

Abstract
Therapeutic progress in neurodegenerative conditions such as Parkinson's disease has been hampered by a lack of detailed knowledge of its molecular etiology. The advancements in genetics and genomics have provided fundamental insights into specific protein players and the cellular processes involved in the onset of disease. In this respect, the autophagy-lysosome system has emerged in recent years as a strong point of convergence for genetics, genomics, and pathologic indications, spanning both familial and idiopathic Parkinson's disease. Most, if not all, genes linked to familial disease are involved, in a regulatory capacity, in lysosome function (e.g., LRRK2, alpha-synuclein, VPS35, Parkin, and PINK1). Moreover, the majority of genomic loci associated with increased risk of idiopathic Parkinson's cluster in lysosome biology and regulation (GBA as the prime example). Lastly, neuropathologic evidence showed alterations in lysosome markers in autoptic material that, coupled to the alpha-synuclein proteinopathy that defines the disease, strongly indicate an alteration in functionality. In this Brief Review article, I present a personal perspective on the molecular and cellular involvement of lysosome biology in Parkinson's pathogenesis, aiming at a larger vision on the events underlying the onset of the disease. The attempts at targeting autophagy for therapeutic purposes in Parkinson's have been mostly aimed at “indiscriminately” enhancing its activity to promote the degradation and elimination of aggregate protein accumulations, such as alpha-synuclein Lewy bodies. However, this approach is based on the assumption that protein pathology is the root cause of disease, while pre-pathology and pre-degeneration dysfunctions have been largely observed in clinical and pre-clinical settings. In addition, it has been reported that unspecific boosting of autophagy can be detrimental. Thus, it is important to understand the mechanisms of specific autophagy forms and, even more, the adjustment of specific lysosome functionalities. Indeed, lysosomes exert fine signaling capacities in addition to their catabolic roles and might participate in the regulation of neuronal and glial cell functions. Here, I discuss hypotheses on these possible mechanisms, their links with etiologic and risk factors for Parkinson's disease, and how they could be targeted for disease-modifying purposes.
Keywords: alpha-synuclein, autophagy, LRRK2, lysosome, neuroprotection, neurotransmission, Parkinson's disease, Rit2, synapse
Introduction
The definitive diagnosis of Parkinson's disease (PD) is based on the two neuropathology hallmarks evident at autopsy: the degeneration of dopamine neurons in the substantia nigra; the accumulation of Lewy bodies positive for pS129-alpha-synuclein (aSyn) in surviving neurons of several brain areas. Together, these categorize PD as a neurodegenerative proteinopathy. Consistently, the catabolism of proteins is of particular importance in neurons, which rely mainly on the ubiquitin-proteasome system and the autophagy-lysosome pathway (ALP) to degrade dangerous aggregates. The latter is an intracellular pathway involving the delivery of cargo to the lysosome for degradation. Such delivery can occur via specialized vesicles (the autophagosomes, which determine unspecialized or “bulk” macroautophagy) or directly via lysosomal membrane-based mechanisms (chaperone-mediated autophagy, microautophagy) (a detailed review on basic cell biology of autophagy and its implication in PD is beyond the scope of this brief Perspective, and, in this regard, the author recommends more specific readings (Mizushima and Komatsu, 2011; Lynch-Day et al., 2012; Hou et al., 2020).
The ALP has been implicated in the etiology of PD by genomic and genetic evidence. Most genes linked to familial disease and, associated with an increased risk of idiopathic PD, are implicated in the ALP. Moreover, lysosomal markers were reported to be reduced in the brains of PD patients (Chu et al., 2009), thus implicating impaired autophagic function in PD progression. As a result, activation of autophagy has been proposed as a therapeutic strategy for neuroprotection, yielding promising results in preclinical models. However, clinical evidence is still needed for autophagy-enhancing agents, with the added difficulty that many of those molecules impact other cellular pathways, hampering their prolonged use. In addition, (macro)autophagy may be detrimental during excessive or prolonged activation (Moors et al., 2017). This is likely due to the lack of specificity in autophagy activation by these compounds, which mostly enhance “bulk” autophagy degradation. Thus, it is important to accurately pinpoint both the molecular mechanisms involved in PD pathogenesis and the exact function of the ALP to be affected within a therapeutic strategy. For example, the lysosome represents the point of convergence of all autophagy pathways (selective and non-selective). Lysosomal activity is regulated and altered by proteins encoded by genes linked to familial PD (and thus to an etiologic role in idiopathic PD) such as aSyn, LRRK2, VPS35, and GBA1 (Volta, 2022). This evidence expands the role of lysosomes in PD beyond the ALP, as genes such as VPS35 mediate endosomal trafficking. Endosomes are a class of vesicles that sort cargo, usually from the extracellular environment and/or the plasma membrane, to different intracellular destinies, including lysosomal degradation (Cullen and Steinberg, 2018). This pathway is extremely important to regulate the abundance of receptors at the membrane, or the removal of dangerous species from the extracellular space. In neurons it gains specific relevance due to the unique arborization and cytoplasmic volume of this cell type, making correct intracellular sorting and trafficking vital for functioning physiologic processes. For this reason, focusing on genes of clear neuronal function could help shed light on neuron-specific processes (Volta et al., 2015). Nevertheless, therapeutic targeting of the endosome system is currently an unexplored avenue and is shadowed by the clear biological implications of the ALP in degrading pathologic aggregate protein species. For these reasons, in this Perspective I focus on the degradative and signaling capacities of lysosomes linked to their role in ALP.
