

Abstract

The cross-linked nature of vulcanized rubbers as used in tire and many other applications prohibits an effective closed-loop mechanical or chemical recycling. Moreover, vulcanization significantly retards the material’s biodegradation. Here, we report a recyclable and biodegradable rubber that is generated by the vulcanization of amorphous, unsaturated polyesters. The elastic material can be broken down via solvolysis into the underlying monomers. After removal of the vulcanized repeat units, the saturated monomers, constituting the major share of the material, can be recovered in overall recycling rates exceeding 90%. Respirometric biodegradation experiments by 13CO2 tracking under environmental conditions via the polyesters’ diol monomer indicated depolymerization and partial mineralization of the vulcanized polyester rubbers.
Keywords: vulcanizates, recycling, biodegradation, elastomers, sustainable materials
Short abstract
Polyesters from saturated and unsaturated monomers can be vulcanized to rubbers and chemically recycled via solvolysis to their monomers.
Introduction
Rubbers, or vulcanizates, are among the polymeric materials with the largest production volume, with an annual output of both natural and synthetic rubber exceeding 29 million metric tons.1 Their performance and ease of production render them well-suited for application in, e.g., tires, seals, and gloves.2−4 The excellent elastomeric properties and durability of rubbers arise from the vulcanization process, which creates a covalent network between the polymer chains through sulfur bonds. However, this stability, while advantageous for material properties, hampers recycling and decelerates their biodegradation.5,6
Due to their cross-linked nature, rubbers are intractable and insoluble, rendering them inherently unamenable to recycling through conventional methods involving material flow.6 Consequently, a large share of the one billion waste tires exceeding their operational lifespan every year, representing the biggest source of waste rubber, are landfilled or burned.7,8 Current reuse practices mainly involve the grinding of waste rubber to particles for subsequent use primarily as fillers in asphalt and in cement production.9,10 To a limited extent, these shredded rubber particles can be added to virgin rubber in the production of new tires or other products.
Chemical recycling of vulcanizates encompasses processes such as pyrolysis, yielding so-called “tire-derived fuel”,11 as well as devulcanization,12−14 which allows the further usage of the rubber as well as potential fillers or reinforcing materials. Devulcanization involves the selective breaking of the sulfur cross-links by chemical, biological, or physical means, such as thermal or mechanical treatment. However, the parallel breaking of rubber polymer chains under the harsh conditions required unavoidably compromises the material properties of the resulting devulcanized rubbers. Therefore, only a minor share of devulcanized rubber can be added to the virgin material without compromising the overall material properties.
Vulcanizates may end up in the environment by different pathways, e.g., mismanagement of waste or shedding of tire wear particles. While natural rubber is readily biodegradable,5 vulcanization significantly retards this process by adding bonds that need to be cleaved to yield bioavailable low-molecular-weight products. Furthermore, the cross-linked nature of vulcanized rubber decreases the accessibility of enzymes to cleavable double bonds in the polymer chains.15
A variety of concepts exist for creating recyclable and/or degradable elastomers, the most prominent example being thermoplastic elastomers, based on the formation of reversible noncovalent cross-links. These materials combine remarkable mechanical properties with (re)processability and the potential of using renewable feedstocks.16 Nonetheless, the lack of covalent cross-links leads among others to lower thermal stability and abrasion resistance, as well as stress softening, limiting their utility for certain applications.17 The creation of reversible covalently cross-linked networks is another widely studied approach,18−20 with vitrimers21 being a prominent example. However, the synthesis of these materials necessitates intricate, multistep procedures, leading so far to costly materials. Instead of reversible cross-links, the reversible bonds enabling recycling or biodegradation can also be located in the polymer backbone itself. Networks of amorphous polyesters synthesized with multifunctional monomers exhibit rubber-like properties and are chemically recyclable, as demonstrated elegantly by the Hillmyer group.22,23 Yet, their processing markedly differs from vulcanization of rubbers.
The vulcanization of unsaturated, amorphous polyesters would ideally combine the processing and material properties of rubbers with the recyclability and degradability of polyesters.24,25 The general feasibility of vulcanizing biobased polyesters was demonstrated among others by Cramail et al.,26 Fuller et al.,27 and Matsumura et al.(28) with unsaturated polyhydroxyalkanoates and ricinoleic acid-based polyesters. The recyclability of these materials or their biodegradation was not addressed, however.
We now report amorphous polyesters generated by A2 + B2 polycondensation of commercially available saturated and unsaturated comonomers and their sulfur vulcanization to elastomeric rubbers and demonstrate their deconstruction via solvolysis for chemical recycling to the monomer, (abiotic) hydrolysis, and environmental biodegradation with isotope-selective CO2 tracking.
Experimental Section
Materials
Ethylene glycol (EG, ≥99.5%) and disodium hydrogen phosphate dihydrate (≥98%) were purchased from Carl Roth, hexenedioic acid (HA, >98%) from TCI, maleic acid (MA, 99%) and sodium dihydrogen phosphate monohydrate (≥99.99%) from Merck, N-tertbutyl benzothiazole sulfonamide (TBBS) and 2,2-dibutyl diethylmalonate (98%) from abcr, methanol (≥99.8%), dibutyl tin oxide (DBTO), 2,6-di-tert-butyl-p-cresol (BHT, 99%), and 13C2–EG (99 atom % 13C) from Sigma-Aldrich, and chloroform (≥99.8%) from VWR. Deuterated solvents were obtained from Eurisotop and dried over molecular sieves from Riedel-de Haën (0.4 nm). Pripol 1009 (dimer acid, DA, 98%) was kindly provided by Croda/Cargill.
Characterization Techniques
Differential scanning calorimetry (DSC) was performed on a Netzsch DSC 204 F1 instrument with the software Netzsch Proteus Thermal Analysis, version 6.1.0. Data reported for melting points (Tm) are from the second heating cycle with a heating/cooling rate of 10 K min–1 and for glass transition temperatures (Tg) from the maximum of the derivative of the second heating cycle using a heating/cooling rate of 30 K min–1.
