ribosome (PDB ID: 4U50) (27) and eIF5A (PDB ID: 6TNU) (28) was rigid body fitted into the postprocessed cryo-EM map using Chimera X (29) and manually modelled into the density using Coot (30,31). The final model was refined using Servalcat (32) with implemented REFMAC5 (33). All structural panels were generated using Chimera X (29).
DMS labeling and reverse transcription assays
Yeast at OD = 0.8 (30°C, 220 rpm) were treated for 10 min with antibiotics prior to addition of DMS to 109 mM and further incubation for 5 min. 2 ml cell suspension were mixed with 7 ml ice-cold Stop-solution (30% β-mercaptoethanol, 25% isoamyl alcohol) and centrifuged (4000 × g, 2 min, 4°C). Pellets were washed in 30% ice-cold β-mercaptoethanol and centrifuged again (4000 × g, 2 min, 4°C). Pellets were resuspended in ice-cold water and spun (6000 × g, 0.5 min, 4°C). Pellets were snap frozen and RNA isolated using a hot acid-phenol extraction. Reverse transcription was performed using the following primers (ES7S: 5′- CAAAGACTTTGATTTCTC-3′; A-site-finger: 5′-TAAGTAACAAGGACTTC-3′, helix 84: 5′-GCTTTTACCCTTTTG-3′) as described (14) and cDNA was separated on a 15%-TBE/7 M urea gel and visualized as described before (14).
Results
ES7S ablation has minor consequences for ribosome levels and biogenesis
Ribosomal RNA is highly conserved as is the general ribosomal architecture (34). ES however deviate from that notion. Structural comparison of yeast and human ribosomes suggests a highly conserved 18S rRNA structure in the platform region of the SSU where ES7S is located (Supplementary Figure S1). In both species, ES7S forms a short helix (Supplementary Figure S1A) that protrudes into the cytosol (Supplementary Figure S1B). The structural similarities and the fact that ES7S stopped expanding in more complex eukaryotes as compared to bacteria suggest a common role of this ES amongst eukaryotes. We, therefore, set out to delete the entire S. cerevisiae ES7S from the yeast 18S rRNA and closed the stem with a tetraloop to obtain a helix resembling the bacterial helix 26 from which ES7S emerged. To study the effects of this truncation in a homogeneous system we employed an rdn1-depleted S. cerevisiae which exclusively transcribes plasmid-born rRNA (35). In this system, all 150 copies of the rdn1 gene which decodes rRNA were removed and expression was restored from a switchable plasmid. We positively selected the leu2-marker on the P373 plasmid and selected against the ura3 marker within the original plasmid. First, we verified successful plasmid exchange and selection by poisoned primer extension assays (Figure 1A) and sequencing of isolated plasmids. Interestingly, we observed a striking growth defect in yeast exclusively expressing 18S rRNA lacking ES7S (ΔES7S) (Figure 1B). Because deletion of certain ES was previously linked to loss of ribosomes due to biogenesis defects (4), we measured steady-state rRNA levels but could not detect large, significant changes in ΔES7S rRNAs as compared to wild-type plasmid-born rRNA (Supplementary Figure S2A, B). This result is paralleled by the identical pattern of 18S rRNA precursors observed in ΔES7S and wild-type cells (Supplementary Figure S2C, D), suggesting that ribosome biogenesis and steady-state levels are not altered dramatically. To further ensure that biogenesis is not affected in the ΔES7S strain, we created another deletion strain; ΔES9S. When we then probed for rRNA precursors, we observed a more than 2-fold enrichment of 20S rRNA precursors, highlighting the potential of ES to contribute to SSU maturation (Supplementary Figure S3A, B). ES7S ablation, however, did not cause maturation defects (Supplementary Figure S2C, D; Supplementary Figure S3A, B). Next, to learn about the ribosomal capacity to produce polypeptides, in vitro translation assays were performed. As indicated by the identical levels of newly synthesized proteins, ΔES7S ribosomes performed indistinguishably from wild-type ribosomes (Figure 1C, D). These findings were supported by polysome profiling revealing indistinguishable amounts of polysomes in vivo (Supplementary Figure S2E). Despite a slight reduction in 40S subunits suggesting a certain biogenesis defect, polysome formation was not affected (Supplementary Figure S2E). Importantly, we did not observe reduced polysomes, suggesting that translation initiation was not markedly perturbed. This result was mirrored in the relative levels of ribosomal proteins in whole cell lysates; only a marginal reduction was observed (median log2FC = −0.17) by mass spectrometry, again suggesting unchanged cellular ribosome levels (Supplementary Figure S2F). Importantly, proteins proximal to ES7S, eS1 and eS17, did not display high fold changes, suggesting that ribosome association of these proteins was not dramatically altered. Lastly, we solved the structure of the 80S-ΔES7S ribosomes using cryo-electron microscopy and observed four distinct functional states (Supplementary Figure S4, Supplementary Table S1). The main population contained the hibernation factor Stm1 and the elongation factor eIF5A (Supplementary Figure S4). Stm1 binds ribosomes to preserve them during starvation and is dynamically regulated by TORC1 (36). The 80S-Stm1-eIF5A structure could be solved to 2.4 Å (Supplementary Figure S5A), while another population containing actively translating ribosomes bearing two tRNAs (Supplementary Figure S4) could be refined to 3.1 Å (Supplementary Figure S5). In the latter population, the tRNAs were located in the P/E and A/A sites, and thus the functional state corresponds to a ratcheted state (during which the SSU is rotated relative to the LSU and the SSU head is swiveled) with incomplete tRNA translocation. Analysis of all the maps and structures for the different states indicated that, with the exception of density for the deleted region of ES7S (Figure 1E), the 80S-ΔES7S ribosomes were fully matured in terms of rRNA and ribosomal protein composition. Moreover, comparison with structures of wild-type yeast 80S ribosomes (27), especially within the decoding center, indicates that deletion of ES7S did not induce any detectable conformational changes within the 18S rRNA (Supplementary Figure S2G).
Figure 1.

Ribosomal activity is unaltered in slow-growing ΔES7S cells. (A) Generation of pure ribosome populations was verified by poisoned primer extension assays. Reverse transcription products stop at different positions due to the successful removal of ES7S. The secondary structures display full-length ES7S on top and the truncated ΔES7S at the bottom. Arrowheads indicate stops due to the poisoned nucleotide. (B) Growth curves of wild-type and ΔES7S cells were measured in a Tecan plate reader and rates were calculated from the exponential growth phase. Stars indicate significant differences in growth rates (n = 4, P ≤ 0.05, Wilcoxon Rank Sum test). (C) In vitro translation was performed for the indicated times with lysates obtained from wild-type and ΔES7S cells. Reactions were electrophoresed and radiograms were imaged. (D) The radioactive signal was quantified and normalized to the protein loaded (n = 4, error bars indicate the standard deviation). (E) The structure of the small ribosomal subunit lacking ES7S was solved using cryo-EM. The rudimentary helix 26 is shown in red and the electron density for the helix is indicated in the zoom and compared to the wild-type structure.
