

Abstract
The latest Ebola virus (EBOV) epidemic spread rapidly through Guinea, Sierra Leone, and Liberia, creating a global public health crisis and accelerating the assessment of experimental therapeutics and vaccines in clinical trials. One of those vaccines is based on recombinant vesicular stomatitis virus expressing the EBOV glycoprotein (VSV-EBOV), a live-attenuated vector with marked preclinical efficacy. Here, we provide the preclinical proof that VSV-EBOV completely protects macaques against lethal challenge with the West African EBOV-Makona strain. Complete and partial protection was achieved with a single dose given as late as 7 and 3 days before challenge, respectively. This indicates that VSV-EBOV may protect humans against EBOV infections in West Africa with relatively short time to immunity, promoting its use for immediate public health responses.
The largest documented outbreak of Ebola hemorrhagic fever (EHF) caused by the Ebola virus (EBOV) started in rural Guinea in late 2013 and spread regionally, predominantly affecting Guinea, Sierra Leone, and Liberia (1, 2). This global health crisis to date accounts for nearly 27,200 cases and more than 11,100 deaths, with a high infection rate among health care workers (2). The global scale of this outbreak is further demonstrated by the impact on public health systems in multiple countries that had to respond to introduction of cases, including evacuations of exposed and/or infected aid workers (2). In addition, the inability to recruit timely and adequate medical, logistic, and financial support became a serious problem. Thus, this outbreak emphasizes the urgent need to develop therapeutics and vaccines for filoviruses to avert future outbreaks, and several promising experimental approaches have been accelerated for clinical trials (3). One of the most promising experimental vaccines is based on the recombinant vesicular stomatitis virus platform expressing the EBOV glycoprotein (GP) (previously referred to as rVSV-ZEBOV, here designated VSV-EBOV) as an immunogen. VSV-EBOV is a live-attenuated vaccine vector that has demonstrated an adequate preclinical safety profile and shown marked efficacy in pre- and postexposure vaccination in rodent and macaque models (4, 5). The mechanism of protection for prophylactic vaccination is thought to be largely antibody-mediated (6).
VSV-EBOV was recently evaluated in phase 1 clinical trials in humans at several worldwide locations. Despite reports of arthritis in about 20% of volunteers at a single study site that used the highest vaccine dose, the vaccine was found in general to be safe and immunogenic, warranting further evaluation for efficacy (7, 8). Unexpectedly, preclinical efficacy data for this or any other EBOV vaccine currently in phase 1 clinical trials against the current West African EBOV-Makona strain is lacking. Because genetic divergence among known EBOV strains is similar, with about 3% whole-genome divergence (1), one may assume that VSV-EBOV will protect against all known EBOV strains. However, analysis of sequence data from May and June 2014 in Sierra Leone suggested a higher mutation rate of EBOV in this ongoing outbreak (9), raising concerns about the applicability of current experimental intervention strategies, including VSV-EBOV, against the emerged EBOV-Makona strain. Morerecent sequence data from Mali (October and November 2014) indicated a mutation rate of EBOV in West Africa similar to observations in past outbreaks (10), lessening those concerns. However, to address this important question, we performed efficacy testing of good manufacturing practice (GMP)–grade VSV-EBOV in a recently established cynomolgus macaque challenge model for the West African EBOV-Makona strain (11). In parallel, we investigated the time to protective immunity after a single high-dose VSV-EBOV vaccination against challenge with EBOV-Makona. All infectious animal work was performed in the maximum containment laboratory at the Rocky Mountain Laboratories, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, USA, applying standard operating protocols approved by the Institutional Biosafety Committee (12).
Fifteen cynomolgus macaques were randomly assigned to groups of two or three animals and immunized with a single intramuscular injection of 5 × 107 plaque-forming units (PFU) of GMP-grade VSV-EBOV at 28, 21, 14, 7, or 3 days before challenge. This vaccine dose was chosen to be equivalent to the highest immunization dose evaluated in the recent phase 1 clinical trials (7). The control group (three animals) was immunized 28 days before challenge by the same route and dose with the VSV-Marburg virus vaccine (VSV-MARV), previously shown not to protect against EBOV challenge (5, 6). Daily monitoring, including weekly blood sampling and clinical examinations of the animals, revealed normal behavior and no detectable adverse effects after vaccination. Challenge was performed with an intramuscular injection into the opposite leg of 1000 PFU of EBOV-Makona, established as a lethal dose (11). Health monitoring and clinical examinations were intensified and continued until the study ended 42 days after challenge or until euthanasia was required owing to the severity of disease. Humane end-point criteria, specified and approved by the Institutional Animal Care and Use Committee, were applied to determine when animals should be humanely euthanized.