Search Strategy
Studies cited in this article, publisehd after the year 2000, where searched using PubMed with the following keywords: lysosome, autophagy, LRRK2, alpha-synuclein, synapse, Parkinson's disease, genetics, genomics.
Lysosomes and Neurodegeneration in Parkinson's Disease
Our group has consistently shown that pharmacological inhibition of the PD-causing G2019S-LRRK2 kinase in cells requires functional lysosomes to reduce the burden of endogenous, pathologic pS129-aSyn (Obergasteiger et al., 2020). This is in line with the role of LRRK2 in lysosome function and maintenance, as previously reported (Eguchi et al., 2018), possibly in a molecular mechanism comprising GBA1 and aSyn themselves (Volta, 2022). In this view, the lysosome is a strong candidate for being a critical modulator of pathogenesis and an efficacious target for specific therapies.
Available evidence points to a role for autophagy pathways in modulating the accumulation of toxic protein species in neurons, with enhanced autophagy proposed as therapeutic, promoting clearance of aggregates. This implies two consequences: (1) that neuropathology is causative of neuronal dysfunction and degeneration; (2) that autophagy is mainly related to the late stages of the disease, when neuronal loss and proteinopathy are prevalent.
Consistent with these views, most preclinical work on autophagy enhancers focused on neurodegeneration and neuropathology as outcome measures of efficacy (Scrivo et al., 2018). However, these are arguably not the most adequate stages for neuroprotective therapies. The clinical manifestations of PD are preceded by a long presymptomatic phase spanning even decades, in which dysfunction in neuronal and synaptic transmissions is suggested to take place (Figure 1; Schirinzi et al., 2016). This view is confirmed in animal studies (both in vivo and ex vivo) where neuropathology is induced via viral delivery of aSyn or treatment with aSyn aggregate species. It is reported that alterations in dopamine release or synaptic plasticity are observed in the absence of overt protein pathology and neuronal loss (Diogenes et al., 2012; Lundblad et al., 2012). In addition, a number of genetic animal models, such as LRRK2 or VPS35 rodents, do not display neuropathology or neurodegeneration at any point in life, while manifesting age-dependent neuronal and synaptic dysfunctions (Lamonaca and Volta, 2020; Pischedda and Piccoli, 2021). Lastly, imaging studies in non-manifesting LRRK2 carriers showed several alterations in brain connectivity (Vilas et al., 2016). While it is not possible to precisely determine whether or not those individuals could have had ongoing aSyn pathology, LRRK2 PD is of incomplete penetrance and of highly variable neuropathological presentation, possibly arguing that those dysfunctions might not be entirely related to aSyn proteinopathy (Taymans et al., 2023). Of importance, autophagy not only affects late-stage proteinopathy but also synaptic function. Indeed, recent evidence demonstrated that autophagosomes also form at presynaptic terminals, and are not exclusively in the cell bodies. In addition, lysosomes have a role in synaptic plasticity in dendrites, and modulation of autophagy impacts neurotransmitter release (Goo et al., 2017; Vijayan and Verstreken, 2017). It is important to note here that lysosome biology plays fundamental roles in non-neuronal cells as well, specifically in glia. Indeed, several genome-wide association studies (GWAS) hits are highly expressed in microglia and astrocytes, and the current view is that they modulate lysosome-dependent phagocytosis. This might have a direct implication on the propagation of aSyn aggregates, which can be “mopped up” by phagocytic cells in the brain and, degraded by their lysosome system thus preventing further spreading. It is hypothesized that dysfunctional lysosomes in these cells (for example due to genetic alterations) could produce insufficient clearing capacity, thus augmenting the risk for aSyn accumulation and propagation. These are extremely interesting and important aspects of PD pathogenesis, which deserve a focused attention (Tremblay et al., 2019).