Nuclear magnetic resonance (NMR) spectroscopy was performed on a Bruker Avance III HD 400 or Bruker Avance III 400 spectrometer. Chemical shifts were referenced to the solvent signals. Mestrenova software Mestrelab Research S.L. (version 14.1.2) was used for data evaluation.
Molecular masses of polymers were determined via size-exclusion chromatography (SEC) in chloroform at 35 °C on a PSS SECcurity2 instrument, equipped with PSS SDV linear M columns (2 × 30 cm, additional guard column) and a refractive index detector (PSS SECcurity2 RI). A flow rate of 1 mL/min was used. Data was evaluated versus polystyrene standards using the software PSS WinGPC, version 8.32.
Tensile tests were performed on a Zwick Z005/1446 Retroline tC II instrument at a crosshead speed of 5 mm min–1 on a tensile testing specimen (ISO 527-2, type 5A) obtained by vulcanization in a mold (vide infra). The tensile testing samples were preconditioned to ambient conditions for a minimum of 1 day prior to the measurement. Young’s modulus was determined at a crosshead speed of 0.5 mm min–1. Cyclic hysteresis tests were performed on individual test specimens for 10 consecutive cycles of loading and unloading to a constant strain of 20 and 50% with a crosshead speed of 5 mm min–1. The software testXpert from Zwick Roell, version 11.0, was used for data evaluation.
Polymerization Experiments
An amorphous polyester consisting only of DA and EG was synthesized according to an established melt polycondensation protocol.29 This protocol was adapted for the synthesis of amorphous and unsaturated polyesters. Exemplarily, the synthesis of the polyester H1 from DA, EG, and HA is described. DA (5.70 g, 10 mmol, 1 equiv), HA (1.44 g, 10 mmol, 1 equiv), EG (2.48 g, 40 mmol, 4 equiv), BHT (96 mg, 1 wt %), and DBTO (50 mg, 0.20 mmol, 2 mol %) were added into a round-bottom flask. The mixture was thoroughly degassed, stirred with a PTFE-coated stirring bar, and heated to 140 °C. Evolving water was distilled off. After 4 h, vacuum was gradually applied to facilitate the removal of condensate and excess EG, reaching 0.05 mbar after 24 h, while the viscosity of the reaction mixture increased. After 24 h at 0.05 mbar and 140 °C, the temperature was increased to 160 °C for 2 h and the reaction subsequently stopped by letting the reaction mixture cool down to room temperature. The uncured amorphous and unsaturated polyester was obtained as yellowish, highly viscous liquid without further workup.
Additional polyesters were prepared using MA as the unsaturated monomer. Various ratios of unsaturated to saturated monomers were utilized to produce polyesters with varying double-bond densities. The synthesis was carried out in a reactor equipped with individual glass inlets and stirring bars, enabling the simultaneous small-scale synthesis (1–2 g) of up to eight polymers. All polymers were fully amorphous. The molecular weight determined via NMR and SEC as well as the exact monomer composition, unreacted double bonds, and theoretical C atom recycling rates are shown in Table S1. The theoretical recycling rate was calculated with the following equation
| 1 |
Vulcanization Experiments
For vulcanization, the uncured, unsaturated polyesters were mixed in PTFE vials at 100–120 °C with 2.75 phr sulfur, 0.8 phr TBBS, and 5 phr ZnO, where phr denotes parts per 100 rubber. The homogeneous white compounds were manually inserted into a PTFE mold and heated for 1 h to 180 °C. Vulcanized materials are indicated with V, e.g., H1-V. These elastomers were obtained as nonsticky, yellow-brownish tensile testing specimens with a faint sulfur odor, akin to vulcanized natural or synthetic rubber (Figure S1).
For determining the vulcanization time of different rubbers, a small amount of uncured polyester (50–100 mg) was added to a glass vial together with the curing agents as outlined above. The materials were mixed at 100 °C and subsequently vulcanized at 180 °C. The vulcanization time was defined as the point in time at which the sample visibly transitioned from a liquid to an elastic state.
Gel Content Determination
Triplicate extractions of the sol fractions were conducted in CHCl3 to assess the gel content of the vulcanized rubber H1-V sample. Approximately 40 mg of the sample was immersed in 8 mL of CHCl3 for 6 h and shaken at 37 °C. The solution was filtered, the solvent was removed in vacuum, and the remaining mass was measured. The sol fraction was calculated from the ratio of the remaining and the initial mass. The insolubility of the vulcanized rubber even in 100 °C toluene is shown in Figure S3.
Recycling Experiments
Methanolysis of the vulcanized rubber H1-V tensile testing specimen was carried out without any pretreatment of the elastomeric material. The rubber (23 g) was immersed in 300 mL of MeOH and stirred at 150 °C for 6 days in a pressure reactor without the addition of a catalyst. The resultant brown solution (Figure S4, left) was filtered to remove solids such as ZnO particles. The solvent was removed from the filtrate under vacuum, and the monomer solution was distilled. The first fraction was collected at a pressure of 5 mbar at 110–130 °C and the second fraction at 0.05 mbar and 220 °C. The last remaining monomers were distilled via short path distillation at 0.05 mbar and 250–300 °C. The first fraction was an EG-rich mixture (Figure S7), the second fraction mainly consisted of the cross-linked or uncross-linked HA, and in the last fraction, >99% pure (determined from NMR, Figures S5 and S6) dimer dimethyl ester was isolated in 97% yield (14.6 g, Figure S4, right).
Hydrolytic Stability
The hydrolytic stability of polyester consisting of DA, EG, and HA was tested in phosphate buffer solution (pH = 7.2, 50 mM). Around 15 mg of uncured polyester H1 was immersed in 10 mL of phosphate buffer solution, heated up to 75 °C, and stirred for 1 day. Subsequently, the polyester was removed from the buffer, dried in vacuum, and analyzed via SEC. A control sample was processed the same way but aged at 25 °C.