The VSV-MARV–vaccinated control animals and one animal in the day-3 vaccination group [non-human primate (NHP) d-3 number 1] developed classical severe EHF with macular cutaneous rash, thrombocytopenia (Fig. 1A), increased liver enzyme levels (Fig. 1, B to D), and viremia (Fig. 1E); these animals had to be euthanized on days 5, 7, 7, and 8 after challenge (Fig. 1F). Another animal in the day-3 vaccination group (NHP d-3 number 2) developed moderate signs of EHF, including viremia (Fig. 1E) and mild rash, but did not reach the clinical score for humane end-point euthanasia, cleared the virus by day 9 (Fig. 1E), and survived. The third animal in this group developed only very mild signs of disease without viremia or rash and recovered quickly. All nine remaining animals in the day-28, −21, −14, and −7 vaccination groups did not develop any clinical signs of disease, showed no EBOV viremia, and did not have abnormal changes in hematology or liver enzymes, indicating complete protection against lethal EBOV-Makona challenge (Fig. 1).
Fig. 1. Clinical parameters of infected NHPs.
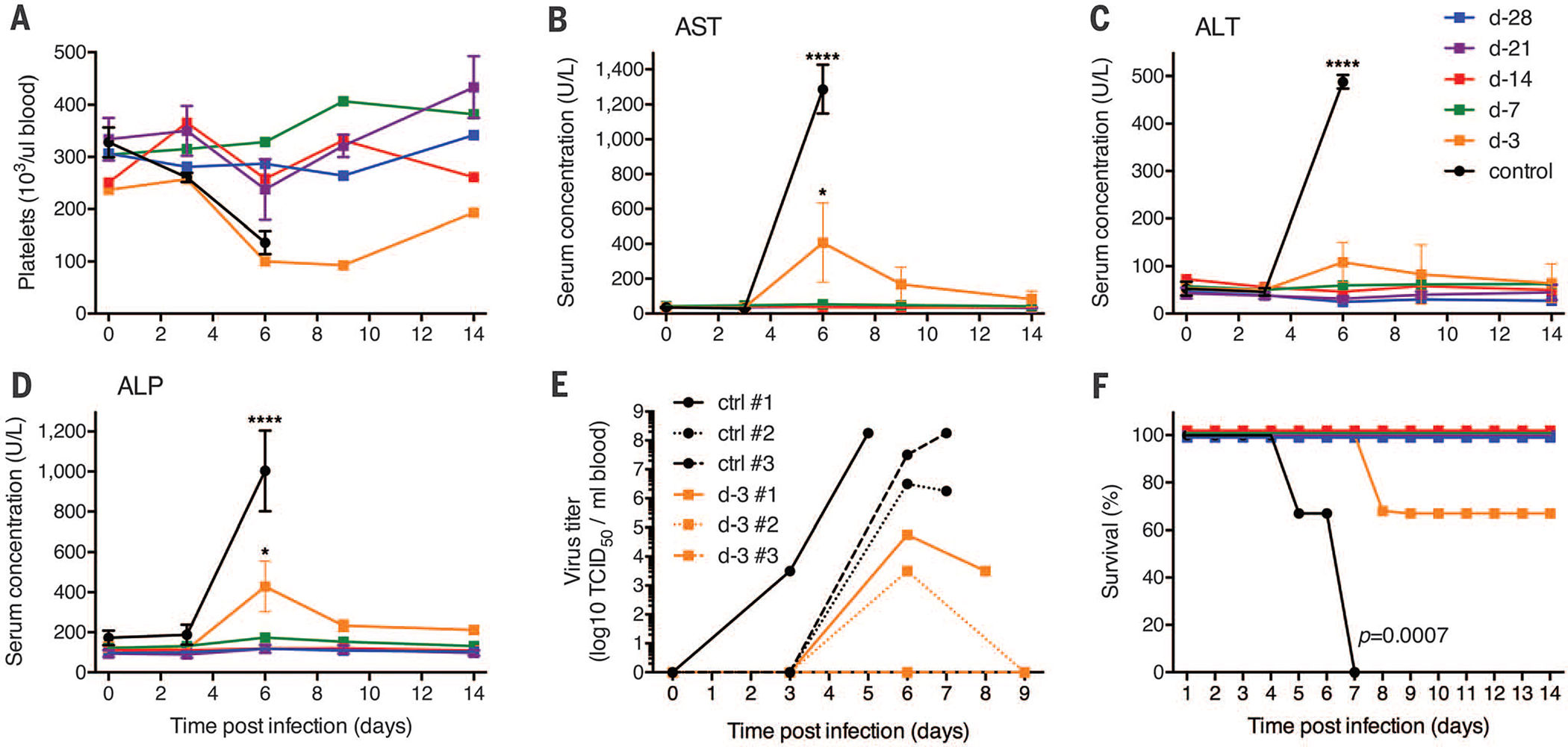
(A) Platelet counts of infected NHPs in EDTA blood at every examination day. Concentrations of aspartate aminotransferase (AST) (B), alanine transaminase (ALT) (C), and alkaline phosphatase (ALP) (D) were determined in serum samples collected on examination days (d) after EBOV-Makona challenge. U, units. Two-way analysis of variance (ANOVA) with Tukey multiple comparison post test was used to determine statistical significance at the level of 0.05 (*) and 0.0001 (****) for data presented in (B) to (D). Error bars indicate SD. (E) Viremia (titers) for individual animals on examination days and time of euthanasia. TCID50, median tissure culture infectious dose. (F) Survival curves showing outcome of the different vaccine groups after challenge. Statistical significance was assessed by using the Kaplan-Meier method.
