

Abstract
Fast-spiking parvalbumin (PV) interneurons are inhibitory interneurons with unique morphological and functional properties that allow them to precisely control local circuitry, brain networks and memory processing. Since the discovery in 1987 that PV is expressed in a subset of fast-spiking GABAergic inhibitory neurons, our knowledge of the complex molecular and physiological properties of these cells has been expanding. In this review, we highlight the specific properties of PV neurons that allow them to fire at high frequency and with high reliability, enabling them to control network oscillations and shape the encoding, consolidation and retrieval of memories. We next discuss multiple studies reporting PV neuron impairment as a critical step in neuronal network dysfunction and cognitive decline in mouse models of Alzheimer’s disease (AD). Finally, we propose potential mechanisms underlying PV neuron dysfunction in AD and we argue that early changes in PV neuron activity could be a causal step in AD-associated network and memory impairment and a significant contributor to disease pathogenesis.
Subject terms: Neuroscience, Physiology
Introduction
Parvalbumin-positive (PV) interneurons are GABAergic neurons that are found throughout the brain and have many distinctive morphological and physiological characteristics. Some PV interneurons have the unique ability to fire at high frequencies. Fast-spiking PV neurons are characterized by their inhibitory feedback and feedforward perisomatic projections onto pyramidal neurons, which allow them to control network synchrony and regulate theta and gamma oscillations that are crucial for learning and memory. Consequently, dysfunctional PV neurons have been associated with several disorders that involve network alterations and cognitive impairment, including Alzheimer’s disease (AD). In this review, we present recent evidence pointing to PV interneuron dysfunction as a key pathogenic mechanism in AD and AD-associated memory impairment. First, we highlight the unique properties of PV interneurons that make them indispensable for the proper control of network dynamics and the generation of functional network oscillations. Next, we focus on the importance of PV interneurons in learning and memory. Finally, we discuss how PV interneuron impairments lead to network abnormalities and memory deficits in AD.
Pv neuron properties
Fast-signaling properties
PV-expressing neurons can be divided to several subpopulations based on morphology and gene expression, including basket cells, axo-axonic cells, bistratified cells, and some oriens-alveus–lacunosum-moleculare (OLM) cells [1]. Moreover, regional differences have been reported in terms of PV-expressing neurons. In the dentate gyrus, for instance, bistratified cells do not express PV and in the prefrontal cortex, some axo-axonic cells are PV-negative [2]. Here, we will review studies that have investigated fast-spiking PV interneurons, which are named as such because of their distinct ability to fire at high frequencies, without going in much detail about the specific subtype of PV neuron that was studied, as this is often not reported.
Due to a remarkably short action potential (AP) duration and refractory period, fast-spiking PV neurons are capable of generating high-frequency trains of APs when artificially stimulated as well as in freely behaving animals [2, 3]. Recordings from PV neurons in brain slices have shown that APs are initiated close to the soma and propagate with high reliability [4]. High frequency AP firing is followed by extremely fast GABA release in the order of a few milliseconds [5], at PV interneuron axon terminals, which is critical for the proper temporal control of microcircuits [5]. These unique firing and signaling properties depend on the expression of specific ion channels and their distinct subcellular localization (Fig. 1A, for an outstanding review on the molecular, cellular, and network properties of PV interneurons see [2]). The Nav1.1 voltage-gated sodium channel subunit is mainly expressed on axons of fast-spiking PV neurons, allowing for rapid AP propagation and enabling high-frequency firing with very little propagation failure [4]. Other key players in PV neuron firing are Kv1 and Kv3 voltage-gated potassium channel subunits [6]. Kv3 channels, expressed predominantly in fast-spiking PV neurons, have a high activation threshold and rapid deactivation kinetics, resulting in fast repolarization and short AP duration [7]. The short duration of the AP and the fast deactivation of Kv3 channels after spike repolarization enable rapid recovery following each AP, thus contributing to the fast-spiking profile [2, 7, 8]. Kv3 channels are found at the soma of PV neurons, but are also highly expressed at axon terminals, facilitating fast GABA release at synaptic sites [7, 9, 10]. Kv1 channels, on the other hand, are present in the axon initial segment and soma of PV neurons, and are responsible for controlling AP firing threshold and are necessary for a persistent potentiation of PV neuron excitability [6, 11]. A more recent study has confirmed the role for the hyperpolarization-activated cyclic nucleotide-gated (HCN) channels in fast AP signaling [12, 13]. In addition to modulating neuronal excitability, this study revealed that HCN channels enhance AP initiation during sustained firing and facilitate the propagation of APs [12]. HCN channels are activated by hyperpolarization and are permeable to K+ and Na+, leading to a positive shift in cell resting membrane potential and thereby enabling faster membrane potential kinetics. HCN channels were also recently linked to fast electrical reactivity of fast-spiking cells in the human neocortex [14].
Fig. 1. PV neurons control network oscillations.

A PV neurons (blue) in the CA1 region of the hippocampus control network activity via feedback inhibition (1), feedforward inhibition (2) and autaptic self-inhibition (3). Their fast-spiking properties and fast GABA release onto pyramidal neurons (gray) are due to the expression of specific ion channels and calcium sensors. Nav1.1 sodium and Kv1 and Kv3 potassium channel subunits enable high frequency firing of PV neuron in response to depolarizing currents while HCN channels facilitate action potential propagation. P/Q-type calcium channel subunits and synaptotagmin 2 allow for precise GABA release resulting in accurately timed inhibitory postsynaptic currents in pyramidal neurons. B CA1 PV and pyramidal neuron spikes are time-locked with theta-nested gamma oscillations during learning and memory. The oscillatory wave depends on PV neurons firing periodically at gamma frequencies in the trough of the theta wave followed by pyramidal neuron firing at the peak of the theta wave. C During rest and sleep, high frequency PV neuron firing is phase-coupled to the oscillatory cycle of sharp-wave ripples oscillations.
PV neurons also express high levels of P/Q-type calcium channels, which contribute to the fast and precise release of GABA from PV axon terminals into the synaptic cleft [5, 15, 16]. Furthermore, the presynaptic calcium sensor synaptotagmin 2, which is predominantly found in PV neurons and exhibits the fastest calcium-binding kinetics of all synaptotagmins, is thought to also contribute to fast GABA release from PV neurons [17, 18]. Another distinctive feature of PV interneurons is their activity-dependent myelination, as other local interneurons are rarely myelinated [19]. This was recently shown to increase AP conduction velocity and facilitate feedforward inhibition in the cortex [20, 21]. Finally, fast-spiking PV neurons are the only interneurons reported to have autapses, synapses made by a neuron onto itself, both in humans and mice. These autapses have been suggested to increase reliability of fast inhibition by PV neurons and to allow modulation of network activity, and they have been reported in various brain regions [22–24]. In conclusion, unique molecular adaptations allow PV neurons to rapidly transmit electrical and chemical signals and control network activity in a very precise manner.
Maturation of synaptic contact and firing properties
PV neurons only reach their full electrophysiological and morphological maturation after a defined critical juvenile period (Fig. 2). Following myelination and the appearance of synaptic contacts in the first postnatal week, extensive refinement of PV neuron inhibitory synapses is observed during the weeks thereafter [25, 26]. PV axons display remodeling, including the removal of distal axonal branches and an increase in uniform axonal path lengths and myelination [26]. Studies have suggested that inhibitory PV synapse maturation and refinement depend on critical communication between microglial cells, PV neurons and their post-synaptic targets [27, 28]. In fact, removal of GABA-B1 receptors from microglia during development lead to a decrease in inhibitory synapses in adulthood and impaired GABAergic transmission in the somatosensory cortex of mice [25]. Also, microglia were found to be necessary for bouton maintenance on PV-expressing axo-axonic cells [29]. Moreover, the unique firing properties of PV neurons develop in an activity-dependent manner [30, 31] and presynaptic input onto PV neurons is critical for their maturation and functional integration into networks [32, 33]. The developmental refinement of PV neuron connectivity underlies important mechanisms of learning and plasticity that persist into adulthood [34–38]. Specifically, PV neuron maturation state has been associated with critical period-type plasticity [34–36, 39–41].
Fig. 2. Timeline of major events in the development and maturation of PV interneurons.
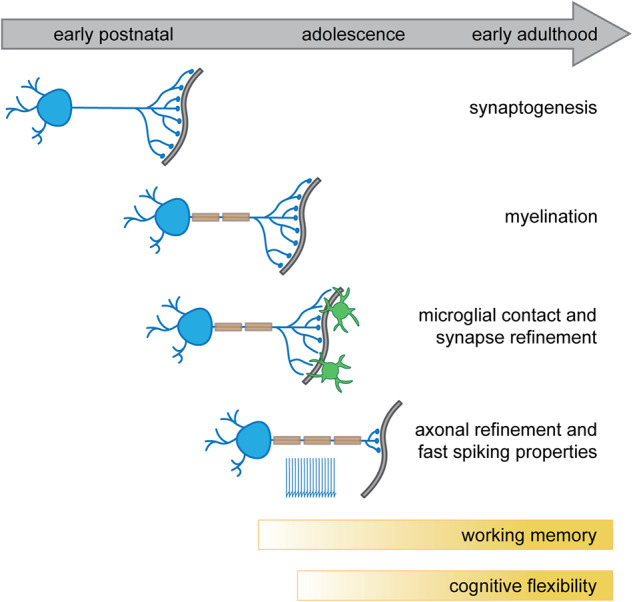
Synaptogenesis and myelination occur in early postnatal development. Microglia subsequently shape inhibitory synapses and strengthen connectivity between PV neurons and their postsynaptic targets. Next, PV neuron axons are further refined and become fully myelinated, and there is increased expression of ion channels required for fast spiking, enabling PV neurons to achieve precise temporal and spatial control of microcircuits. This postnatal pattern of PV neuron maturation is causally linked to an increase in working memory and cognitive flexibility that is observed until early adulthood.
Full electrophysiological maturation of PV neurons further depends on the extent of expression of the above-mentioned ion channels. Kv3 channels, for instance, double their expression level from P18 to P30 [42]. Importantly, between postnatal week 2 and 4, PV neurons reduce their input resistance, resting membrane potential, AP duration and latency, and AP propagation time and release period by more than 50%, while rheobase (i.e., the minimum amount of current required to induce AP firing), sag ratio (i.e., the ratio between the steady state decrease in the voltage and the largest decrease in voltage following a hyperpolarizing current step) and firing frequency are drastically increased [42, 43]. Notably, aberrant PV neuron maturation has been suggested as a key player in the pathogenesis of neurodevelopmental disorders such as schizophrenia (see review [19]).
