

Advances in clinical care and recent research achievements: Primary lateral sclerosis (PLS) has traditionally been regarded as a pure upper motor neuron condition, a view perpetuated by most medical textbooks. Recent research has challenged the classical view that only the motor cortex and the descending corticospinal tracts are affected in PLS and the clinical and radiological profile of the condition has now been characterized by more nuanced descriptions. Research in PLS has gained unprecedented momentum in recent years, resulting in the painstaking characterization of disease burden patterns both post mortem and in vivo. Post mortem studies have confirmed distinguishing characteristics between amyotrophic lateral sclerosis (ALS) and PLS cohorts, namely the scarcity of TAR DNA-binding protein 43 pathology in the spinal anterior horns (Mackenzie and Briemberg, 2020). Genetic studies of larger cohorts have helped to delineate PLS from ALS and hereditary spastic paraplegia. While the core imaging signature of PLS is associated with primary motor cortex, corpus callosum and pyramidal tract degeneration (Finegan et al., 2019a), recent imaging studies have demonstrated frontotemporal changes, brainstem-cortex disconnection (Tahedl et al., 2023b), cerebellar involvement (Finegan et al., 2022) and subcortical grey matter degeneration (Finegan et al., 2019b). The pathognomonic clinical features of PLS such as spasticity, bulbar impairment, and pseudobulbar affect (Tahedl et al., 2023a) have been complemented by reports of language deficits, memory impairment, and executive dysfunction (de Vries et al., 2019). Two important practical developments have also taken place. The new consensus diagnostic criteria published in 2020 reduced the minimum symptom duration criterion to diagnose patients with PLS and introduced the categories of “definite” and “probable” PLS. The validity of the new criteria has already been demonstrated by cohorts of “probable” PLS patients harboring clinical and imaging features consistent with PLS and transitioning to “definite” PLS over time (Finegan et al., 2020). Up to recently, the revised ALS functional rating scale was widely administered to cohorts of PLS patients as a composite measure of motor disability. The recent development of the PLS functional rating scale is an important new tool to accurately appraise and monitor PLS-associated disability. Developed by Mitsumoto et al. and published in 2020, this new instrument has excellent intra-rater, inter-rater, and test-retest reliability and is increasingly utilized worldwide. The introduction of the new diagnostic criteria and the development of the PLS-specific functional rating scale have important practical ramifications for an earlier diagnosis and more accurate clinical monitoring. While no disease-modifying treatment has been developed, considerable advances have taken place in the management of common, PLS-associated symptoms such as pseudobulbar affect. The departure from isolated, single-center research efforts, to consortium-lead collaborative initiatives is likely to give PLS research further momentum. Recent research has also contributed to the development of precision clinical, biofluid, and radiological markers, which are indispensable for successful pharmacological trials.
Controversies and misconceptions: The notable academic advances are in striking contrast with the absence of clinical trials and candidate disease-modifying therapies. Therapy development efforts in motor neuron diseases have been dominated by ALS and SMA studies and restricted phenotypes, such as PLS or SBMA, have attracted considerably less attention. The most commonly cited barrier to therapy development in PLS is its relatively low incidence, but a number of other factors need to be also considered. PLS is often erroneously regarded as a relatively “benign” condition because of the considerably longer survival compared to ALS. There is a fallacious perception that non-invasive ventilation and feeding tube insertions are hardly ever required in PLS adding to the notion that PLS is a less sinister condition. Furthermore, while cognitive and behavioral deficits are well recognized in ALS, extensively studied and their practical ramifications are well understood, there is much less awareness of neuropsychological deficits in PLS which have only been recently described in larger case series (de Vries et al., 2019). The consideration of PLS as a relatively “benign” condition, however, is a grave misconception and is certainly not supported by emerging clinical or radiological evidence. Longitudinal