Previous work had established antibody responses as the main mechanism of protection for prophylactic use of VSV-EBOV in macaques (6). The role of cellular immune responses for protection with this vaccine has never been clearly established, and CD4+ and CD8+ T cell responses are nearly undetectable during immunization and at time of challenge (4, 6). Therefore, we focused on antibody responses in the vaccinated animals. At the time of challenge, animals in the day-28, −21, and −14 vaccination groups showed potent EBOV-GP–specific responses with immunoglobulin (IgG) titers >104 (range of 51,200 to 102,400) (Fig. 2A and fig. S1A). EBOV-GP–specific antibody titers >104 were also associated with protection during testing of the adenovirus 5 vaccine (rAd5-EBOV) platform (13, 14), as well as for a second-generation VSV-based vector not yet in phase 1 clinical trials (15). In contrast, titers of animals in the day-7 and −3 vaccination groups approximated background level at the time of challenge (Fig. 2A and fig. S1A). As expected, the VSV-MARV–vaccinated control animals did not develop any EBOV-GP–specific IgG responses (Fig. 2A). In vitro titers of neutralizing antibody after intramuscular VSV-EBOV immunization have previously been shown to reach relatively low levels before challenge (5, 6, 16). In this study, only the day-28, day-21, and day-14 vaccinated animals had neutralizing antibodies at the time of challenge, with similar titers against three distinct EBOV strains: EBOV-Makona, the challenge virus (Fig. 2B, bar graph); EBOV-Kikwit, the source of the vaccine immunogen (Fig. 2C, bar graph); and EBOV-Mayinga, the prototype of the species (Fig. 2D, bar graph). For the day-28 group, the neutralizing activity of sera was similar to that reported previously (5, 6, 16). However, increased neutralizing antibody titers were measured in serum of all surviving animals 42 days after EBOV-Makona infection (Fig. 2, B to D), a phenomenon that has been reported previously in response to this vaccine (6). There was no significant difference in the neutralizing activity of the day-0 and −42 serum samples against the three distinct EBOV strains (EBOV-Makona, EBOV-Kikwit, and EBOV-Mayinga) (Fig. 2, B to D). These results indicate that the VSV-EBOV vaccine will be protective with comparable efficacy against different EBOV strains, including the currently circulating West African strain.