Biodegradation
Polycondensation and vulcanization of a rubber equivalent in composition to H1-V containing DA, HA, and 13C labeled EG as well as vulcanization agents were carried out as described above. 13C-labeled samples are indicated below by *. After the vulcanization, the final material consisted of 66.95 wt % carbon based on calculations of the components used in the preparation. Of all carbon atoms present in the material, 8.61% theoretically belonged to the utilized EG, corresponding to an estimated calculated 13C-content of 9.62 atom %. The vulcanized rubber was manually cut into small particles (approximately 1 × 1 × 1 mm = 1 mm3) for addition to the soil. Additionally, 13C-enriched cellulose [prepared as a mixture of fully 13C-labeled cellulose (IsoLife, Netherlands) and unlabeled cellulose (Merck, Germany) to yield a cellulose with overall 13C-content of approximately 5 atom %] was prepared as a reference biodegradable polymer.
Soil mineralization experiments were conducted using a flow-through incubation system coupled to an isotope-selective cavity ring-down spectroscopy analyzer (model G2201-i; Picarro, USA) that has been previously described in detail.30 The biodegradation of each material was conducted in triplicate incubation bottles and compared to triplicate control incubation bottles to which no material was added, all held at 25 °C in the dark. The data on biodegradation of cellulose as a reference biodegradable polymer in the same soil has been published previously.31 The soil used (standard soil type 6S from LUFA Speyer, Germany) was characterized as a clayey loam (DIN classification based on particle size) and contained 1.5 wt % organic C, 0.17 wt % N, and had a pH of 7.3. Upon collection, the soil was sieved to 2 mm and stored at 4 °C between receiving and preparing it for incubations. Before the addition of H1-V or cellulose, the soils were brought to 25 °C, and the water content in each bottle was adjusted to 50% of the maximum soil water holding capacity (WHCmax = 40.1 g H2O g–1 dry soil), and the soils were preincubated for 1 week in order for the background soil respiration rate to equilibrate to the new conditions. Throughout the incubation, the bottles were intermittently removed from the flow-through incubation system and stored open in boxes with humidified air. The water contents of the soils were periodically readjusted gravimetrically to account for evaporative losses. Concentrations of 12CO2 and 13CO2 in the efflux gas of the bottles were measured for 15 min at a time, with automatic switching between bottles, and the data from the last 5 min of each interval averaged to serve as single time points. This sample sequence was operated continuously, with periodic measuring of synthetic air with known concentrations and isotopic signatures of CO2 to serve as drift correction during continuous instrument operation. Using the measured concentrations of 12CO2 and 13CO2 along with the total carbon and 13C contents of the added polymers, polymer–13C mineralization rates and subsequently, extents, were calculated according to a previously published protocol.30
Results and Discussion
Material and Recycling
The polycondensation of readily available biobased hydrogenated DA (C36) monomers with EG led to hydrophobic, amorphous polyesters with high molecular weight using a protocol previously used to produce high-molecular-weight semicrystalline polyesters from linear monomers (Figures S13 and S14). In a terpolymerization with comparably low-molecular-weight unsaturated diacids (with C4 being the smallest possible configuration) maleic acid (MA, C4) or hexenedioic acid (HA, C6), amorphous, unsaturated polyesters were synthesized. The melt polycondensations catalyzed with DBTO were stabilized with BHT and conducted at lower temperature than for saturated polyesters (140–160 °C, Figure 1, depicted for HA-containing polyesters) to minimize cross-linking of the double bonds. Still, reasonably high molar masses could be achieved (Mn = 48 kg/mol and Mw = 126 kg/mol, Figure 2a). An exemplary 1H NMR spectrum is shown in Figure S9. The saturated and unsaturated diacids were utilized in various ratios, all resulting in fully amorphous polyesters with Tg values between −45 and −40 °C (Figure 2, right, Figure S11, and Table S1). The undesired reaction of the double bonds at the elevated temperatures of polycondensation occurred only in a negligible amount for HA, as concluded from 1H NMR analysis of the polymers and indicated by a narrow PDI (Table S1). The double bond of MA, however, exhibited a higher reactivity, leading to some cross-linking visible in the SEC chromatograms (Figure S10). The polyester synthesized from 1 equiv of DA, 1 equiv HA, and 2 equiv EG was used as a model system for further vulcanization and is hereinafter termed polyester H1. The SEC trace of an upscaled polymerization with MA (polyester M1) is shown in Figure S10 and DSC traces are shown in Figure S11.
Figure 1.

Polycondensation of saturated (DA and EG) and unsaturated (in this case: HA) monomers to amorphous, unsaturated polyesters (top), their vulcanization to rubbers (left), and their solvolysis and purification (bottom) back to the monomers. The green dots represent ester groups as predetermined breaking points in the chain. The unsaturated monomers usually comprise around 10% of the carbon atoms in the rubber polymer.
Figure 2.

(a) SEC and (b) DSC traces of polyester H1.
To this end, polyester H1, which was a highly viscous, nonsticky substance at room temperature, was mixed with elemental sulfur, zinc oxide, and TBBS as vulcanization accelerators according to a “conventional vulcanization” protocol.32,33 Stearic acid, typically employed in this protocol to solubilize ZnO particles in the hydrophobic matrix, was not used here due to the assumption that the already present carboxylate and hydroxyl chain ends as well as polar ester groups would be sufficient in that regard. Indeed, manual mixing of the components at 100 °C yielded a homogeneous, white material. Vulcanizing for 1 h at 180 °C led to an elastic rubber (Materials section, Figure S1). The additional polyesters were vulcanized according to the same protocol, and their vulcanization time depended on the identity and relative amount of unsaturated comonomer used, with MA displaying the highest reactivity (Figure S2). The resulting polyester-based vulcanized rubber H1-V was intractable and insoluble, exhibiting a gel content of ≥90% (Table S2). Tensile testing of H1-V showed elastic behavior, comparable to the properties of a commercial rubber band, and an elongation at break of ca. 150% (Figures 3 and S12).
