

Abstract
Objective.
Haemophagocytic lymphohistiocytosis (HLH) and macrophage activation syndrome (MAS) are life-threatening systemic hyperinflammatory syndromes that can develop in most inflammatory contexts. They can progress rapidly, and early identification and management are critical for preventing organ failure and mortality. This effort aimed to develop evidence-based and consensus-based points to consider to assist clinicians in optimizing decision-making in the early stages of diagnosis, treatment and monitoring of HLH/MAS.
Methods.
A multinational, multidisciplinary task force of physician experts, including adult and paediatric rheumatologists, haematologist/oncologists, immunologists, infectious disease specialists, intensivists, allied healthcare professionals and patients/parents, formulated relevant research questions and conducted a systematic literature review (SLR). Delphi methodology, informed by SLR results and questionnaires of experts, was used to generate statements aimed at assisting early decision-making and optimising the initial care of patients with HLH/MAS.
Results.
The task force developed 6 overarching statements and 24 specific points to consider relevant to early recognition of HLH/MAS, diagnostic approaches, initial management and monitoring of HLH/MAS. Major themes included the simultaneous need for prompt syndrome recognition, systematic evaluation of underlying contributors, early intervention targeting both hyperinflammation and likely contributors, careful monitoring for progression/complications and expert multidisciplinary assistance.
Conclusion.
These 2022 EULAR/American College of Rheumatology points to consider provide up-to-date guidance, based on the best available published data and expert opinion. They are meant to help guide the initial evaluation, management and monitoring of patients with HLH/MAS in order to halt disease progression and prevent life-threatening immunopathology.
INTRODUCTION
Haemophagocytic lymphohistiocytosis (HLH) and macrophage activation syndrome (MAS) are life-threatening systemic hyperinflammatory syndromes characterised by fever, elevated ferritin and other markers of systemic inflammation, inappropriately low blood cell counts, disseminated intravascular coagulopathy, hepatitis, central nervous system (CNS) inflammation and high risk for progression to multiple organ dysfunction, shock and often death (1). The term HLH originated as a pathological description in young children, and although it predates the discovery of PRF1 deficiency or other causal genes, it is often still used to imply a ‘primary’ genetic defect. MAS typically arises as a complication of rheumatic diseases like systemic juvenile idiopathic arthritis (sJIA) or systemic lupus erythematosus (SLE). The current, more broad HLH definition includes MAS among the causes of ‘secondary’ HLH (2). The task force (TF) agreed to refer to the whole spectrum of primary and secondary HLH as ‘HLH/MAS’. HLH/MAS can occur in any age group, and typically develops in the setting of infectious, malignant or rheumatological diseases, or less commonly as a manifestation of underlying genetic inborn errors of immunity (IEI) that predispose to hyperinflammation. Early identification and intervention can prevent organ failure and death. Nevertheless, practice patterns in recognizing and managing these conditions vary widely (3).
The scope of terms such as HLH, MAS, ‘cytokine storm syndrome’, ‘hyperinflammation’, cancer immunotherapy–related ‘cytokine release syndrome (CRS)’, ‘hyperferritinemic sepsis–induced multiorgan dysfunction’ or SARS-CoV2–associated ‘multisystem inflammatory syndrome of children or adults’ may overlap such that multiple may reasonably apply to the same patient (4). Confusion regarding these terms and the proper boundaries of their application can have unintended consequences (e.g., primary HLH treatment protocols are rarely indicated in the MAS subset of secondary HLH).
The TF agreed to define and use the term HLH/MAS to encompass a recognisable pattern of clinical findings associated with these syndromes (as discussed in Table 1). The TF defined systemic hyperinflammation as a state of excessive immune activation at risk of progression to HLH/MAS. Additionally, the TF identified three categories of contributors to the development of HLH/MAS: genetic causes of HLH/MAS, predisposing conditions (e.g., sJIA, lymphoma, certain metabolic conditions) that increase susceptibility and acute triggers (e.g., infections, immunotherapies).
Table 1.
Recognisable clinical features/patterns in HLH/MAS*
| Features | Criteria† | |
|---|---|---|
|
| ||
| 1 | Systemic inflammation (elevated or rising) | |
| Fever | All | |
| CRP | – | |
| LDH | – | |
| 2 | Hyperferritinemia (elevated or rising) | All |
| 3 | Cytopenias (low or dropping) | |
| Platelet count† | All | |
| Leucocyte count (particularly neutrophil count) | HLH04, Hscore | |
| Haemoglobin | HLH04, Hscore | |
| 4 | Disseminated intravascular coagulopathy | |
| Increased D-dimer | – | |
| Low/dropping fibrinogen‡ | All | |
| Prolonged PT/INR, PTT | – | |
| 5 | Liver dysfunction | |
| Hepatomegaly | Hscore | |
| Increased ALT, AST, bilirubin | MAS-2016, Hscore | |
| Increased triglycerides | All | |
| 6 | Splenomegaly | HLH04, Hscore |
| 7 | CNS dysfunction | |
| Encephalitis, encephalopathy, altered mental status, seizure | – | |
| CSF pleocytosis, elevated CSF protein, increased ICP | – | |
| Radiological evidence of inflammation | – | |
HLH = haemophagocytic lymphohistiocytosis; MAS = macrophage activation syndrome; CRP = C-reactive protein; LDH = lactate dehydrogenase; PT/INR = prothrombin time/international normalised ratio; PTT = partial thromboplastin time; ALT = alanine aminotransferase; AST = aspartate aminotransferase; CNS = central nervous system; CSF = cerebrospinal fluid; ICP = intracranial pressure.
HLH04 (2), MAS-2016 (6), or Hscore (7), or the feature was not included in any of these criteria (indicated with ‘–’). For more details, see Supplemental Table 2, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42636).
As acute-phase reactants, low-normal values in the face of systemic inflammation may signify consumption.
Several important collaborative efforts have shaped the current approach to the diagnosis and management of HLH and MAS. The Histiocyte Society, and later rheumatology consortia, developed and refined classification criteria to define HLH (2,5) or MAS (6). Subsequent diagnostic tools like the Hscore (7) and MS score (8) provided more continuous measures. Several groups have developed and published consensus-based management documents that provide context-specific recommendations (see Table 2).
Table 2.
Preceding HLH/MAS consensus publications*
| Genetic HLH | Ehl et al (ref. 92) |
| Malignancy-associated HLH | Lehmberg et al (paediatric) (ref. 93) |
| Malignancy-associated HLH | Daver et al (adult) (ref. 109) |
| HLH diagnosis | Jordan et al (ref. 94) |
| MAS diagnosis | Ravelli et al (ref. 6,19) |
| HLH in adults | La Rosée et al (ref. 67) |
| HLH in intensive care | Hines et al (ref. 74) |
Consensus publications gathered at https://www.histiocytesociety.org/HLH-Consensus. HLH = haemophagocytic lymphohistiocytosis; MAS = macrophage activation syndrome.
There remains an unmet need for guidance during the early stages of HLH/MAS: a period between when suspicion first arises and when an underlying aetiology has been established. Early HLH/MAS can be highly variable between patients, and often involves rapid changes within the same patient. Patients may not fully meet relevant criteria, their diagnostic workup may be evolving and their condition may be rapidly deteriorating. Nevertheless, it is precisely at these early timepoints where appropriate interventions may have the best chance of preventing the worst outcomes. To address this need, an international multidisciplinary TF developed consensus-based and evidence-based guidance statements. Although collectively HLH/MAS is not rare, these guidance statements are termed points to consider (PTC) to recognise the limitations of the evidence supporting them. These PTC target a broad range of frontline, primary care and subspecialty providers and are meant to assist them in recognising HLH/MAS, identifying its contributors, intervening despite diagnostic ambiguity and monitoring for progression and organ damage.
METHODS
The American College of Rheumatology (ACR) and EULAR standardised operating procedures were followed during the project (9). With approval from the EULAR executive committee, and in parallel with two EULAR/ACR consensus guidance efforts in autoinflammatory diseases (10,11), an international, multidisciplinary TF was convened to develop PTC at the earliest stages in the recognition and management of HLH/MAS. The conveners (SWC and FdB) invited North American and European TF members with established expertise in the management of HLH/MAS to contribute. The TF consisted of 14 paediatric and adult rheumatologists, 4 haematologists/oncologists, 2 immunologists, 2 infectious disease specialists and 3 intensivists. In addition, the TF included a nurse experienced in caring for patients with HLH/MAS, two patient representatives and three methodologists.
At an initial face-to-face meeting in August 2019, the team defined the goals of the project, the target population and relevant questions using the Population, Intervention, Comparison, Outcome (PICO) format. The target audience was defined as healthcare professionals, policy makers, health insurance companies and patients and their caregivers. A systematic literature review (SLR) was performed by three team members (BS, MW, AG), with support from a librarian (DH), epidemiologist (DP) and senior methodologists (AR, EDe, DA) to identify relevant literature using PubMed, Embase and the Cochrane Library published before November 2020. Initial search terms for the SLR consisted of the full spectrum of names used to signify the syndrome of HLH/MAS. The resulting articles were filtered based on quality and relevance to PICO questions. SLR themes are discussed in brief throughout and in the Supplemental Methodology (available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42636), and detailed in a separate ‘SLR manuscript’ (12).
In response to the PICO questions and informed by the synthesis of SLR results and expert opinion, the TF drafted and refined overarching and specific PTC in the form of statements. Individual statements were suggested, edited and refined in two rounds of preconsensus meeting questionnaires using a secure web-based system (Jotform). These statements addressed early identification, diagnosis, monitoring and early management of HLH/MAS as described in the SLR manuscript (12) and Tables 3 and 4. The response rate for each questionnaire was 100%. The TF members were asked to indicate their agreement with each statement or item with yes or no. A free text option was provided to capture every member’s comment or suggestion for modification. A request was also included for members to add items to be addressed, edited or altered. Responses to this questionnaire were reformulated as draft statements. Comments and suggestions provided in the questionnaires were used to modify the draft statements and to add additional items. The revised and amended statements were then circulated through a second round of questionnaires. After the two rounds, the draft statements were revised to incorporate all suggestions and reviewed by the TF members. These draft statements were then included for discussion at the consensus meetings. The TF reviewed, discussed and voted on these statements in a consensus meeting held virtually over 3 days in March/April 2021. Prior to each of these consensus meetings, the results of the SLR and the draft statements were distributed to all TF members. During the meetings, statements that achieved at least 80% agreement were accepted; statements with <80% were discussed a final time in a nominal group, round robin format and were only accepted if the statement reached an 80% agreement at that point. Delphi technique was used to achieve consensus throughout the process.
Table 3.
Overarching principles*
| LoE/GoR | LoA (scale 0–10) mean ± SD | ||
|---|---|---|---|
|
| |||
| A | Systemic hyperinflammation is an immunopathological continuum; the characteristic clinical and laboratory findings are individually non-specific, but when viewed collectively and longitudinally are recognisable and warrant prompt diagnostic evaluation. Therapeutic intervention may be warranted even when not satisfying specific classification/diagnostic criteria for HLH/MAS. | 5D | 9.4 ± 1.2 |
| B | Systemic hyperinflammation can be associated with hyperferritinemia and can progress to life-threatening HLH/MAS. | 5D | 9.6 ± 0.7 |
| C | Systemic hyperinflammation and HLH/MAS can occur in nearly any inflammatory state, but certain predisposing conditions and/or inflammatory triggers warrant a high index of suspicion.† | 2B | 9.1 ± 1.3 |
| D | Investigating and treating modifiable contributors to systemic hyperinflammation and HLH/MAS are essential in the management of every patient. | 4C | 9.7 ± 0.6 |
| E | HLH/MAS should be treated with an urgency based on both the degree of inflammation and extent of organ dysfunction; the goals of therapy are to prevent/limit immunopathology, preserve integrity of the ongoing diagnostic workup and minimise toxicity. | 5D | 9.7 ± 0.7 |
| F | The evaluation and management of patients with systemic hyperinflammation suspected of having or progressing to HLH/MAS may benefit from consultation with experts in these disorders. | 5D | 9.2 ± 1.8 |
Level of evidence (LoE): 1a = systematic review of randomised controlled trials (RCTs); 1b = individual RCT; 2a = systematic review of cohort studies; 2b = individual cohort study (including low-quality RCT); 3a = systematic review of case–control studies; 3b = individual case–control study; 4 = case-series (and poor-quality cohort and case–control studies); 5 = expert opinion without explicit critical appraisal, or based on physiology, bench research or ‘first principles’. Grade of recommendation (GoR): A = based on consistent level 1 studies; B = based on consistent level 2 or 3 studies or extrapolations from level 1 studies; C = based on level 4 studies or extrapolations from level 2 or 3 studies; D = based on level 5 studies or on troublingly inconsistent or inconclusive studies of any level. For level of agreement (LoA), a 0–10 Likert scale was used. HLH = haemophagocytic lymphohistiocytosis; MAS = macrophage activation syndrome.