Control of microcircuits and E/I balance
In addition to their specific molecular attributes, PV interneurons also have distinctive morphological features that allow them to control microcircuits. Their long dendrites cross multiple layers in the hippocampus and in the cortex [2] and they receive excitatory input from many different areas, such as from entorhinal cortex and medial septum in the hippocampus or from thalamic inputs in the cortex, as well as from a vast number of local excitatory neurons across different hippocampal areas or cortical layers [1, 44–47]. While pyramidal neurons in the hippocampus have on average 1.6 spines/μm, PV neurons receive 3.3 excitatory inputs/μm of dendritic shaft [48]. In total, PV neurons receive ten times more excitatory than inhibitory inputs. Conversely, paired recordings from PV and pyramidal neurons revealed that a single PV interneuron projects to almost all local pyramidal neurons within its vicinity [49]. In the hippocampus, one PV neuron contacts approximately 1100 pyramidal neurons with on average six synaptic contacts per neuron [48]. These very specific input and output characteristics allow PV neurons to provide precise feedforward and feedback inhibitory control in hippocampal and cortical microcircuits (Fig. 1A). Accordingly, PV interneurons are crucial for the maintenance of excitation/inhibition (E/I) balance [11, 50–56]. In addition to controlling homeostatic balance at baseline, the precise firing of pyramidal neurons following sensory stimulation also depends on PV neuron-mediated feed-forward inhibition [50, 57–59]. Hippocampal place cell activity, for instance, is generated through an interaction between dendritic excitation by pyramidal cells and perisomatic inhibition by PV interneurons, and optogenetic silencing of PV interneurons increases the firing rate of pyramidal neurons in their place fields during behavior [60]. Entorhinal cortex stellate excitatory cells were shown to be mainly connected via PV interneurons, and this inhibitory microcircuit was found to shape grid-cell firing patterns [61]. By dynamically controlling the firing activity of excitatory cells, PV neurons also play a role in defining the exact population size of pyramidal neurons activated in a specific circuit [37, 62, 63]. This property of PV neurons seems indispensable in the recruitment of defined neuronal ensembles that underlie memory formation and storage (further discussed below).
Control of network synchrony and generation of network oscillations
Given their ability to control microcircuits and reliably fire at high frequencies, PV neurons play a crucial role in network synchrony and the generation of network oscillations (Fig. 1B, C). Many studies have demonstrated that PV neurons are necessary for the generation of gamma oscillations [22, 50, 64–67] (see review [68]), which range from 30 to 120 Hz and are important for information processing and memory formation [69]. Ex vivo slice and whole-cell recordings demonstrated that PV interneuron-mediated inhibition is crucial to generate gamma rhythms [68, 70]. Recently, autaptic inhibition was shown to facilitate the tuning of PV neuron firing to gamma oscillations [22]. In vivo recordings of neuronal activity further showed that PV neuron APs are phase-locked to gamma oscillations [1, 65, 71], and optogenetic manipulation of PV neurons significantly modulates gamma oscillations [72–74].
In vivo experiments have also demonstrated that PV neuron activity is important for theta oscillations and hippocampal sharp-wave ripple activity, which are both synchronized with cortical oscillations named spindles (12–15 Hz) and are important for memory consolidation [67, 75–78]. Royer et al. found that PV interneurons fire maximally during sharp-wave ripples and preferentially before the trough of the theta wave in head-fixed mice during treadmill running, and that optogenetic silencing of PV interneurons impaired the spike-theta phase relationship of pyramidal neurons during behavior [60]. Optogenetic inhibition of CA1 PV neurons was also shown to disrupt the phase-locking and firing coherence of pyramidal neurons during ripples in CA1 [59]. Optogenetic stimulation of PV neurons, on the other hand, increased theta coherence as well as the firing rate of pyramidal cells during theta oscillations [59, 74, 79]. Stark et al. activated PV interneurons in the hippocampus and neocortex of freely behaving mice and noted that PV neuron activation induced theta-band-limited spiking in pyramidal cells, providing direct evidence for the role of active PV interneurons in theta frequency-favored spiking in these cells [46, 59]. On a network level, chemogenetic inhibition of PV neurons in either CA1 or mPFC blocked the increase in ripple-spindle coupling between these two regions, a phenomenon known to be important in memory consolidation, and prevented the increase in sleep-associated delta, theta and ripple CA1 LFP spectral power [67, 78]. Selective removal of the GABA-A receptor γ2 subunit from PV neurons, which impairs GABA-A receptors function and accordingly fast synaptic inhibition onto PV neurons, was also found to impair theta oscillations and theta-gamma coupling in the CA1, confirming that these network properties critically depend on synaptic inhibition of PV neurons [80]. More recently, PV neuron myelination was shown to significantly contribute to the synchrony of cortical oscillations and the behavioral state-dependent modulation of network activity [20].
PV neurons in plasticity, learning and memory
Learning and memory
In view of the importance of PV neurons in network synchrony and in the precise control of microcircuits, their functional involvement in learning and memory is not surprising. Indeed, removal of PV interneurons selectively from the hippocampal CA1 area disrupts spatial working memory [81], and impairing excitatory inputs onto PV neurons has profound effects on learning and memory and associated network alterations [73, 82, 83]. PV interneurons also play a key role in aversive and appetitive associative memory paradigms across brain regions [62, 84–89]. More recently it was shown that when hippocampal PV neuron activity is inhibited during sleep directly following fear-conditioning (FC), memory consolidation is blocked and fear memory impaired [67, 75, 78]. Furthermore, inhibition of PV neurons in the mPFC after FC disrupted long-term fear memory consolidation by impairing ripple-spindle coupling between the hippocampus and the PFC. Inhibiting PV neurons after consolidation or during retrieval had no effect on fear memory [78].
As neuronal excitability determines the recruitment of pyramidal neurons to memory ensembles or engrams [90], PV neurons are likely to also play a role in controlling which neurons are recruited to these ensembles and which are not (Fig. 3). It is hypothesized that PV neurons shape neuronal ensembles by preventing undesired activation of excitatory neurons (see review [91]). Thereby, they may be involved in controlling the size of an engram or its reactivation, and thus contribute to the storage and recall of specific memories [38, 67, 92, 93]. Morrison and colleagues found that silencing PV neurons in the lateral amygdala during FC increases the size of the engram, as indicated by the expression of the activity-dependent gene Arc [94]. PV interneurons are able, via their global and local inhibitory connections, to both shape neuronal ensembles and control the firing rate of neurons involved in such ensembles [92–95]. PV neurons also regulate the spatial tuning of place cells and the maintenance of place fields [96]. Taken together, multiple roles of PV interneurons in contextual memory consolidation have been described. PV cells control the firing rate of excitatory neurons during memory consolidation, shape theta and gamma oscillations, and ensure that only context-specific activities are retained in a local ensemble. It is important to note that other interneurons have also been suggested to control the size of neuronal ensembles involved in memory processing. Somatostatin (SST)-positive interneurons in the hippocampus are activated following FC and suppress surrounding dentate gyrus granule cell activity [97, 98]. Inhibiting SST neurons in the dentate gyrus during FC training enhanced freezing behavior during recall and increased the number of cFos-positive granule cells. Conversely, activation of SST neurons during training led to an impairment in FC memory and a reduced number of cFos-positive granule cells compared to control mice.
Fig. 3. A potential role for PV neurons in shaping memory-specific neuronal ensembles.
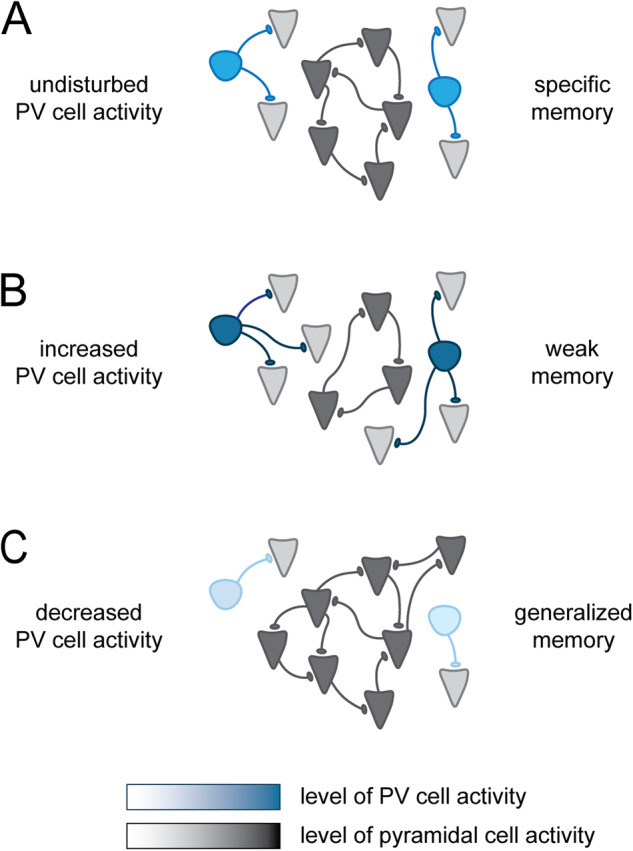
A PV neurons shape memory engrams by preventing excessive activation of pyramidal cells. They thus control the size of an engram and contribute to the storage and recall of specific memories. B When PV neuron activity is increased, pyramidal cells receive too much inhibition during memory allocation, and smaller engrams are formed. This would hypothetically result in weaker memories. C When PV neuron activity is decreased, too many pyramidal cells are recruited in memory engrams, which would hypothetically lead to larger engrams and generalization of memories. It is important to note that in addition to PV cells, other interneurons have also been shown to play a crucial role in shaping neuronal memory ensembles (see main text for details).
PV neuron plasticity and perineuronal nets
Donato and colleagues put forward a mechanism of PV neuron plasticity by which PV neuron activity is regulated through learning. They showed that PV expression levels in PV neurons correlate with GAD67 levels, and thus may reflect GABAergic output. Environmental enrichment as well as Morris water maze training induced an increase in the relative proportion of what they defined as low-PV neurons, i.e., late-born PV neurons that are marked by relatively low levels of PV. These low-PV neurons also showed an increase in plasticity as measured by a higher turn-over of synapses onto PV neurons and more inhibitory synaptic puncta. Accordingly, chemogenetic inhibition of PV neurons also enhanced plasticity and learning and improved memory formation in familiar object recognition and Morris water maze tasks. On the other hand, contextual FC increased the relative proportion of high-PV neurons, i.e., early-born PV neurons that express relatively high levels of PV, and decreased PV neuron plasticity. Chemogenetic activation of PV neurons disrupted learning and memory in familiar object recognition and Morris water maze tasks by impairing PV plasticity [34, 35] (see review [99]). Interestingly, using chondroitinase ABC, an enzyme that digests chondroitin sulfate proteoglycans in perineuronal nets (PNNs), the authors were able to induce a shift towards a low-PV state [35]. PNNs are net-like structures around specific types of neurons formed by extracellular matrix (ECM) proteins. They are critically involved in brain development and may also control plasticity in the adult brain [100]. In the cortex and the CA1 region of the hippocampus, PNNs are predominantly found around PV interneurons. They have been extensively studied in the visual cortex, where they are necessary for the onset and end of a critical period of brain development [100]. Many studies have now shown that PNNs can affect PV neuron activity and plasticity, and in turn play an important role in learning and memory (see reviews [100, 101]).