Fig. 2. Humoral immune responses of NHPs.
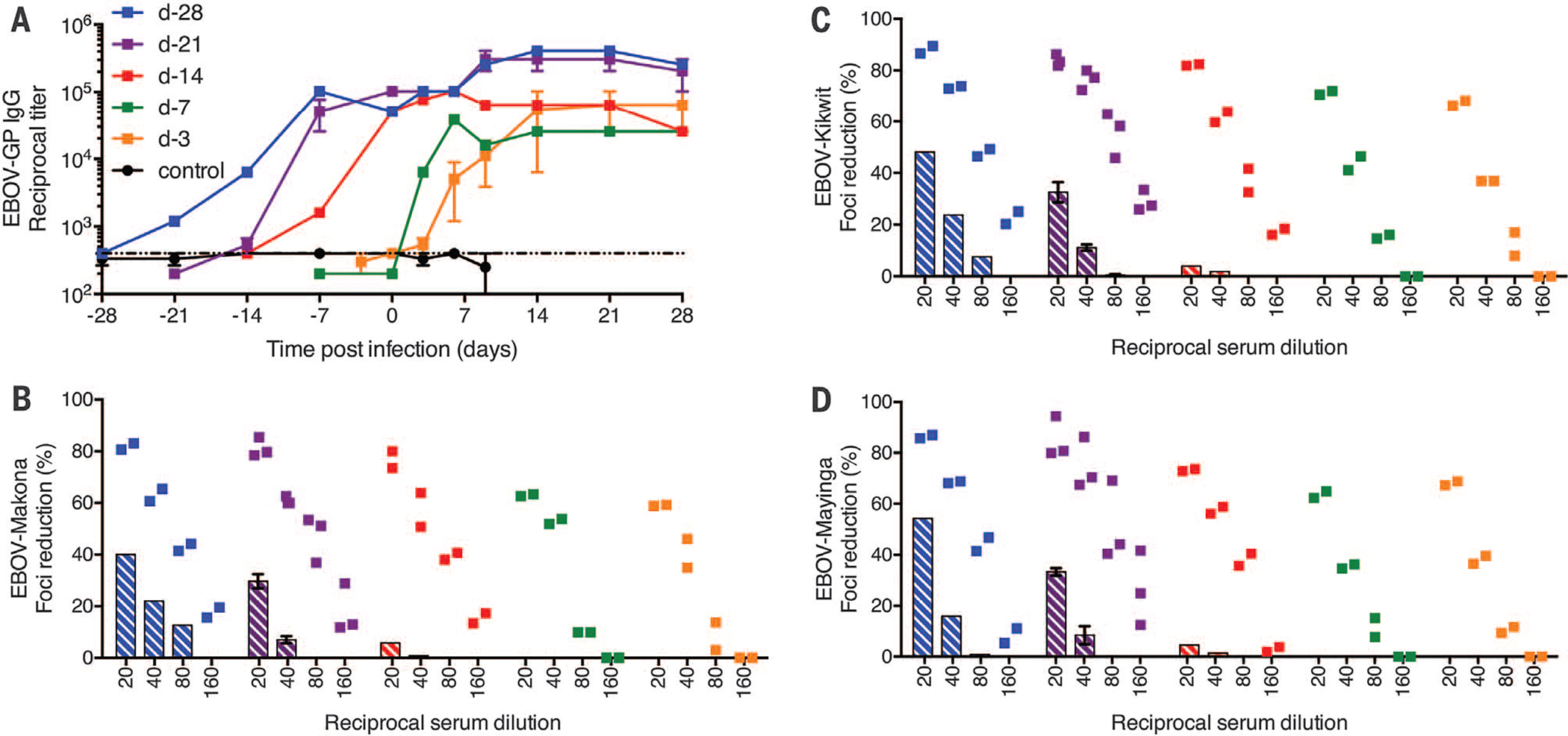
EBOV-GP–specific IgG titers were determined in the serum of animals at examination days. (A) Summary of EBOV-GP-IgG titers of all groups. The dotted line marks the enzyme-linked immunosorbent assay (ELISA) cut-off based on titers obtained from negative control animals. Neutralizing antibody titers were determined by using the focus reduction assay against three distinct EBOV strains: (B) EBOV-Makona, the challenge strain; (C) EBOV-Kikwit, the source of the vaccine immunogen; and (D) EBOV-Mayinga, the prototype strain of the species. Data are presented as percent foci reduction in relation to a negative control serum. Bar graphs (average of animals per group) represent the neutralizing activity on day 0 (day of challenge); individual animal serum-neutralizing activity is shown for day 42 after challenge.