See text and other tables for examples/clarification.
Table 4.
Consensus statements*
| LoE/GoR | LoA (scale 0–10) mean ± SD | ||
|---|---|---|---|
|
| |||
| Recognition, screening and early diagnosis | |||
| 1.1 | The following unexplained or unusually severe clinical and laboratory features, particularly if co-occurring, may represent a systemic hyperinflammatory syndrome and should prompt consideration of HLH/MAS in appropriate clinical contexts: | HLH: 1A MAS: 3B |
9.5 ± 1.1 |
| • Persistent fever. | |||
| • Elevated and/or rising ferritin or other markers of inflammation/damage (CRP, LDH). | |||
| • Inappropriately low or declining haemoglobin, platelet counts or white blood cells (neutrophils and lymphocytes). | |||
| • Hepatic dysfunction (increased ALT, AST, bilirubin). | |||
| • Coagulopathy (low fibrinogen, increased PT/INR, increased d-dimers). | |||
| • Splenomegaly. | |||
| • CNS dysfunction. | |||
| 1.2 | Patients with features of a systemic hyperinflammatory syndrome that could represent or progress to HLH/MAS should have a ferritin level checked. | 1A | 10.0 ± 0.2 |
| 1.3 | Patients with a normal ferritin but ongoing clinical suspicion for HLH/MAS should have serial ferritin testing. | 5D | 9.4 ± 1.0 |
| 1.4 | In addition to ferritin, clinicians should obtain the following routine laboratory evaluations: CBC with differential, liver panel, fibrinogen, d-dimer, LDH and CRP. | 1A | 9.5 ± 0.7 |
| 1.5 | Following initial laboratory evaluations, assessment of specialised biomarkers of inflammation (e.g., IL-2Rα [CD25], CD163, IL-18, CXCL9, neopterin, if available) may further aid in the diagnosis of HLH/MAS. These tests should be interpreted in consultation with a specialist with expertise in HLH/MAS. | 4C | 9.2 ± 1.2 |
| Criteria | |||
| 2.0 | Existing classification or diagnostic criteria perform well in specific settings, but no single set of criteria is sufficient to diagnose a syndrome of HLH/MAS across all contexts. | 5D | 9.4 ± 1.2 |
| Evaluating contributors | |||
| 3.1 | Certain underlying infections, rheumatic diseases, malignancies, metabolic diseases and genetic inborn errors of immunity are frequently associated with HLH/MAS and clinicians should consider evaluations for these in appropriate contexts. | 2B | 9.6 ± 0.6 |
| 3.2 | Genetic testing in patients with probable HLH/MAS can dramatically affect diagnosis and management and should be considered early. | 3B | 9.0 ± 1.8 |
| 3.3 | Decision-making about genetic testing in patients with probable HLH/MAS is complex, should integrate age, clinical features and laboratory/functional test results, and should involve specialists with expertise in HLH/MAS. | 4C | 9.4 ± 0.9 |
| 3.4 | In patients for whom genetic testing is indicated, next-generation sequencing (e.g., targeted gene panel, whole exome or whole genome sequencing) to screen for pathogenic variants, rather than single gene Sanger sequencing, is recommended. | 5D | 9.2 ± 1.5 |
| 3.5 | Genetic counselling to assist with consenting and interpretation of results should be offered to patients being considered for genetic testing. | 5D | 9.1 ± 1.4 |
| Prognostic factors and CNS involvement | |||
| 4.1 | Underlying malignancy, CNS involvement, liver failure, multiple organ dysfunction and prolonged active disease are associated with a poor prognosis in patients with probable HLH/MAS; these should prompt urgency in establishing the diagnosis of HLH/MAS, identifying triggering conditions and initiating appropriate treatment. | 2B | 9.5 ± 1.1 |
| 4.2 | All individuals with probable HLH/MAS should undergo a complete neurological examination. Patients with any of the following should be assessed for CNS involvement: age <1 year, known genetic HLH disorder, encephalopathy, seizures, altered mental status, irritability, meningism, headache, vision changes or focal deficits. | 4C | 9.2 ± 1.5 |
| 4.3 | Assessment for CNS involvement should include brain MRI and evaluation of cerebrospinal fluid glucose, protein and cell count with differential (with pathological review of cytology) when safe to do so. | 4C | 9.5 ± 1.0 |
| 4.4 | In patients with probable HLH/MAS, assessment for CNS involvement should not delay initiation of systemic immunomodulatory therapy. | 4C | 9.7 ± 0.7 |
| Treatment | |||
| 5.1 | For patients with probable HLH/MAS and persistent, severe or worsening inflammation or organ dysfunction, initiation of immunomodulatory treatment should be considered while diagnostic testing is ongoing. | 5D | 9.7 ± 0.7 |
| 5.2 | Choice of initial immunomodulatory treatment is complex and requires balancing an assessment of urgent risk due to rapid HLH/MAS progression with potential for obscuring diagnosis of malignancy or worsening active infection. | 2B | 9.6 ± 0.9 |
| 5.3 | Initial empiric immunomodulatory therapy in patients with rapidly progressive HLH/MAS could include high-dose glucocorticoids, anakinra and/or IVIg based on local access. | GC: 2B Anakinra: 2B IVIg: 4C |
8.9 ± 2.1 |
| 5.4 | In addition to supportive care and immunomodulatory HLH/MAS treatment, patients should receive appropriate antimicrobial and antiviral therapies and treatment of any underlying triggers or disorders. | 4C | 9.8 ± 0.5 |
| 5.5 | In patients for whom prolonged immunomodulatory regimens are anticipated, consideration should be given to the use of antimicrobial and/or antiviral prophylaxis in consultation with an infectious disease expert. | 2B | 9.3 ± 1.9 |
| Monitoring | |||
| 6.1 | In patients with probable HLH/MAS, worsening or lack of improvement in laboratory parameters of systemic inflammation (particularly ferritin), DIC, hepatitis or cytopenias may indicate disease progression and a need to re-assess diagnosis and/or treatment. | 2B | 9.6 ± 0.8 |
| 6.2 | Patients with systemic hyperinflammation suspected of having or progressing to HLH/MAS require continuous clinical monitoring and frequent reassessment of organ dysfunction, which may necessitate ICU care. | 4C | 9.8 ± 0.4 |
| 6.3 | Clinicians should monitor initial response to treatment by assessing clinical and laboratory markers of organ involvement at least daily and markers of systemic inflammation at least twice weekly.† | 2B | 9.3 ± 1.0 |
| Multidisciplinary teams | |||
| 7.0 | A multidisciplinary approach is preferred and can optimise the diagnostic workup and management of patients with systemic hyperinflammation and HLH/MAS. | 5D | 9.6 ± 1.0 |
Level of evidence (LoE): 1a = systematic review of randomised controlled trials (RCTs); 1b = individual RCT; 2a = systematic review of cohort studies; 2b = individual cohort study (including low-quality RCT); 3a = systematic review of case–control studies; 3b = individual case–control study; 4 = case-series (and poor-quality cohort and case–control studies); 5 = expert opinion without explicit critical appraisal, or based on physiology, bench research or ‘first principles’. Grade of recommendation (GoR): A = based on consistent level 1 studies; B = based on consistent level 2 or 3 studies or extrapolations from level 1 studies; C = based on level 4 studies or extrapolations from level 2 or 3 studies; D = based on level 5 studies or on troublingly inconsistent or inconclusive studies of any level. For level of agreement (LoA), a 0–10 Likert scale was used. HLH = haemophagocytic lymphohistiocytosis; MAS = macrophage activation syndrome; CRP = C-reactive protein; LDH = lactate dehydrogenase; ALT = alanine aminotransferase; AST = aspartate aminotransferase; PT/INR = prothrombin time/international normalised ratio; CNS = central nervous system; CBC = complete blood cell count; IL-2Rα = interleukin-2 receptor α; MRI = magnetic resonance imaging; GC = glucocorticoid; IVIg = intravenous immunoglobulin; DIC = disseminated intravascular coagulation; ICU = intensive care unit.
See text and Table 5 for examples/clarification.
Oxford levels of evidence and a grade of recommendation were assigned for each statement (12,13). Each TF member then assigned their level of agreement for each statement using an 11-point Likert scale from 0 to 10 (0 = completely disagree, 10 = completely agree).
RESULTS
Systematic literature review
Briefly, original research articles of any study design with diagnosis, treatment and monitoring of HLH/MAS that reported more than six cases were included. Of the 18,020 articles from PubMed, Embase and Cochrane, 258 were selected for full-text review and 167 articles were included for data extraction. Based on the expertise of TF members, SLR results (12) and discussion at consensus conferences, the TF generated 6 overarching principles (Table 3) and 24 disease-specific PTC pertaining to HLH/MAS (Table 4).
Overarching principles
Recognising the complexity and urgency of management decisions in systemic hyperinflammation and HLH/MAS, the TF generated six overarching principles (Table 3) that provide guidance on the early recognition of characteristic clinical features, the systematic evaluation of contributors (including genetic causes, predisposing conditions and acute triggers), the implementation of early therapies and the monitoring of HLH/MAS progression.
Points to consider
The TF generated specific statements intended to offer practical consensus-based and evidence-based guidance for clinicians making decisions at the earliest stages of HLH/MAS consideration, recognition and management (Table 4).
PTC 1.1–1.5: recognition, screening and early diagnosis.
Given the variety of genetic causes, predisposing conditions and acute triggers from which HLH/MAS arises, recognising the presenting features and making a diagnosis are often challenging. Existing diagnostic criteria lack both sensitivity and specificity, especially in the context of confounding conditions like lymphoma or sepsis.
Based on existing criteria, current literature and expert experience, the TF agreed on clinical and laboratory abnormalities that together establish a recognisable pattern of potentially life-threatening HLH/MAS (PTC 1.1). Individual findings are non-specific and must be evaluated collectively and longitudinally. However, recognising the pattern of clinical and laboratory abnormalities that constitute HLH/MAS is critical for prompting an aetiological workup, considering treatments and initiating a monitoring strategy before serious complications or death occur.
Ferritin is a sensitive test for HLH/MAS, and there was broad consensus that ferritin levels should be checked in all patients with new, ongoing or heightened suspicion for HLH/MAS even if prior measurements have been normal (PTC 1.1–1.3). Essentially all patients with HLH/MAS with systemic disease have elevated ferritin levels (14,15), and hyperferritinemia is part of all existing HLH/MAS criteria (Table 1). Levels >500 ng/mL were 84% sensitive in paediatric patients with HLH (2), and served as the cut-off in clinical trials conducted by the Histiocyte Society, but this level is associated with poor specificity in other contexts and higher ferritin cut-off values have been used (12). The ferritin cut-off values used in paediatric HLH/MAS studies (500–2,000 ng/mL) tend to be lower than in adult studies (often >10,000 ng/mL) (2,6,7), where infectious and malignant contributors predominate (16). Other conditions such as iron overload, malignancy and hepatitis commonly induce high ferritin levels even in the absence of HLH/MAS (17).