Figure 1.
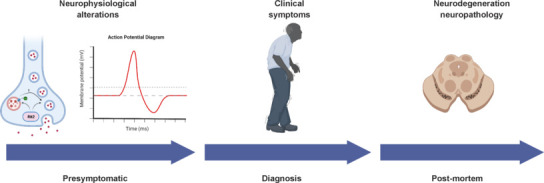
Natural history of Parkinson's disease.
Patients with Parkinson's disease experience a long phase, which can last decades, characterized by sub-clinical symptoms in non-motor domains (with rapid eye movement sleep behavior disorders being the most reported). In this phase, it is hypothesized that neurons become dysfunctional, with alterations in neuronal and synaptic communications accounting for those sub-clinical signs. Importantly, persistent synaptic dysfunction can lead to neuronal demise and death. With the progression of the disease, patients will begin manifesting the cardinal motor features of Parkinson's disease, which will eventually lead to clinical diagnosis. At this point, neurons are already lost, and addressing dysfunction would not restore ongoing neurodegeneration but could slow the process or even halt it, ideally preserving a pool of functional neurons. Lastly, patients will pass away and the autoptic analysis will provide the definitive diagnosis of the disease based on the neuropathological presentation. Candidate therapeutics to be developed, efficacious in modifying the course of the disease, will have to demonstrate capacity for neuroprotection as well, related to these last stages. Created with BioRender.com.
In this perspective, I argue that autophagy-lysosome processes could be the link between early synaptic dysfunction and late degeneration and pathology, representing a common cellular process involved in both disease stages (see Figure 2 for complementary roles of the lysosomes). In support of this view, it has been reported that several PD-linked proteins participate in synaptic regulation, and many of them have dual roles at the synapse and in autophagy. Thus, two scenarios can be hypothesized based on these premises:
Figure 2.
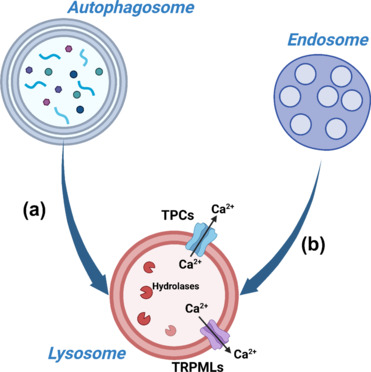
Downstream roles of lysosomes.
Lysosomes are the final effectors of both the ALP and the endosome pathway, mediating the degradation of the cargo that is delivered to them. In ALP, autophagosomes engulf cytoplasmic contents (molecules and organelles), and fuse with lysosomes where the hydrolases degrade the cargo into essential constituents (a). Endosomes, on the other hand, collect cargo from the plasma membrane and/or the extracellular environment. Then, they sort cargoes into different possible cellular destinations, one of which is the lysosomes for the degradation of the endosomal content (b). In addition to these catabolic roles, lysosomes also function as signaling organelles. They constitute important intracellular Ca2+ stores, which can be released in the cytoplasm via specialized channels on their membrane (the most characterized so far being TPC1/2 and TRPML1/3). These characteristics make lysosomes versatile organelles which are very likely to play different roles in cellular physiology. Created with BioRender.com. ALP: Autophagy-lysosome pathway; Ca2+: calcium; TPCs: two-pore channels; TRPMLs: transient receptor potential mucolipin.
-ALP-based alterations in synaptic efficacy trigger neuronal demise, with accumulation of toxic protein species occurring in parallel;
-The same impairment in ALP function first causes functional (synaptic) alterations in neurons, and later results in proteinopathy, possibly as a consequence of persistent, long-term alteration(s).
Considering the reports currently present in the literature, I personally find the second scenario to be more feasible, as there is direct experimental evidence for the two components to occur separately.