Figure 3.
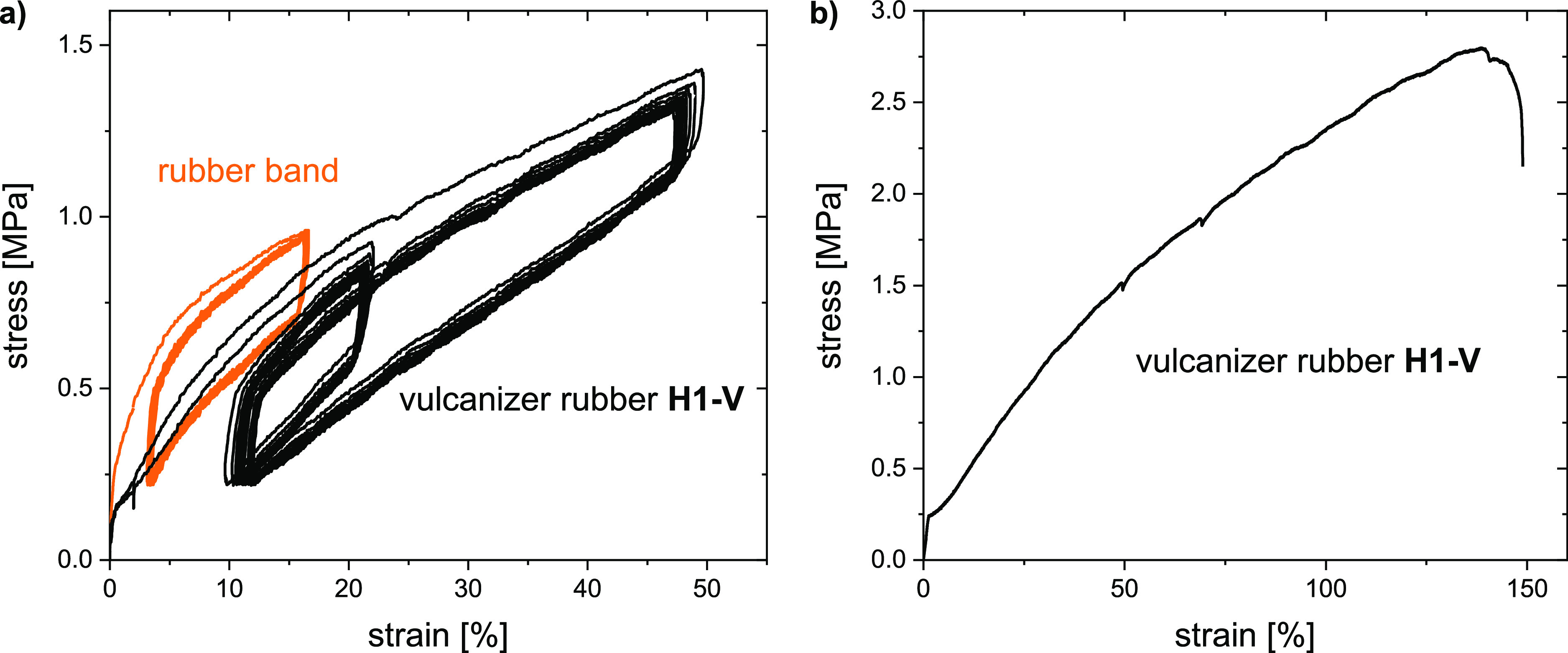
(a) Cyclic hysteresis tests of vulcanized rubber H1-V compared to a conventional rubber band and (b) stress–strain curves of vulcanized rubber H1-V.
Depolymerization as a means of chemical recycling was performed via methanolysis without additional catalysts, as previously reported for polyethylene-like polyesters29,34 or PET.35 After 6 days at 150 °C, the cross-linked rubber underwent complete depolymerization into its constituent monomers. The resulting brownish solution (Figure S4, left) was filtered to remove solids such as vulcanizing agents. Subsequent separation by distillation yielded the individual monomers. The main component of the polymer, dimer dimethyl ester, was recovered via short-path distillation at high temperatures and high vacuum in 97% yield and a purity >99% (concluded from 1H NMR, Figures S5 and S6). Additionally, an EG-rich fraction was recovered (Figure S7). Notably, the unsaturated monomers (subject to the cross-linking process) and their reaction products were effectively separated, particularly from the dimer acid fraction. While the recycling conditions studied here are certainly not optimum for larger-scale operations due to the long reaction time, they can be easily improved by the use of a catalyst.34 Methanolysis of PET, for example, is usually performed with KOH as the catalyst at 180–280 °C36 and is still the topic of numerous ongoing studies.37,38 Note that both the methanolysis of PET39 and dimer acid purification via distillation40 are established procedures on an industrial scale.
As the saturated monomers do not undergo any changes during vulcanization, the recovered monomer is suitable for reutilization by polycondensation. The introduction of fresh unsaturated monomers would allow for the synthesis of a new virgin polyester. Isolation of the saturated dicarboxylate monomer, which accounts for the largest part of the material, is facilitated by its relatively high molecular weight and consequently boiling point compared to the other components. Relatively low-molecular-weight unsaturated monomers were judiciously chosen, as these are not recycled when cross-linked, to enable a high C atom recycling rate.