studies have demonstrated relentlessly progressive multi-focal degeneration and a cerebral disease-burden comparable to ALS (Tahedl et al., 2021). Imaging studies have also shown that not only the primary motor cortex is affected, but pre- and supplementary motor regions, cerebellar lobules, and the brainstem also exhibit progressive degenerative change (Tahedl et al., 2023b). In contrast to ALS, the epidemiology literature of PLS is less robust, and key variables, such as incidence, prevalence, and the average symptom onset to diagnosis interval are relatively poorly characterized. Another misconception is the complete absence of neurophysiological signs of lower motor neuron dysfunction in PLS, as studies have consistently captured some alterations in motor unit morphology and changes in recruitment rate (de Carvalho et al., 2020). Furthermore, transcranial magnetic stimulation is surprisingly underutilized in the clinical setting, despite being the most reliable technique to appraise upper motor neuron dysfunction in PLS (de Carvalho et al., 2020). Clinical experience suggests that the diagnostic journey of patients with PLS is particularly circuitous. Patients often face lengthy investigations for myelopathies, spinal canal stenosis, hereditary spastic paraplegia, neurovascular syndromes, etc., before PLS is even considered. Rare PLS variants such as Mills' disease may be misrecognized or mistaken for neurovascular syndromes. During the diagnostic phase of PLS, there is often considerable anxiety about incipient ALS, and the focus of investigations in the clinical setting is reliably distinguishing suspected PLS from upper-motor-neuron-predominant ALS. Due to shared clinical and radiological features, the distinction of PLS from early-stage upper motor neuron-predominant ALS can be challenging, and often protracted clinical observation and repeated neurophysiological testing is required to confidently delineate the two conditions. The uncertainty while awaiting the confirmation of PLS and reassuringly demonstrating that the clinical trajectory is not consistent with ALS is a source of distress for the patients and their caregivers. Accordingly, any early biomarker that would reliably differentiate PLS from ALS would be of huge pragmatic value. Initiatives to distinguish ALS from PLS based on multiparametric imaging data have not been particularly successful (Bede et al., 2021). From a wet biomarker perspective, phosphorylated neuro-filament heavy chain, chitotriosidase, and CDF chitinase profiles were investigated, but their discriminatory potential from ALS has not been clearly demonstrated.
Evidence for extra-motor dysfunction: Recent case-series suggest that the most affected neuropsychological domains in PLS are language, social cognition, and executive function (de Vries et al., 2019). Impaired verbal fluency is often detected and apathy is also commonly described. Behavioral changes are not well characterized, but obsessive-compulsive behavior and alterations in personality have been sporadically observed. While frank frontotemporal dementia has also been described in association with PLS, its incidence is thought to be relatively low and not linked to hexanucleotide repeat expansions in C9orf72. Experience from ALS suggests that neuropsychological deficits in PLS may have a number of adverse clinical ramifications including impaired compliance with rehabilitation efforts, difficulty with fall prevention strategies, adherence to medications, usages of assistive devices, and may negatively impact on participation in future clinical trials. Extra-pyramidal manifestations are another understudied facet of the disease. Imaging studies have consistently highlighted basal ganglia degeneration (Finegan et al., 2019b) and there have been sporadic reports of parkinsonism, postural instability, and freezing. In the presence of widespread upper motor neuron signs, subtle extrapyramidal manifestations may be difficult to appreciate clinically without dedicated investigations; such as computational gait analyses or functional imaging. Subclinical extra-pyramidal and cerebellar dysfunction (Finegan et al., 2022) may contribute to impaired mobility, bulbar dysfunction, and increased fall risk. While imaging remains a useful tool to evaluate cerebral changes ante mortem, it is not without practical caveats and the most comprehensive way of characterizing extra-motor disease burden at a macroscopic, cellular, and molecular level is detailed neuropathological evaluation post mortem which may be particularly informative in rare phenotypes such as PLS-FTD or PSP-PLS.