Towards a Biological View of the Natural History of Parkinson's Disease
The next challenge in PD research, and likely in neurodegeneration in general, would be to causally link these different temporal stages at both the preclinical and clinical settings, ultimately delineating a natural history of the disease with specific neuronal processes and molecular players (Figure 1). To do so, research needs “hints” on molecular players involved in both processes, with an etiologic link to PD. As mentioned earlier, genes linked to familial PD represent a valuable pathogenic resource as they are direct causes of disease. Nevertheless, these can only explain a limited percentage of PD cases, as familial PD patients are ~10% of the total, idiopathic ones (a figure which includes all mutations and genes reported so far). GWAS contribute valuable knowledge, as they indicate factors (albeit not causative ones) that increase the risk of the more common idiopathic disease. More than 90 associated loci have been reported so far, and several of them cluster in autophagy-lysosome function (Nalls et al., 2019), further highlighting the importance of this process in disease onset mechanisms. These nominated loci can represent the “hints” needed in cell biology and neuronal physiology to begin reconstructing molecular mechanisms of pathogenesis, providing possible players to be investigated. Indeed, the extremely large amount of genomic data has not been paralleled by an equally large functional validation of the identified risk factors, mostly due to technical limitations inherent in biological science with respect to the rapidity of the work. Thus, it will be of extreme utility to keep characterizing the physiological functions of the associated factors, and possibly their interactions with familial PD-related proteins, in the modulation of ALP and synaptic function. Ideally, these could be molecular linkers enabling the reconstruction of a molecular timeline of pathogenesis and disease progression. This understanding would constitute the ideal background for target validation and development. Not only will it provide etiologically relevant targets, but also targets involved in the early stages of disease, making it possible to design and develop therapies for early treatment to maximize the chances for disease modification.
Our recent work based on this rationale, for example, allowed the identification of an unknown function in lysosome biology for the GWAS-associated risk factor Rit2 (Obergasteiger et al., 2023). In this work, we found that the small GTPase Rit2 is required for lysosome activity in cells and neurons. The expression of Rit2 is reduced in PD models and brains, and forced expression stimulates lysosomal function, reduces aSyn accumulation, and is neuroprotective in vivo. Of note, and consistent with the need to reconcile molecular PD players in coherent mechanisms, we also found that Rit2 interacts with LRRK2, limiting its pathogenic kinase hyperactivation.
I believe this is a valuable example for other similar studies, moving from GWAS hits to cell biology and target validation across complementary models (as a further example see recent excellent work on the GWAS hit TMEM175 (Jinn et al., 2017)). This approach would provide unprecedented amounts of functional data directly useful for much-needed therapies capable of halting or slowing disease progression.
Additional file: Open peer review report 1 (83.4KB, pdf) .
Acknowledgments:
The author thanks Dr. Dayne Beccano-Kelly (Cardiff University, UK) for critical proofreading and comments.
Funding Statement
Funding: This work was supported by grants from Parkinson Canada, The Weston Brain Foundation and the Euregio Science Fund (to MV).
Footnotes
Conflicts of interest: The author declares no conflicts of interest.
Data availability statement: The data are available from the corresponding author on reasonable request.
Open peer reviewer: Mingxue Gu, Baylor College of Medicine, USA.
P-Reviewer: Gu M; C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
References
- Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH. Alterations in lysosomal and proteasomal markers in Parkinson's disease: relationship to alpha-synuclein inclusions. Neurobiol Dis. 2009;35:385–398. doi: 10.1016/j.nbd.2009.05.023. [DOI] [PubMed] [Google Scholar]
- Cullen PJ, Steinberg F. To degrade or not to degrade: mechanisms and significance of endocytic recycling. Nat Rev Mol Cell Biol. 2018;19:679–696. doi: 10.1038/s41580-018-0053-7. [DOI] [PubMed] [Google Scholar]