Pv interneuron dysfunction in AD
PV neuron dysfunction has been extensively linked to brain diseases that involve network alterations, decreased plasticity and memory impairment, including Alzheimer’s disease (AD). AD is marked by memory impairment and aberrant neuronal network activity, including alterations in the activation and deactivation of specific brain regions, impairments in oscillatory rhythmic activity and network hypersynchrony. Recent studies show that these changes in network activity are causally linked to memory deficits in AD. Importantly, in AD patients as well as in animal models of AD, restoring network function can improve cognitive abilities, indicating that network imbalance and its underlying neuronal mechanisms, including the involvement of PV neurons, provide attractive substrates for clinical intervention.
GABAergic dysfunction in AD
APOE4 expression and amyloid-beta (Aβ) accumulation, the former being an important risk factor and the latter a classical hallmark of AD, have both been linked to impaired GABAergic transmission and E/I imbalance, causing hyperexcitability of neuronal networks [102–105]. (For an excellent review on risk factors and protein aggregation hallmarks in AD we refer the reader to [106]). AD patients have an increased incidence and prevalence of seizures (15% and 21% respectively), which is seven times higher than people without dementia [107, 108]. Interestingly, epileptiform activity has also been linked to increased Aβ production and the accelerated deposition of amyloid plaques, hence contributing to AD progression [109, 110]. Subtle changes in hippocampal network activity during the preclinical stages of AD have been proposed in many studies as an early biomarker for the disease [111].
In line with these findings, studies in mouse models of AD have demonstrated that increased expression of amyloid precursor protein (APP) or APOE4 and accumulation of Aβ or tau, all can cause network hypersynchrony (i.e., an aberrant increase in network activity), spontaneous epileptiform activity and inhibitory dysfunction [69, 110]. Palop and colleagues first reported a compensatory remodeling of inhibitory hippocampal circuits as a consequence of aberrant excitation and hypersynchrony [112]. In a follow-up study, they showed that hypersynchrony and epileptiform activity are caused by inhibitory neuron dysfunction [113]. Several studies then confirmed that GABAergic dysfunction is at the core of neuronal network impairment in AD [102, 105, 114–118]. Busche and colleagues reported brain region connectivity disturbances in APP23xPS45 transgenic mice, in particular disturbed long-range coherence among distant cortical areas and cortico-hippocampal and thalamo-cortical areas. Interestingly, they showed that these disturbances are due to impaired GABAergic transmission leading to E/I imbalance. Increasing GABAergic transmission using benzodiazepam treatment, but not decreasing excitatory transmission by blocking glutamate receptors, restored long-range coherence and network dynamics in these mice [119]. Together, these findings provide evidence of a hyperactive neuronal circuitry in AD that is caused primarily by GABAergic dysfunction (see review [102]).
Impairment of neuronal oscillations in AD
As discussed above, E/I balance is crucial for network oscillations and a loss of inhibitory control can shift E/I balance and induce significant changes in theta and gamma oscillations. Accordingly, various studies observed alterations in oscillatory activity in AD patients in the theta [120–122] and gamma ranges [123–127]. It is important to note that learning has been associated with changes in gamma oscillations and the subsequent encoding of memories has been linked to an increase in gamma power [128–130]. Gamma power in the hippocampus and the frontal cortex decrease with normal aging, suggesting that they are an important substrate for age-related cognitive decline in general [131]. Furthermore, EEG and MEG measurements have revealed AD-associated alterations of brain oscillatory activity at other frequencies, i.e., delta, alpha and beta [132]. Additional studies have reported alterations in hippocampal network activation in AD patients, as well as an increased activation of the default mode network (DMN), known to be involved in high-level cognitive function, during memory recall [69]. Changes in DNM activation also occur during healthy aging as well as in populations at risk for AD, including people with mild cognitive impairment (MCI) and APOE-4 carriers [133]. As such, alterations in hippocampal and cortical oscillatory activity have been proposed as early biomarkers of AD [111, 134]. Notably, all AD mouse models to our knowledge present, to different degrees, alterations in network oscillations and many studies have confirmed specific changes in gamma and theta oscillations in vivo and ex vivo (Table 1), making oscillatory network changes one of the most robust endophenotypes in mouse models of AD that matches clinical observations in humans.
Table 1.
Altered neuronal oscillations and PV neuron impairments in mouse models of AD.
| AD model | Oscillatory changes | PV neuron dysfunction | Rescue experiments |
|---|---|---|---|
| hAPP |
◄ Theta (ex vivo) [211] |
◄ Functional impairment of PV neurons [113, 147, 149] ◄ PV neuron excitability decreased [211] |
◄ Nav1.1 overexpression [113] ◄ 40 Hz PV stimulation [147] ◄ MGE-derived neuron transplant [149] ◄ Erb4 deletion from PV neurons [212] |
| 5xFAD | ◄ Gamma [148] |
◄ Functional impairment of PV neurons [148] |
◄ 40 Hz PV stimulation [148] |
| APP/PS1 |
◄ Gamma [154] ◄ Theta [215] ◄ Theta and Gamma [216] ◄ Gamma (ex vivo) [217] |
◄ Functional impairment of PV neurons [142] |
◄ Restoring PV activity [142] ◄ Restoring GABAergic activity [216] ◄ Transplantation of GABAergic interneuron progenitor [150] ◄ Rescue with Citalopram [218] |
| TgCRND8 |
◄ Gamma [219] ◄ Theta and Gamma (ex vivo) [220] |
◄ Decreased PV expression [135, 138, 139, 219] | ◄ γ-secretase (semagacestat) inhibition recued Nav1.1 levels and spatial memory[219] |
| P301L | ◄ Theta and Gamma [221] | ◄ Decreased PV expression [167] | ◄ Gamma entrainment [222, 223] |
| Tg2576 | ◄ Theta [224, 225] |
◄ Input onto PV [151] |
◄ EE [136] ◄ Optogenetic activation of ECIIPN–CA1PV synapses with a theta burst stimulation [151] |
| App KI | ◄ Gamma [226] |
◄ Functional impairment ◄ Decreased PV expression [140] |
◄ GABA agonist [140] |
| APP(SL)/PS1 KI | N/A | ◄ Decreased PV expression [141] | N/A |
| rTg4510 |
◄ Theta [227] ◄ Gamma (ex vivo) [228] |
◄ No change in PV expression [228] | N/A |
| TAS10 | ◄ Gamma (ex vivo) [230] | N/A | N/A |
| 3xTg-AD | N/A | ◄ Decreased PV expression [231] | N/A |
| VLW | N/A | ◄ Input onto PV [232] | N/A |
| TauPS2APP | N/A | ◄ Decreased PV expression [233] | N/A |
| App/PS1-CB1−/− | N/A | ◄ Decreased PV expression [234] | N/A |
| APP/PS1/Nptx2−/− | N/A | ◄ PV output onto PYR [235] | N/A |
| Aβ application |
◄ Theta [236] |
◄ PV output onto PYR [153] ◄ Decreased PV expression [242] |
N/A |
Impaired PV neuron activity in AD
Since many studies have placed PV neurons at the center of network oscillations and memory processes, as described in the sections above, it is not surprising that the observed changes in AD patients and mouse models, including changes in gamma and theta oscillations and cognitive deficits, are potentially linked to PV neuron dysfunction. Verret and colleagues were the first to show that PV neurons are impaired in hAPP mice. This was accompanied by alterations in gamma power and increased epileptic activity. Interestingly, the authors demonstrated that PV neurons show decreased expression of the Nav1.1 sodium channel subunit and reported a similar decrease in post-mortem brain tissue of AD patients. Overexpressing Nav1.1 in hAPP mice rescued network imbalance, gamma power and memory impairment in these mice [113]. In addition, a significant decrease in the number of PV neurons, as measured by a decrease in PV levels, has been reported in several AD mouse lines [135–141], although several other studies have reported no alterations in PV neuron numbers [142–146]. Given the fact that restoring GABAergic transmission, via either chemogenetics, optogenetics, genetic manipulations or even interneuron transplants, can rescue network function and behavior [82, 113, 119, 140, 142, 147–151], the reported decrease in PV neurons number may reflect a decrease in PV expression rather than an actual loss of neurons. Additional studies exploring the correlation between PV expression and GABAergic activity in AD and investigating PV neuron survival are necessary to test whether PV neurons are more prone to neuronal loss in AD. One consistent finding among all these studies, however, is a decrease in inhibitory transmission and activity in AD that is likely due to a specific impairment of PV neurons, even though other interneurons may also be contributing to this dysfunction [98, 152, 153], resulting in a failure of inhibitory control, oscillatory changes, an overall increased excitation and epileptic activity, and cognitive decline.
Restoring or mimicking PV neuron function in AD
In the last decade, many studies have focused on restoring PV neuron function in order to rescue AD pathology and cognitive impairment (Table 1). Data from our lab showed that restoring PV neuron activity in the hippocampus of APP/PS1 mice has beneficial effects on hippocampal networks and spatial memory on the short and long-term. Iaccarino and colleagues showed that in 5xFAD mice, gamma activity is significantly lower than in wild type control mice. Stimulating PV interneurons optogenetically at 40 Hz for 1 h restored gamma activity and led to a significant microglia-mediated decrease in soluble and insoluble Aβ levels, hence delaying AD pathology [148]. Moreover, other studies have validated that either direct or indirect stimulation of PV interneurons can restore theta and gamma oscillations as well as memory performance in different AD mouse models, as summarized in Table 1. Taken together, these studies provide a strong case for PV neuron hypofunction being causally involved in network and memory problems in AD and providing a substrate for non-invasive interventions.
Mechanisms of PV neuron dysfunction in AD
While the above studies highlight PV neurons as an attractive and highly relevant target for restoring network function and memory in AD, an important question remains whether PV neuron impairment is a cause or a consequence of AD pathogenesis. One possibility is that PV neuron dysfunction is due to Aβ-dependent intrinsic changes in channel properties or expression. Alternatively, PV neurons may become impaired because of changes in afferent synaptic inputs and impaired synaptic transmission onto PV neurons, or due to a high vulnerability in general resulting from their above-mentioned unique fast-spiking properties. Further insight into the earliest PV neuron alterations in AD might help to shed light on this issue.