Taken together, these data established, that the absence of ES7S from yeast 18S rRNA did neither strongly affect ribosome levels, structure nor the ability to produce polypeptides despite the manifested severe growth defect.
The proteostasis network is activated by ΔES7S-induced protein instability
Since translation quality is essential for proteomic integrity (37), we wondered whether proteostasis was altered in ΔES7S cells. To test this, a 35S-methionine pulse/chase experiment was performed, where we labeled newly synthesized proteins and followed their decay over time. First, we observed decreased in vivo incorporation of radioactivity into newly synthesized proteins in ΔES7S cells (Figure 2A). Second, and more strikingly, an overall ∼50% reduction in protein half-live in ΔES7S cells was observed. In particular, larger proteins decayed at higher rates, suggesting reduced cellular stability. Intriguingly, we were able to mimic this effect by treating wild-type cells with paromomycin, a ribosome-targeted and miscoding-inducing antibiotic (9). We observed very similar decreased protein half-lives in the presence of the drug (Figure 2B) and again, larger proteins were more strongly affected. This data suggests that the destabilizing effect observed for ΔES7S proteomes is as substantial as treating wild-type cells with ∼IC50 concentration of paromomycin (Figure 2C) (13), which is paralleled by the doubled generation time (Figure 1B). Since we also saw decreased pulse intensity in ΔES7S cells (Figure 2A; t = 0) we wondered if the slow growth caused the destabilization of the proteome or vice versa. To test these two options, we grew wild-type cells in YP-medium supplemented with glycerol as a sole carbon source. The cells doubled markedly slower which is due to the loss of ribosome synthesis (38). Performing pulse/chase experiments in YP-glycerol, we observed reduced incorporation of radioactivity as observed for ΔES7S (Supplementary Figure S6A, B), however, we did not observe a decrease in half-life (Supplementary Figure S6A, C). Importantly, when treating wild-type cells with paromomycin, the capacity to incorporate methionine was not altered as compared to untreated cells, yet the half-life was reduced (Supplementary Figure S6B, C). From these experiments we conclude, that reduced protein half-life is caused by the effects of ΔES7S ribosomes directly during translation rather than being a result of slow growth.
Figure 2.

ΔES7S cells display proteomic instability. Wild-type, ΔES7S cells (A) and wild-type cells pre-treated with 1 mg/ml paromomycin (B) were grown to OD = 0.8, pulsed with radioactive methionine for 10 min and chased with cold methionine for the indicated times. Samples were electrophoresed and radioactivity measured. (C) Decay rates were calculated from the radioactive signal. (D) Proteins constituting the proteasome were investigated via shotgun proteomics. Data are represented as a log2-ratio of ES7S over wild-type cells after quantification with the MaxQuant algorithm (Cox et al., 2014). Stars indicate the significance of foldchanges as determined by Student's t-test and corrected for multiple testing (** P ⇐ 0.01, n = 3). Dark grey bars indicate subunits of the 20S proteasome and light grey those of the 19S cap. (E) Proteins involved in different chaperone networks were investigated via shotgun proteomics. Data are represented as in A (* P ≤ 0.05, ** P ≤ 0.01, n = 3). (F) Cellular aggregates were isolated by repeated rounds of sonication and centrifugation of whole-cell lysates. Aggregates were electrophoresed in 10% polyacrylamide-sodium dodecyl sulfate gels and normalized to whole-cell lysates after staining with colloidal Coomassie. Lines indicate the size range normalized in (F) and arrowheads highlight unique intense bands in ΔES7S aggregates. (G) Intensities of bands in F were quantified and normalized to the intensity of whole-cell lysates. Stars indicate the significance of differences as determined by Student's t-test (n = 3, * P ≤ 0.05). (H) Shotgun proteomics was performed on proteins separated by density-gradient centrifugation. Whole-cell lysates and different fractions corresponding in density to 80S ribosomes and heavy polysomes (as indicated) were analyzed and compared for wild-type and ΔES7S cells. Data are represented as a log2-ratio of ES7S over wild-type cells after quantification with the MaxQuant algorithm. (I) Whole-cell lysates from wild-type and ΔES7S cells were separated by density-gradient centrifugation and fractions were probed for their Hsp104 content via western blotting. Intensities were normalized to uL3 levels and differences were tested for significance by Student t-tests. Data is shown as foldchange ±95% confidence interval.
Substantial amino acid misincorporation causes destabilized, aggregation-prone and possibly toxic misfolding products which must be cleared by the proteostasis network to maintain viability. To test whether cells adjust their proteome to the deletion of ES7S we performed mass spectrometric quantification of proteins from whole cell lysates. In agreement with the observed increase in protein degradation (Figure 2A), subunits of the proteasome were consistently upregulated in cells harboring ΔES7S ribosomes further supporting the previous observations (Figure 2D). Furthermore, deregulation of the chaperone network was evident, characterized by altered steady-state levels of specific chaperones (Figure 2E). Strikingly, the Hsp104 disaggregation network was consistently upregulated, suggesting that cells face aberrant protein aggregation, attempt to resolve aggregates and finally degrade terminally misfolded proteins. To test the proteomes’ aggregation tendency, we biochemically isolated aggregated, insoluble proteins from wild-type and ΔES7S whole-cell lysates. These assays demonstrated the presence of qualitatively different aggregates in the ΔES7S strain when compared to the wild-type (Figure 2F). First, two intense discrete bands in the 40–60 kDa range were exclusively present in aggregates isolated from ΔES7S cells and, second, larger proteins (60–200 kDa) were more abundant in the ΔES7S aggregates (Figure 2F, G). To resolve aggregates, Hsp104 associates with protein aggregates to extract individual misfolded peptide chains (39). This association with aggregates alters the sedimentation behavior of Hsp104 in density gradients (40). Performing mass spectrometry and western blot analysis on different fractions of density gradients indeed revealed increased sedimentation of Hsp104 and its co-chaperone into denser fractions of sucrose gradients in ΔES7S lysates as compared to wild-type (Figure 2H, I). Our mass spectrometry analysis revealed an up to 180-fold enrichment of Hsp104 in heavy fractions of the gradient suggesting substantial association to sedimenting aggregates in ΔES7S cells (Figure 2H) which was confirmed by western blot analysis (Figure 2I).
These findings revealed a substantially increased protein aggregation propensity if synthesized by ribosomes lacking ES7S and demonstrated cellular attempts to resolve and degrade proteins generated by the likely error-prone ΔES7S ribosomes.