- Diogenes MJ, Dias RB, Rombo DM, Vicente Miranda H, Maiolino F, Guerreiro P, Nasstrom T, Franquelim HG, Oliveira LM, Castanho MA, Lannfelt L, Bergstrom J, Ingelsson M, Quintas A, Sebastiao AM, Lopes LV, Outeiro TF. Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J Neurosci. 2012;32:11750–11762. doi: 10.1523/JNEUROSCI.0234-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi T, Kuwahara T, Sakurai M, Komori T, Fujimoto T, Ito G, Yoshimura SI, Harada A, Fukuda M, Koike M, Iwatsubo T. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc Natl Acad Sci U S A. 2018;115:E9115–9124. doi: 10.1073/pnas.1812196115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goo MS, Sancho L, Slepak N, Boassa D, Deerinck TJ, Ellisman MH, Bloodgood BL, Patrick GN. Activity-dependent trafficking of lysosomes in dendrites and dendritic spines. J Cell Biol. 2017;216:2499–2513. doi: 10.1083/jcb.201704068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Watzlawik JO, Fiesel FC, Springer W. Autophagy in Parkinson's disease. J Mol Biol. 2020;432:2651–2672. doi: 10.1016/j.jmb.2020.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinn S, Drolet RE, Cramer PE, Wong AH, Toolan DM, Gretzula CA, Voleti B, Vassileva G, Disa J, Tadin-Strapps M, Stone DJ. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases alpha-synuclein aggregation. Proc Natl Acad Sci U S A. 2017;114:2389–2394. doi: 10.1073/pnas.1616332114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamonaca G, Volta M. Alpha-synuclein and LRRK2 in synaptic autophagy: linking early dysfunction to late-stage pathology in Parkinson's disease. Cells. 2020;9:1115. doi: 10.3390/cells9051115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad M, Decressac M, Mattsson B, Bjorklund A. Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc Natl Acad Sci U S A. 2012;109:3213–3219. doi: 10.1073/pnas.1200575109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autophagy in Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a009357. doi: 10.1101/cshperspect.a009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Moors TE, Hoozemans JJ, Ingrassia A, Beccari T, Parnetti L, Chartier-Harlin MC, van de Berg WD. Therapeutic potential of autophagy-enhancing agents in Parkinson's disease. Mol Neurodegener. 2017;12:11. doi: 10.1186/s13024-017-0154-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Blauwendraat C, Vallerga CK, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, Bras J, Young E, von Coelln R, Simón-Sánchez J, Schulte C, Sharma M, Krohn L, Pihlstrøm L, Siitonen A, Iwaki H, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18:1091–1102. doi: 10.1016/S1474-4422(19)30320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obergasteiger J, Frapporti G, Lamonaca G, Pizzi S, Picard A, Lavdas AA, Pischedda F, Piccoli G, Hilfiker S, Lobbestael E, Baekelandt V, Hicks AA, Corti C, Pramstaller PP, Volta M. Kinase inhibition of G2019S-LRRK2 enhances autolysosome formation and function to reduce endogenous alpha-synuclein intracellular inclusions. Cell Death Discov. 2020;6:45. doi: 10.1038/s41420-020-0279-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pischedda F, Piccoli G. LRRK2 at the pre-synaptic site: a 16-years perspective. J Neurochem. 2021;157:297–311. doi: 10.1111/jnc.15240. [DOI] [PubMed] [Google Scholar]
- Schirinzi T, Madeo G, Martella G, Maltese M, Picconi B, Calabresi P, Pisani A. Early synaptic dysfunction in Parkinson's disease: Insights from animal models. Mov Disord. 2016;31:802–813. doi: 10.1002/mds.26620. [DOI] [PubMed] [Google Scholar]
- Scrivo A, Bourdenx M, Pampliega O, Cuervo AM. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018;17:802–815. doi: 10.1016/S1474-4422(18)30238-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taymans JM, Fell M, Greenamyre T, Hirst WD, Mamais A, Padmanabhan S, Peter I, Rideout H, Thaler A. Perspective on the current state of the LRRK2 field. NPJ Parkinsons Dis. 2023;9:104. doi: 10.1038/s41531-023-00544-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME, Cookson MR, Civiero L. Glial phagocytic clearance in Parkinson's disease. Mol Neurodegener. 2019;14:16. doi: 10.1186/s13024-019-0314-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayan V, Verstreken P. Autophagy in the presynaptic compartment in health and disease. J Cell Biol. 2017;216:1895–1906. doi: 10.1083/jcb.201611113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilas D, Segura B, Baggio HC, Pont-Sunyer C, Compta Y, Valldeoriola F, Jose Marti M, Quintana M, Bayes A, Hernandez-Vara J, Calopa M, Aguilar M, Junque C, Tolosa E, the Barcelona LSG. Nigral and striatal connectivity alterations in asymptomatic LRRK2 mutation carriers: a magnetic resonance imaging study. Mov Disord. 2016;31:1820–1828. doi: 10.1002/mds.26799. [DOI] [PubMed] [Google Scholar]
- Volta M, Milnerwood AJ, Farrer MJ. Insights from late-onset familial parkinsonism on the pathogenesis of idiopathic Parkinson's disease. Lancet Neurol. 2015;14:1054–1064. doi: 10.1016/S1474-4422(15)00186-6. [DOI] [PubMed] [Google Scholar]
- Volta M. Lysosomal pathogenesis of Parkinson's disease: insights from LRRK2 and GBA1 rodent models. Neurotherapeutics. 2022;20:127–139. doi: 10.1007/s13311-022-01290-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.