Early and transient hyperexcitability of PV neurons
In a recent series of experiments from our lab we investigated PV neuron intrinsic properties and associated microcircuit adaptations in the hippocampus of APP/PS1 mice at an early disease stage, i.e., at 3 months of age, when memory deficits are first detected. We showed that hippocampal PV neurons, but not pyramidal neurons, are initially and transiently hyperexcitable at this age. At a later disease stage, i.e., at 7 months of age, PV neurons appeared to be hypoactive while pyramidal neurons were hyperexcitable. Importantly, inhibiting PV neuron activity at an early stage or stimulating PV neuron activity at a later stage, both restored Morris water maze performance in APP/PS1 mice, suggesting that both states, hyperexcitable first and hypoexcitable later, are causally linked to memory impairment in AD [142]. [140]Interestingly, early hyperexcitability of PV neurons is paralleled by an increase in PNNs surrounding PV cells, which may serve to support the increased energy demand of these cells, but at the same time also limits plasticity and causes memory impairment [146]. Together, these findings suggest biphasic alterations in PV neuron activity during amyloidosis in APP/PS1 mice and potentially different mechanisms of memory impairment at different disease stages. Interestingly, biphasic alterations in inhibitory transmission were also reported in other studies [154, 155] and biphasic alterations in network connectivity have been observed during AD progression in patients [156, 157]. It would be relevant to test whether an aberrant early increase in PV neuron activity contributes to the silencing of the (prospective) engram neurons and impairing memory retrieval (Fig. 3). In fact, Roy et al. showed a decrease in FC-activated excitatory engram neurons in the dentate gyrus of AD mice. Optogenetic reactivation of these cells restored fear memory in AD mice [158]. Future studies could investigate whether silencing of PV neurons in the dentate gyrus could also restore fear memory in these mice.
To test whether early PV neuron hyperexcitability causally contributes to AD pathogenesis, we artificially induced PV neuron hyperexcitability in wild type mice and used selective chemogenetic activation to trigger a persistent AD-like hippocampal network state [159]. Under these conditions PV neurons not only became persistently hyperexcitable, but they were also more sensitive to a low dose of amyloid-beta (Ab) injected directly into the hippocampus. While infusing the same concentration of Aβ in healthy mice had no cellular nor behavioral effect, under conditions of induced PV neuron hyperexcitability, this low concentration of Aβ was able to cause PV neuron hypo-excitability, increase pyramidal neuron firing frequency, disrupt synaptic transmission onto pyramidal neurons and significantly impair spatial memory. These findings suggest that early PV neuron hyperexcitability could be a first and causal step in later PV neuron hypofunction, but only under AD-like conditions of increased Aβ levels, which may be a key mechanism in triggering network and memory impairments in AD. Given the biphasic nature of PV neuron dysfunction in APP/PS1 mice, we hypothesize that the reported decrease in PV expression and reduced activity of PV neurons observed at later stages in various AD mouse models are a consequence of early PV neuron hyperexcitability (Fig. 4). Indeed, reducing PV neuron activity at an early stage, using selective chemogenetic, inhibition restored synaptic transmission and intrinsic properties of both PV neurons and pyramidal neurons as well as spatial memory up to eight weeks after the end of the intervention [142].
Fig. 4. A hypothetical pathogenic mechanism for neuronal network changes in AD based on PV neuron dysfunction.

Neuronal hyperexcitability and network dysfunction are commonly observed in AD patients and in mouse models of AD. Taken together, published data seem to support a model in which interneurons, in particular PV interneurons, play a key role. Early in disease pathogenesis, before any significant aggregation of Aβ into plaques, soluble Aβ causes an aberrant increase in the excitability of PV cells and a corresponding increase in network inhibition. This is accompanied by an increase in perineuronal nets around PV cells, possibly to support the increased energy demand due to higher activity. Pyramidal cells at this stage seem unaffected, possibly because they are less vulnerable or less sensitive to Aβ toxicity. At a later stage, pyramidal cells also show increased excitability, possibly as a homeostatic response to restore excitation/inhibition balance. Increasing Aβ concentrations and Aβ aggregation may contribute to the increased excitability of pyramidal cells. Finally, PV cells become hypoactive while pyramidal cells remain hyperexcitable due to increasing Aβ pathology, resulting in an overall network hyperexcitability. The reduced activity of PV neurons is linked to a reduced expression and/or function of Nav1.1 and Kv3 channels. This stage is supported by both patient and mouse data showing reduced gamma oscillations and increased epileptic activity.
PV neuron hyperexcitability may also contribute to amyloid pathology, as early restoration of PV neuron activity led to a decrease in amyloid pathology, and blocking Aβ production specifically in GABAergic neurons significantly reduced plaque pathology [142, 148, 160]. Indeed, APP is prominently expressed in a subset of hippocampal interneurons; 53% of PV cells in the CA1 region of the hippocampus are APP-positive and hyperphosphorylated tau as well as Aβ were found to accumulate in PV neurons in different AD mouse models [160–162].
Selective vulnerability of PV neurons and associated microcircuits
The observation that early PV neuron hyperexcitability is causal to several AD-like cellular, network and behavioral adaptations is intriguing, but a question that remains is what elicits PV neuron hyperexcitability in the first place? One possibility is that these neurons are more vulnerable to Ab-induced stress than other neurons. Treating APP/PS1 mice early with a β-site APP cleaving enzyme 1 (BACE1) inhibitor completely restored PV neuron excitability and function, and when hippocampal slices from wild type mice were exposed to Aβ ex vivo, PV neurons quickly became hyperexcitable while pyramidal neurons did not [142]. Moreover, Aβ significantly impaired PV neuron function in an induced-PV hyperexcitability model [159]. Multiples studies have highlighted an increased vulnerability of PV neurons to Aβ toxicity (Table 1), suggesting that PV neuron dysfunction in AD is, at least in part, Ab-dependent. Little is known about the direct molecular interactions of Aβ with receptors or channels on GABAergic neurons, or on PV neurons specifically. As mentioned previously, PV neurons express specific ion channel subunits, such as Nav1.1 and Kv3, which are required for their fast-spiking properties. Interestingly, BACE1 expression is linked to Nav1.1 expression as BACE1-null mice have reduced Nav1.1 protein levels [163]. Studies have also demonstrated that Aβ affects various synaptic inputs from and onto PV neurons [164, 165]. Exploring further the direct effect of Aβ on PV neuron channels and synapses would help elucidate the mechanisms underlying PV neuron vulnerability in AD. The combined observation of early Ab-induced PV neuron hyperexcitability and selective Ab-dependent PV cell vulnerability suggest a pathogenic feedback loop in which increasing Aβ levels cause PV neuron hyperexcitability and hyperactive PV neurons in turn contribute to increased Aβ production.
An interplay between tau and amyloid?
Tau mouse models also display aberrant network activity and inhibitory network impairments. Tau pathology was found to cause an increase in interneuron activity, grid cell dysfunction and loss of excitatory neurons in mice expressing tau-P301L in the entorhinal cortex. This was accompanied by an enhancement of theta power and a disruption of spatial memory [166]. In another study, P301L mice showed a loss of interneurons, which was associated with impaired synaptic plasticity as well as cognitive deficits. Moreover, a GABA-A receptor agonist was effective in restoring both the cognitive and synaptic impairments, supporting the role of inhibitory neurons in tau-dependent neuronal network dysfunction [167]. The question remains if these changes in interneuron function reflect a direct effect of tau toxicity or network adaptations in response to tau pathology.
Interestingly, Aβ seems to play a role in tau pathology. Aβ was shown to increase tau spreading and toxicity in a study where rTgTauEC mice were crossed with APP/PS1 mice [168]. Several studies have confirmed that tau and Aβ pathology are usually comorbid [169, 170]. A recent study by Busche et al. puts forward a new mechanism of network dysfunction where Aβ promotes neuronal hyperactivity while tau suppresses activity. Whereas in APP/PS1 mice a significant percentage of neurons were clearly hyperactive, in rTg4510 mice many neurons were silent. When the two lines were crossed, tau blocked Ab-dependent hyperactivity, resulting in mainly silent neurons. Intriguingly, when the authors suppressed tau expression, they could rescue circuit activity in tau mice but not in mice with both tau and Aβ pathology [171]. As Aβ is undeniably involved in neuronal hyperactivity, it might be that an early Ab-dependent hyperactivity of PV neurons paves the way to an imbalanced activity and circuit dysfunction, which worsens with increasing tau levels and eventually leads to neurodegeneration.
Glial cells and inhibitory dysfunction
Glial cells have been at the center of some recent AD studies [172–174]. Their involvement may go beyond a simple response to protein aggregation and neurodegeneration, as their roles in maintaining E/I balance [175], synaptic plasticity [176], network activity [177] and memory processing [178, 179] have been firmly established. However, as regulators of neuronal network balance and synaptic transmission, their contribution to AD needs further exploration.
Microglia are thought to be important for Aβ clearance [180–183]. Activated microglia are present in close proximity of amyloid plaques [184–186]. Genome-wide association studies for AD have up to now identified 75 risk loci for AD and pathway enrichment analysis of risk genes clearly confirmed microglial involvement [187]. For instance, a mutation in the triggering receptor expressed on myeloid cells 2 (TREM2), a receptor specific to microglia, significantly increases risk for AD [47, 188]. Importantly, this mutation was demonstrated to reduce the role of TREM2 in the activation of microglia, hence leading to reduced Aβ clearance [189, 190]. In a study by Iaccarino et al., optogenetic 40 Hz stimulation of PV neurons induced morphological changes in microglia of 5xFAD mice and reduced the levels of both soluble and insoluble Aβ [148]. This suggests that PV neuron activity is causally linked to microglia activation and Aβ clearance. The mechanism behind this link remains unclear, and a remaining question is whether microglia also play a role in regulating PV neuron activity. Several studies have reported a microglia-dependent loss of synapses is early in AD, leading to neurodegeneration and memory loss [191–193]. Moreover, microglia are able to selectively prune inhibitory synapses, although this was only observed during postnatal development [25]. Thus, microglia could be contributing to inhibitory dysfunction and network alterations in AD by weakening inhibitory synapses and through a defective clearance of Aβ around specific synapses of interneurons.
Astrocytes have similarly been implicated in network imbalance in AD because of their role in synaptic, specifically GABAergic, transmission [194–200]. GABA levels in reactive hippocampal astrocytes are increased in AD mouse models and in AD patients [201–203]. It was further revealed, in a mouse model of AD, that GABA released from astrocytes activates GABA-A receptors and impairs LTP induction in the hippocampus. Notably, when astrocytic GABA synthesis or release were inhibited, both LTP and memory were restored [202]. Several other studies have confirmed the role of reactive astrocytes in network imbalance and impaired synaptic plasticity in AD [196, 204]. Interestingly, astrogliosis has been reported at a very early stage of AD [174, 205]. Taken together, network imbalance in AD, and specifically the dysfunction of inhibitory neurons, might in part be due to alterations in astrocyte activity at early disease stages. Further studies are needed to confirm a potential role of astrocytes specifically in PV neuron dysfunction in AD.