Abnormalities in other widely available clinical and laboratory indicators of inflammation, coagulopathy or organ damage/dysfunction also raise the level of suspicion for HLH/MAS (PTC 1.4) (Table 5). However, many HLH/MAS-associated biomarkers may also indicate parallel inflammatory processes (e.g., elevated LDH in thrombotic microangiopathy) (18). More specialised biomarkers measuring key HLH/MAS pathways (PTC 1.5) (Table 5; Supplemental Table 1, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42636) are increasingly available from reference laboratories. These include measures of activation of T cells (soluble interleukin-2 [IL-2] receptor-α/CD25, T cell human leukocyte antigen – DR isotype (HLA–DR isotype expression), macrophages (CD163, neopterin), inflammasomes (IL-18) and the interferon-γ (IFNγ) pathway (CXCL9). Their relative specificity (compared with other inflammatory parameters listed in Table 5) is helpful in confirming an HLH/MAS diagnosis and in monitoring. The TF recommended assessment of specialized inflammatory biomarkers, interpreted with the aid of consultants, when available (PTC 1.5). Longitudinal assessment of both routine and specialised HLH/MAS biomarkers improves their diagnostic utility and is essential for monitoring for progression or resolution (as discussed below) (19).
Table 5.
Laboratory and biomarker testing in HLH/MAS*
| Test | In HLH/MAS | Biology | Criteria† | Monitoring frequency | Prognostic utility refs‡ | Caveats |
|---|---|---|---|---|---|---|
|
| ||||||
| Complete blood cell count | ||||||
| Neutrophil count | ↓ | Affected by marrow production, proliferation, tissue sequestration, consumption | 1, 3 | F | ✓31, 116 | Glucocorticoid demargination |
| Lymphocyte count | ↓ | 1, 3 | F | – | ||
| Haemoglobin | ↓ | – | F | ✓3, 31, 70, 114 | ||
| Platelet count | ↓ | 1, 2, 3 | F | ✓26, 31, 35, 42, 47 | ||
| Inflammatory biomarkers | ||||||
| CRP | ↑ | Hepatic release in response to IL-6 | – | F | ✓53 | Blunted by IL-6 blocking drugs |
| Ferritin | ↑ | Macrophage/hepatocyte activation | 1, 2, 3 | F | ✓35 114–117 | ↑ By iron overload |
| ESR | ↑↓ | Falls with fibrinogen consumption | – | I | ✓107 | ↑ By IVIg, dialysis |
| LDH | ↑ | Cellular death/injury | – | I | ✓114 123 | ↑ With haemolysis, TMA |
| IL-2Rα | ↑ | T cell activation | 1 | I, R | ✓30 | |
| CXCL9 | ↑ | Chemokine induced by IFNy | – | I, R | ||
| IL-18 | ↑ | Inflammasome-activated, induces IFNy | – | PRN | ✓124 | |
| Liver function tests | ||||||
| ALT, AST, bilirubin | ↑ | Hepatocyte injury | 2, 3 | F | ✓31, 35, 47 | |
| Triglycerides | ↑ | Cytokine inhibition of lipoprotein lipase | 1, 2, 3 | R, PRN | ✓114 | Fasting |
| Albumin | ↓ | Vascular leak/third-spacing | – | F | ✓70, 107, 114, 116 | |
| Coagulopathy tests | ||||||
| Fibrinogen | ↓ | Fibrinogen consumption/fibrin degradation | 1, 2, 3 | F | ✓26, 47, 116 | |
| d-dimer | ↑ | F/I | ✓26, 47 | |||
| PT/INR/PTT | ↑ | Factor consumption | – | F | ✓ | Heparin effects |
| CNS tests | ||||||
| Brain imaging | Abnormal | Inflammation of white or grey matter, meninges, hypoxia | – | PRN | ✓3, 42, 76, 77, 81 | |
| CSF studies | ↑ | Pleocytosis and/or high protein~CNS inflammation | – | PRN | ✓3, 42, 76, 77, 81 | |
HLH = haemophagocytic lymphohistiocytosis; MAS = macrophage activation syndrome; F = frequent (e.g., daily); CRP = C- reactive protein; IL-6 = interleukin-6; ESR = erythrocyte sedimentation rate; I = intermittent (e.g., weekly); IVIg = intravenous immunoglobulin; LDH = lactate dehydrogenase; TMA = thrombotic microangiopathy; IL-2Rα = interleukin-2 receptor α; R = rarely (e.g., monthly); ALT = alanine aminotransferase; AST = aspartate aminotransferase; PRN = as needed; PT/INR/PTT = prothrombin time/international normalised ratio/partial thromboplastin time; CSF = cerebrospinal fluid; CNS = central nervous system.
Degree of abnormality and/or failure to improve correlated with worse outcomes.
PTC 2.0: existing criteria.
Several criteria have been developed to identify patients with syndromes that may represent HLH or MAS (Table 1; Supplemental Table 2 at https://onlinelibrary.wiley.com/doi/10.1002/art.42636). The HLH-94 criteria (refined in HLH-04) were developed to classify infants and children for treatment trials targeting paediatric patients with genetic causes of HLH/MAS (2,5). The MAS-2016 criteria were developed to classify MAS in patients with known or strongly suspected sJIA (6). The HScore was developed in adults with primarily malignancy or infection-associated HLH (7), and the MS score to distinguish MAS from underlying sJIA (8). The HScore and HLH-2004 criteria have been validated in some additional contexts (20–23). The TF agreed that each set of criteria were useful within the context in which they were developed, but that no existing set of criteria was a sufficient diagnostic tool in all settings and populations (PTC 2). There is substantial feature overlap between criteria (Table 1; see also Supplemental Table 2 at https://onlinelibrary.wiley.com/doi/10.1002/art.42636).
PTC 3.1: evaluating contributors.
The TF emphasised the critical importance of timely identification of underlying contributors (genetic causes, predisposing conditions and acute triggers as described in ‘Introduction’ section), often in a rapidly evolving and ill patient (PTC 3.1). A thorough workup should begin immediately on suspicion for HLH/MAS and should be tailored to the most likely contributors, paying particular attention to the patient’s age, family history, infectious exposures/risks, recent treatments and underlying conditions. Although HLH/MAS is thought to result from the interaction of multiple host and environmental contributors, available data typically implicate a single aetiology (as reflected in Table 6 and more thoroughly in the SLR manuscript [12]). Additionally, >2,000 case reports and series demonstrate that HLH/MAS can occur in most settings that provoke an immune or inflammatory response (12). Genetic causes of HLH/MAS represent a minority of all cases (particularly in adults), but they have made essential contributions to diagnostic and treatment advances. The IEI include nearly 500 genetically defined disorders (24), and for most of these HLH/MAS is a rare complication. The canonical high-penetrance genetic causes of HLH are those that profoundly impair granule-mediated cytotoxicity as well as the X linked lymphoproliferative syndromes (Table 7). The distinction between genetic causes and variants conferring susceptibility has grown less clear with time. Nevertheless, the identification of a genetic cause/contributor has profound implications (as discussed below). Among predisposing conditions, malignancy (especially lymphoma) is a major contributor to HLH/MAS. Investigation for underlying malignancy should be considered in all patients with HLH/MAS, particularly in adults where it occurs in nearly half of cases (7, 20,25–37). Although MAS is most recognised and best studied in sJIA and adult-onset Still disease (AOSD), SLE may be a more common cause of HLH/MAS in adults in part due to its higher prevalence (38–40).
Table 6.
Proportion of attributable HLH/MAS cases by primary contributor*
| HLH/MAS cases, median % (min–max); refs |
||
|---|---|---|
| Paediatric | Adult | |
|
| ||
| Genetic causes | ||
| Genetic HLH disorders | 12 (3–46); refs 20, 27, 42, 44–47, 49, 50, 66, 125 | Rare; ref 27 |
| Other inborn error of immunity | 6 (2–18); refs 27, 49, 66, 125 | Rare; ref 27 |
| Predisposing conditions | ||
| Rheumatological | 10 (2–26); refs 20, 27, 42–44, 46–48, 50, 66, 125 | 8 (2–26); refs 7, 20, 25–36, 41 |
| Malignancies | 5 (2–19); refs 20, 27, 42, 43, 46, 50, 66, 125 | 46 (26–73); refs 7, 20, 25–36 |
| Acute triggers† | ||
| All-cause infections | 57 (9–88); refs 20, 27, 42, 43, 46, 48–50, 66, 125 | 27 (9–75); refs 7, 20, 26–36, 41 |
| Viral infections | 57 (18–80) | 14 (2–33) |
| Bacterial infections | 10 (3–58) | 14 (2–45) |
| Idiopathic | ||
| Unknown aetiology | 42 (17–49); refs 27, 42, 44, 47, 125 | 18 (4–40); refs 20, 27, 28, 31–33, 35, 36 |
Summary of individual cohort studies identified in the systematic literature review (SLR) that attempted to capture all cases of haemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS) over a study period and aimed to identify an underlying predisposing condition or acute trigger. Results were divided by age <18 or ≥18 years. Only series with ≥30 patients, and that attributed a single contributing aetiology per patient, were included. Attributable cases are presented as the median percentage with range (minimum [min] to maximum [max]) for all cohort studies included. As such, columns sum to >100% (see the SLR publication [12] for details). Rare = <1.5%.
The SLR did not identify studies that quantified the proportion of HLH/MAS attributable to iatrogenic triggers (e.g., chimeric antigen receptor T cell cytokine release syndrome).
Table 7.
Genes associated with HLH/MAS susceptibility*
| Gene | Protein | Disease acronym | Frequency of HLH/MAS† | Clinical associations‡ | Specialised testing§ | OMIM |
|---|---|---|---|---|---|---|
|
| ||||||
| Impaired lymphocyte granule-mediated cytotoxicity | ||||||
| PRF1 | Perforin | FHL2 | High | Early onset, isolated CNS involvement | FL, NK | 603553 |
| UNC13D | Munc13-4 | FHL3 | High | Isolated CNS involvement | NK, Degran | 608898 |
| STX11 | Syntaxin11 | FHL4 | High | Variable age at onset, possible risk MDS/leukaemia | NK, Degran | 603552 |
| STXBP2 | Munc 18 to 2 | FHL5 | High | IBD, SNHL, hypogammaglobulinaemia | NK, Degran | 613101 |
| RAB27A | Rab27a | GS2 | High | Albinism, infection | NK, Degran | 607624 |
| LYST | LYST | CHS | Moderate | Albinism, infection | NK, Degran | 214500 |
| AP3B1 | AP3 | HPS2 | Moderate/Low | Albinism, infection, bleeding | NK, Degran | 608233 |
| RHOG | RhoG | – | Unknown | NK, Degran | – | |
| Impaired EBV control | ||||||
| SH2D1A | SAP | XLP1 | High | Lymphoma | FL | 308240 |
| ITK | ITK | LPFS1 | Moderate | Lymphoma | 613011 | |
| CD27 | CD27 | LPFS2 | Moderate | Lymphoma | 615122 | |
| CD70 | CD70 | LPFS3 | Moderate | Lymphoma | 618261 | |
| MAGT1 | MAGT1 | XMEN | Moderate | Lymphoma | 300853 | |
| CTPS1 | CTPS1 | – | Moderate | Lymphoma | 615897 | |
| RASGRP1 | RASGRP1 | – | Moderate | Lymphoma | 618534 | |
| Impaired inflammasome regulation | ||||||
| XIAP/BIRC4 | XIAP | XLP2 | Moderate | IBD | FL, IL-18 | 300635 |
| NLRC4 | NLRC4 | AIFEC, MAS | Moderate | Early onset, IBD | IL-18 | 616050 |
| CDC42¶ | CDC42 | NOCARH | High | Early onset, rash | IL-18 | – |
| Other immune dysregulation | ||||||
| NCKAP1L | HEM1 | – | Unknown | Infection, autoimmunity | 618982 | |
| RC3H1 | ROQUIN | – | Unknown | 618998 | ||
| HAVCR2 | TIM3 | – | Moderate | SPTCL | 606652 | |
| Dysregulated metabolism | ||||||
| SLC7A7 | SLC7A7 | LPI | Moderate | Enteral protein intolerance | 222700 | |
List is not comprehensive; consult with appropriate specialists for appropriate breadth of testing. FHL = familial haemophagocytic lymphohistiocytosis 2; CNS = central nervous system; MDS = myelodysplastic syndrome; GS2, Griscelli syndrome type 2; IBD = inflammatory bowel disease; SNHL = sensory neural hearing loss; CHS = Chediak-Higashi syndrome; HPS2 = Hermansky-Pudlak syndrome type 2; XLP1 = X linked lymphoproliferative syndrome; LPFS1 = lymphoproliferative syndrome; XMEN = X linked immunodeficiency with magnesium defect, Epstein-Barr virus infection and neoplasia; AIFEC = autoinflammation with infantile enterocolitis; NOCARH = neonatal-onset pancytopenia, autoinflammation, rash and episodes of HLH; SPTCL = subcutaneous panniculitis-like T cell lymphoma; LPI = lysinuric protein intolerance.