By recycling only the dimer acid component of vulcanized rubber H1-V, a theoretical recycling rate of 78% C atoms is attainable. Here, a recycling rate of 76% from monomer to monomer was demonstrated. By recovering and purifying the EG monomers as well, the theoretical recycling rate increases to 87% and, with the same monomer ratio, to 91% for MA-based materials, respectively. Lower amounts of unsaturated monomers correspond to even higher theoretical recycling rates. To this end, in preliminary experiments even with a higher molar ratio of DA to HA of 4.7, (weight ratio ca. 18) vulcanizable materials were obtained, corresponding to a theoretical recycling rate of up to 97% of the carbon atoms (Table S1). As in our concept, the unsaturated monomers are not reused, it is important to note that especially MA is a platform chemical,41 thus affordable, and HA can be produced biobased as well.42 Also other available unsaturated monomers such as itaconic acid or muconic acid could be used as alternative cross-linkers.
Biodegradation and Hydrolytic Stability
The in-chain ester groups as predetermined breaking points not only facilitate chemical recycling of the polyester rubbers; in principle, they can also be hydrolyzed by abiotic or biotic means. On the other hand, such processes are expected to be impeded by the hydrophobic nature of the material. Indeed, exposing the uncured polyester H1 to neutral aqueous buffer even at elevated temperature (phosphate buffer, pH 7.2, 75 °C, 24 h) did not result in any significant change of molecular weight (Table S3). The abiotic hydrolysis of the uncured polyester is not expected to be significantly faster than that of the vulcanized material.
One likely receiving environment of rubber-derived micro- or nanoplastics is soil. The soil biodegradability of the polyester-based vulcanized rubber was elucidated via stable isotope-selective CO2 tracking, a method previously established for quantitatively and unambiguously following mineralization of polymers.43,44 A small-scale batch of polyester H1* was synthesized employing 13C2–EG and subsequently vulcanized and manually cut into small particles (approximately 1 mm3). Monitoring of biodegradation in agricultural soil at 25 °C showed substantial and continual conversion of the EG monomer to 13CO2, with the mineralization extent reaching around 60% of the added EG–13C after 240 days (Figure 4). As not all monomers were 13C labeled and investigated, these experiments were not suited to classify the material as biodegradable according to, e.g., ISO norms.45,46 Nonetheless, the mineralization of the EG units indicates an environmental hydrolysis of the polyester to its monomers, which is widely accepted as the rate-limiting step of polymer biodegradation in soil,47,48 followed by microbial utilization of EG-containing oligomers or free EG. Neither the cross-linked nature of the material nor the vulcanization additives prevented the substantial degradation of this rubber under mild conditions. While also conventional natural and synthetic rubbers can be degraded by certain microorganisms,5 most studies have demonstrated this using isolated strains under idealized conditions,49,50 while the degradation behavior of rubbers in nonspecific, natural environments is less well investigated. The limited evidence available indicates that tire wear particles, mainly consisting of conventional synthetic rubbers, are nearly inert in aquatic environments, showing close to zero CO2 evolution over 80 days.51 Further experiments are needed to investigate the biodegradation of the rubbers presented here, especially to investigate the microbial utilization of the different components, such as dimer acid and sulfur-containing monomer units. Furthermore, experiments comparing the biodegradability of polyester-based rubbers and conventional rubbers in the natural environment would be instructive. Note that remediation of micro- or nanoplastics will be facilitated by the particles’ high surface area,52,53 which facilitates surface degradation processes (as opposed to bulk degradation mechanisms) like enzymatic degradation in particular.54
Figure 4.

Soil biodegradation extents at 25 °C of 13C-labeled vulcanized rubber H1-V* compared to those of cellulose (calculated from rates shown in Figure S8). Labeling was achieved by employing 13C2–EG in the polyester synthesis. Incubations were performed in the same bulk agricultural soil and in triplicate for each material. Measured data for individual replicate incubations are shown as points, with dashed lines as linear interpolation between measurements, to guide the eye. The data on biodegradation of cellulose as a reference biodegradable polymer in the same soil has been published previously.31
Conclusions
The polyester-based rubbers reported here are chemically recyclable and biodegradable while maintaining the characteristics of covalently cross-linked elastomers formed via conventional vulcanization procedures. Upscaling of the materials and processes shown herein is promising as the synthetic procedures are well-established, and readily available monomers were used exclusively. In particular, the theoretical chemical recycling rates exceeding 90% constitute a substantial advancement from the status quo of chemical recycling of conventional rubbers. As the monomers used herein are biobased or can be synthesized from biobased resources, these materials would also contribute to a shift of the chemical industry to renewable feedstocks.24,25
The recyclability and biodegradability of rubbers are particularly crucial for tires, as one billion tires exceed their operational lifetime every year generating massive amounts of waste, and tire wear particles are inevitably introduced into the environment in a million ton range every year,52,55 making them the major source of microplastics in some environments.55 Considering the potential impact of both the particles and leaching additives on human health and wildlife56,57 and with the anticipated increase in tire runoff due to increased volume of traffic, the development of environmentally benign rubber materials is desirable. Notably, chemical recycling of tires enables the recovery of not only the rubber but also valuable fillers and reinforcement materials such as carbon black and steel. Tires are highly engineered products optimized over decades with key metrics like wet traction, rolling resistance and abrasion resistance.58 As they are subject to safety requirements, new materials would need extensive testing. Anticipated next steps comprise the upscaling of production for applying more advanced processing methods, such as compression molding. Detailed studies of the viscosity behavior of the uncured rubber and curing kinetics as well as long-term stability experiments to determine the biodegradability of a final compound and the stability under stress are required to establish the processability of this material with existing equipment and applications, such as tires.
Acknowledgments
We thank Sebastian Schwab, Leonie Bühler, Marie Fritschi, and Thierry Dezest for assistance with experiments and Lars Bolk for DSC measurements. We thank Prof. Michael Sander (ETH Zürich) for access to incubation infrastructure and isotope-selective cavity ring-down spectroscopy instrumentation. We further thank the University of Konstanz scientific workshop team for support. Support of our studies on nonpersistent materials by the ERC (Advanced Grant DEEPCAT, no. 832480) is gratefully acknowledged. T.F.N. gratefully acknowledges support by a Postdoctoral Research Fellowship from the Alexander von Humboldt Foundation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssuschemeng.3c08435.