Research priorities: On the basis of the considerable disability associated with PLS, and the compelling radiological evidence of the progressive nature of the condition, it is clear that translational research focusing on the development of disease-modifying therapies is an urgent priority. In addition to disease-modifying therapy initiatives, research efforts to develop effective symptom control for PLS-associated clinical manifestations, such as spasticity, pseudobulbar affect, and spastic dysarthria need to be multiplied. Other key research priorities include biomarker development with particular attention to markers that reliably distinguish PLS from early-stage ALS. Detailed, prospective epidemiology studies are needed to delineate the role of demographic, geographical, and environmental factors (Figure 1). Instead of targeted screening for genetic variants associated with hereditary spastic paraplegia and ALS, routine whole genome sequencing may help to identify relevant genetic variants. Only well-designed, longitudinal, multi-timepoint imaging studies with detailed clinical assessments can decipher anatomical propagation patterns and describe the natural history of PLS. Instead of administering brief screening instruments, comprehensive neuropsychological assessments exploring several cognitive and behavioural domains are required to characterize the cognitive profile of PLS. Alterations in respiratory patterns and changes in the respiratory drive are strikingly poorly characterized in PLS, therefore the comprehensive study of respiratory dysfunction with pulmonary function tests, neurophysiological methods, and oximetry is an important research priority. Given the relatively low incidence of PLS, there is an imperative for effective international collaboration, data-sharing, and protocol harmonization.
Figure 1.
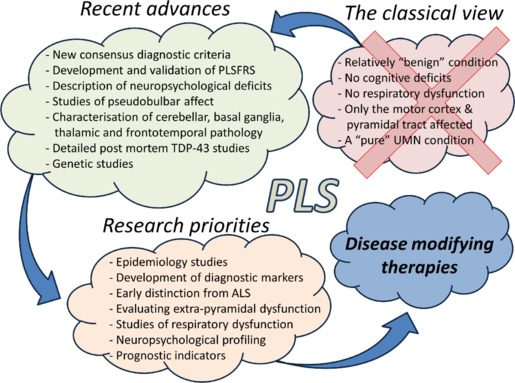
Recent advances and research priorities in PLS: the re-conceptualization of PLS as a multi-system condition.
ALS: Amyotrophic lateral sclerosis; PLS: primary lateral sclerosis; PLSFRS: primary lateral sclerosis functional rating scale; TDP-43: TAR DNA-binding protein 43; UMN: upper motor neuron.
Cause for optimism: The publication of the new diagnostic criteria and the validation of new clinical instruments have important practical ramifications to expedite the diagnosis and monitor disease progression clinically. Computational neuroimaging studies have contributed to the recognition that PLS is not merely an upper motor neuron condition solely affecting the primary motor cortex, but a relentlessly progressive neurodegenerative condition with detectable frontotemporal, subcortical, and cerebellar involvement. Radiological observations have been increasingly complemented by insightful neuropsychology studies capturing considerable extra-motor dysfunction. The sample size limitations of single-center initiatives are increasingly overcome by international collaborations and research efforts are increasingly spearheaded by disease-specific consortia paving the way to the first clinical trials in PLS.
Conclusions: Once conceptualized as a “pure” upper-motor-neuron condition, recent studies have led to the reconsideration of PLS as a genuine “multi-system” condition. Carefully designed neurophysiology, neuropsychology, neuroradiology, and neuropathology studies have contributed to the nuanced characterization of disease burden across the entire neuraxis in PLS.
This work was sponsored by the Spastic Paraplegia Foundation (SPF) (to PB). Professor PB is also supported by the Health Research Board (HRB EIA-2017-019 & JPND-Cofund-2-2019-1), the Irish Institute of Clinical Neuroscience (IICN), the EU Joint Programme – Neurodegenerative Disease Research (JPND), the Andrew Lydon Scholarship, The Hayes Family Charitable Fund and the Iris O'Brien Foundation.