In the appropriate inheritance pattern. Genes with moderate/low haemophagocytic lymphohistiocytosis (HLH) frequency have other phenotypes.
Beyond those common in HLH/macrophage activation syndrome (HLH/MAS), see Table 1.
FL = flow cytometry can assess protein abundance; NK = NK function should be abnormal even in remission; Degran = CD107a mobilisation should be abnormal; IL-18 = chronically and highly elevated serum interleukin-18 (IL-18).
Only C-terminal CDC42 mutations have been associated with HLH/MAS.
Infection is the most common acute trigger of HLH/MAS. In children, infection is the most common aetiology, with a specific pathogen identified in over 50% of new HLH/MAS presentations. Broad testing for infection (e.g., blood and other cultures, viral PCR, etc) should be pursued based on clinical scenario. Some infections warrant special attention for their role in HLH/MAS. Epstein-Barr virus (EBV) is a well-known trigger of HLH, particularly in individuals with genetic (Table 7) or acquired immunodeficiency or certain malignancies. It is unclear why the incidence of EBV-HLH appears higher in Asia, but this is consistent with other EBV-triggered phenotypes (31–34,41–49). Region-specific and season-specific infections should also be considered as causes of HLH/MAS in endemic areas, including dengue virus in tropical/subtropical climates (46,50,51), histoplasmosis in the mid-western and southern US (31), and less frequently malaria (51,52), tuberculosis (7,25,26,32,33,36,48), scrub typhus (33,47,50), typhoid fever (42,47,50,51), tickborne diseases (42,43,53) and leishmaniasis (25,42,54). Although inflammation in patients with COVID-19, multisystem inflammatory syndrome in children (MIS-C) or adults (MIS-A) rarely rises to meet HLH or MAS criteria, the pandemic normalised the need to identify and treat (SARS-CoV2) infection-associated immunopathology. Testing for genetic contributors should be considered regardless of the type of infection, particularly in young children.
The increasing use of immune effector cell cancer therapies, including chimeric antigen receptor T cells, has elevated their recognition as iatrogenic triggers of HLH/MAS. The incidence of CRS and immune effector cell–associated neurological syndrome (ICANS) is highest in leukemias and lymphomas. The distinctions between CRS/ICANS and HLH are unclear and their ideal treatment is an evolving target beyond the scope of this effort (55–58).
For many patients, multiple contributors interact to drive HLH/MAS. Acute infections are identified in the majority of patients with HLH/MAS with predisposing rheumatological conditions (>65%) or IEIs (>80%) (54,59–62). In adults, the presence of multiple pathogens at the time of HLH/MAS diagnosis may increase risk of mortality (63). Thus, concern for HLH/MAS should prompt consideration of multiple potential contributors regardless of known underlying conditions or aetiologies.
PTC 3.2–3.5: genetic testing.
Genetic causes for HLH/MAS are likely under-recognised and their identification profoundly affects treatment, prognosis and genetic counselling (PTC 3.2) (Table 7). For example, screening for CNS involvement is particularly important in genetic HLH (47). Early recognition of familial haemophagocytic lymphohistiocytosis may accelerate allogeneic haematopoietic stem cell transplantation (HSCT) and can support HSCT in affected presymptomatic siblings (64,65). Some HLH/MAS therapeutic trials include or exclude specific genetic causes (ClinicalTrials.gov identifiers NCT04641442 and NCT03113760).
When to perform genetic testing, on whom, what test(s) to send and how to interpret detected variants are complex and evolving decisions (PTC 3.3). Features suggestive of a genetic cause include young age at presentation, positive family history, consanguinity and prominent CNS disease. HLH/MAS due to cytotoxicity defects tends to present in infancy and early childhood, whereas HLH/MAS in other IEI (particularly those with EBV immunodeficiency) (Table 7) present in a broader age range including older children (66). Although genetic HLH has presented in adulthood, actionable results of genetic testing in adult HLH/MAS are rare (25,67,68). Other relevant clinical features/contexts like albinism, inflammatory bowel disease, isolated CNS involvement and EBV-immunodeficiency suggest specific genetic causes (PTC 3.3) (Table 7).
Given the high prevalence of genetic causes in children and the large clinical impact of a positive finding, the TF supported early genetic testing in children and high-risk adults, preferably using multigene panels or whole exome/genome sequencing (PTC 3.4). Single-gene sequencing remains appropriate with family history of a known genetic HLH disorder, characteristic clinical features (e.g., albinism), positive protein or functional testing (e.g., perforin flow cytometry) or in resource-limited settings. Genetic counselling is warranted for all patients undergoing genetic testing (PTC 3.5).
PTC 4.1–4.4: prognostic factors and CNS involvement.
Prognosis in HLH/MAS is dependent on multiple factors, including the nature of the underlying contributors, degree of organ dysfunction and duration of active disease (PTC 4.1). HLH/MAS can be fatal in any context, but relative to other causes malignancy-associated HLH/MAS is associated with worse survival (33,66,69,70) and HLH/MAS in rheumatic diseases has a more favourable prognosis (3,40,66,71–73). EBV is associated with poor prognosis when present in patients with genetic immunodeficiency or predisposing (rheumatic or malignant) conditions (42,47,69), but prognosis appears better in patients with EBV as the sole contributor (45,46). Specific patterns of organ injury also predict poor outcomes (PTC 4.1). Liver involvement is common and can progress to life-threatening liver failure (47). Patients with multiorgan dysfunction often require treatment in an intensive care unit (ICU) setting, a strong predictor of poor outcome (50,53,74,75).
CNS involvement is sufficiently common, insidious and dangerous to warrant specific attention. It is associated with both mortality and long-term neurological sequelae in survivors regardless of underlying contributors (3,42,47,76–80). It should be suspected in all patients being evaluated for HLH/MAS (PTC 4.2). CNS manifestations of HLH/MAS can be broad (Tables 1 and 5), and cerebrospinal fluid (CSF) and imaging findings usually demonstrate evidence of inflammation in affected patients (76). Incidence of CNS involvement varies by age and aetiology (45,47,76), and children with HLH/MAS are at higher risk than adults, especially children with genetic causes (47,76,81,82). Some degree of CNS involvement is present in a sizeable percentage of children with EBV-HLH (83), sJIA-MAS (71) and adults with secondary HLH/MAS (80,84,85).
CNS involvement should be considered in all patients, and all should undergo a complete neurological examination. Patients presenting under 1 year of age, those otherwise suspected of having familial disease (47) and any patient with symptoms or signs concerning for CNS dysfunction (including an unreliable exam) should undergo assessment for CNS involvement (PTC 4.2). Assessment for CNS involvement may include CSF evaluation (glucose, protein, cell counts and often cytological review) and contrast-enhanced brain MRI as well as other testing (electroencephalogram, MR angiography, spinal imaging) as clinically indicated (PTC 4.3). Full evaluation often must await stabilisation of cardiorespiratory function, coagulopathy or intracranial pressure. Providers should not delay empiric or context-specific treatments in order to complete the CNS workup (PTC 4.4).
PTC 5.1–5.5: treatment considerations.
Treatment of patients with suspected HLH/MAS requires a dynamic risk–benefit assessment. Consideration of HLH/MAS-directed immunomodulation should occur simultaneously with diagnostic evaluations (PTC 5.1–3), treatment of contributing factors (PTC. 5.4) and prevention of complications (PTC 5.4–5.5) (49,77,86). Figure 1 is intended to depict how these PTC on early diagnosis, monitoring and management may function in practice, in relation to each other and relative to the goal of context-specific treatment. Age-appropriate supportive care should follow accepted guidelines, such as the Surviving Sepsis Campaign (87,88), and its provision, as well as the frequency of monitoring (as discussed below), may require intensive care. ICU admission was required in over a third of children with HLH/MAS and nearly half of adults with MAS (61). In children and adults requiring ICU admission for HLH/MAS, nearly 70% required mechanical ventilation or vasopressors/inotropes and nearly half required renal replacement therapy (32,50). Use of intensive care appears higher in HLH/MAS occurring in context with worse outcomes, like malignancy (34,71,89).
Figure 1.
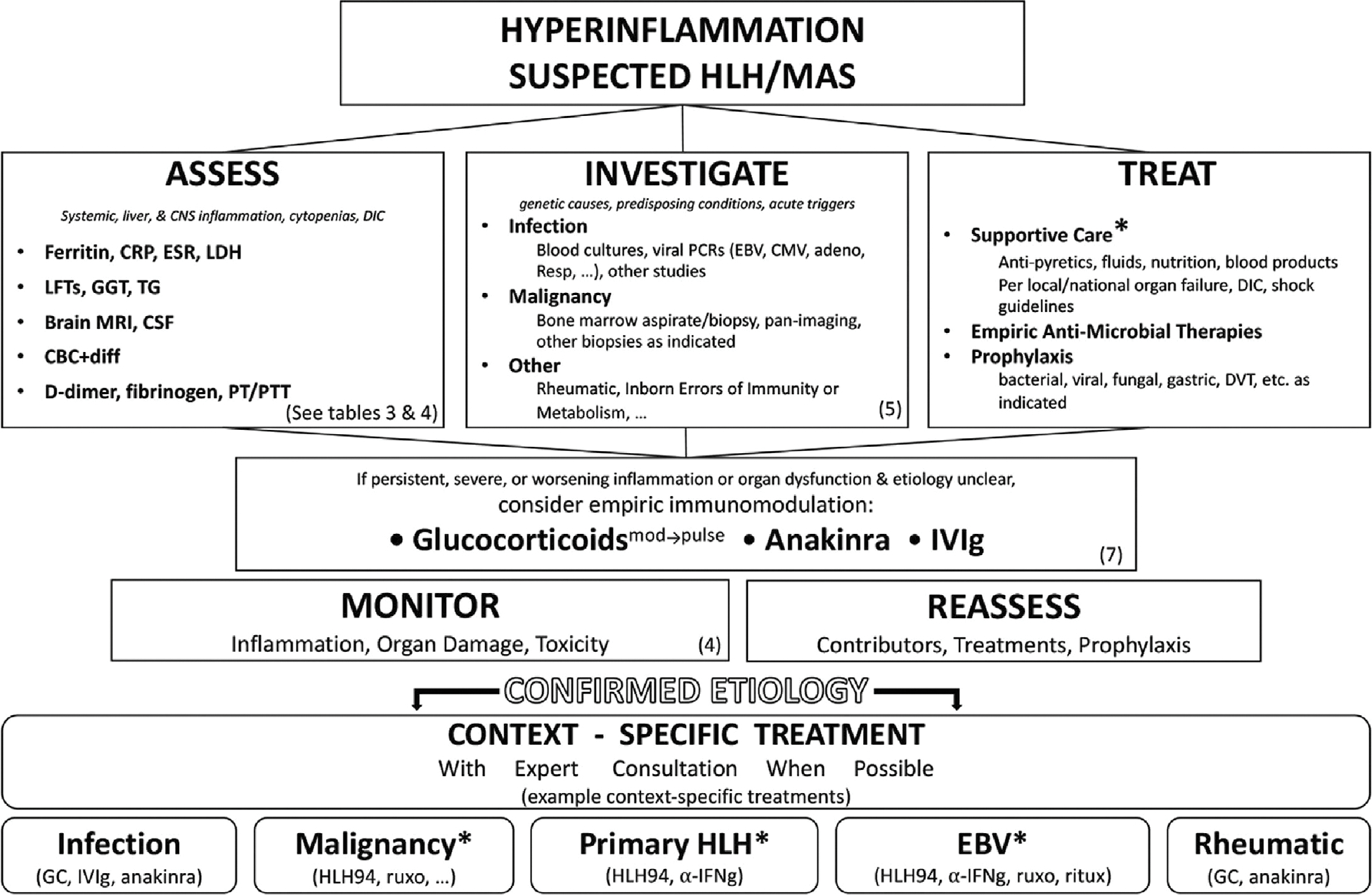
Summary of the approach to early or suspected haemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS). When HLH/MAS is suspected, providers should (in parallel and as clinically appropriate) assess for the key features of HLH/MAS, investigate suspected contributors and treat with supportive care, with empiric and prophylactic antimicrobials, with other prophylaxis regimens, and possibly with empiric immunomodulation. Ongoing monitoring and reassessment should prompt re-evaluation of treatments being given. Patients should transition to context-specific treatment immediately on identification of a confirmed aetiology. *Addressed in separate guidance documents, see www.histiocytesociety.org/HLH-consensus. α-IFNγ = interferon-γ neutralising antibody; CBC+diff = complete blood cell count with leucocyte differential; CMV = cytomegalovirus; CRP = C-reactive protein; CSF = cerebrospinal fluid; DIC = disseminated intravascular coagulopathy; DVT = deep vein thrombosis; EBV = Epstein-Barr virus; ESR = erythrocyte sedimentation rate; GC = glucocorticoid; GGT = γ-glutamyl transferase; HLH94 = HLH-94 treatment protocol 1 or current standard of care; IVIg = intravenous immunoglobulin; LDH = lactate dehydrogenase; LFTs = liver function tests; PT/PTT = prothrombin time/partial thromboplastin time; TG = triglycerides; ruxo = ruxolitinib; ritux = rituximab.