Supplementary methods and data, including NMR, SEC, DSC, and tensile testing data of different amorphous polyesters and vulcanizates and additional data concerning vulcanization times and hydrolytic stability (PDF)
Author Contributions
S.T.S. and S.M. jointly devised the experimental program. S.T.S. prepared and characterized the material and conducted recycling experiments. T.F.N. conducted the degradation experiments. S.T.S., T.F.N., and S.M. wrote the manuscript.
The authors declare no competing financial interest.
Notes
A patent has been filed by S.T.S. and S.M. on the findings reported here.
Supplementary Material
References
- Statista . Natural and synthetic rubber production globally 2022|Statista. https://www.statista.com/statistics/618804/total-global-natural-and-synthetic-rubber-production/ (accessed November 15, 2023).
- Gent A. N.; Walter J. D.. Pneumatic Tire; Mechanical Engineering Faculty Research, 2006. [Google Scholar]
- Shulman V. L.Tire Recycling. In Waste: A Handbook for Management, 2nd ed.; Letcher T. M., Vallero D. A., Eds.; Academic Press: London, 2019; pp 489–515. [Google Scholar]
- Coran A. Y.Vulcanization. In The Science and Technology of Rubber, 4th ed.; Erman B., Mark J. E., Roland C. M., Eds.; Elsevier Academic Press: Amsterdam, Heidelberg, 2013; pp 337–381. [Google Scholar]
- Ali Shah A.; Hasan F.; Shah Z.; Kanwal N.; Zeb S. Biodegradation of natural and synthetic rubbers: A review. Int. Biodeterior. Biodegrad. 2013, 83, 145–157. 10.1016/j.ibiod.2013.05.004. [DOI] [Google Scholar]
- Formela K. Sustainable development of waste tires recycling technologies - recent advances, challenges and future trends. Adv. Ind. Eng. Polym. Res. 2021, 4, 209–222. 10.1016/j.aiepr.2021.06.004. [DOI] [Google Scholar]
- RubberWorld . Global tire recycling market forecast at $18.1 billion by 2032. https://rubberworld.com/global-tire-recycling-market-forecast-at-18-1-billion-by-2032/ (accessed October 11, 2023).
- U.S. Tire Manufacturers Association . 2021 US Scrap Tire Management Summary, 2022.
- Chatziaras N.; Psomopoulos C. S.; Themelis N. J. Use of waste derived fuels in cement industry: a review. Manag. Environ. Qual. 2016, 27, 178–193. 10.1108/MEQ-01-2015-0012. [DOI] [Google Scholar]
- Shu X.; Huang B. Recycling of waste tire rubber in asphalt and portland cement concrete: An overview. Constr. Build. Mater. 2014, 67, 217–224. 10.1016/j.conbuildmat.2013.11.027. [DOI] [Google Scholar]
- Urrego-Yepes W.; Cardona-Uribe N.; Vargas-Isaza C. A.; Martínez J. D. Incorporating the recovered carbon black produced in an industrial-scale waste tire pyrolysis plant into a natural rubber formulation. J. Environ. Manage. 2021, 287, 112292. 10.1016/j.jenvman.2021.112292. [DOI] [PubMed] [Google Scholar]
- Saputra R.; Walvekar R.; Khalid M.; Mubarak N. M.; Sillanpää M. Current progress in waste tire rubber devulcanization. Chemosphere 2021, 265, 129033. 10.1016/j.chemosphere.2020.129033. [DOI] [PubMed] [Google Scholar]
- Dorigato A.; Rigotti D.; Fredi G. Recent advances in the devulcanization technologies of industrially relevant sulfur-vulcanized elastomers. Adv. Ind. Eng. Polym. Res. 2023, 6, 288–309. 10.1016/j.aiepr.2022.11.003. [DOI] [Google Scholar]
- Sun Y.; Yan X.; Liang H.; Böhm G.; Jia L. Rubber Recycling: Mending the Interface between Ground Rubber Particles and Virgin Rubber. ACS Appl. Mater. Interfaces 2020, 12, 47957–47965. 10.1021/acsami.0c13722. [DOI] [PubMed] [Google Scholar]
- Tsuchii A.; Hayashi K.; Hironiwa T.; Matsunaka H.; Takeda K. The effect of compounding ingredients on microbial degradation of vulcanized natural rubber. J. Appl. Polym. Sci. 1990, 41, 1181–1187. 10.1002/app.1990.070410527. [DOI] [Google Scholar]
- Petersen S. R.; Prydderch H.; Worch J. C.; Stubbs C. J.; Wang Z.; Yu J.; Arno M. C.; Dobrynin A. V.; Becker M. L.; Dove A. P. Ultra-Tough Elastomers from Stereochemistry-Directed Hydrogen Bonding in Isosorbide-Based Polymers. Angew. Chem., Int. Ed. 2022, 61, e202115904 10.1002/anie.202115904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobny J. G.Introduction. In Handbook of Thermoplastic Elastomers, 2nd ed.; Drobny J. G., Ed.; Plastics Design Library; William Andrew Publishing: Norwich, NY, 2014; pp 1–11. [Google Scholar]
- Trovatti E.; Lacerda T. M.; Carvalho A. J. F.; Gandini A. Recycling tires? Reversible crosslinking of poly(butadiene). Adv. Mater. 2015, 27, 2242–2245. 10.1002/adma.201405801. [DOI] [PubMed] [Google Scholar]
- Fortman D. J.; Brutman J. P.; De Hoe G. X.; Snyder R. L.; Dichtel W. R.; Hillmyer M. A. Approaches to Sustainable and Continually Recyclable Cross-Linked Polymers. ACS Sustain. Chem. Eng. 2018, 6, 11145–11159. 10.1021/acssuschemeng.8b02355. [DOI] [Google Scholar]