ES7S ablation accelerates translation of slow codons by accepting non-cognate tRNAs
Misincorporation of amino acids could be a general feature of ΔES7S ribosomes. Alternatively, this phenomenon could depend on the mRNA codon identity present within the decoding A-site. To analyze whether codons (some or all) were differentially occupied between wild-type and ΔES7S cells, ribosome profiling was performed. We confirmed the established footprint length of 28 nucleotides in our libraries (Supplementary Figure S7A) and observed a very clear 3-nucleotide periodicity (Supplementary Figure S7B), a hallmark of actively translating ribosomes. mSeq libraries, as expected, did neither display enrichment for 28nt fragments (Supplementary Figure S7C) nor periodicity (Supplementary Figure S7D). Importantly, the transcriptome appears unaltered in ΔES7S cells suggesting that the global codon usage is comparable to wild-type cells (Supplementary Figure S7E). Based on the high-quality RiboSeq reads, A-site occupancies were calculated for wild-type and ΔES7S ribosomes (Figure 3A). While only a small number of codons showed slightly increased ribosomal A-site occupancy a substantial subset displayed reduced occupancy. These included codons encoding the amino acids proline, arginine, threonine and glutamine. The strongest reduction in ribosome occupancy was observed for the CGA codon. There is no cognate tRNA pairing to this codon, instead, it is translated by the tRNAArg(ICG). This codon has been described to induce ribosome stalling (41); several CGA codons in a row induce disome formation which triggers the ribosome quality control (RQC, a process resolving stalled ribosomes by splitting ribosomes and degrading the nascent chain) to rescue trapped ribosomes (41,42). To understand the extent to which these codons are read faster by ribosomes lacking ES7S, we extracted CGA positions transcriptome-wide and compared ribosome accumulation in their vicinity. In wild-type cells, clear slow-down on individual CGA codons is evident by a strong enrichment of these codons within the ribosomal A-site as compared to upstream regions (Figure 3B). In contrast, ΔES7S ribosomes show no pausing, with ribosome occupancies at the same level as in upstream regions. Next, we investigated A-site occupancies of consecutive CGA codons, very rare combinations with little coverage that introduce noise in the readout (Figure 3C). Again, ΔES7S ribosomes displayed lower enrichment at CGA dicodons suggesting faster translation of these classic roadblocks of translation. To test the hypothesis that CGA codon patches are translated at higher rates by ΔES7S ribosomes, we created a ribosome-stalling reporter featuring a stretch of 12 Arg-codons, six of which were CGA codons (41). This established RQC reporter induces robust stalling in wild-type cells as evidenced by the very low mKate/GFP ratio (Figure 3D), which is in agreement with earlier studies (41). In ΔES7S cells, however, this ratio is roughly two-fold increased, suggesting that more mKate was synthesized relative to GFP than in wild-type cells. This data further suggests that CGA codons are read faster; to an extent that translation does not pause anymore at individual CGA codons and is decelerated to a lesser extent in poly-CGA patches.
Figure 3.

Slow codons are translated at a higher pace due to misincorporations of amino acids. (A) Ribosome profiling was performed on wild-type and ΔES7S cells and A-site occupancies were determined as described in (Nedialkova & Leidel, 2015). Data are shown as the ratio of ΔES7S over wild-type occupancies. Dots indicate the mean ratio and arrows indicate the standard deviation from three biological replicates. (B) CGA codon positions were extracted transcriptome-wide and occupancies surrounding the codon were measured for wild-type and ΔES7S ribosomes. Local intensities within the region were normalized to total intensities. Light grey bars indicate the position of the A-site at position 0 and the A-site of a potentially stalled ribosome at position −10. (C) CGA dicodons were extracted transcriptome-wide and surroundings were analyzed as in (B). Dark grey bars indicate the P-site and E-site of ribosomes located on dicodons. (D) CGA stalling assays were performed in wild-type and ΔES7S cells and mKate/GFP ratios were calculated. Bars indicate the mean ± SD. Individual points show the underlying data. (E) The relative A-site occupancy (ratio of A-site occupancy based on calibrated ribosome profiling data from ΔES7S over wild-type; a ratio of 1 indicates no changes in A-site occupancy) is plotted as a function of the tRNA adaptation index (Chan & Lowe, 2016). Data are shown as mean ± SD. The grey dashed line indicates no difference between wild-type and ΔES7S occupancies. Stars indicate the significance of differences as determined by Student's t-test (n = 6, * P ≤ 0.05). (F) Double-luciferase assays were performed to quantify the mistranslation of specific codons in the active-site residues K529 and H245 of Firefly Luciferase. Relative error rates (ratio of ΔES7S over wild-type; mean ± 95% confidence interval) are shown as a function of the tRNA adaptation index. The red, dashed line indicates no difference in error rates.
Translation elongation is a diffusion-limited process with cognate tRNA entering the A-site as the rate-limiting step (43,44). Since our results indicated that a feature of ΔES7S ribosomes is incorrect codon recognition, as suggested by the increased vulnerability to paromomycin (Figure 2A, B), we hypothesized that mistranslation can globally shorten the time spent by the ribosome until a tRNA is accommodated. In such a scenario, codons that encounter high numbers of usually rejected noncognate tRNAs, due to scarce cognate tRNAs, would be particularly vulnerable to mistranslation. To test this hypothesis, we correlated the tRNA adaptation index (an estimate for the abundance of a given tRNA pairing to each codon) with the change in A-site occupancy that we observed in ΔES7S cells (Figure 3E). Indeed, we found a high correlation (r = 0.6731) between the two measures, which indicated that codons decoded by very rare tRNAs are generally translated faster by ΔES7S ribosomes - possibly due to high rates of misincorporation. In contrast, codons with abundant cognate tRNAs did not change in speed dramatically. There are two possibilities: Either high translation rates are achieved by high misincorporation rates or by more efficient codon-specific translation. To distinguish between these two scenarios we next performed a series of double luciferase assays (Figure 3F). In these assays, the firefly luciferase gene carried either an active site lysine (K529) or histidine (H245) mutation. Hence, bioluminescence can only be restored in case of a miscoding event at these exact codons. We sampled codons over the full range of tRNA adaptation indexes (tAI) and observed a dependence of error rates on tRNA abundance for both constructs (Figure 3F). Both assays featured the two codons with the lowest tAI, CGA and CGG encoding arginine. These codons showed significantly higher levels of misincorporation, irrespective of whether a lysine or histidine is misincorporated, suggesting that misincorporation is not amino acid dependent (Figure 3F). Further codons with very low tAI also displayed significantly increased misincorporation rates. Importantly, these codons also displayed decreased A-site occupancy in the RiboSeq experiment in ΔES7S cells, suggesting faster translation. We observed one more codon displaying increased misreading frequency the notoriously hard-to-translate CCA proline codon (Figure 3F) (45,46). Investigating codons with intermediate tAI and unchanged A-site occupancy (Figure 3A), such as the ACC codon, did not reveal differences in the misreading frequencies. Lastly, codons with an increased A-site occupancy such as the AAG codon (Figure 3A), which features the highest tAI, possessed very slightly, but statistically significantly, reduced error rates in the ΔES7S ribosomes (Figure 3F).