Choosing and adjusting empiric immunomodulation for suspected HLH/MAS can be challenging. Decision-making must integrate HLH/MAS severity and rate of progression, specific organ involvement, likely contributors, comorbid conditions and concurrent medications (Figure 1). Ideally, targeted immunomodulation would be initiated as early as possible (PTC 5.1) and neither induce immunosuppression nor compromise the aetiological workup. In practice, determining the target and balancing these risks are essential, patient-specific challenges. Although no studies have evaluated empiric treatment of HLH/MAS prior to/regardless of aetiology, immunomodulatory treatment has dramatically improved survival in most aetiologies of HLH/MAS (12,49,90,91). In patients with high-risk features or progressive HLH/MAS, the TF strongly recommends considering empiric immunomodulation during the initial evaluation and management period (PTC 5.1–3). Once there is sufficient understanding of a patient’s underlying contributors, management should shift to context-specific treatments and recent context-specific guidance documents may be helpful in this transition (67,74,92–94) (Figure 1, asterisks).
The TF currently endorses use of glucocorticoids (GCs), the recombinant IL-1 receptor antagonist (IL-1RA) anakinra and/or intravenous immunoglobulin (IVIg) for empiric immunomodulation in suspected HLH/MAS (PTC 5.3) (Figure 1 and Table 8). Multiple treatments may be initiated concurrently depending on clinical context and availability. Published treatment data demonstrate the strongest support for GCs across all forms of HLH/MAS (12). The choice of GC formulation (most commonly prednisone, prednisolone, dexamethasone [DEX] or methylprednisolone [MP]) and route of administration (oral versus intravenous) should be tailored to the patient and care setting (Table 8). ‘Pulse’ doses of intravenous MP (10–30 mg/kg/day up to 1 gm, given daily) are effective in severe rheumatic and neuro-inflammatory diseases (95–97), and have been used successfully in HLH/MAS (61). DEX is used in HLH treatment protocols due to better CNS penetration at an initial dose of 10 mg/m2/day (~2–4 mg/kg/day of MP). Given DEX’s long half-life, shorter-acting GCs may be preferable in rapidly evolving diagnostic scenarios.
Table 8.
Empiric HLH/MAS immunomodulatory treatment dosing*
| Paediatric | Adult |
|---|---|
|
| |
| Glucocorticoids† | |
| a. Prednisone/prednisolone per os or methylprednisolone intravenous 1–2 mg/kg/day. | |
| b. Dexamethasone intravenous per os 10 mg/m2/day. | |
| c. High-dose methylprednisolone intravenous 10–30 mg/kg/day (maximum 1 gm/day) for 1–3 days, followed by strategies a or b. | |
| Intravenous immunoglobulin | |
| 1 gm/kg/day ×2 days | 0.4–1 gm/kg/day ×2–5 days |
| Anakinra‡ | |
| Intravenous (preferred) or subcutaneous 5–10 mg/kg/day | |
This table reflects the summary of reported immunomodulatory pharmacotherapy in the literature and is not intended as a substitute for clinical judgment. Dosages are derived from the systematic literature review (12) unless otherwise noted. HLH = haemophagocytic lymphohistiocytosis; MAS = macrophage activation syndrome; per os = by mouth.
Dosing schedules and substitution with other glucocorticoids and/or other (intravenous or oral) preparations can be based on preference, availability and patient need.
Daily dose often divided every 6–12 hours.
Importantly, GC administration may obscure pathological diagnosis and/or staging of leukaemia or lymphoma (98). Therefore, definitive testing for malignancy (typically biopsy/aspirate of bone marrow, lymph node and/or other indicated tissues) should be attempted prior to GC administration when possible. GC-related immunosuppression depends on dose, duration of exposure and relevant pathogens. Although GC treatment (alongside appropriate antimicrobial treatment) prevents immunopathology in many localised infections (99,100), large studies have not supported its utility for immunomodulation in sepsis. Thus, the role of GC in infection-associated HLH/MAS remains patient-dependent and pathogen-dependent. Providers should monitor for other dose-dependent GC side effects like hyperglycaemia, hypertension, myopathy and psychosis.
Empiric use of anakinra and/or IVIg in early, evolving or undifferentiated HLH/MAS may provide immunomodulation without significant immunosuppression and without impairing malignancy workup. The TF supported their inclusion despite sparse data due to good pharmacological and safety profiles, strong efficacy in other systemic inflammatory diseases and significant clinical experience. Anakinra is a safe and effective treatment for many autoinflammatory and rheumatic disorders. Its rapid onset and short half-life may be desirable in rapidly evolving patients. Even used at high doses in adults with bacterial sepsis (up to 48 mg/kg/day), it showed no signal for immunosuppression and appeared to limit mortality in patients with sepsis with hepatobiliary dysfunction and coagulopathy (90,101). A retrospective study in secondary HLH supported the safety and possible efficacy of early anakinra use in controlling inflammation (102). IVIg has demonstrated efficacy in Kawasaki disease, and it neither obstructs cancer workup nor suppresses immune function. Notably, serological testing should be sent from samples taken prior to IVIg when feasible. Reports of its efficacy in HLH/MAS are restricted to case series. High-dose IVIg is also a substantial colloid load that can compromise cardiac function and worsen oedema. It rarely causes haemolysis or aseptic meningitis.
Clinical context is essential when considering escalation or context-specific treatment(s), and clinicians are encouraged to consult with local, regional or national experts on a case-specific basis. B cell depletion may be useful in some patients with EBV-HLH (103,104). Early initiation of treatment regimens centred around the chemotherapeutic etoposide (guidelines in Ehl et al [92]) have been life-saving for patients with primary HLH and severe EBV-HLH (49,77). Evidence for high-dose etoposide is less favourable for HLH/MAS in the context of sJIA/AOSD, though lower doses may be useful (105). It is not indicated for most non-EBV infections (34,106–108). The utility of etoposide in malignancy-associated HLH is currently unclear (93,109).
For patients with increasing inflammation and/or worsening organ damage despite early immunomodulation, treatment escalation with higher doses of GC and/or alternative agents (Table 9) should be considered in consultation with HLH/MAS experts. Increasing evidence supports the involvement of the IFNγ pathway in HLH/MAS. The IFNγ neutralising antibody emapalumab was recently approved in the US for the treatment of refractory, recurrent, or progressive HLH (110). Ruxolitinib (and other JAK inhibitors) broadly targets cytokine signalling, including IFNγ, and has shown promising early results in HLH/MAS (111–113). Alongside HLH/MAS-directed immunomodulation, treatment of contributing factors is critical for optimizing outcomes (PTC 5.4). This will often include empiric antimicrobial and sometimes antiviral agents, accounting for exposures/geography, comorbidities (e.g., renal failure) and chronic immunosuppression. Like other aspects of HLH/MAS treatment, infectious prophylaxis should be considered early and revisited as the patient and workup evolve (PTC 5.5). Secondary infections can complicate both the inpatient and outpatient course of HLH/MAS. Antifungal and Pneumocystis jirovecii pneumonia prophylaxis are recommended, and are part of the HLH-94 protocol (5). Antifungal and antiviral prophylaxis were administered in more recent trials with newer agents like emapalumab (110). In addition to assistance with empiric treatment, consultation with immunocompromised infectious disease specialists may aid prophylaxis planning (PTC 5.5).
Table 9.
Other immunomodulatory therapies used in HLH/MAS*
| Dosing (refs) |
|||||
|---|---|---|---|---|---|
| Route | Paediatric | Adult | Adverse events† | Notes | |
|
| |||||
| Etoposide (chemotherapy) | Per os; intravenous | 50–150 mg/m2/dose 1–2 doses/week (45, 49, 61, 77, 105, 108) | 50–150 mg/m2/dose 1–2 doses/week (31, 105, 108, 126–129) | BM suppression, hepatotoxicity, hypotension (infusion-related), mucositis/alopecia, nausea/vomiting, secondary malignancy | Infectious screening and PPx |
| Ciclosporin (calcineurin inhibition)‡ | Per os; intravenous | 3–5 mg/kg/day, two times per day (45, 49, 61, 77, 130, 131) | 2–7 mg/kg/day twice daily (31, 108, 113, 26–129) | Nephrotoxicity/HTN, hepatotoxicity, hirsutism, gingival hypertrophy, neurotoxicity | Monitor levels |
| Ruxolitinib (JAK inhibition) | Per os | 2.5–20 mg/dose (112, 113, 132)§ or 25 mg/m2/dose, two times per day | Dyslipidaemia, cytopenias, hepatotoxicity, immunosuppression (herpes viruses) | Infectious screening and PPx | |
| Emapalumab (IFNγ neutralisation) | Intravenous | Refractory HLH (110): 1 → 10 mg/kg/dose sJIA-MAS¶: 6, then 3 mg/kg/dose every 3 days | Immunosuppression (mycobacteria, herpes viruses, and Histoplasma capsulatum), HTN, infusion reactions | Infectious screening and PPx | |
| Rituximab (B cell depletion) | Intravenous | 375 mg/m2/dose (maximum 1 gm), two times per day, separated by 2 weeks (103, 104) | Infusion reactions, HTN, hepatotoxicity, immunosuppression (hepatitis B), cytopenias, ↓IgG, mucocutaneous reaction, progressive multifocal leukoencephalopathy (rarely) | Specifically for EBV-HLH | |
Consultation with providers experienced in managing haemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS) is strongly advised prior to administration. BM = bone marrow; HTN = hypertension; IFNγ = interferon-γ; PPx = prophylaxis.
List of common and important adverse events (shown in boldface) with short-term (up to 3 months) use of therapies, based on those described in https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm and https://www.uptodate.com/contents/table-of-contents/drug-information/general-drug-information.
Primarily used in MAS. Can be substituted with tacrolimus (initial dose 0.1 mg/kg/day by mouth [per os] divided every 12 hours, targeting trough 8–20 ng/mL).
Weight-based dosing per indicated references. Body surface area dosing used in ongoing HLHRUXO trial (ClinicalTrials.gov identifier NCT04551131).
Dosing in ongoing systemic juvenile idiopathic arthritis (sJIA)–MAS trial (ClinicalTrials.gov identifier NCT05001737).
PTC 6.1–6.3: monitoring.
Monitoring for disease progression, new organ involvement and damage and response to treatment begins on suspicion for HLH/MAS. Monitoring plans should be tailored to severity, organ involvement and likely contributors of HLH/MAS. Many of the biomarkers useful for diagnosing HLH/MAS also have prognostic relevance (Table 5; Supplemental Table 1 [PTC 6.1] at https://onlinelibrary.wiley.com/doi/10.1002/art.42636). For example, both higher initial ferritin levels and failure of ferritin to improve during therapy associate with worse outcomes (71,114–118).