Finally, while AD has been mainly considered as a gray matter disorder, recent evidence points to myelin impairment as a potential disease mechanism. It has been proposed that myelin damage might occur earlier than Aβ and tau pathology in AD [206], and restoring myelin deficits in APP/PS1 mice rescues cognition and hippocampal physiology [207]. Interestingly, activity-dependent myelination driven by absence seizures was shown to contribute to epilepsy progression [208]. In a recent study, myelinated axons of PV neurons were found to contain a significant number of clustered mitochondria. Mitochondrial calcium transients were shown to be modulated by myelination on PV axons and myelin loss caused deficits in mitochondrial calcium responses at branch points [209]. These findings highlight an intricate link between myelination and metabolic support that could specifically affect PV cells in the early stages of AD. Whether deficits in PV neuron myelination could also be involved in the increased risk for epilepsy or the cognitive decline observed in AD requires further investigation, especially with the emergence of glial cells as important players in inhibitory dysfunction and network abnormalities.
Conclusion
PV neuron dysfunction is clearly related to network impairments and cognitive deficits in AD. What makes these neurons so vulnerable? One hypothesis has been that their ability to fire at high frequencies, and the corresponding high energy consumption, contribute to PV neuron vulnerability. However, a recent paper by Hu and Jonas showed that high-frequency firing of PV cells is very efficient. The energy required for one AP was only 1.6 times the theoretical minimum. This efficiency was due to the fast inactivation kinetics of sodium channels combined with a delayed activation of Kv3-type potassium channels in PV neurons [210]. Hence, any alteration in the expression or activity of these channels in AD could make PV neurons vulnerable. In this review, we propose that PV neurons, because of their distinct molecular and morphological characteristics, are selectively vulnerable to Aβ toxicity, and that their selective vulnerability is at the core of early network dysfunction in AD. More studies are needed to investigate how Aβ selectively affects the excitability of PV neurons, and which specific ion channels are involved. Aβ may also impair afferent inputs onto PV neurons, which on the long term could lead to a hypofunction of PV neurons. Finally, specific AD-related genetic risk factors, such as APOE4, may be contributing to early changes in PV neuron excitability in the adult brain, which renders the neuronal circuitry more vulnerable to Aβ toxicity, initiating network dysfunction, hyperactivity of principal networks and cognitive deficits.
Author contributions
SH drafted the manuscript. SH and REvK designed the figures and tables. All authors reviewed and edited the final version of the manuscript.
Funding
SH is funded by the Blaschko Trust (Department of Pharmacology and Linacre College, University of Oxford). REvK received funding from the Netherlands Organization for Health Research and Development (ZonMw; grant 91218018).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321:53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu H, Gan J, Jonas P. Fast-spiking, parvalbumin + GABAergic interneurons: From cellular design to microcircuit function. Science. 2014;345:1255263. doi: 10.1126/science.1255263. [DOI] [PubMed] [Google Scholar]
- 3.Lapray D, Lasztoczi B, Lagler M, Viney TJ, Katona L, Valenti O, et al. Behavior-dependent specialization of identified hippocampal interneurons. Nat Neurosci. 2012;15:1265–1271. doi: 10.1038/nn.3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu H, Jonas P. A supercritical density of Na(+) channels ensures fast signaling in GABAergic interneuron axons. Nat Neurosci. 2014;17:686–693. doi: 10.1038/nn.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bucurenciu I, Kulik A, Schwaller B, Frotscher M, Jonas P. Nanodomain coupling between Ca2+ channels and Ca2+ sensors promotes fast and efficient transmitter release at a cortical GABAergic synapse. Neuron. 2008;57:536–45. doi: 10.1016/j.neuron.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 6.Goldberg EM, Clark BD, Zagha E, Nahmani M, Erisir A, Rudy B. K+ channels at the axon initial segment dampen near-threshold excitability of neocortical fast-spiking GABAergic interneurons. Neuron. 2008;58:387–400. doi: 10.1016/j.neuron.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rudy B, McBain CJ. Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 2001;24:517–26. doi: 10.1016/s0166-2236(00)01892-0. [DOI] [PubMed] [Google Scholar]
- 8.Hu H, Martina M, Jonas P. Dendritic mechanisms underlying rapid synaptic activation of fast-spiking hippocampal interneurons. Science. 2010;327:52–8. doi: 10.1126/science.1177876. [DOI] [PubMed] [Google Scholar]
- 9.Goldberg EM. Specific functions of synaptically localized potassium channels in synaptic transmission at the neocortical GABAergic fast-spiking cell synapse. J Neurosci. 2005;25:5230–5. doi: 10.1523/JNEUROSCI.0722-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richardson A, Ciampani V, Stancu M, Bondarenko K, Newton S, Steinert JR, et al. Kv3.3 subunits control presynaptic action potential waveform and neurotransmitter release at a central excitatory synapse. ELife. 2022;11:e75219. doi: 10.7554/eLife.75219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campanac E, Gasselin C, Baude A, Rama S, Ankri N, Debanne D. Enhanced intrinsic excitability in basket cells maintains excitatory-inhibitory balance in hippocampal circuits. Neuron. 2013;77:712–22. doi: 10.1016/j.neuron.2012.12.020. [DOI] [PubMed] [Google Scholar]
- 12.Roth FC, Hu H. An axon-specific expression of HCN channels catalyzes fast action potential signaling in GABAergic interneurons. Nat Commun. 2020;11:2248. doi: 10.1038/s41467-020-15791-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elgueta C, Köhler J, Bartos M. Persistent discharges in dentate gyrus perisoma-inhibiting interneurons require hyperpolarization-activated cyclic nucleotide-gated channel activation. J Neurosci. 2015;35:4131–9. doi: 10.1523/JNEUROSCI.3671-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szegedi V, Bakos E, Furdan S, Kovács BH, Varga D, Erdélyi M, et al. HCN channels at the cell soma ensure the rapid electrical reactivity of fast-spiking interneurons in human neocortex. PLoS Biol. 2023;21:e3002001. doi: 10.1371/journal.pbio.3002001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bucurenciu I, Bischofberger J, Jonas P. A small number of open Ca2+ channels trigger transmitter release at a central GABAergic synapse. Nat Neurosci. 2010;13:19–21. doi: 10.1038/nn.2461. [DOI] [PubMed] [Google Scholar]
- 16.Zaitsev AV, Povysheva NV, Lewis DA, Krimer LS. P/Q-type, but not N-type, calcium channels mediate GABA release from fast-spiking interneurons to pyramidal cells in rat prefrontal cortex. J Neurophysiol. 2007;97:3567–73. doi: 10.1152/jn.01293.2006. [DOI] [PubMed] [Google Scholar]
- 17.Chen C, Jonas P. Synaptotagmins: that’s why so many. Neuron. 2017;94:694–6. doi: 10.1016/j.neuron.2017.05.011. [DOI] [PubMed] [Google Scholar]
- 18.Xu J, Mashimo T, Sudhof TC. Synaptotagmin-1, -2, and -9: Ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007;54:567–81. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Stedehouder J, Kushner SA. Myelination of parvalbumin interneurons: a parsimonious locus of pathophysiological convergence in schizophrenia. Mol Psychiatry. 2017;22:4–12. doi: 10.1038/mp.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dubey M, Pascual-Garcia M, Helmes K, Wever DD, Hamada MS, Kushner SA, et al. Myelination synchronizes cortical oscillations by consolidating parvalbumin-mediated phasic inhibition. Elife. 2022;11:e73827. doi: 10.7554/eLife.73827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Micheva KD, Kiraly M, Perez MM, Madison DV. Conduction velocity along the local axons of parvalbumin interneurons correlates with the degree of axonal myelination. Cereb Cortex. 2021;31:3374–92. doi: 10.1093/cercor/bhab018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deleuze C, Bhumbra GS, Pazienti A, Lourenço J, Mailhes C, Aguirre A, et al. Strong preference for autaptic self-connectivity of neocortical PV interneurons facilitates their tuning to γ-oscillations. PLoS Biol. 2019;17:e3000419. doi: 10.1371/journal.pbio.3000419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deleuze C, Pazienti A, Bacci A. Autaptic self-inhibition of cortical GABAergic neurons: synaptic narcissism or useful introspection? Curr Opin Neurobiol. 2014;26:64–71. doi: 10.1016/j.conb.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 24.Szegedi V, Paizs M, Baka J, Barzó P, Molnár G, Tamas G, et al. Robust perisomatic GABAergic self-innervation inhibits basket cells in the human and mouse supragranular neocortex. Elife. 2020;9:e51691. doi: 10.7554/eLife.51691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Favuzzi E, Huang S, Saldi GA, Binan L, Ibrahim LA, Fernández-Otero M, et al. GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell. 2021;184:5686. doi: 10.1016/j.cell.2021.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Micheva KD, Kiraly M, Perez MM, Madison DV. Extensive structural remodeling of the axonal arbors of parvalbumin basket cells during development in mouse neocortex. J Neurosci. 2021;41:9326–39. doi: 10.1523/JNEUROSCI.0871-21.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benamer N, Vidal M, Balia M, Angulo MC. Myelination of parvalbumin interneurons shapes the function of cortical sensory inhibitory circuits. Nat Commun. 2020;11:5151. doi: 10.1038/s41467-020-18984-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang L-P, Zhao N, Caudal LC, Chang H-F, Zhao R, Lin C-H, et al. Impaired bidirectional communication between interneurons and oligodendrocyte precursor cells affects social cognitive behavior. Nat Commun. 2022;13:1394. doi: 10.1038/s41467-022-29020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gallo NB, Berisha A, Van Aelst L. Microglia regulate chandelier cell axo-axonic synaptogenesis. Proc Natl Acad Sci USA. 2022;119:e2114476119. doi: 10.1073/pnas.2114476119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller MN, Okaty BW, Kato S, Nelson SB. Activity-dependent changes in the firing properties of neocortical fast-spiking interneurons in the absence of large changes in gene expression. Dev Neurobiol. 2011;71:62–70. doi: 10.1002/dneu.20811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dehorter N, Ciceri G, Bartolini G, Lim L, del Pino I, Marín O. Tuning of fast-spiking interneuron properties by an activity-dependent transcriptional switch. Science. 2015;349:1216–20. doi: 10.1126/science.aab3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pouchelon G, Dwivedi D, Bollmann Y, Agba CK, Xu Q, Mirow AMC, et al. The organization and development of cortical interneuron presynaptic circuits are area specific. Cell Rep. 2021;37:109993. doi: 10.1016/j.celrep.2021.109993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daw MI, Ashby MC, Isaac JTR. Coordinated developmental recruitment of latent fast spiking interneurons in layer IV barrel cortex. Nat Neurosci. 2007;10:453–61. doi: 10.1038/nn1866. [DOI] [PubMed] [Google Scholar]