In summary, our data suggest that ribosomes were more error-prone in the absence of ES7S. Amino acid misincorporations were more frequent at codons which were slowly translated in wild-type cells, due to the increased frequency of accepting noncognate tRNAs during sampling events. This resulted in a net increase in translation velocity at these codons at the cost of decreasing decoding accuracy. This effect likely destabilizes the proteome. The frequencies at which amino acids are misincorporated are not extremely high in our reporter assays. Yet the speed at which ribosomes translate can be altered dramatically in crucial areas of a given transcript.
Altered local translation rates modulate levels and stability of ATP-cassette transmembrane proteins
Protein folding fidelity relies on the incorporation of correct amino acids into the nascent chain, however, fine-tuned translation rates further contribute to cotranslational folding, especially of multi-domain proteins (23,47,48). Since we established translational acceleration of slowly translated codons upon ES7S deletion, we tested whether aberrant translation profiles in multi-domain proteins affected protein stability and levels. Therefore, we calculated the absolute difference in global translation speed based on A-site occupancies proteome-wide (Figure 4A). This number represents an estimate on how much faster or slower a protein would be synthesized in its entirety. The larger that number, the slower a protein would be translated in the ES7S deletion strain. These alterations in translation rates were not correlated to steady-state protein levels (r2 = 0.006). This is not surprising because only a subset of proteins depends on accurate translation speed for correct folding.
Figure 4.

ΔES7S-induced changes in local translation rates modulate ABC-transporter properties. Global changes in translation speed were calculated based on each transcript's coding sequence. (A) Distribution of absolute changes in translation speed transcriptome-wide. (B) Translational profile of the ABC-transporter Vmr1. Grey bars indicate the positions of transmembrane domains. (C) Translational profile of the ABC-transporter Yol075C. Grey bars indicate the positions of transmembrane domains. (D) Translational profile of the ABC-transporter Yor1. Dark and light grey bars indicate the positions of transmembrane and nucleotide-binding domains respectively. (B–D) The lower bar indicates positions with significantly increased translation velocity in red and unchanged velocity in grey. The numbers give the number of accelerated positions as compared to the total number of amino acids. The significance was calculated based on differences in A-site occupancies from individual experiments (n = 3). (E) Western blot of HA-tagged Yor1 protein. Steady-state protein levels of Yor1 (upper panel) and uL3 as a loading control are shown. (F) Quantification of western blot signal from Yor1 normalized to uL3 signal (n = 4; * P ≤ 0.05, Wilcoxon Rank Sum test). (G) Western blot analysis of Yor1 protein isolated from cycloheximide chased cells for the indicated times. uL3 served as a control. (H) Relative Yor1 decay rates of wild-type and ΔES7S cells were calculated from immunoblot signals of cycloheximide chased cells in (G).
Translation rates seem to play a more important role in local contexts supporting domain folding or membrane insertion (47,49). We thus focused on the translation of ABC-cassette transporters, for which dependence on local translation rates has been reported (50,51). We calculated single transcript translation profiles based on A-site occupancies and observed regions of fast and slow local translation suggesting that translation occurs at uneven rates along these transcripts (Figure 4B–D). Many of the ABC-cassette transporters display significantly altered local translation rates when comparing wild-type and ΔES7S cells. These positions are scarce along the transcripts and, more intriguingly, they cluster together (Figure 4B–D). These usually slowly translated regions are faster translated in ΔES7S cells, disturbing the trajectory of cotranslational protein folding. Yor1, the yeast homolog of human CFTR, is known to rely on translation rates for correct folding (52). For this protein, we observed regions of particularly slow translation in wild-type profiles that were absent in ΔES7S profiles (Figure 4D). We assumed that altered local translation could alter protein stability and steady-state levels. To test whether steady-state levels were affected, we genomically tagged YOR1 and determined its steady-state levels by western blot analysis. Strikingly we observed a reduction of ∼50% in Yor1 steady-state levels in the ES7S deletions strain (Figure 4E-F), suggesting that local translation rates indeed modulated Yor1 abundance. Next, to test whether Yor1 was indeed destabilized, cycloheximide chase experiments that block novel protein biosynthesis were performed (Figure 4G). We observed a slight but significant increase in decay rates (Figure 4H), suggesting that the reduction in the steady-state levels was due to the lower stability of Yor1.
Taken together, this data suggested a two-layered effect of ES7S deletion; misincorporation of amino acids at typically slowly translated codons, and alteration of local translation rates. While here, we cannot separate the two effects, both factors ultimately destabilized the proteome and are likely at the heart of causing the striking growth defect observed in ES7S knock-out cells.
ES7S modulates antibiotic sensitivity and ribosome dynamics
Intrigued by the striking similarities in protein decay rates between paromomycin-treated wild-type cells and ΔES7S cells (Figure 2A), we determined the IC50 levels of several ribosome-targeting antibiotics and the reducing agent dithiothreitol (DTT). While the IC50 of DTT was identical between mutant and wild-type cells (Figure 5A), IC50 values for most ribosome-targeted antibiotics were reduced in ΔES7S cells. This suggested that truncated ribosomes were more vulnerable to these drugs. An exception to that pattern is anisomycin (Figure 5B), which inhibits peptide-bond formation on the LSU. The IC50 was identical suggesting a robust capability to form peptide bonds in the absence of ES7S, a result compatible with the in vitro translation assays (Figure 1C, D). The most dramatic changes in IC50 levels were observed for paromomycin and geneticin (G418) (Figure 5C, D), two aminoglycoside-antibiotics that perturb the decoding step of translation by preactivating the A-site for tRNA accommodation (53,54). They were further described to slow down the counterclockwise rotation and clockwise head swiveling of the SSU during translocation (55). Cycloheximide, interfering with translocation by blocking the E-site (Figure 5E) and blasticidin S which interferes with P-site tRNA hybrid-state formation (Figure 5F) (54,56) showed lesser IC50 reductions comparing ΔES7S and wild-type ribosomes.
Figure 5.

ES7S ablation alters ribosomal dynamics. Cells were grown in the presence of different concentrations of antibiotics and the reducing agent DTT. Growth curves were recorded and based on the doubling times for each concentration, IC50 was calculated. The reagents used were (A) DTT, (B) anisomycin, (C) geneticin, (D) paromomycin, (E) cycloheximide and (F) blasticidin S. Cells were treated with 110 mM DMS for 5 min and total RNA isolated. RT assays were performed to test the dynamics of the A-site finger (G, H) and helix 84 (I, J). Lines indicate (G) the tip of the A-site finger. Arrowheads in G indicate the DMS reactive nucleotides C982 and C961 and in I the residues C2680 and C2675 and the asterisk indicates the DMS independent band used for normalization. The # in I represents the nucleotide A2678.