We also measured systemic levels of various cytokines, chemokines, and other soluble factors in the serum of infected animals. An aberrant cytokine and chemokine response is considered a hallmark of severe or lethal EHF in the macaque model and in humans (11, 17). Consistent with these reports, all VSV-MARV–vaccinated control animals showed marked increases of interleukin-1β (IL-1β), IL-6, IL-15, interferon-γ (IFN-γ), IL-10, and monocyte chemotactic protein (MCP)-1 after EBOV-Makona challenge, indicative of a cytokine storm (Fig. 3). In contrast, VSV-EBOV–vaccinated animals (day-28, −21, −14, and −7 groups) showed low to undetectable levels of these cytokines and chemokines, reflecting effective control of EBOV replication (Fig. 3). The animals in the day-3 vaccination group displayed intermediate levels of these cytokines and chemokines on days 3, 6, and 9 after challenge (Fig. 3). The one animal that succumbed to challenge in this group showed similar cytokine or chemokine levels as the control animals, whereas both animals that survived in the day-3 group converted on day 14 after challenge to levels of cytokines and chemokines seen in all other vaccinated and protected animals (Fig. 3).
Fig. 3. Serum cytokine and chemokine levels of NHPs.
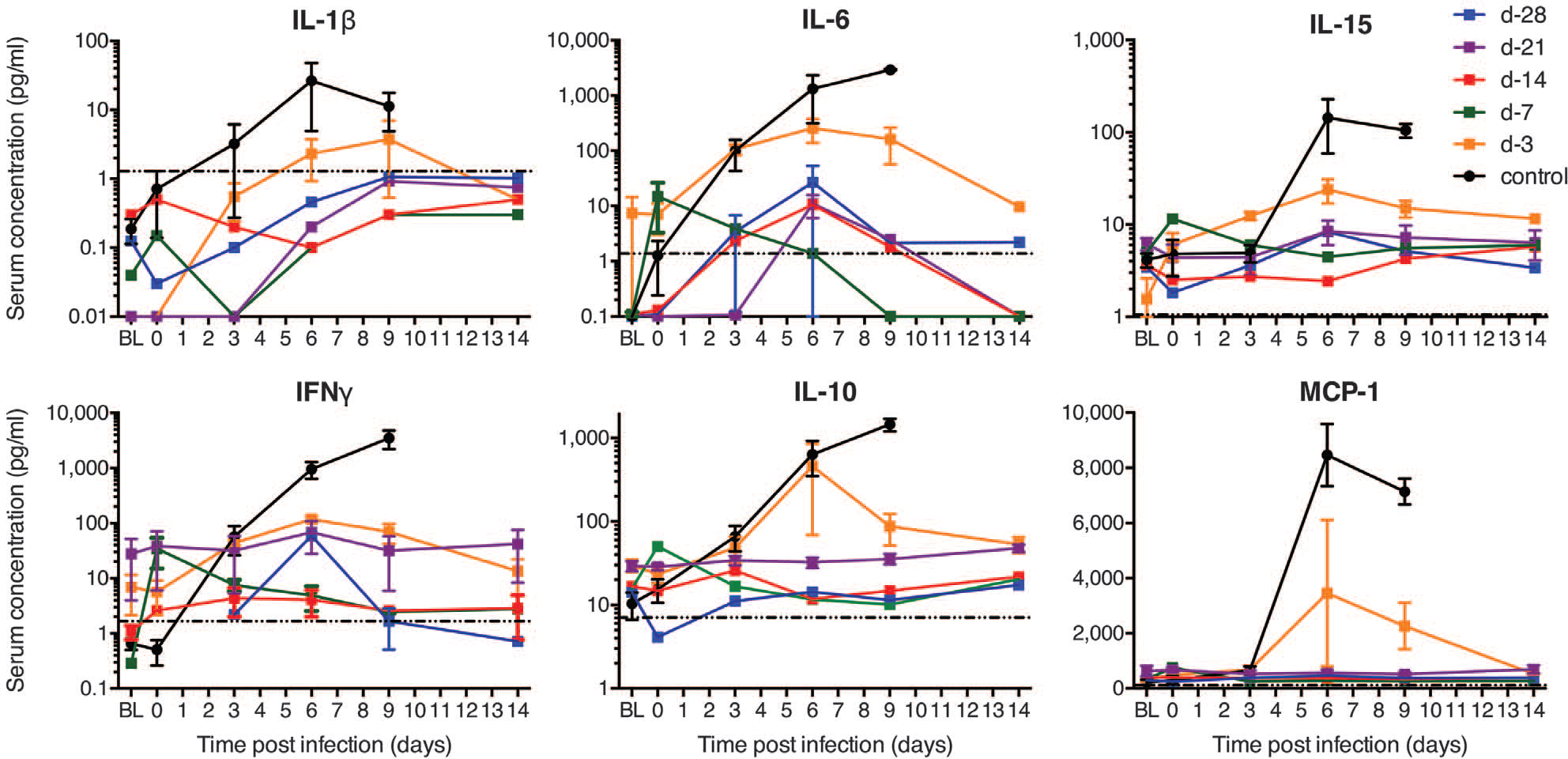
Kinetics of IL-1β, IL-6, IL-15, IFN-γ, IL-10, and MCP-1 were analyzed in serum samples of each animal collected on examination days and time of euthanasia. The dotted line in each panel marks the assay detection limit. Terminal values for the control animal euthanized on day 5 were added to the day-6 time point in this group; similarly, the day-9 time point in the control and day-3 vaccination groups represents all values, including terminal samples obtained from NHPs euthanized on days 7 and 8. BL indicates baseline (before vaccination).
Protection of animals vaccinated 7 and 3 days before challenge occurred despite the lack of detectable EBOV-GP–specific IgG at time of challenge (Fig. 2A). We therefore tested the sera of the VSV-MARV–vaccinated control and the day-7 and day-3 vaccinated animals for EBOV-GP–specific IgM responses. Vaccine-specific IgM antibodies at the time of challenge were undetectable for the day-3 vaccinated animals and weakly positive for the day-7 vaccinated animals (fig. S1B). EBOV-GP–specific IgM responses developed in both vaccine groups within a week after vaccination and increased in titer over time (fig. S1B). This observation, combined with the elevated cytokine and chemokine profiles, prompted us to analyze further early innate responses to better understand the mechanism by which these