- 34.Donato F, Chowdhury A, Lahr M, Caroni P. Early- and late-born parvalbumin basket cell subpopulations exhibiting distinct regulation and roles in learning. Neuron. 2015;85:770–86. doi: 10.1016/j.neuron.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 35.Donato F, Rompani SB, Caroni P. Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature. 2013;504:272–6. doi: 10.1038/nature12866. [DOI] [PubMed] [Google Scholar]
- 36.Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–88. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- 37.Mendez P, Bacci A. Assortment of GABAergic plasticity in the cortical interneuron melting pot. Neural Plast. 2011;2011:976856. doi: 10.1155/2011/976856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramsaran AI, Wang Y, Golbabaei A, Yeung BA, de Snoo ML, Rashid AJ, et al. A shift in the mechanisms controlling hippocampal engram formation during brain maturation. Neuroscience. 2023;380:543–51. doi: 10.1126/science.ade6530. [DOI] [PubMed] [Google Scholar]
- 39.Di Cristo G, Chattopadhyaya B, Kuhlman SJ, Fu Y, Bélanger M-C, Wu CZ, et al. Activity-dependent PSA expression regulates inhibitory maturation and onset of critical period plasticity. Nat Neurosci. 2007;10:1569–77. doi: 10.1038/nn2008. [DOI] [PubMed] [Google Scholar]
- 40.Kuhlman SJ, Olivas ND, Tring E, Ikrar T, Xu X, Trachtenberg JT. A disinhibitory microcircuit initiates critical-period plasticity in the visual cortex. Nature. 2013;501:543–6. doi: 10.1038/nature12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Southwell DG, Froemke RC, Alvarez-Buylla A, Stryker MP, Gandhi SP. Cortical plasticity induced by inhibitory neuron. Transplant Sci. 2010;327:1145–8. doi: 10.1126/science.1183962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldberg EM, Jeong H-Y, Kruglikov I, Tremblay R, Lazarenko RM, Rudy B. Rapid developmental maturation of neocortical FS cell intrinsic excitability. Cereb Cortex. 2011;21:666–82. doi: 10.1093/cercor/bhq138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyamae T, Chen K, Lewis DA, Gonzalez-Burgos G. Distinct physiological maturation of parvalbumin-positive neuron subtypes in mouse prefrontal cortex. J Neurosci. 2017;37:4883–902. doi: 10.1523/JNEUROSCI.3325-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hafner G, Witte M, Guy J, Subhashini N, Fenno LE, Ramakrishnan C, et al. Mapping brain-wide afferent inputs of parvalbumin-expressing GABAergic neurons in barrel cortex reveals local and long-range circuit motifs. Cell Rep. 2019;28:3450–61.e8. doi: 10.1016/j.celrep.2019.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawaguchi Y, Otsuka T, Morishima M, Ushimaru M, Kubota Y. Control of excitatory hierarchical circuits by parvalbumin-FS basket cells in layer 5 of the frontal cortex: insights for cortical oscillations. J Neurophysiol. 2019;121:2222–36. doi: 10.1152/jn.00778.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swanson OK, Maffei A. From hiring to firing: activation of inhibitory neurons and their recruitment in behavior. Front Mol Neurosci. 2019;12:168. doi: 10.3389/fnmol.2019.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tremblay R, Lee S, Rudy B. GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron. 2016;91:260–92. doi: 10.1016/j.neuron.2016.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Booker SA, Vida I. Morphological diversity and connectivity of hippocampal interneurons. Cell Tissue Res. 2018;373:619–41. doi: 10.1007/s00441-018-2882-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Packer AM, Yuste R. Dense, unspecific connectivity of neocortical parvalbumin-positive interneurons: a canonical microcircuit for inhibition? J Neurosci. 2011;31:13260–71. doi: 10.1523/JNEUROSCI.3131-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Atallah BV, Scanziani M. Instantaneous modulation of gamma oscillation frequency by balancing excitation with inhibition. Neuron. 2009;62:566–77. doi: 10.1016/j.neuron.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kapfer C, Glickfeld LL, Atallah BV, Scanziani M. Supralinear increase of recurrent inhibition during sparse activity in the somatosensory cortex. Nat Neurosci. 2007;10:743–53. doi: 10.1038/nn1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Le Roux N, Amar M, Moreau A, Baux G, Fossier P. Impaired GABAergic transmission disrupts normal homeostatic plasticity in rat cortical networks. Eur J Neurosci. 2008;27:3244–56. doi: 10.1111/j.1460-9568.2008.06288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Le Roux N, Amar M, Baux G, Fossier P. Homeostatic control of the excitation-inhibition balance in cortical layer 5 pyramidal neurons. Eur J Neurosci. 2006;24:3507–18. doi: 10.1111/j.1460-9568.2006.05203.x. [DOI] [PubMed] [Google Scholar]
- 54.Nanou E, Lee A, Catterall WA. Control of excitation/inhibition balance in a hippocampal circuit by calcium sensor protein regulation of presynaptic calcium channels. J Neurosci. 2018;38:4430–40. doi: 10.1523/JNEUROSCI.0022-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Poo C, Isaacson JS. Odor representations in olfactory cortex: ‘sparse’ coding, global inhibition, and oscillations. Neuron. 2009;62:850–61. doi: 10.1016/j.neuron.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu GK, Arbuckle R, Liu BH, Tao HW, Zhang LI. Lateral sharpening of cortical frequency tuning by approximately balanced inhibition. Neuron. 2008;58:132–43. doi: 10.1016/j.neuron.2008.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wehr M, Zador AM. Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature. 2003;426:442–6. doi: 10.1038/nature02116. [DOI] [PubMed] [Google Scholar]
- 58.Wilent WB, Contreras D. Dynamics of excitation and inhibition underlying stimulus selectivity in rat somatosensory cortex. Nat Neurosci. 2005;8:1364–70. doi: 10.1038/nn1545. [DOI] [PubMed] [Google Scholar]
- 59.Stark E, Eichler R, Roux L, Fujisawa S, Rotstein HG, Buzsaki G. Inhibition-induced theta resonance in cortical circuits. Neuron. 2013;80:1263–76. doi: 10.1016/j.neuron.2013.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Royer S, Zemelman BV, Losonczy A, Kim J, Chance F, Magee JC, et al. Control of timing, rate and bursts of hippocampal place cells by dendritic and somatic inhibition. Nat Neurosci. 2012;15:769–75. doi: 10.1038/nn.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Couey JJ, Witoelar A, Zhang SJ, Zheng K, Ye J, Dunn B, et al. Recurrent inhibitory circuitry as a mechanism for grid formation. Nat Neurosci. 2013;16:318–24. doi: 10.1038/nn.3310. [DOI] [PubMed] [Google Scholar]
- 62.Lucas EK, Clem RL. GABAergic interneurons: the orchestra or the conductor in fear learning and memory? Brain Res Bull. 2018;141:13–19. doi: 10.1016/j.brainresbull.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McKenzie S. Inhibition shapes the organization of hippocampal representations. Hippocampus. 2018;28:659–71. doi: 10.1002/hipo.22803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Csicsvari J, Jamieson B, Wise KD, Buzsaki G. Mechanisms of gamma oscillations in the hippocampus of the behaving rat. Neuron. 2003;37:311–22. doi: 10.1016/s0896-6273(02)01169-8. [DOI] [PubMed] [Google Scholar]
- 65.Tukker JJ, Fuentealba P, Hartwich K, Somogyi P, Klausberger T. Cell type-specific tuning of hippocampal interneuron firing during gamma oscillations in vivo. J Neurosci. 2007;27:8184–9. doi: 10.1523/JNEUROSCI.1685-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vida I, Bartos M, Jonas P. Shunting inhibition improves robustness of gamma oscillations in hippocampal interneuron networks by homogenizing firing rates. Neuron. 2006;49:107–17. doi: 10.1016/j.neuron.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 67.Ognjanovski N, Schaeffer S, Wu J, Mofakham S, Maruyama D, Zochowski M, et al. Parvalbumin-expressing interneurons coordinate hippocampal network dynamics required for memory consolidation. Nat Commun. 2017;8:15039. doi: 10.1038/ncomms15039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- 69.Palop JJ, Mucke L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci. 2016;17:777–92. doi: 10.1038/nrn.2016.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mann EO, Mody I. Control of hippocampal gamma oscillation frequency by tonic inhibition and excitation of interneurons. Nat Neurosci. 2010;13:205–12. doi: 10.1038/nn.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buzsáki G, Wang X-J. Mechanisms of gamma oscillations. Annu Rev Neurosci. 2012;35:203–25. doi: 10.1146/annurev-neuro-062111-150444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cardin JA, Carlen M, Meletis K, Knoblich U, Zhang F, Deisseroth K, et al. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–7. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FE, Bannerman DM, et al. Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron. 2007;53:591–604. doi: 10.1016/j.neuron.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 74.Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ognjanovski N, Broussard C, Zochowski M, Aton SJ. Hippocampal network oscillations rescue memory consolidation deficits caused by sleep loss. Cereb Cortex. 2018;28:3711–23. doi: 10.1093/cercor/bhy174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gan J, Weng SM, Pernia-Andrade AJ, Csicsvari J, Jonas P. Phase-locked inhibition, but not excitation, underlies hippocampal ripple oscillations in awake mice in vivo. Neuron. 2017;93:308–14. doi: 10.1016/j.neuron.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Klausberger T, Magill PJ, Marton LF, Roberts JD, Cobden PM, Buzsaki G, et al. Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature. 2003;421:844–8. doi: 10.1038/nature01374. [DOI] [PubMed] [Google Scholar]