To better understand the requirements for precise ES7S-supported translation in our yeast system we modulated its sequence more carefully. Therefore, we compared the ES7S sequences and secondary structures across different taxa. Interestingly, the structure of yeast ES7S featured an open loop conformation unlike the human ES7S and E. coli helix 26 (Supplementary Figure S8A). Since helix 26 is shorter in E.coli, we created two partial truncations in yeast ES7S. We removed two base pairs (2 bpdel) or four base pairs (4 bpdel) from the stem of ES7S (our prior ES7S deletion encompassed the full expansion segment) (Supplementary Figure S8B). Subsequently, we determined IC50 for paromomycin and found that reducing ES7S length by as little as 2 bp, and thus making it more bacteria-like, resulted in strongly (∼10×) increased susceptibility to paromomycin and decreased growth rates (Supplementary Figure S8C) similar to the complete deletion (Figure 2F). Similarly, a humanized ES7S was constructed in yeast which had the identical length as compared to the yeast ES but consisted of the human sequence (Supplementary Figure S8B). In contrast to the truncations, the human-resembling ES7S within the yeast 18S rRNA increased the resistance to paromomycin more than two-fold (Supplementary Figure S8C) but also lowered growth rates. This data suggested that ES7S expansion and sequence modalities increased translation accuracy.
Mechanistically, tRNA accommodation and translocation is an energy-driven process involving two highly conserved GTPases (eEF-1 and eEF2) and is achieved by the motions of the SSU in respect to the LSU. We wondered whether ES7S contributes to or is in fact part of these ribosomal dynamics during translation. To test that hypothesis, we subjected wild-type cells to different antibiotics, thus enriching translating ribosomes in specific translational substates. These cells were DMS treated to probe the accessibility of unpaired cytosines and adenines (Supplementary Figure S9). Indeed, when treating cells with G418, which reduces the rate of back SSU rotation and clockwise head swiveling (55), the labeling efficiency of two adenines within the ES7S loop slightly but significantly increased as compared to untreated cells (Supplementary Figure S9). Anisomycin and Blasticidin S treatment however did not show an effect on ES7S accessibility. This data suggests that during different translation states, blocked by antibiotics, ES7S obtains slightly altered conformations that allow for different DMS labeling.
The aminoglycosides paromomycin and G418 stabilize classical tRNA conformations within ribosomes and thus hinder translocation and have been shown to induce miscoding (54). We, therefore, asked if perturbed inter-subunit motions connected to translocation in ΔES7S ribosomes were at the base of the observed increased error rates (Figure 3F) and the strong growth defect (Figure 1B). During the translation elongation cycle, ribosomal subunits traverse a sequence of motions relative to each other involving the reversible formation and dissolution of inter-subunit bridges. Most prominent is the change within bridge B1a that is formed between the A-site finger (helix 38 of the 25S rRNA) and the head of the SSU. These contacts are transiently lost and reformed in the ratcheted state of the ribosome which is characterized by SSU rotation and head swiveling (57). To test for altered dynamics of these bridges, wild-type and ΔES7S cells were subjected to in vivo DMS treatment and the accessibility of the LSU helices contacting the SSU head during elongation (B1a bridge) was probed (57). We found increased labeling intensities for the adenines forming the tip of the A-site finger in ΔES7S ribosomes (Figure 5G-H). These findings suggested increased accessibility of the A-site finger in the ratcheted state, during which the B1a bridge is broken and established with new interaction partners (57). To ensure that this increase in DMS-labeling efficiency was specific, we probed the adjacent LSU helix 84. This helix shelters the E-site but does not contribute to the B1a bridge. We could not detect altered accessibility to DMS (Figure 5I-J) supporting the specific loss of the B1a bridge in the ratcheted state.
Taken together, our in vivo structural probing experiments suggest a scenario in which ES7S ablation causes altered SSU rotations, specifically subunit ratcheting, thus modulating the translocation trajectories during the elongation cycle and consequently A-site tRNA decoding accuracy.
Discussion
Published studies to date, focused on the large ES in the LSU (12–15,58), thus functional insight into the potential roles of ES within the SSU remained sparse. These earlier studies highlighted the great potential of ES to recruit interaction partners necessary for translation in the cytosol (13,14,59). As of yet, none of the investigated ES was shown to play a direct role in any of the core functions of translation per se. Our new findings, thus, provide the first evidence that a small ES, ES7S which faces toward the cytosol, directly affects the speed and accuracy of translation. In particular, yeast ribosomes lacking ES7S showed accelerated and more error-prone decoding at slowly translated codons (e.g. CGA) (Figure 4). Consequently, the proteome in these cells was vulnerable to degradation and aggregation (Figure 3) thus explaining the severe growth retardation phenotype (Figure 1).
Ribosomal motions along the elongation cycle are often linked to crucial translational events. Helix 26, the rRNA moiety from which ES7S expanded, is highly mobile during tRNA recognition and accommodation in E. coli (5). While helix 26 mobility during tRNA recognition in eukaryotes remains less clearly resolved, removal of ES7S in yeast causes a decrease in codon recognition accuracy (Figure 3F). In accordance with data from bacteria (5), our structural probing suggests certain dynamics of ES7S that can be captured with ribosome-targeted antibiotics (Supplementary Figure S9). Other functional elements have been described that decrease or increase translational accuracy (60). Mutations in ribosomal proteins uS12 and uS4 create hyper-accurate (SmD and SmP) or error-prone (ram) ribosomes. In both scenarios, the A-site is distorted (up to 10 Å) by mutations within the ribosomal proteins leading to increased or decreased accuracy (61,62). In our cryo-EM structure of the yeast ΔES7S 80S ribosome, no alterations in the global ribosome structure and in particular within the A-site were apparent, suggesting that the architecture of the decoding center in the SSU is unchanged (Figure 1E, Supplementary Figure S2G). Intriguingly, we observed as a main population the ratcheted state, featuring an A:A and a P:E tRNA, a result paralleled by our in vivo structural probing assays (Figure 5G, H). This state does not correspond to the main state observed in the literature (57) and therefore likely represents a slow step in ΔES7S translation. The otherwise observed structural integrity of ribosomes lacking ES7S places the ribosomal structural dynamics into focus. We assessed dynamics with different antibiotics and structural probing (Figure 5). Paromomycin and geneticin but also blasticidin S and cycloheximide interfere with translocation by blocking specific ribosomal motions (27,53,55,63). Higher susceptibility to antibiotics could be a consequence of slowed translocation where ES7S ablation perturbs one step and the antibiotic a second one which yields the large cumulative effect on growth rates observed here (Figure 5). This notion is further supported by our in vivo structural probing data, showing enhanced rRNA accessibility of intersubunit bridge B1a and thus an enrichment of ratcheted states in ΔES7S ribosomes (Figure 5). Prior studies highlighted the role of non-ribosomal proteins in translation accuracy (64). Based on our mass spectrometry data, no differences in r-protein levels or additional proteins associated with the ribosome were detected (Supplementary Figure S2F). This further suggests that alterations observed here occur exclusively due to the ES rRNA truncation.