animals, particularly the day-3 vaccinated NHPs, were protected. VSV is a potent inducer of type I IFN responses (18, 19), and transient, low-level VSV viremia is known to be associated with VSV-EBOV vaccination in NHPs and humans (5, 7, 8, 20, 21). With the exception of the day-3 vaccinated animals that had VSV-specific RNA levels of 1 to 6 PFU equivalents, all vaccinated NHPs had cleared VSV at the time of challenge (day 0). The day-3 vaccinated animals expressed higher levels of IFN-α at both day 0 and day 3 after challenge compared with the other groups (Fig. 4). Elevated IFN-α was associated with increased IL-15 and IFN-γ at day 3 after challenge (Fig. 4). Day-7 vaccinated animals also showed elevated levels of IFN-α, IL-15, and IFN-γ at time of challenge that diminished by day 3 (Fig. 4). This cytokine signature is highly suggestive of natural killer (NK) cell activation, which requires type I IFN and IL-15 production by activated macrophages, resulting in high levels of IFN-γ expression by NK cells and direct killing of virus-infected cells. More comprehensive comparison of serum cytokines in all vaccinated groups revealed up-regulation of other potentially macrophage-derived cytokines (IL-6, transforming growth factor–α, and MCP-1) in day-3 vaccinated animals by 3 days after challenge (fig. S2). These findings suggest that VSV vaccination induced activation of macrophages and NK cells, the latter of which has previously been implicated in survival from EBOV infections (22, 23). Although EBOV-VP35 and -VP24 proteins have potent IFN antagonist activity (24, 25), induction of innate immunity by VSV vaccination likely establishes an early antiviral state in the host that limits EBOV replication upon challenge (Fig. 1E, day 6). However, it is unlikely that innate immune responses alone can protect from lethal EBOV infection, because VSV-MARV–vaccinated control animals also showed elevated levels of IFN-γ on day 3 after challenge, likely triggered by EBOV-Makona infection, and were not protected (Fig. 4). In contrast to these control animals, the day-3 immunized animals had measurable EBOV-GP–specific IgM and IgG antibodies by days 3 and 6 after challenge, respectively, leading to protection (Fig. 2A and fig. S1). Thus, innate immune activation by VSV-EBOV may provide a window of protection that limits virus replication in the critical period needed for the development of specific adaptive responses, most importantly antibodies. Future studies will specifically compare naïve versus control vaccinated versus VSV-EBOV–vaccinated animals, including detailed analysis of innate and adaptive immune responses.