- 78.Xia F, Richards BA, Tran MM, Josselyn SA, Takehara-Nishiuchi K, Frankland PW. Parvalbumin-positive interneurons mediate neocortical-hippocampal interactions that are necessary for memory consolidation. Elife. 2017;6:e27868. doi: 10.7554/eLife.27868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Amilhon B, Huh CY, Manseau F, Ducharme G, Nichol H, Adamantidis A, et al. Parvalbumin interneurons of hippocampus tune population activity at theta frequency. Neuron. 2015;86:1277–89. doi: 10.1016/j.neuron.2015.05.027. [DOI] [PubMed] [Google Scholar]
- 80.Wulff P, Ponomarenko AA, Bartos M, Korotkova TM, Fuchs EC, Bähner F, et al. Hippocampal theta rhythm and its coupling with gamma oscillations require fast inhibition onto parvalbumin-positive interneurons. Proc Natl Acad Sci USA. 2009;106:3561–6. doi: 10.1073/pnas.0813176106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Murray AJ, Sauer JF, Riedel G, McClure C, Ansel L, Cheyne L, et al. Parvalbumin-positive CA1 interneurons are required for spatial working but not for reference memory. Nat Neurosci. 2011;14:297–9. doi: 10.1038/nn.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen YJ, Zhang M, Yin DM, Wen L, Ting A, Wang P, et al. ErbB4 in parvalbumin-positive interneurons is critical for neuregulin 1 regulation of long-term potentiation. Proc Natl Acad Sci USA. 2010;107:21818–23. doi: 10.1073/pnas.1010669107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.He X, Li J, Zhou G, Yang J, McKenzie S, Li Y, et al. Gating of hippocampal rhythms and memory by synaptic plasticity in inhibitory interneurons. Neuron. 2021;109:1013–.e9. doi: 10.1016/j.neuron.2021.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Çaliskan G, Müller I, Semtner M, Winkelmann A, Raza AS, Hollnagel JO, et al. Identification of parvalbumin interneurons as cellular substrate of fear memory persistence. Cerebral Cortex. 2016;26 2325–40. [DOI] [PMC free article] [PubMed]
- 85.Letzkus JJ, Wolff SB, Luthi A. Disinhibition, a circuit mechanism for associative learning and memory. Neuron. 2015;88:264–76. doi: 10.1016/j.neuron.2015.09.024. [DOI] [PubMed] [Google Scholar]
- 86.Sans-Dublanc A, Razzauti A, Desikan S, Pascual M, Monyer H, Sindreu C. Septal GABAergic inputs to CA1 govern contextual memory retrieval. Sci Adv. 2020;6:eaba5003. doi: 10.1126/sciadv.aba5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Trouche S, Koren V, Doig NM, Ellender TJ, El-Gaby M, Lopes-dos-Santos V, et al. A hippocampus-accumbens tripartite neuronal motif guides appetitive memory in space. Cell. 2019;176:1393–.e16. doi: 10.1016/j.cell.2018.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wolff SB, Grundemann J, Tovote P, Krabbe S, Jacobson GA, Muller C, et al. Amygdala interneuron subtypes control fear learning through disinhibition. Nature. 2014;509:453–8. doi: 10.1038/nature13258. [DOI] [PubMed] [Google Scholar]
- 89.Yiannakas A, Kolatt Chandran S, Kayyal H, Gould N, Khamaisy M, Rosenblum K. Parvalbumin interneuron inhibition onto anterior insula neurons projecting to the basolateral amygdala drives aversive taste memory retrieval. Curr Biol. 2021;31:2770–.e6. doi: 10.1016/j.cub.2021.04.010. [DOI] [PubMed] [Google Scholar]
- 90.Rao-Ruiz P, Yu J, Kushner SA, Josselyn SA. Neuronal competition: microcircuit mechanisms define the sparsity of the engram. Curr Opin Neurobiol. 2019;54:163–70. doi: 10.1016/j.conb.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Raven F, Aton SJ. The Engram’s dark horse: how interneurons regulate state-dependent memory processing and plasticity. Front Neural Circuits. 2021;15:750541. doi: 10.3389/fncir.2021.750541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chowdhury A, Caroni P. Time units for learning involving maintenance of system-wide cFos expression in neuronal assemblies. Nat Commun. 2018;9:4122. doi: 10.1038/s41467-018-06516-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Karunakaran S, Chowdhury A, Donato F, Quairiaux C, Michel CM, Caroni P. PV plasticity sustained through D1/5 dopamine signaling required for long-term memory consolidation. Nat Neurosci. 2016;19:454–64. doi: 10.1038/nn.4231. [DOI] [PubMed] [Google Scholar]
- 94.Morrison DJ, Rashid AJ, Yiu AP, Yan C, Frankland PW, Josselyn SA. Parvalbumin interneurons constrain the size of the lateral amygdala engram. Neurobiol Learn Mem. 2016;135:91–99. doi: 10.1016/j.nlm.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 95.Agetsuma M, Hamm JP, Tao K, Fujisawa S, Yuste R. Parvalbumin-positive interneurons regulate neuronal ensembles in visual cortex. Cereb Cortex. 2018;28:1831–45. doi: 10.1093/cercor/bhx169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jeong N, Singer AC. Learning from inhibition: functional roles of hippocampal CA1 inhibition in spatial learning and memory. Curr Opin Neurobiol. 2022;76:102604. doi: 10.1016/j.conb.2022.102604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stefanelli T, Bertollini C, Luscher C, Muller D, Mendez P. Hippocampal somatostatin interneurons control the size of neuronal memory ensembles. Neuron. 2016;89:1074–85. doi: 10.1016/j.neuron.2016.01.024. [DOI] [PubMed] [Google Scholar]
- 98.Delorme J, Wang L, Kuhn FR, Kodoth V, Ma J, Martinez JD, et al. Sleep loss drives acetylcholine- and somatostatin interneuron–mediated gating of hippocampal activity to inhibit memory consolidation. Proc Natl Acad Sci USA. 2021;118:e2019318118. doi: 10.1073/pnas.2019318118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Caroni P. Regulation of Parvalbumin Basket cell plasticity in rule learning. Biochem Biophys Res Commun. 2015;460:100–3. doi: 10.1016/j.bbrc.2015.02.023. [DOI] [PubMed] [Google Scholar]
- 100.Fawcett JW, Oohashi T, Pizzorusso T. The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nat Rev Neurosci. 2019;20:451–65. doi: 10.1038/s41583-019-0196-3. [DOI] [PubMed] [Google Scholar]
- 101.Spijker S, Koskinen MK, Riga D. Incubation of depression: ECM assembly and parvalbumin interneurons after stress. Neurosci Biobehav Rev. 2020;118:65–79. doi: 10.1016/j.neubiorev.2020.07.015. [DOI] [PubMed] [Google Scholar]
- 102.Ambrad Giovannetti E, Fuhrmann M. Unsupervised excitation: GABAergic dysfunctions in Alzheimer’s disease. Brain Res. 2019;1707:216–26. doi: 10.1016/j.brainres.2018.11.042. [DOI] [PubMed] [Google Scholar]
- 103.Jiménez-Balado J, Eich TS. GABAergic dysfunction, neural network hyperactivity and memory impairments in human aging and Alzheimer’s disease. Semin Cell Dev Biol. 2021;116:146–59. doi: 10.1016/j.semcdb.2021.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li G, Bien-Ly N, Andrews-Zwilling Y, Xu Q, Bernardo A, Ring K, et al. GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell. 2009;5:634–45. doi: 10.1016/j.stem.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li Y, Sun H, Chen Z, Xu H, Bu G, Zheng H. Implications of GABAergic neurotransmission in Alzheimer’s Disease. Front Aging Neurosci. 2016;8:31. doi: 10.3389/fnagi.2016.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s Disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, et al. Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia. 2006;47:867–72. doi: 10.1111/j.1528-1167.2006.00554.x. [DOI] [PubMed] [Google Scholar]
- 108.Scarmeas N, Honig LS, Choi H, Cantero J, Brandt J, Blacker D, et al. Seizures in Alzheimer disease: who, when, and how common? Arch Neurol. 2009;66:992–7. doi: 10.1001/archneurol.2009.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chin J, Scharfman HE. Shared cognitive and behavioral impairments in epilepsy and Alzheimer’s disease and potential underlying mechanisms. Epilepsy Behav. 2013;26:343–51. doi: 10.1016/j.yebeh.2012.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–8. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Goutagny R, Krantic S. Hippocampal oscillatory activity in Alzheimer’s disease: toward the identification of early biomarkers? Aging Dis. 2013;4:134–40. [PMC free article] [PubMed] [Google Scholar]
- 112.Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012;149:708–21. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hazra A, Gu F, Aulakh A, Berridge C, Eriksen JL, Ziburkus J. Inhibitory neuron and hippocampal circuit dysfunction in an aged mouse model of Alzheimer’s disease. PLoS One. 2013;8:e64318. doi: 10.1371/journal.pone.0064318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Katsuki F, Gerashchenko D, Brown RE. Alterations of sleep oscillations in Alzheimer’s disease: a potential role for GABAergic neurons in the cortex, hippocampus, and thalamus. Brain Res Bull. 2022;187:181–98. doi: 10.1016/j.brainresbull.2022.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Melgosa-Ecenarro L, Doostdar N, Radulescu CI, Jackson JS, Barnes SJ. Pinpointing the locus of GABAergic vulnerability in Alzheimer’s disease. Semin Cell Dev Biol. 2022. 10.1016/j.semcdb.2022.06.017. [DOI] [PubMed]
- 117.Nava-Mesa MO, Jimenez-Diaz L, Yajeya J, Navarro-Lopez JD. GABAergic neurotransmission and new strategies of neuromodulation to compensate synaptic dysfunction in early stages of Alzheimer’s disease. Front Cell Neurosci. 2014;8:167. doi: 10.3389/fncel.2014.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Perez C, Ziburkus J, Ullah G. Analyzing and modeling the dysfunction of inhibitory neurons in Alzheimer’s Disease. PLoS One. 2016;11:e0168800. doi: 10.1371/journal.pone.0168800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Busche MA, Kekus M, Adelsberger H, Noda T, Forstl H, Nelken I, et al. Rescue of long-range circuit dysfunction in Alzheimer’s disease models. Nat Neurosci. 2015;18:1623–30. doi: 10.1038/nn.4137. [DOI] [PubMed] [Google Scholar]