No comparative studies evaluate the ideal laboratory monitoring protocol. Given the propensity for rapid clinical changes, initial monitoring may include daily assessment of inflammatory biomarkers (e.g., CRP, ferritin), indicators of organ damage (e.g., CBC, fibrinogen, ALT) and any drug-specific monitoring. More frequent monitoring may be needed for evolving or critically ill patients and may require ICU care (74) (PTC 6.2) (Table 5). Lack of response to initial therapy should prompt a careful re-examination of both underlying diagnoses and therapeutic approach. When available, more specific HLH/MAS biomarkers like sIL-2Ra, IL-18 and CXCL9 should be monitored less frequently than conventional disease activity measures like ferritin and CRP (PTC 6.5) (Table 4). CXCL9 may be particularly useful for monitoring response to IFNγ-blocking therapies (110). Specialised tests may also be helpful in distinguishing HLH/MAS relapse from acquired infection or drug reaction. Treatment response and dose-escalation criteria used in HLH/MAS trials also reflect the overlap between diagnostic and monitoring tests (12).
PTC 7.0: multidisciplinary teams.
Mounting evidence suggests that a multidisciplinary team experienced in managing HLH/MAS may improve recognition, reduce immunosuppression and improve outcomes (PTC 7.0) (119,120). Such response teams often include representatives from infectious diseases, haematology/oncology, rheumatology, immunology, pharmacy and other relevant specialties, although their optimal composition and function has not been established. Their goals include improving early identification, streamlining communication and improving consistency of care. These groups may also be able to better incorporate new findings, conduct quality improvement and engage in collaborative research.
DISCUSSION
HLH/MAS is a life-threatening immunopathological state requiring a systematic evaluation of aetiological factors and prompt intervention. It occurs in many contexts, can present to various providers and its contributors are often unclear. Thus, the TF has targeted these PTC at a broad audience to aid in recognising the clinical and laboratory features of HLH/MAS, investigating underlying contributors, initiating appropriate (empiric, targeted, and prophylactic) treatments and monitoring for response, progression and complications.
While generating the PTCs for the earliest stages of HLH/MAS, the multisubspecialty TF was also charged with identifying areas of substantial unmet need (box 1). Among these, TF members identified a need to standardise and harmonise the terminology used to describe and categorise patients with HLH/MAS. This nomenclature should be based on both clinical manifestations and underlying contributor(s), account for diagnostic uncertainty, apply across a range of sites and specialties and associate with validated criteria. Given the breadth of providers this change would affect, it may require a distinct, objective and international collaboration.
Box 1.
Identified HLH/MAS research priorities1*
| 1. Unify nomenclature and validate criteria for diagnosis of HLH/MAS across specialties. |
| 2. Better characterise epidemiology, contributors, management practices and outcomes in resource-limited settings. |
| 3. Institute/Expand multicentre prospective HLH/MAS registries and biobanks. |
| 4. Study the effect of real-time results of specialised biomarkers. |
| 5. Study the utility of early and broad exome/genome-wide testing. |
| 6. Study the utility of multidisciplinary HLH/MAS response teams. |
| 7. Expand and improve prospective trials in HLH/MAS: |
| a. Timing of immunomodulation undifferentiated disease; |
| b. Active comparator groups (e.g., adaptive designs); |
| c. Novel early treatments. |
HLH/MAS = haemophagocytic lymphohistiocytosis/macrophage activation syndrome.
There is also an urgent need to expand access to, and clarify the role of routine and specialised testing for patients with suspected HLH/MAS. The TF has highlighted the importance of trending ferritin levels in recognising, diagnosing and monitoring HLH/MAS, but results are not quickly available in many locations. Specialised biomarkers (e.g., sIL-2Rα, IL-18, CXCL9) may be more specific for HLH/MAS, but results often do not return quickly enough to be useful in early decision-making. These tests are often unavailable outside of academic centres. Studies are needed that systematically evaluate the impact of real-time biomarker assessments on treatment decisions and patient outcomes, and that determine optimal diagnostic cut-offs.
The TF also highlighted a need to better study how rapid genetic diagnostics affects treatment stratification and outcomes for (particularly paediatric) patients presenting with HLH/MAS. Mounting data demonstrate that rapid whole-genome sequencing in high-risk populations (e.g., hospitalised infants) may shorten time to diagnosis, both improving care and decreasing overall medical costs (121). Despite dramatic improvements in sequencing cost and speed, identification of actionable genetic contributors to HLH/MAS is often delayed by availability and/or restrictive payer policies. The results of these studies may encourage hospital systems and payers to support improved access and rapid results. Given the large (and rising) number of identifiable genetic variants with important management consequences, the TF encouraged broad genetic testing particularly in paediatric patients with HLH/MAS.
Therapeutically, the TF identified the need for expanded clinical research to better understand the effectiveness of existing therapies, and the need for long-term investments in basic/translational research to identify novel, targetable pathways. These studies are needed both in specific contexts as well as in HLH/MAS broadly. Specifically, studies are needed that address the efficacy of early immunomodulation (analogous to time-to-antibiotics in sepsis) and protocolised assessment of CNS involvement. Trials of treatment efficacy are needed that use active comparators and more proximate outcomes than survival (e.g., steroid exposure, length of stay, durable functional impairment and quality of life). To this end, ongoing clinical trials to test the safety and effectiveness of agents such as ruxolitinib (ClinicalTrials.gov identifier NCT04551131), alemtuzumab (ClinicalTrials.gov identifier NCT02472054), tadekinig alfa (ClinicalTrials.gov identifier NCT03113760), emapalumab (ClinicalTrials.gov identifier NCT05001737) and MAS825 (ClinicalTrials.gov identifier NCT04641442) in a variety of HLH/MAS settings are of vital interest.
To meet these testing and therapeutic challenges, there is also a need to improve multicentre, prospective HLH/MAS registries and biobanks. HLH/MAS overall is not particularly rare, and our SLR identified and screened over 12 000 published articles, but very few of these were prospective or controlled (12). Studies in resource-limited countries/environments were particularly lacking. As our community builds research infrastructure and advocates for expanded access, improved turnaround times and targeted therapeutics, it must prioritise inclusion of resource-limited settings and implementation in underserved areas.
The HLH/MAS paradigm has evolved rapidly in response to genetic, biomarker, clinical and therapeutic insights. These insights reflect the diversity and intersection of contributors and suggest convergence on a shared HLH/MAS physiology and phenotype. These insights have also led to more diagnostic and therapeutic options while highlighting the wide spectrum of primary care and subspecialty providers who care for patients with early features of HLH/MAS. With these advances, the challenge of identifying and managing at-risk patients and patients with early HLH/MAS has grown. These PTC aim to translate these insights into practical guidance that will hasten recognition, streamline diagnosis and improve early management as the essential tasks needed to limit immunopathology, mitigate organ dysfunction and achieve the best outcomes for patients with HLH/MAS.
Supplementary Material
ACKNOWLEDGMENTS
This study was reviewed and approved by all task force members (see Appendix A for collaborators) and the EULAR and ACR Executive Committees prior to submission. The task force is grateful to the librarian Darren Hamilton (London Health Sciences Center, London, Ontario, Canada) for his contribution to the systematic literature search, Brian Feldman and Joan Moore for their support in conducting the Delphi process and EULAR and the ACR for financial and logistical support. This project is part of a series of ‘points to consider’ consensus efforts (overseen by EDe and RG-M) to standardise the diagnosis and care of patients within major groups of known autoinflammatory diseases including (1) the IL-1–mediated diseases CAPS, TRAPS, MKD and DIRA; (2) the autoinflammatory interferonopathies chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE), STING-associated vasculopathy with onset in infancy (SAVI) and Aicardi-Goutières syndrome; and (3) the early diagnosis and management of inflammatory conditions with the potential progression to HLH/MAS. This research was supported in part by the intramural research programme of the National Institute for Allergy and Infectious Diseases. We are grateful for the invaluable financial and organizational support from the Autoinflammatory Alliance and the Systemic JIA Foundation, who substantially contributed to an international meeting and workgroup organisation in August 2019 that developed the outline of the points to consider project. The funds for that meeting came largely from patient fundraisers, online fundraising and the work of countless volunteers who made this project possible.
APPENDIX A: THE HLH/MAS TASK FORCE COLLABORATORS
The HLH/MAS task force collaborators are as follows: Bita Shakoory, Ashley Geerlinks, Marta Wilejto, Kate F. Kernan, Melissa Hines, Angelo Ravelli, Rashmi Sinha, Daniel Aletaha, Carl E. Allen, Hamid Bassiri, Edward M. Behrens, Joseph Carcillo, Linda Carl, W. Winn Chatham, Jeffrey I. Cohen, Randall Q. Cron, Erik Drewniak, Alexei A. Grom, Lauren A. Henderson, AnnaCarin Horne, Michael Jordan, Kim E. Nichols, Grant S. Schulert, Sebastiaan Vastert, Raphaela Goldbach-Mansky, Fabrizio de Benedetti, Rebecca A. Marsh, and Scott W. Canna.
REFERENCES
- 1.Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood 2020;135:1332–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124–31. [DOI] [PubMed] [Google Scholar]
- 3.Minoia F, Davì S, Horne A, et al. Dissecting the heterogeneity of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Rheumatol 2015;42:994–1001. [DOI] [PubMed] [Google Scholar]
- 4.Henderson LA, Canna SW, Friedman KG, et al. American College of Rheumatology clinical guidance for multisystem inflammatory syndrome in children associated with SARS-CoV-2 and hyperinflammation in pediatric COVID-19: version 1. Arthritis Rheumatol 2020;72:1791–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henter JI, Samuelsson-Horne A, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood 2002;100:2367–73. [DOI] [PubMed] [Google Scholar]
- 6.Ravelli A, Minoia F, Davì S, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European League against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation collaborative initiative. Arthritis Rheumatol 2016;68:566–76. [DOI] [PubMed] [Google Scholar]
- 7.Fardet L, Galicier L, Lambotte O, et al. Development and validation of the Hscore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol 2014;66:2613–20. [DOI] [PubMed] [Google Scholar]
- 8.Minoia F, Bovis F, Davì S, et al. Development and initial validation of the MS score for diagnosis of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Ann Rheum Dis 2019;78:1357–62. [DOI] [PubMed] [Google Scholar]
- 9.Van der Heijde D, Aletaha D, Carmona L, et al. 2014 Update of the EULAR standardised operating procedures for EULAR-endorsed recommendations. Ann Rheum Dis 2015;74:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gedik KC, Lamot L, Romano M, et al. The 2021 European Alliance of Associations for Rheumatology/American College of Rheumatology points to consider for diagnosis and management of autoinflammatory type I interferonopathies: CANDLE/PRAAS, SAVI and AGS. Ann Rheum Dis 2022;81:601–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Romano M, Arici ZS, Piskin D, et al. The 2021 EULAR/American College of Rheumatology points to consider for diagnosis, management and monitoring of the interleukin-1 mediated autoinflammatory diseases: cryopyrin-associated periodic syndromes, tumour necrosis factor receptor-associated periodic syndrome, mevalonate kinase deficiency, and deficiency of the interleukin-1 receptor antagonist. Ann Rheum Dis 2022;81:907–21. [DOI] [PubMed] [Google Scholar]
- 12.Shakoory B, Poddighe D, Cinar OK, et al. The early stages of diagnosis and management of suspected hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS): a systematic literature review. Abstracts from the ISSAID 2021 Periodic Congress. Pediatr Rheumatol 2023;21(Suppl 2). [Google Scholar]
- 13.University of Oxford Centre for Evidence-Based Medicine. Oxford Centre for Evidence-Based Medicine: levels of evidence (March 2009). URL: https://www.cebm.net/2009/06/oxford-centre-evidence-based-medicine-levels-evidence-march-2009/.