Amino acid misincorporations are rare events that can cause protein dysfunction, aggregation and toxicity (47,65). The cellular machinery is thus under pressure to maintain high synthesis accuracy and to efficiently degrade erroneous proteins. Here, we describe the specific proteostasis components which are activated during the constant stress of error-prone translation as a consequence of ES7S removal (Figure 3). The classic heat shock response activates Hsf1, which in turn induces the transcription of heat shock proteins (Hsps), especially Hsp70 and Hsp90 (66). Here, however, we observe a more refined response to frequent amino acid misincorporation which is dominated by the Hsp104 machinery. The misincorporation of amino acids induces protein aggregation which is resolved by the Hsp104 machinery and proteins are degraded by the proteasome. Translation errors have further been described to occur in clusters in E. coli with one mistranslation event increasing the likelihood for the adjacent codon to be mistranslated (65). If that trend is present in the ΔES7S strain it could explain the reduced ribosomal density downstream of CGA dicodons (Figure 3C). Faster mistranslation triggered by an initial mistranslation event at CGA codons would lower A-site occupancy in adjacent downstream codons as observed here (Figure 3C). Clusters of misincorporated amino acids are potent triggers of aggregation (65) as observed in our experiments (Figure 2F). The translational acceleration at slow codons further impacts the local speed at which mRNAs are translated with possible consequences for protein folding. Slow codons appear to be conserved to maintain local slow translation rates and allow cotranslational folding of nascent chains (67). When calculating alterations in translation profiles we observed that acceleration usually occur in clusters of codons, rather than being spread along a sequence (Figure 4B–D), which is in agreement with the clustering of slow codons along coding sequences. Indeed disruption of local translation velocities has been linked to altered protein stability and function (23,51,67,68). The dependence of protein folding on translation velocity is well-documented for ABC transporters (50,52). Here we observe aberrant translation trajectories for several transporters, which in the case of Yor1 contribute to reduced protein stability and steady-state levels (Figure 4E–H). This finding is consistent with earlier reports linking the expression of Yor1 to uL11 levels and codon-specific translation elongation changes (52).
While ES7S in ophistokonds modulates the elongation step of translation, ES7S in far distant clades, namely in Trypanosoma (Discobids), is extended and bifurcated (69). In that configuration, it appears to be essential for eIF3 binding to the small subunit during 43S formation. These two different strategies of enhancement of substeps of the translation process by expanding helix 26 highlight how one region within the ribosome can play completely different roles in different evolutionary contexts. During eukaryotic evolution, the average length of cellular proteins increased compared to bacteria, thus increasing the necessity to reduce the translation error rate to an acceptable level (8,10). Given the normal error rate of 6.9 × 10−4 a protein of 1449 amino acids could be faithfully translated. Any increase in error rate lowers this value suggesting that substantial parts of the ΔES7S proteome cannot be translated without errors (10). The ribosomal core harboring the active sites, however, remained unaltered during evolution, suggesting that other ribosomal adaptations must be in place to enhance accuracy. ES7S appears to be such an element since removing or even truncating it without altering the overall ribosome structure (Figure 1E, Supplementary Figure S2G) causes increased velocities at typically slowly translated codons and decoding defects (Figure 3F). Our experiments further suggest that modulating the ES7S sequence towards that of more complex, multicellular eukaryotes (such as humans) increases the resistance to miscoding-inducing antibiotics substantially (Supplementary Figure S7C) without altering the ribosomal composition or factor binding. ES7S in eukaryal systems is thus sufficient to modulate translation velocities and enhance decoding accuracy to the extent required for the precise synthesis of longer proteins in a fashion that guides translocation trajectories and SSU motions.
Supplementary Material
Acknowledgements
We thank Max Wolfensberger and Marc Landolfo for experimental support. We thank Jiri Novacek (CEITEC, Masaryk University, Brno, Czech Republic) for data collection. We acknowledge Cryo-electron microscopy and tomography core facility CEITEC MU of CIISB, Instruct-CZ Centre supported by MEYS CR (LM2018127). Anne-Christine Uldry from the Proteomics & Mass Spectrometry Core Facility (University of Bern) is thanked for help during MS data analyses. Our thanks are extended to Marina Rodnina and Ingo Wohlgemuth (Max‐Planck‐Institut für Biophysikalische Chemie, Göttingen) for MS data analyses.
Author contributions: N.P. and R.R. conceived the project idea. V.S. created the deletion strain. R.R. performed all experiments, and data analysis. A.R. created the ΔES9S deletions strain. C.E. and S.A.L. were involved in the RiboSeq experiments and analysis. L.D-P., H.P. and D.N.W. performed electron microscopic analysis of mutant ribosomes. M.C. performed in vitro translation experiments. All authors agreed to all the analyses and commented on the manuscript. N.P. supervised the overall project and N.P and R.R. wrote the final manuscript.
Contributor Information
Robert Rauscher, Department for Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland.
Cristian Eggers, Department for Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland; Graduate School for Cellular and Biomedical Sciences, University of Bern, Bern, Switzerland.
Lyudmila Dimitrova-Paternoga, Institute for Biochemistry and Molecular Biology, University of Hamburg, Martin-Luther-King-Platz 6, 20146 Hamburg, Germany.
Vaishnavi Shankar, Department for Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland; Graduate School for Cellular and Biomedical Sciences, University of Bern, Bern, Switzerland.
Alessia Rosina, Department for Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland; Graduate School for Cellular and Biomedical Sciences, University of Bern, Bern, Switzerland.
Marina Cristodero, Department for Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland.
Helge Paternoga, Institute for Biochemistry and Molecular Biology, University of Hamburg, Martin-Luther-King-Platz 6, 20146 Hamburg, Germany.
Daniel N Wilson, Institute for Biochemistry and Molecular Biology, University of Hamburg, Martin-Luther-King-Platz 6, 20146 Hamburg, Germany.
Sebastian A Leidel, Department for Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland.
Norbert Polacek, Department for Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland.
Data availability
mRNA and RiboSeq data was deposited in the Gene Expression Omnibus database (GSE220642) and structural data into the Protein Data Bank (8BN3) and the Electron Microscopy Data Bank (EMD-16127).
Supplementary data
Supplementary Data are available at NAR Online.
Funding
Swiss National Science Foundation [188969 to N.P. and 184947 to S.A.L.]; Deutsche Forschungsgemeinschaft [WI3285/11-1 to D.N.W.]. Funding for open access charge: Swiss National Science Foundation [188969 to N.P.].
Conflict of interest statement. None declared.