- Martin R.; Rekondo A.; Ruiz de Luzuriaga A.; Cabañero G.; Grande H. J.; Odriozola I. The processability of a poly(urea-urethane) elastomer reversibly crosslinked with aromatic disulfide bridges. J. Mater. Chem. A 2014, 2, 5710. 10.1039/c3ta14927g. [DOI] [Google Scholar]
- Montarnal D.; Capelot M.; Tournilhac F.; Leibler L. Silica-like malleable materials from permanent organic networks. Science 2011, 334, 965–968. 10.1126/science.1212648. [DOI] [PubMed] [Google Scholar]
- Brutman J. P.; De Hoe G. X.; Schneiderman D. K.; Le T. N.; Hillmyer M. A. Renewable, Degradable, and Chemically Recyclable Cross-Linked Elastomers. Ind. Eng. Chem. Res. 2016, 55, 11097–11106. 10.1021/acs.iecr.6b02931. [DOI] [Google Scholar]
- De Hoe G. X.; Zumstein M. T.; Tiegs B. J.; Brutman J. P.; McNeill K.; Sander M.; Coates G. W.; Hillmyer M. A. Sustainable Polyester Elastomers from Lactones: Synthesis, Properties, and Enzymatic Hydrolyzability. J. Am. Chem. Soc. 2018, 140, 963–973. 10.1021/jacs.7b10173. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Romain C.; Williams C. K. Sustainable polymers from renewable resources. Nature 2016, 540, 354–362. 10.1038/nature21001. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Fevre M.; Jones G. O.; Waymouth R. M. Catalysis as an Enabling Science for Sustainable Polymers. Chem. Rev. 2018, 118, 839–885. 10.1021/acs.chemrev.7b00329. [DOI] [PubMed] [Google Scholar]
- Dworakowska S.; Le Coz C.; Chollet G.; Grau E.; Cramail H. Cross Linking of Polyesters Based on Fatty Acids. Eur. J. Lipid Sci. Technol. 2019, 121, 1900264. 10.1002/ejlt.201900264. [DOI] [Google Scholar]
- Gagnon K.; Lenz R.; Farris R.; Fuller R. Chemical modification of bacterial elastomers: 2. Sulfur vulcanization. Polymer 1994, 35, 4368–4375. 10.1016/0032-3861(94)90094-9. [DOI] [Google Scholar]
- Ebata H.; Yasuda M.; Toshima K.; Matsumura S. Poly (ricinoleic acid) based novel thermosetting elastomer. J. Oleo Sci. 2008, 57, 315–320. 10.5650/jos.57.315. [DOI] [PubMed] [Google Scholar]
- Eck M.; Schwab S. T.; Nelson T. F.; Wurst K.; Iberl S.; Schleheck D.; Link C.; Battagliarin G.; Mecking S. Biodegradable High Density Polyethylene like Material. Angew. Chem. 2023, 135, e202213438 10.1002/ange.202213438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson T. F.; Baumgartner R.; Jaggi M.; Bernasconi S. M.; Battagliarin G.; Sinkel C.; Künkel A.; Kohler H.-P. E.; McNeill K.; Sander M. Biodegradation of poly(butylene succinate) in soil laboratory incubations assessed by stable carbon isotope labelling. Nat. Commun. 2022, 13, 5691. 10.1038/s41467-022-33064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson T. F.; Rothauer D.; Sander M.; Mecking S. Degradable and Recyclable Polyesters from Multiple Chain Length Bio- and Waste-Sourceable Monomers. Angew. Chem., Int. Ed. 2023, 62, e202310729 10.1002/anie.202310729. [DOI] [PubMed] [Google Scholar]
- Vélez J. S.; Velásquez S.; Giraldo D. Mechanical and rheometric properties of gilsonite/carbon black/natural rubber compounds cured using conventional and efficient vulcanization systems. Polym. Test. 2016, 56, 1–9. 10.1016/j.polymertesting.2016.09.005. [DOI] [Google Scholar]
- Junkong P.; Morimoto R.; Miyaji K.; Tohsan A.; Sakaki Y.; Ikeda Y. Effect of fatty acids on the accelerated sulfur vulcanization of rubber by active zinc/carboxylate complexes. RSC Adv. 2020, 10, 4772–4785. 10.1039/C9RA10358A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häußler M.; Eck M.; Rothauer D.; Mecking S. Closed-loop recycling of polyethylene-like materials. Nature 2021, 590, 423–427. 10.1038/s41586-020-03149-9. [DOI] [PubMed] [Google Scholar]
- Sinha V.; Patel M. R.; Patel J. V. Pet Waste Management by Chemical Recycling: A Review. J. Polym. Environ. 2010, 18, 8–25. 10.1007/s10924-008-0106-7. [DOI] [Google Scholar]
- Ghosal K.; Nayak C.; Ghosal K.; Nayak C. Recent advances in chemical recycling of polyethylene terephthalate waste into value added products for sustainable coating solutions – hope vs. hype. Mater. Adv. 2022, 3, 1974–1992. 10.1039/d1ma01112j. [DOI] [Google Scholar]
- Barnard E.; Rubio Arias J. J.; Thielemans W. Chemolytic depolymerisation of PET: a review. Green Chem. 2021, 23, 3765–3789. 10.1039/D1GC00887K. [DOI] [Google Scholar]
- Clark R. A.; Shaver M. P. Depolymerization within a Circular Plastics System. Chem. Rev. 2024, 124, 2617–2650. 10.1021/acs.chemrev.3c00739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullo A. H.Can recyclers break the polyester barrier? Chem. Eng. News [Online], Nov 27, 2023. https://cen.acs.org/environment/recycling/recyclers-break-polyester-barrier/101/i39 (accessed January 30, 2024).