Footnotes
C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
References
- Bede P, Murad A, Lope J, Li Hi Shing S, Finegan E, Chipika RH, Hardiman O, Chang KM. Phenotypic categorisation of individual subjects with motor neuron disease based on radiological disease burden patterns: a machine-learning approach. J Neurol Sci. 2021;432:120079. doi: 10.1016/j.jns.2021.120079. [DOI] [PubMed] [Google Scholar]
- de Carvalho M, Kiernan MC, Pullman SL, Rezania K, Turner MR, Simmons Z. Neurophysiological features of primary lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21:11–17. doi: 10.1080/21678421.2020.1837174. [DOI] [PubMed] [Google Scholar]
- de Vries BS, Spreij LA, Rustemeijer LMM, Bakker LA, Veldink JH, van den Berg LH, Nijboer TCW, van Es MA. A neuropsychological and behavioral study of PLS. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:376–384. doi: 10.1080/21678421.2019.1620284. [DOI] [PubMed] [Google Scholar]
- Finegan E, Chipika RH, Li Hi Shing S, Doherty MA, Hengeveld JC, Vajda A, Donaghy C, McLaughlin RL, Pender N, Hardiman O, Bede P. The clinical and radiological profile of primary lateral sclerosis: a population-based study. J Neurol. 2019a;266:2718–2733. doi: 10.1007/s00415-019-09473-z. [DOI] [PubMed] [Google Scholar]
- Finegan E, Li Hi Shing S, Chipika RH, Doherty MA, Hengeveld JC, Vajda A, Donaghy C, Pender N, McLaughlin RL, Hardiman O, Bede P. Widespread subcortical grey matter degeneration in primary lateral sclerosis: a multimodal imaging study with genetic profiling. Neuroimage Clin. 2019b;24:102089. doi: 10.1016/j.nicl.2019.102089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finegan E, Li Hi Shing S, Siah WF, Chipika RH, Chang KM, McKenna MC, Doherty MA, Hengeveld JC, Vajda A, Donaghy C, Hutchinson S, McLaughlin RL, Hardiman O, Bede P. Evolving diagnostic criteria in primary lateral sclerosis: The clinical and radiological basis of “probable PLS”. J Neurol Sci. 2020;417:117052. doi: 10.1016/j.jns.2020.117052. [DOI] [PubMed] [Google Scholar]
- Finegan E, Siah WF, Li Hi Shing S, Chipika RH, Hardiman O, Bede P. Cerebellar degeneration in primary lateral sclerosis: an under-recognized facet of PLS. Amyotroph Lateral Scler Frontotemporal Degener. 2022;23:542–553. doi: 10.1080/21678421.2021.2023188. [DOI] [PubMed] [Google Scholar]
- Mackenzie IRA, Briemberg H. TDP-43 pathology in primary lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21:52–58. doi: 10.1080/21678421.2020.1790607. [DOI] [PubMed] [Google Scholar]
- Mitsumoto H, Chiuzan C, Gilmore M, Zhang Y, Simmons Z, Paganoni S, Kisanuki YY, Zinman L, Jawdat O, Sorenson E, Floeter MK, Pioro EP, Fernandes Filho JAM, Heitzman D, Fournier CN, Oskarsson B, Heiman-Patterson T, Maragakis N, Joyce N, Hayat G, et al. Primary lateral sclerosis (PLS) functional rating scale: PLS-specific clinimetric scale. Muscle Nerve. 2020;61:163–172. doi: 10.1002/mus.26765. [DOI] [PubMed] [Google Scholar]
- Tahedl M, Li Hi Shing S, Finegan E, Chipika RH, Lope J, Hardiman O, Bede P. Propagation patterns in motor neuron diseases: individual and phenotype-associated disease-burden trajectories across the UMN-LMN spectrum of MNDs. Neurobiol Aging. 2021;109:78–87. doi: 10.1016/j.neurobiolaging.2021.04.031. [DOI] [PubMed] [Google Scholar]
- Tahedl M, Tan EL, Siah WF, Hengeveld JC, Doherty MA, McLaughlin RL, Hardiman O, Finegan E, Bede P. Radiological correlates of pseudobulbar affect: Corticobulbar and cerebellar components in primary lateral sclerosis. J Neurol Sci. 2023a;451:120726. doi: 10.1016/j.jns.2023.120726. [DOI] [PubMed] [Google Scholar]
- Tahedl M, Tan EL, Shing SLH, Chipika RH, Siah WF, Hengeveld JC, Doherty MA, McLaughlin RL, Hardiman O, Finegan E, Bede P. Not a benign motor neuron disease: longitudinal imaging captures relentless motor connectome disintegration in primary lateral sclerosis. Eur J Neurol. 2023b;30:1232–1245. doi: 10.1111/ene.15725. [DOI] [PubMed] [Google Scholar]