References
- 1. Clark C.G., Tague B.W., Ware V.C., Gerbi S.A. Xenopus laevis 28S ribosomal RNA: a secondary structure model and its evolutionary and functional implications. Nucleic Acids Res. 1984; 12:6197–6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gerbi S.A. The evolution of eukaryotic ribosomal DNA. Biosystems. 1986; 19:247–258. [DOI] [PubMed] [Google Scholar]
- 3. Alkemar G., Nygård O. Secondary structure of two regions in expansion segments ES3 and ES6 with the potential of forming a tertiary interaction in eukaryotic 40S ribosomal subunits. RNA. 2004; 10:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ramesh M., Woolford J.L. Eukaryote-specific rRNA expansion segments function in ribosome biogenesis. RNA. 2016; 22:1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rodnina M.v., Fischer N., Maracci C., Stark H. Ribosome dynamics during decoding. Philos. Trans. Roy. Soc. B: Biol. Sci. 2017; 372:20160182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Behrmann E., Loerke J., Budkevich T.v., Yamamoto K., Schmidt A., Penczek P.A., Vos M.R., Bürger J., Mielke T., Scheerer P. et al. Structural snapshots of actively translating Human ribosomes. Cell. 2015; 161:845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Flis J., Holm M., Rundlet E.J., Loerke J., Hilal T., Dabrowski M., Bürger J., Mielke T., Blanchard S.C., Spahn C.M.T. et al. tRNA translocation by the eukaryotic 80S ribosome and the impact of GTP hydrolysis. Cell Rep. 2018; 25:2676–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kramer E.B., Vallabhaneni H., Mayer L.M., Farabaugh P.J. A comprehensive analysis of translational missense errors in the yeast Saccharomyces cerevisiae. RNA. 2010; 16:1797–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kramer E.B., Farabaugh P.J. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA. 2007; 13:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Balchin D., Hayer-Hartl M., Hartl F.U. In vivo aspects of protein folding and qualitycontrol. Science. 2016; 353:aac4354. [DOI] [PubMed] [Google Scholar]
- 11. Jeeninga R.E., van Delft Y., de Graaff-Vincent M., Dirks-Mulder A., Venema J., Raue H.A. Variable regions V13 and V3 of Saccharomyces cerevisiae contain structural features essential for normal biogenesis and stability of 5.8S and 25S rRNA. RNA. 1997; 3:476–488. [PMC free article] [PubMed] [Google Scholar]
- 12. Knorr A.G., Schmidt C., Tesina P., Berninghausen O., Becker T., Beatrix B., Beckmann R. Ribosome-NatA architecture reveals that rRNA expansion segments coordinate N-terminal acetylation. Nat. Struct. Mol. Biol. 2019; 26:35–39. [DOI] [PubMed] [Google Scholar]
- 13. Fujii K., Susanto T.T., Saurabh S., Barna M. Decoding the function of expansion segments in ribosomes. Mol. Cell. 2018; 72:1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shankar V., Rauscher R., Reuther J., Gharib W.H., Koch M., Polacek N. rRNA expansion segment 27Lb modulates the factor recruitment capacity of the yeast ribosome and shapes the proteome. Nucleic Acids Res. 2020; 48:3244–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leppek K., Fujii K., Quade N., Susanto T.T., Boehringer D., Lenarčič T., Xue S., Genuth N.R., Ban N., Barna M. Gene- and species-specific hox mRNA translation by ribosome expansion segments. Mol. Cell. 2020; 80:980–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Akirtava C., May G.E., Joel McManus C. False-positive IRESes from Hoxa9 and other genes resulting from errors in mammalian 50 UTR annotations. Proc. Natl. Acad. Sci. U.S.A. 2022; 119:e2122170119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Petrov A.S., Gulen B., Norris A.M., Kovacs N.A., Bernier C.R., Lanier K.A., Fox G.E., Harvey S.C., Wartell R.M., Hud N.v. et al. History of the ribosome and the origin of translation. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:15396–15401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sigmund C.D., Ettayebi M., Borden A., Morgan E.A. [46]Antibiotic resistance mutations in ribosomal RNA genes of Escherichia coli. Methods Enzymol. 1988; 164:673–690. [DOI] [PubMed] [Google Scholar]
- 19. Neuhoff V., Arold N., Taube D., Ehrhardt W. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis. 1988; 9:255–262. [DOI] [PubMed] [Google Scholar]
- 20. Cox J., Hein M.Y., Luber C.A., Paron I., Nagaraj N., Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics. 2014; 13:2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ingolia N.T., Brar G.A., Rouskin S., McGeachy A.M., Weissman J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012; 7:1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nedialkova D.D., Leidel S.A. Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell. 2015; 161:1606–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kimanius D., Dong L., Sharov G., Nakane T., Scheres S.H.W. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem. J. 2021; 478:4169–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng S.Q., Palovcak E., Armache J.P., Verba K.A., Cheng Y., Agard D.A. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017; 14:331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wagner T., Merino F., Stabrin M., Moriya T., Antoni C., Apelbaum A., Hagel P., Sitsel O., Raisch T., Prumbaum D. et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol. 2019; 2:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garreau De Loubresse N., Prokhorova I., Holtkamp W., Rodnina M.V., Yusupova G., Yusupov M. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 2014; 513:517–522. [DOI] [PubMed] [Google Scholar]
- 28. Buschauer R., Matsuo Y., Sugiyama T., Chen Y.H., Alhusaini N., Sweet T., Ikeuchi K., Cheng J., Matsuki Y., Nobuta R. et al. The Ccr4-not complex monitors the translating ribosome for codon optimality. Science. 2020; 368:eaay6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goddard T.D., Huang C.C., Meng E.C., Pettersen E.F., Couch G.S., Morris J.H., Ferrin T.E. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 2018; 27:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr.D Biol.Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 32. Yamashita K., Palmer C.M., Burnley T., Murshudov G.N. Cryo-EM single-particle structure refinement and map calculation using Servalcat. Acta Crystallogr. D Struct. Biol. 2021; 77:1282–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murshudov G.N., Skubák P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F., Vagin A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Melnikov S., Ben-Shem A., Garreau De Loubresse N., Jenner L., Yusupova G., Yusupov M. One core, two shells: bacterial and eukaryotic ribosomes. Nat. Struct. Mol. Biol. 2012; 19:560–567. [DOI] [PubMed] [Google Scholar]
- 35. Rakauskaite R., Dinman J.D. An arc of unpaired “hinge bases” facilitates information exchange among functional centers of the ribosome. Mol. Cell. Biol. 2006; 26:8992–9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shetty S., Hofstetter J., Battaglioni S., Ritz D., Hall M.N. TORC1 phosphorylates and inhibits the ribosome preservation factor Stm1 to activate dormant ribosomes. EMBO J. 2023; 42:e112344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schwanhäusser B., Gossen M., Dittmar G., Selbach M. Global analysis of cellular protein translation by pulsed SILAC. Proteomics. 2009; 9:205–209. [DOI] [PubMed] [Google Scholar]
- 38. Kuhn K.M., DeRisi J.L., Brown P.O., Sarnow P. Global and specific translational regulation in the genomic response of saccharomyces cerevisiae to a rapid transfer from a fermentable to a nonfermentable carbon source. Mol. Cell. Biol. 2001; 21:916–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lum R., Tkach J.M., Vierling E., Glover J.R. Evidence for an unfolding/threading mechanism for protein disaggregation by saccharomyces cerevisiae Hsp104. J. Biol. Chem. 2004; 279:29139–29146. [DOI] [PubMed] [Google Scholar]