- 14.Demirkol D, Yildizdas D, Bayrakci B, et al. Hyperferritinemia in the critically ill child with secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction syndrome/macrophage activation syndrome: what is the treatment? Crit Care 2012;16:R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen CE, Yu X, Kozinetz CA, et al. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2008;50:1227–35. [DOI] [PubMed] [Google Scholar]
- 16.Lehmberg K, McClain KL, Janka GE, et al. Determination of an appropriate cut-off value for ferritin in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2014;61:2101–3. [DOI] [PubMed] [Google Scholar]
- 17.Otrock ZK, Daver N, Kantarjian HM, et al. Diagnostic challenges of hemophagocytic lymphohistiocytosis. Clin Lymphoma Myeloma Leuk 2017;17S:S105–10. [DOI] [PubMed] [Google Scholar]
- 18.Minoia F, Tibaldi J, Muratore V, et al. Thrombotic microangiopathy associated with macrophage activation syndrome: a multinational study of 23 patients. J Pediatr 2021;235:196–202. [DOI] [PubMed] [Google Scholar]
- 19.Ravelli A, Minoia F, Davì S, et al. Expert consensus on dynamics of laboratory tests for diagnosis of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. RMD Open 2016;2:e000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Debaugnies F, Mahadeb B, Ferster A, et al. Performances of the H-score for diagnosis of hemophagocytic lymphohistiocytosis in adult and pediatric patients. Am J Clin Pathol 2016;145:862–70. [DOI] [PubMed] [Google Scholar]
- 21.Batu ED, Erden A, Seyhoğlu E, et al. Assessment of the HScore for reactive haemophagocytic syndrome in patients with rheumatic diseases. Scand J Rheumatol 2017;46:44–8. [DOI] [PubMed] [Google Scholar]
- 22.Knaak C, Nyvlt P, Schuster FS, et al. Hemophagocytic lymphohistiocytosis in critically ill patients: diagnostic reliability of HLH-2004 criteria and HScore. Crit Care 2020;24:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valade S, Monseau G, Mariotte E, et al. Diagnostic performance of hemophagocytic lymphohistiocytosis criteria and hscore in critically ill patients with severe hemophagocytic syndrome. Crit Care Med 2021;49:e874–9. [DOI] [PubMed] [Google Scholar]
- 24.Tangye SG, Al-Herz W, Bousfiha A, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 2022;42:1473–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carvelli J, Piperoglou C, Farnarier C, et al. Functional and genetic testing in adults with HLH reveals an inflammatory profile rather than a cytotoxicity defect. Blood 2020;136:542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tseng YT, Sheng WH, Lin BH, et al. Causes, clinical symptoms, and outcomes of infectious diseases associated with hemophagocytic lymphohistiocytosis in Taiwanese adults. J Microbiol Immunol Infect 2011;44:191–7. [DOI] [PubMed] [Google Scholar]
- 27.Otrock ZK, Hock KG, Riley SB, et al. Elevated serum ferritin is not specific for hemophagocytic lymphohistiocytosis. Ann Hematol 2017;96:1667–72. [DOI] [PubMed] [Google Scholar]
- 28.Lee H, Kim HS, Lee JM, et al. Natural killer cell function tests by flowcytometry-based cytotoxicity and IFN-γ production for the diagnosis of adult hemophagocytic lymphohistiocytosis. Int J Mol Sci 2019;20:5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan L, Kan Y, Meeks JK, et al. 18F-FDG PET/CT for identifying the potential causes and extent of secondary hemophagocytic lymphohistiocytosis. Diagn Interv Radiol 2016;22:471–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayden A, Lin M, Park S, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv 2017;1:2529–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bubik RJ, Barth DM, Hook C, et al. Clinical outcomes of adults with hemophagocytic lymphohistiocytosis treated with the HLH-04 protocol: a retrospective analysis. Leuk Lymphoma 2020;61:1592–600. [DOI] [PubMed] [Google Scholar]
- 32.Barba T, Maucort-Boulch D, Iwaz J, et al. Hemophagocytic lymphohistiocytosis in intensive care unit: a 71-case strobe-compliant retrospective study. Medicine (Baltimore) 2015;94:e2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoon SE, Eun Y, Huh K, et al. A comprehensive analysis of adult patients with secondary hemophagocytic lymphohistiocytosis: a prospective cohort study. Ann Hematol 2020;99:2095–104. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Q, Li L, Zhu L, et al. Adult onset haemophagocytic lymphohistiocytosis prognosis is affected by underlying disease: analysis of a single-institution series of 174 patients. Swiss Med Wkly 2018;148:w14641. [DOI] [PubMed] [Google Scholar]
- 35.Zhou J, Zhou J, Wu ZQ, et al. A novel prognostic model for adult patients with hemophagocytic lymphohistiocytosis. Orphanet J Rare Dis 2020;15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang HY, Yang CF, Chiou TJ, et al. Risk of hemophagocytic lymphohistiocytosis in adults with fevers of unknown origin: the clinical utility of a new scoring system on early detection. Hematol Oncol 2017;35:835–44. [DOI] [PubMed] [Google Scholar]
- 37.Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al. Adult haemophagocytic syndrome. Lancet 2014;383:1503–16. [DOI] [PubMed] [Google Scholar]
- 38.Pringe A, Trail L, Ruperto N, et al. Macrophage activation syndrome in juvenile systemic lupus erythematosus: an under-recognized complication? Lupus 2007;16:587–92. [DOI] [PubMed] [Google Scholar]
- 39.Borgia RE, Gerstein M, Levy DM, et al. Features, treatment, and outcomes of macrophage activation syndrome in childhood-onset systemic lupus erythematosus. Arthritis Rheumatol 2018;70:616–24. [DOI] [PubMed] [Google Scholar]
- 40.Liu AC, Yang Y, Li MT, et al. Macrophage activation syndrome in systemic lupus erythematosus: a multicenter, case-control study in China. Clin Rheumatol 2018;37:93–100. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Wang D, Zhang Q, et al. The significance of pre-therapeutic F-18-FDG PET–CT in lymphoma-associated hemophagocytic lymphohistiocytosis when pathological evidence is unavailable. J Cancer Res Clin Oncol 2016;142:859–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu XJ, Wang HS, Ju XL, et al. Clinical presentation and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in China: a retrospective multicenter study. Pediatr Blood Cancer 2017;64. [DOI] [PubMed] [Google Scholar]
- 43.Yang SL, Xu XJ, Tang YM, et al. Associations between inflammatory cytokines and organ damage in pediatric patients with hemophagocytic lymphohistiocytosis. Cytokine 2016;85:14–7. [DOI] [PubMed] [Google Scholar]
- 44.Ammann S, Lehmberg K, Stadt UZ, et al. Primary and secondary hemophagocytic lymphohistiocytosis have different patterns of T-cell activation, differentiation and repertoire. Eur J Immunol 2017;47:364–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yanagisawa R, Nakazawa Y, Matsuda K, et al. Outcomes in children with hemophagocytic lymphohistiocytosis treated using HLH-2004 protocol in Japan. Int J Hematol 2019;109:206–13. [DOI] [PubMed] [Google Scholar]
- 46.My LT, Lien LB, Hsieh WC, et al. Comprehensive analyses and characterization of haemophagocytic lymphohistiocytosis in Vietnamese children. Br J Haematol 2010;148:301–10. [DOI] [PubMed] [Google Scholar]
- 47.Koh KN, Im HJ, Chung NG, et al. Clinical features, genetics, and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in Korea: report of a nationwide survey from Korea Histiocytosis Working Party. Eur J Haematol 2015;94:51–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cui Y, Zhang YC, Kang YL, et al. High-volume hemofiltration in critically ill patients with secondary hemophagocytic lymphohistiocytosis/macrophage activation syndrome: a prospective study in the PICU. Pediatr Crit Care Med 2016;17:e437–43. [DOI] [PubMed] [Google Scholar]
- 49.Bergsten E, Horne A, Aricó M, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood 2017;130:2728–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parajuli B, Angurana SK, Awasthi P, et al. Hemophagocytic lymphohistiocytosis in a PICU of a developing economy: clinical profile, intensive care needs, outcome, and predictors of mortality. Pediatr Crit Care Med 2021;22:e44–57. [DOI] [PubMed] [Google Scholar]
- 51.Veerakul G, Sanpakit K, Tanphaichitr VS, et al. Secondary hemophagocytic lymphohistiocytosis in children: an analysis of etiology and outcome. J Med Assoc Thai 2002;85 Suppl:S530–41. [PubMed] [Google Scholar]
- 52.Muthu V, Dhooria S, Sehgal IS, et al. Malaria-associated secondary haemophagocytic lymphohistiocytosis: report of two cases & a review of literature. Indian J Med Res 2017;145:399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dao D, Xoay TD, Galeano BK, et al. Etiologies and clinical outcomes of patients with secondary hemophagocytic lymphohistiocytosis at a tertiary PICU. Pediatr Crit Care Med 2019;20:e311–8. [DOI] [PubMed] [Google Scholar]
- 54.Bode SF, Ammann S, Al-Herz W, et al. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica 2015;100:978–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities [review]. Nat Rev Clin Oncol 2018;15:47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santomasso BD, Nastoupil LJ, Adkins S, et al. Management of immune-related adverse events in patients treated with chimeric antigen receptor T-cell therapy: ASCO guideline. J Clin Oncol 2021;39:3978–92. [DOI] [PubMed] [Google Scholar]
- 57.Lichtenstein DA, Schischlik F, Shao L, et al. Characterization of HLH-like manifestations as a CRS variant in patients receiving CD22 CAR T cells. Blood 2021;138:2469–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Diorio C, Shraim R, Myers R, et al. Comprehensive serum proteome profiling of cytokine release syndrome and immune effector cell–associated neurotoxicity syndrome patients with B-cell all receiving CAR t19. Clin Cancer Res 2022;28:3804–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin CI, Yu HH, Lee JH, et al. Clinical analysis of macrophage activation syndrome in pediatric patients with autoimmune diseases. Clin Rheumatol 2012;31:1223–30. [DOI] [PubMed] [Google Scholar]
- 60.Pal P, Bathia J, Giri PP, et al. Macrophage activation syndrome in pediatrics: 10 years data from an Indian center. Int J Rheum Dis 2020;23:1412–6. [DOI] [PubMed] [Google Scholar]
- 61.Buda P, Gietka P, Książyk JB, et al. The influence of various therapeutic regimens on early clinical and laboratory response and outcome of children with secondary hemophagocytic lymphohistiocytosis. Arch Med Sci 2018;14:138–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Naveen R, Jain A, Muhammed H, et al. Macrophage activation syndrome in systemic lupus erythematosus and systemic-onset juvenile idiopathic arthritis: a retrospective study of similarities and dissimilarities. Rheumatol Int 2021;41:625–31. [DOI] [PubMed] [Google Scholar]
- 63.Brito-Zerón P, Kostov B, Moral-Moral P, et al. Prognostic factors of death in 151 adults with hemophagocytic syndrome: etiopathogenically driven analysis. Mayo Clin Proc Innov Qual Outcomes 2018;2:267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bergsten E, Horne A, Myrberg IH, et al. Stem cell transplantation for children with hemophagocytic lymphohistiocytosis: results from the HLH-2004 study. Blood Adv 2020;4:3754–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lucchini G, Marsh R, Gilmour K, et al. Treatment dilemmas in asymptomatic children with primary hemophagocytic lymphohistiocytosis. Blood 2018;132:2088–96. [DOI] [PubMed] [Google Scholar]
- 66.Chinn IK, Eckstein OS, Peckham-Gregory EC, et al. Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis. Blood 2018;132:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.La Rosée P, Horne A, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood 2019;133:2465–77. [DOI] [PubMed] [Google Scholar]
- 68.Miao Y, Zhu HY, Qiao C, et al. Pathogenic gene mutations or variants identified by targeted gene sequencing in adults with hemophagocytic lymphohistiocytosis. Front Immunol 2019;10:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li F, Yang Y, Jin F, et al. Clinical characteristics and prognostic factors of adult hemophagocytic syndrome patients: a retrospective study of increasing awareness of a disease from a single-center in China. Orphanet J Rare Dis 2015;10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pan H, Huo Y, Sun L. Comparison between clinical features and prognosis of malignancy- and non-malignancy-associated pediatric hemophagocytic lymphohistiocytosis. BMC Pediatr 2019;19:468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Minoia F, Davì S, Horne A, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis Rheumatol 2014;66:3160–9. [DOI] [PubMed] [Google Scholar]