- Abraham T. W.; Höfer R.. Lipid-Based Polymer Building Blocks and Polymers. In Polymer Science: A Comprehensive Reference, New ed.; Matyjaszewski K., Möller M., Eds.; Elsevier Science: Amsterdam, London, 2012; Vols. 1–10, pp 15–58. [Google Scholar]
- Wojcieszak R.; Santarelli F.; Paul S.; Dumeignil F.; Cavani F.; Gonçalves R. V. Recent developments in maleic acid synthesis from bio-based chemicals. Sustainable Chem. Processes 2015, 3, 9. 10.1186/s40508-015-0034-5. [DOI] [Google Scholar]
- Dell’Anna M. N.; Laureano M.; Bateni H.; Matthiesen J. E.; Zaza L.; Zembrzuski M. P.; Paskach T. J.; Tessonnier J.-P. Electrochemical hydrogenation of bioprivileged cis, cis -muconic acid to trans −3-hexenedioic acid: from lab synthesis to bench-scale production and beyond. Green Chem. 2021, 23, 6456–6468. 10.1039/D1GC02225C. [DOI] [Google Scholar]
- Zumstein M. T.; Schintlmeister A.; Nelson T. F.; Baumgartner R.; Woebken D.; Wagner M.; Kohler H.-P. E.; McNeill K.; Sander M. Biodegradation of synthetic polymers in soils: Tracking carbon into CO2 and microbial biomass. Sci. Adv. 2018, 4, eaas9024 10.1126/sciadv.aas9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batiste D. C.; De Hoe G. X.; Nelson T. F.; Sodnikar K.; McNeill K.; Sander M.; Hillmyer M. A. Site-Specific Mineralization of a Polyester Hydrolysis Product in Natural Soil. ACS Sustain. Chem. Eng. 2022, 10, 1373–1378. 10.1021/acssuschemeng.1c07948. [DOI] [Google Scholar]
- International Organization for Standardization . Plastics—Determination of the Ultimate Aerobic Biodegradability of Plastic Materials in Soil by Measuring the Oxygen Demand in a Respirometer or the Amount of Carbon Dioxide Evolved, 2019. (ISO 17556).
- International Organization for Standardization . Determination of the Ultimate Aerobic Biodegradability of Plastic Materials under Controlled Composting Conditions—Method by Analysis of Evolved Carbon Dioxide, 2018. (ISO 14855).
- Wang G.-X.; Huang D.; Ji J.-H.; Völker C.; Wurm F. R. Seawater-Degradable Polymers-Fighting the Marine Plastic Pollution. Adv. Sci. 2020, 8, 2001121. 10.1002/advs.202001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander M. Biodegradation of Polymeric Mulch Films in Agricultural Soils: Concepts, Knowledge Gaps, and Future Research Directions. Environ. Sci. Technol. 2019, 53, 2304–2315. 10.1021/acs.est.8b05208. [DOI] [PubMed] [Google Scholar]
- Berekaa M. M.; Linos A.; Reichelt R.; Keller U.; Steinbüchel A. Effect of pretreatment of rubber material on its biodegradability by various rubber degrading bacteria. FEMS Microbiol. Lett. 2000, 184, 199–206. 10.1016/s0378-1097(00)00048-3. [DOI] [PubMed] [Google Scholar]
- Aboelkheir M. G.; Bedor P. B.; Leite S. G.; Pal K.; Toledo Filho R. D.; Gomes de Souza F. Biodegradation of Vulcanized SBR: A Comparison between Bacillus subtilis, Pseudomonas aeruginosa and Streptomyces sp. Sci. Rep. 2019, 9, 19304. 10.1038/s41598-019-55530-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klun B.; Rozman U.; Kalčíková G. Environmental aging and biodegradation of tire wear microplastics in the aquatic environment. J. Environ. Chem. Eng. 2023, 11, 110604. 10.1016/j.jece.2023.110604. [DOI] [Google Scholar]
- Kole P. J.; Löhr A. J.; van Belleghem F. G. A. J.; Ragas A. M. J. Wear and Tear of Tyres: A Stealthy Source of Microplastics in the Environment. Int. J. Environ. Res. Public Health 2017, 14, 1265. 10.3390/ijerph14101265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pew Charitable Trusts and SYSTEMIQ . Breaking the plastic wave: A comprehensive assessment of pathways towards stopping ocean plastic pollution. https://www.pewtrusts.org/-/media/assets/2020/10/breakingtheplasticwave_mainreport.pdf (accessed November 27, 2023).
- Laycock B.; Nikolić M.; Colwell J. M.; Gauthier E.; Halley P.; Bottle S.; George G. Lifetime prediction of biodegradable polymers. Prog. Polym. Sci. 2017, 71, 144–189. 10.1016/j.progpolymsci.2017.02.004. [DOI] [Google Scholar]
- Knight L. J.; Parker-Jurd F. N. F.; Al-Sid-Cheikh M.; Thompson R. C. Tyre wear particles: an abundant yet widely unreported microplastic?. Environ. Sci. Pollut. Res. 2020, 27, 18345–18354. 10.1007/s11356-020-08187-4. [DOI] [PubMed] [Google Scholar]
- Tian Z.; Zhao H.; Peter K. T.; Gonzalez M.; Wetzel J.; Wu C.; Hu X.; Prat J.; Mudrock E.; Hettinger R.; et al. A ubiquitous tire rubber-derived chemical induces acute mortality in coho salmon. Science 2021, 371, 185–189. 10.1126/science.abd6951. [DOI] [PubMed] [Google Scholar]
- Bhuyan M. S. Effects of Microplastics on Fish and in Human Health. Front. Environ. Sci. 2022, 10, 827289. 10.3389/fenvs.2022.827289. [DOI] [Google Scholar]
- Grosch K. A. The Rolling Resistance, Wear and Traction Properties of Tread Compounds. Rubber Chem. Technol. 1996, 69, 495–568. 10.5254/1.3538383. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.