- 40. Morales R., Hu P.P., Duran-Aniotz C., Moda F., Diaz-Espinoza R., Chen B., Bravo-Alegria J., Makarava N., Baskakov I.v., Soto C. Strain-dependent profile of misfolded prion protein aggregates. Sci. Rep. 2016; 6:20526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sitron C.S., Park J.H., Brandman O. Asc1, Hel2, and Slh1 couple translation arrest to nascent chain degradation. RNA. 2017; 23:798–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu C.C.C., Zinshteyn B., Wehner K.A., Green R. High-resolution ribosome profiling defines discrete ribosome elongation states and translational regulation during cellular stress. Mol. Cell. 2019; 73:959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shah P., Ding Y., Niemczyk M., Kudla G., Plotkin J.B. Rate-limiting steps in yeast protein translation. Cell. 2013; 153:1589–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang G., Fedyunin I., Miekley O., Valleriani A., Moura A., Ignatova Z. Global and local depletion of ternary complex limits translational elongation. Nucleic Acids Res. 2010; 38:4778–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Krafczyk R., Qi F., Sieber A., Mehler J., Jung K., Frishman D., Lassak J. Proline codon pair selection determines ribosome pausing strength and translation efficiency in bacteria. Commun. Biol. 2021; 4:589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tesina P., Lessen L.N., Buschauer R., Cheng J., Wu C.C.-C., Berninghausen O., Buskirk A.R., Becker T., Beckmann R., Green R. Molecular mechanism of translational stalling by inhibitory codon combinations and poly(A) tracts. EMBO J. 2020; 39:e103365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stein K.C., Kriel A., Frydman J. Nascent polypeptide domain topology and elongation rate direct the cotranslational hierarchy of Hsp70 and TRiC/CCT. Mol. Cell. 2019; 75:1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang G., Hubalewska M., Ignatova Z. Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat. Struct. Mol. Biol. 2009; 16:274–280. [DOI] [PubMed] [Google Scholar]
- 49. Chartron J.W., Hunt K.C.L., Frydman J. Cotranslational signal-independent SRP preloading during membrane targeting. Nature. 2016; 536:224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kimchi-Sarfaty C., Oh J.M., Kim I.W., Sauna Z.E., Calcagno A.M., Ambudkar S.V., Gottesman M.M. A ‘silent’ polymorphism in the MDR1 gene changes substrate specificity. Science. 2007; 315:525–528. [DOI] [PubMed] [Google Scholar]
- 51. Kirchner S., Cai Z., Rauscher R., Kastelic N., Anding M., Czech A., Kleizen B., Ostedgaard L.S., Braakman I., Sheppard D.N. et al. Alteration of protein function by a silent polymorphism linked to tRNA abundance. PLoS Biol. 2017; 15:e2000779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Oliver K.E., Rauscher R., Mijnders M., Wang W., Wolpert M.J., Maya J., Sabusap C.M., Kesterson R.A., Kirk K.L., Rab A. et al. Slowing ribosome velocity restores folding and function of mutant CFTR. J. Clin. Invest. 2019; 129:5236–5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lin J., Zhou D., Steitz T.A., Polikanov Y.S., Gagnon M.G. Ribosome-targeting antibiotics: modes of action, mechanisms of resistance, and implications for drug design. Annu. Rev. Biochem. 2018; 87:451–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yusupova G., Yusupov M. Crystal structure of eukaryotic ribosome and its complexes with inhibitors. Philos. Trans. Roy. Soc. B: Biol. Sci. 2017; 372:20160184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Belardinelli R., SHARMA H., PESKE F., RODNINA M.V. Perturbation of ribosomal subunit dynamics by inhibitors of tRNA translocation. RNA. 2021; 27:981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Svidritskiy E., Ling C., Ermolenko D.N., Korostelev A.A. Blasticidin S inhibits translation by trapping deformed tRNA on the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:12283–12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Spahn C.M.T., Gomez-Lorenzo M.G., Grassucci R.A., Jørgensen R., Andersen G.R., Beckmann R., Penczek P.A., Ballesta J.P.G., Frank J. Domain movements of elongation factor eEF2 and the eukaryotic 80S ribosome facilitate tRNA translocation. EMBO J. 2004; 23:1008–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Krauer N., Rauscher R., Polacek N. tRNA synthetases are recruited to yeast ribosomes by rRNA expansion segment 7L but do not require association for functionality. Non-Coding RNA. 2021; 7:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee K., Sharma R., Shrestha O.K., Bingman C.A., Craig E.A. Dual interaction of the Hsp70 J-protein cochaperone Zuotin with the 40S and 60S ribosomal subunits. Nat. Struct. Mol. Biol. 2016; 23:1003–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Allen P.N., Noller H.F. Mutations in ribosomal proteins S4 and S12 influence the higher order structure of 16 S ribosomal RNA. J. Mol. Biol. 1989; 208:457–468. [DOI] [PubMed] [Google Scholar]
- 61. Agarwal D., Gregory S.T., O’Connor M Error-prone and error-restrictive mutations affecting ribosomal protein S12. J. Mol. Biol. 2011; 410:1–9. [DOI] [PubMed] [Google Scholar]
- 62. Demirci H., Wang L., Murphy F.v. IV, Murphy E.L., Carr J.F., Blanchard S.C., Jogl G., Dahlberg A.E., Gregory S.T. The central role of protein S12 in organizing the structure of the decoding site of the ribosome. RNA. 2013; 19:1791–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fourmy D., Yoshizawa S., Puglisi J.D. Paromomycin binding induces a local conformational change in the A-site of 16 s rRNA. J. Mol. Biol. 1998; 277:333–345. [DOI] [PubMed] [Google Scholar]
- 64. Fleischer T.C., Weaver C.M., McAfee K.J., Jennings J.L., Link A.J. Systematic identification and functional screens of uncharacterized proteins associated with eukaryotic ribosomal complexes. Genes Dev. 2006; 20:1294–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wohlgemuth I., Garofalo R., Samatova E., Günenç A.N., Lenz C., Urlaub H., Rodnina M.v. Translation error clusters induced by aminoglycoside antibiotics. Nat. Commun. 2021; 12:1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Morimoto R.I. Cell-nonautonomous regulation of proteostasis in aging and disease. Cold Spring Harb. Perspect. Biol. 2020; 12:a034074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Perach M., Zafrir Z., Tuller T., Lewinson O. Identification of conserved slow codons that are important for protein expression and function. RNA Biol. 2021; 18:2296–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rauscher R., Bampi G.B., Guevara-Ferrer M., Santos L.A., Joshi D., Mark D., Strug L.J., Rommens J.M., Ballmann M., Sorscher E.J. et al. Positive epistasis between disease-causing missense mutations and silent polymorphism with effect on mRNA translation velocity. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2010612118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bochler A., Querido J.B., Prilepskaja T., Soufari H., Simonetti A., Del Cistia M.L., Kuhn L., Ribeiro A.R., Valášek L.S., Hashem Y. Structural differences in translation initiation between pathogenic trypanosomatids and their mammalian hosts. Cell Rep. 2020; 33:108534. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
mRNA and RiboSeq data was deposited in the Gene Expression Omnibus database (GSE220642) and structural data into the Protein Data Bank (8BN3) and the Electron Microscopy Data Bank (EMD-16127).