Fig. 4. Serum cytokine levels early after challenge.
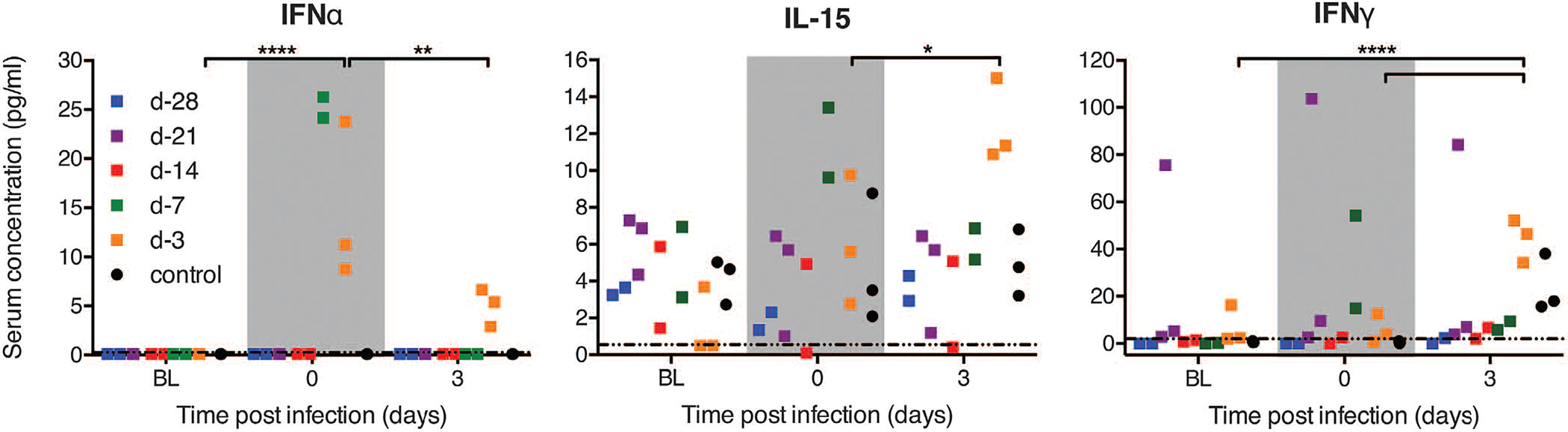
Serum concentrations for IFN-α, IL-15, and IFN-γ were determined before vaccination (baseline, BL), on day of challenge (gray region, day 0), and early after EBOV-Makona challenge (day 3). Each square represents an individual animal at the indicated time point. The dotted line in each panel marks the assay detection limit. Two-way ANOVA with Tukey multiple comparison post test was used to determine statistical significance at the level of 0.05 (*), 0.01 (**), and 0.0001 (****) for the day-3 vaccination group.