- 72.Bae CB, Jung JY, Kim HA, et al. Reactive hemophagocytic syndrome in adult-onset still disease: clinical features, predictive factors, and prognosis in 21 patients. Medicine (Baltimore) 2015;94:e451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ke Y, Lv C, Xuan W, et al. Clinical analysis of macrophage activation syndrome in adult rheumatic disease: a multicenter retrospective study. Int J Rheum Dis 2020;23:1488–96. [DOI] [PubMed] [Google Scholar]
- 74.Hines MR, Greenwood TV, Beutel G, et al. Consensus-based guidelines for the recognition, diagnosis, and management of hemophagocytic lymphohistiocytosis in critically ill children and adults. Crit Care Med 2022;50:860–72. [DOI] [PubMed] [Google Scholar]
- 75.Mannion ML, Cron RQ. Intensive care requirement, rather than degree of serum ferritin elevation, predicts mortality in macrophage activation syndrome. Pediatr Crit Care Med 2012;13:616. [DOI] [PubMed] [Google Scholar]
- 76.Horne A, Trottestam H, Aricò M, et al. Frequency and spectrum of central nervous system involvement in 193 children with haemophagocytic lymphohistiocytosis. Br J Haematol 2008;140:327–35. [DOI] [PubMed] [Google Scholar]
- 77.Trottestam H, Horne A, Aricò M, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood 2011;118:4577–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim MM, Yum MS, Choi HW, et al. Central nervous system (CNS) involvement is a critical prognostic factor for hemophagocytic lymphohistiocytosis. Korean J Hematol 2012;47:273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhao YZ, Zhang Q, Li ZG, et al. Central nervous system involvement in 179 Chinese children with hemophagocytic lymphohistiocytosis. Chin Med J (Engl) 2018;131:1786–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Song Y, Pei RJ, Wang YN, et al. Central nervous system involvement in hemophagocytic lymphohistiocytosis in adults: a retrospective analysis of 96 patients in a single center. Chin Med J (Engl) 2018;131:776–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Deiva K, Mahlaoui N, Beaudonnet F, et al. CNS involvement at the onset of primary hemophagocytic lymphohistiocytosis. Neurology 2012;78:1150–6. [DOI] [PubMed] [Google Scholar]
- 82.Yang S, Zhang L, Jia C, et al. Frequency and development of CNS involvement in Chinese children with hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2010;54:408–15. [DOI] [PubMed] [Google Scholar]
- 83.Xue H, Chen C, Li W, et al. Analysis of prognostic risk factors in children with Epstein-Barr virus-associated hemophagocytic syndrome. Minerva Pediatr 2015;67:251–61. [PubMed] [Google Scholar]
- 84.Cai G, Wang Y, Liu X, et al. Central nervous system involvement in adults with haemophagocytic lymphohistiocytosis: a single-center study. Ann Hematol 2017;96:1279–85. [DOI] [PubMed] [Google Scholar]
- 85.Benson LA, Li H, Henderson LA, et al. Pediatric CNS-isolated hemophagocytic lymphohistiocytosis. Neurol Neuroimmunol Neuroinflamm 2019;6:e560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Birndt S, Schenk T, Heinevetter B, et al. Hemophagocytic lymphohistiocytosis in adults: collaborative analysis of 137 cases of a nationwide German registry. J Cancer Res Clin Oncol 2020;146:1065–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weiss SL, Peters MJ, Alhazzani W, et al. Surviving sepsis campaign international guidelines for the management of septic shock and sepsis-associated organ dysfunction in children. Intensive Care Med 2020;46:10–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Evans L, Rhodes A, Alhazzani W, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock 2021. Crit Care Med 2021;49:e1063–143. [DOI] [PubMed] [Google Scholar]
- 89.Otrock ZK, Eby CS. Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol 2015;90:220–4. [DOI] [PubMed] [Google Scholar]
- 90.Shakoory B, Carcillo JA, Chatham WW, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med 2016;44:275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant 2019;25:625–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ehl S, Astigarraga I, Greenwood TV, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract 2018;6:1508–17. [DOI] [PubMed] [Google Scholar]
- 93.Lehmberg K, Nichols KE, Henter JI, et al. Consensus recommendations for the diagnosis and management of hemophagocytic lymphohistiocytosis associated with malignancies. Haematologica 2015;100:997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jordan MB, Allen CE, Greenberg J, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer 2019;66:e27929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 2010;363:221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fisler RE, Liang MG, Fuhlbrigge RC, et al. Aggressive management of juvenile dermatomyositis results in improved outcome and decreased incidence of calcinosis. J Am Acad Dermatol 2002;47: 505–11. [DOI] [PubMed] [Google Scholar]
- 97.Akaishi T, Takeshita T, Himori N, et al. Rapid administration of high-dose intravenous methylprednisolone improves visual outcomes after optic neuritis in patients with AQP4-IGG-positive NMOSD. Front Neurol 2020;11:932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Borenstein SH, Gerstle T, Malkin D, et al. The effects of prebiopsy corticosteroid treatment on the diagnosis of mediastinal lymphoma. J Pediatr Surg 2000;35:973–6. [DOI] [PubMed] [Google Scholar]
- 99.McGee S, Hirschmann J. Use of corticosteroids in treating infectious diseases. Arch Intern Med 2008;168:1034–46. [DOI] [PubMed] [Google Scholar]
- 100.Group RC, Horby P, Lim WS. Dexamethasone in hospitalized patients with Covid-19. N Engl J Med 2021;384:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fisher CJ, Dhainaut JF, Opal SM, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome: results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA 1994;271:1836–43. [PubMed] [Google Scholar]
- 102.Eloseily EM, Weiser P, Crayne CB, et al. Benefit of anakinra in treating pediatric secondary hemophagocytic lymphohistiocytosis. Arthritis Rheumatol 2020;72:326–34. [DOI] [PubMed] [Google Scholar]
- 103.Balamuth NJ, Nichols KE, Paessler M, et al. Use of rituximab in conjunction with immunosuppressive chemotherapy as a novel therapy for Epstein Barr virus-associated hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol 2007;29:569–73. [DOI] [PubMed] [Google Scholar]
- 104.Chellapandian D, Das R, Zelley K, et al. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol 2013;162:376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Horne A, Greenwood TV, Chiang SC, et al. Efficacy of moderately dosed etoposide in macrophage activation syndrome–hemophagocytic lymphohistiocytosis. J Rheumatol 2021;48:1596–602. [DOI] [PubMed] [Google Scholar]
- 106.Hot A, Toh ML, Coppéré B, et al. Reactive hemophagocytic syndrome in adult-onset still disease: clinical features and long-term outcome: a case-control study of 8 patients. Medicine (Baltimore) 2010;89:37–46. [DOI] [PubMed] [Google Scholar]
- 107.Ahn SS, Yoo BW, Jung SM, et al. Application of the 2016 EULAR/-ACR/PRINTO classification criteria for macrophage activation syndrome in patients with adult-onset still disease. J Rheumatol 2017;44:996–1003. [DOI] [PubMed] [Google Scholar]
- 108.Wang Y, Wang Z, Wu L, et al. Recombinant human thrombopoietin is an effective treatment for thrombocytopenia in hemophagocytic lymphohistiocytosis. Ann Hematol 2013;92:1695–9. [DOI] [PubMed] [Google Scholar]
- 109.Daver N, McClain K, Allen CE, et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer 2017;123:3229–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Locatelli F, Jordan MB, Allen C, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med 2020;382:1811–22. [DOI] [PubMed] [Google Scholar]
- 111.Boonstra PS, Ahmed A, Merrill SA, et al. Ruxolitinib in adult patients with secondary hemophagocytic lymphohistiocytosis. Am J Hematol 2021;96:E103–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang Q, Zhao YZ, Ma HH, et al. A study of ruxolitinib response–based stratified treatment for pediatric hemophagocytic lymphohistiocytosis. Blood 2022;139:3493–504. [DOI] [PubMed] [Google Scholar]
- 113.Zhou L, Liu Y, Wen Z, et al. Ruxolitinib combined with doxorubicin, etoposide, and dexamethasone for the treatment of the lymphoma-associated hemophagocytic syndrome. J Cancer Res Clin Oncol 2020;146:3063–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhou J, Zhou J, Wu ZQ, et al. Ferritin index is a strong prognostic marker in adult hemophagocytic lymphohistiocytosis. Int J Clin Pract 2021;75:e13704. [DOI] [PubMed] [Google Scholar]
- 115.Lachmann G, Knaak C, Vorderwülbecke G, et al. Hyperferritinemia in critically ill patients. Crit Care Med 2020;48:459–65. [DOI] [PubMed] [Google Scholar]
- 116.Lee JY, Kim JH, Lee JS, et al. Initial characteristics and clinical severity of hemophagocytic lymphohistiocytosis in pediatric patients admitted in the emergency department. Pediatr Emerg Care 2021;37:204–7. [DOI] [PubMed] [Google Scholar]
- 117.Kyriazopoulou E, Leventogiannis K, Norrby-Teglund A, et al. Macrophage activation-like syndrome: an immunological entity associated with rapid progression to death in sepsis. BMC Med 2017;15:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Naymagon L, Tremblay D, Mascarenhas J. Reevaluating the role of ferritin in the diagnosis of adult secondary hemophagocytic lymphohistiocytosis. Eur J Haematol 2020;104:344–51. [DOI] [PubMed] [Google Scholar]
- 119.Halyabar O, Chang MH, Schoettler ML, et al. Calm in the midst of cytokine storm: a collaborative approach to the diagnosis and treatment of hemophagocytic lymphohistiocytosis and macrophage activation syndrome. Pediatr Rheumatol Online J 2019;17:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Patel S, Bibi A, Eisenberg R, et al. The role of early subspeciality consultation in the timing of hemophagocytic lymphohistiocytosis diagnosis and management. J Clin Rheumatol 2022;28:e462–6. [DOI] [PubMed] [Google Scholar]
- 121.Dimmock D, Caylor S, Waldman B, et al. Project Baby Bear: rapid precision care incorporating rWGS in 5 California children’s hospitals demonstrates improved clinical outcomes and reduced costs of care. Am J Hum Genet 2021;108:1231–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ravelli A, Grom AA, Behrens EM, et al. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun 2012;13:289–98. [DOI] [PubMed] [Google Scholar]
- 123.Jia J, Song Y, Lin N, et al. Clinical features and survival of extranodal natural killer/T cell lymphoma with and without hemophagocytic syndrome. Ann Hematol 2016;95:2023–31. [DOI] [PubMed] [Google Scholar]
- 124.Takada H, Ohga S, Mizuno Y, et al. Oversecretion of IL-18 in haemophagocytic lymphohistiocytosis: a novel marker of disease activity. Br J Haematol 1999;106:182–9. [DOI] [PubMed] [Google Scholar]
- 125.Popko K, Górska E, Wołowiec M, et al. Disturbances in NK cells in various types of hemophagocytic lymphohistiocytosis in a population of Polish children. J Pediatr Hematol Oncol 2019;41:e277–83. [DOI] [PubMed] [Google Scholar]
- 126.Imashuku S, Kuriyama K, Sakai R, et al. Treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis (EBV-HLH) in young adults: a report from the HLH study center. Med Pediatr Oncol 2003;41:103–9. [DOI] [PubMed] [Google Scholar]
- 127.Hu Y, Xu J, Wang L, et al. Treatment of hemophagocytic lymphohistiocytosis with cyclophosphamide, vincristine, and prednisone. Swiss Med Wkly 2012;142:w13512. [DOI] [PubMed] [Google Scholar]
- 128.Song Y, Wang Y, Wang Z. Requirement for etoposide in the initial treatment of Epstein-Barr virus-associated haemophagocytic lymphohistiocytosis. Br J Haematol 2019;186:717–23. [DOI] [PubMed] [Google Scholar]
- 129.Song Y, Wang Z, Hao Z, et al. Requirement for etoposide in the treatment of pregnancy related hemophagocytic lymphohistiocytosis: a multicenter retrospective study. Orphanet J Rare Dis 2019;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jovanovic A, Kuzmanovic M, Kravljanac R, et al. Central nervous system involvement in hemophagocytic lymphohistiocytosis: a single-center experience. Pediatr Neurol 2014;50:233–7. [DOI] [PubMed] [Google Scholar]
- 131.Cabler SS, Hogan PG, Fritz SA, et al. Incidence and treatment of hemophagocytic lymphohistiocytosis in hospitalized children with Ehrlichia infection. Pediatr Blood Cancer 2020;67:e28436. [DOI] [PubMed] [Google Scholar]
- 132.Keenan C, Nichols KE, Albeituni S. Use of the JAK inhibitor ruxolitinib in the treatment of hemophagocytic lymphohistiocytosis. Front Immunol 2021;12:614704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.