This study provides the preclinical efficacy data for GMP-grade VSV-EBOV in the gold standard macaque disease model for EHF and thus fills a gap in our understanding of appropriate responses to West African EHF. Vaccination with a single high dose of GMP-grade VSV-EBOV (previously rVSV-ZEBOV), the same preparation that was recently evaluated in phase 1 clinical trials and is currently being administered in vaccine trials in West Africa, did not result in any adverse effects in macaques, which largely is in line with reports of minor adverse effects in human volunteers after VSV-EBOV vaccination at phase 1 clinical study sites in Europe, Africa, and the Americas (7, 8). Total EBOV-GP–specific IgG titers >104 seem to be a correlate of protection for prophylactic use of VSV-EBOV (6), similar to the rAd5-EBOV (13, 14) and a second-generation rVSV/ZEBOV (15) vaccine platform. However, the time to immunity for VSV-EBOV is short, providing partial protection against lethal disease even when administered 3 days before challenge. The initial mechanism here appears to be a strong innate immune response to VSV-EBOV vaccination that may include macrophage activation and NK cell–mediated control of EBOV replication before specific adaptive responses develop. Thus, our data warrant the continuation of safety and immunogenicity trials with GMP-grade VSV-EBOV in humans and the continuation of the recently initiated clinical trials in West Africa (26), in particular the ring vaccination trial in Guinea (27).
Supplementary Material
ACKNOWLEDGMENTS
We thank the Rocky Mountain Veterinary Branch [NIH, National Institute of Allergy and Infectious Diseases (NIAID)] for its support of this study. This work was jointly funded by the Division of Intramural Research, NIAID, NIH; the Public Health Agency of Canada; and the Center for Security Science Program, Canada. Use of the GMP-produced vaccine material is regulated through the Public Health Agency of Canada. The authors declare no competing financial interests. H.F. is a co-applicant on two patents regarding VSV-EBOV (U.S. patent application nos. PCT/CA03/01125US and 61/014,626). The data are available via figshare: http://dx.doi.org/10.6084/m9.figshare.1456239.
REFERENCES AND NOTES
- 1.Baize S et al. , N. Engl. J. Med. 371, 1418–1425 (2014). [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization (WHO), “Ebola situation report, 3 June 2015,” http://apps.who.int/ebola/ebola-situation-reports (2015). [Google Scholar]
- 3.U.S. National Institutes of Health, www.ClinicalTrials.gov (2015). [PubMed]
- 4.Geisbert TW, Feldmann H, J. Infect. Dis. 204 (suppl. 3), S1075–S1081 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones SM et al. , Nat. Med. 11, 786–790 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Marzi A et al. , Proc. Natl. Acad. Sci. U.S.A. 110, 1893–1898 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agnandji ST et al. , N. Engl. J. Med. 10.1056/NEJMoa1502924 (2015). [DOI] [Google Scholar]
- 8.Regules JA et al. , N. Engl. J. Med. 10.1056/NEJMoa1414216 (2015). [DOI] [Google Scholar]
- 9.Gire SK et al. , Science 345, 1369–1372 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoenen T et al. , Science 348, 117–119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marzi A et al. , Emerg. Infect. Dis. 10.3201/eid2110.150259 (2015). [DOI] [Google Scholar]
- 12.See supplementary materials on Science Online.
- 13.Sullivan NJ et al. , Nature 408, 605–609 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Sullivan NJ et al. , PLOS Med. 3, e177 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mire CE et al. , Nature 520, 688–691 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiu X et al. , PLOS ONE 4, e5547 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feldmann H, Geisbert TW, Lancet 377, 849–862 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts A et al. , J. Virol. 72, 4704–4711 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts A et al. , J. Virol. 73, 3723–3732 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai L et al. , JAMA 313, 1249–1255 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Günther S et al. , J. Infect. Dis. 204 (suppl. 3), S785–S790 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Feldmann H et al. , PLOS Pathog. 3, e2 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warfield KL et al. , J. Exp. Med. 200, 169–179 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basler CF et al. , Proc. Natl. Acad. Sci. U.S.A. 97, 12289–12294 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mateo M et al. , J. Virol. 84, 1169–1175 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.U.S. National Institutes of Health, www.clinicaltrials.gov/ct2/show/NCT02344407?term=NCT02344407&rank=1 and www.clinicaltrials.gov/ct2/show/NCT02378753?term=NCT02378753&rank=1 (2015). [PubMed]
- 27.WHO, “Ebola vaccine efficacy trial to launch in Guinea,” www.who.int/mediacentre/news/releases/2015/ebola-vaccine-trial/en/ (2015).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.