- 120.Czigler B, Csikos D, Hidasi Z, Anna Gaal Z, Csibri E, Kiss E, et al. Quantitative EEG in early Alzheimer’s disease patients - power spectrum and complexity features. Int J Psychophysiol. 2008;68:75–80. doi: 10.1016/j.ijpsycho.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 121.Moretti DV, Frisoni GB, Fracassi C, Pievani M, Geroldi C, Binetti G, et al. MCI patients’ EEGs show group differences between those who progress and those who do not progress to AD. Neurobiol Aging. 2011;32:563–71. doi: 10.1016/j.neurobiolaging.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 122.van der Hiele K, Vein AA, van der Welle A, van der Grond J, Westendorp RG, Bollen EL, et al. EEG and MRI correlates of mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging. 2007;28:1322–9. doi: 10.1016/j.neurobiolaging.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 123.Herrmann CS, Demiralp T. Human EEG gamma oscillations in neuropsychiatric disorders. Clin Neurophysiol. 2005;116:2719–33. doi: 10.1016/j.clinph.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 124.Stam CJ, Jones BF, Manshanden I, van Cappellen van Walsum AM, Montez T, Verbunt JP, et al. Magnetoencephalographic evaluation of resting-state functional connectivity in Alzheimer’s disease. Neuroimage. 2006;32:1335–44. doi: 10.1016/j.neuroimage.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 125.Stam CJ, van Cappellen van Walsum AM, Pijnenburg YA, Berendse HW, de Munck JC, Scheltens P, et al. Generalized synchronization of MEG recordings in Alzheimer’s Disease: evidence for involvement of the gamma band. J Clin Neurophysiol. 2002;19:562–74. doi: 10.1097/00004691-200212000-00010. [DOI] [PubMed] [Google Scholar]
- 126.van Deursen JA, Vuurman EF, Verhey FR, van Kranen-Mastenbroek VH, Riedel WJ. Increased EEG gamma band activity in Alzheimer’s disease and mild cognitive impairment. J Neural Transm. 2008;115:1301–11. doi: 10.1007/s00702-008-0083-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang J, Fang Y, Wang X, Yang H, Yu X, Wang H. Enhanced gamma activity and cross-frequency interaction of resting-state electroencephalographic oscillations in patients with Alzheimer’s Disease. Front Aging Neurosci. 2017;9:243. doi: 10.3389/fnagi.2017.00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fell J, Fernandez G, Klaver P, Axmacher N, Mormann F, Haupt S, et al. Rhinal-hippocampal coupling during declarative memory formation: dependence on item characteristics. Neurosci Lett. 2006;407:37–41. doi: 10.1016/j.neulet.2006.07.074. [DOI] [PubMed] [Google Scholar]
- 129.Howard MW, Rizzuto DS, Caplan JB, Madsen JR, Lisman J, Aschenbrenner-Scheibe R, et al. Gamma oscillations correlate with working memory load in humans. Cereb Cortex. 2003;13:1369–74. doi: 10.1093/cercor/bhg084. [DOI] [PubMed] [Google Scholar]
- 130.van Vugt MK, Schulze-Bonhage A, Litt B, Brandt A, Kahana MJ. Hippocampal gamma oscillations increase with memory load. J Neurosci. 2010;30:2694–9. doi: 10.1523/JNEUROSCI.0567-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Samson RD, Barnes CA. Impact of aging brain circuits on cognition. Eur J Neurosci. 2013;37:1903–15. doi: 10.1111/ejn.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Buchan RJ, Nagata K, Yokoyama E, Langman P, Yuya H, Hirata Y, et al. Regional correlations between the EEG and oxygen metabolism in dementia of Alzheimer’s type. Electroencephalogr Clin Neurophysiol. 1997;103:409–17. doi: 10.1016/s0013-4694(97)00015-5. [DOI] [PubMed] [Google Scholar]
- 133.Mevel K, Chételat G, Eustache F, Desgranges B. The default mode network in healthy aging and Alzheimer’s Disease. Int J Alzheimer’s Dis. 2011;2011:e535816. doi: 10.4061/2011/535816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Poil SS, de Haan W, van der Flier WM, Mansvelder HD, Scheltens P, Linkenkaer-Hansen K. Integrative EEG biomarkers predict progression to Alzheimer’s disease at the MCI stage. Front Aging Neurosci. 2013;5:58. doi: 10.3389/fnagi.2013.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Albuquerque MS, Mahar I, Davoli MA, Chabot JG, Mechawar N, Quirion R, et al. Regional and sub-regional differences in hippocampal GABAergic neuronal vulnerability in the TgCRND8 mouse model of Alzheimer’s disease. Front Aging Neurosci. 2015;7:30. doi: 10.3389/fnagi.2015.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cattaud V, Bezzina C, Rey CC, Lejards C, Dahan L, Verret L. Early disruption of parvalbumin expression and perineuronal nets in the hippocampus of the Tg2576 mouse model of Alzheimer’s disease can be rescued by enriched environment. Neurobiol Aging. 2018;72:147–58. doi: 10.1016/j.neurobiolaging.2018.08.024. [DOI] [PubMed] [Google Scholar]
- 137.Huh S, Baek SJ, Lee KH, Whitcomb DJ, Jo J, Choi SM, et al. The reemergence of long-term potentiation in aged Alzheimer’s disease mouse model. Sci Rep. 2016;6:29152. doi: 10.1038/srep29152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Krantic S, Isorce N, Mechawar N, Davoli MA, Vignault E, Albuquerque M, et al. Hippocampal GABAergic neurons are susceptible to amyloid-beta toxicity in vitro and are decreased in number in the Alzheimer’s disease TgCRND8 mouse model. J Alzheimers Dis. 2012;29:293–308. doi: 10.3233/JAD-2011-110830. [DOI] [PubMed] [Google Scholar]
- 139.Mahar I, Albuquerque MS, Mondragon-Rodriguez S, Cavanagh C, Davoli MA, Chabot JG, et al. Phenotypic alterations in hippocampal NPY- and PV-expressing interneurons in a presymptomatic transgenic mouse model of Alzheimer’s Disease. Front Aging Neurosci. 2016;8:327. doi: 10.3389/fnagi.2016.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Petrache AL, Rajulawalla A, Shi A, Wetzel A, Saito T, Saido TC, et al. Aberrant excitatory-inhibitory synaptic mechanisms in entorhinal cortex microcircuits during the pathogenesis of Alzheimer’s Disease. Cereb Cortex. 2019;29:1834–50. doi: 10.1093/cercor/bhz016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Takahashi H, Brasnjevic I, Rutten BP, Van Der Kolk N, Perl DP, Bouras C, et al. Hippocampal interneuron loss in an APP/PS1 double mutant mouse and in Alzheimer’s disease. Brain Struct Funct. 2010;214:145–60. doi: 10.1007/s00429-010-0242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hijazi S, Heistek TS, Scheltens P, Neumann U, Shimshek DR, Mansvelder HD, et al. Early restoration of parvalbumin interneuron activity prevents memory loss and network hyperexcitability in a mouse model of Alzheimer’s disease. Mol Psychiatry. 2020;25:3380–98. doi: 10.1038/s41380-019-0483-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lemmens MAM, Sierksma ASR, Rutten BPF, Dennissen F, Steinbusch HWM, Lucassen PJ, et al. Age-related changes of neuron numbers in the frontal cortex of a transgenic mouse model of Alzheimer’s disease. Brain Struct Funct. 2011;216:227–37. doi: 10.1007/s00429-011-0305-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sanchez-Mejias E, Nunez-Diaz C, Sanchez-Varo R, Gomez-Arboledas A, Garcia-Leon JA, Fernandez-Valenzuela JJ, et al. Distinct disease-sensitive GABAergic neurons in the perirhinal cortex of Alzheimer’s mice and patients. Brain Pathol. 2020;30:345–63. doi: 10.1111/bpa.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Trujillo-Estrada L, Dávila JC, Sánchez-Mejias E, Sánchez-Varo R, Gomez-Arboledas A, Vizuete M, et al. Early neuronal loss and axonal/presynaptic damage is associated with accelerated amyloid-β accumulation in AβPP/PS1 Alzheimer’s Disease mice subiculum. J Alzheimers Dis. 2014;42:521–41. doi: 10.3233/JAD-140495. [DOI] [PubMed] [Google Scholar]
- 146.Vegh MJ, Heldring CM, Kamphuis W, Hijazi S, Timmerman AJ, Li K, et al. Reducing hippocampal extracellular matrix reverses early memory deficits in a mouse model of Alzheimer inverted question marks disease. Acta Neuropathol Commun. 2014;2:76. doi: 10.1186/s40478-014-0076-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Etter G, van der Veldt S, Manseau F, Zarrinkoub I, Trillaud-Doppia E, Williams S. Optogenetic gamma stimulation rescues memory impairments in an Alzheimer’s disease mouse model. Nat Commun. 2019;10:5322. doi: 10.1038/s41467-019-13260-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Iaccarino HF, Singer AC, Martorell AJ, Rudenko A, Gao F, Gillingham TZ, et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature. 2016;540:230–5. doi: 10.1038/nature20587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Martinez-Losa M, Tracy TE, Ma K, Verret L, Clemente-Perez A, Khan AS, et al. Nav1.1-overexpressing interneuron transplants restore brain rhythms and cognition in a mouse model of Alzheimer’s Disease. Neuron. 2018;98:75–89.e5. doi: 10.1016/j.neuron.2018.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lu MH, Zhao XY, Xu DE, Chen JB, Ji WL, Huang ZP, et al. Transplantation of GABAergic interneuron progenitor attenuates cognitive deficits of Alzheimer’s Disease model mice. J Alzheimers Dis. 2020. 10.3233/JAD-200010. [DOI] [PubMed]
- 151.Yang X, Yao C, Tian T, Li X, Yan H, Wu J, et al. A novel mechanism of memory loss in Alzheimer’s disease mice via the degeneration of entorhinal-CA1 synapses. Mol Psychiatry. 2016. 10.1038/mp.2016.151. [DOI] [PMC free article] [PubMed]
- 152.Schmid LC, Mittag M, Poll S, Steffen J, Wagner J, Geis HR, et al. Dysfunction of somatostatin-positive interneurons associated with memory deficits in an Alzheimer’s Disease model. Neuron. 2016;92:114–25. doi: 10.1016/j.neuron.2016.08.034. [DOI] [PubMed] [Google Scholar]
- 153.Chung H, Park K, Jang HJ, Kohl MM, Kwag J. Dissociation of somatostatin and parvalbumin interneurons circuit dysfunctions underlying hippocampal theta and gamma oscillations impaired by amyloid β oligomers in vivo. Brain Struct Funct. 2020;225:935–54. doi: 10.1007/s00429-020-02044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hollnagel JO, Elzoheiry S, Gorgas K, Kins S, Beretta CA, Kirsch J, et al. Early alterations in hippocampal perisomatic GABAergic synapses and network oscillations in a mouse model of Alzheimer’s disease amyloidosis. PLoS One. 2019;14:e0209228. doi: 10.1371/journal.pone.0209228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kiss E, Gorgas K, Schlicksupp A, Gross D, Kins S, Kirsch J, et al. Biphasic alteration of the inhibitory synapse scaffold protein gephyrin in early and late stages of an Alzheimer Disease model. Am J Pathol. 2016;186:2279–91. doi: 10.1016/j.ajpath.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 156.Nakamura A, Cuesta P, Kato T, Arahata Y, Iwata K, Yamagishi M, et al. Early functional network alterations in asymptomatic elders at risk for Alzheimer’s disease. Sci Rep. 2017;7:6517. doi: 10.1038/s41598-017-06876-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pusil S, López ME, Cuesta P, Bruña R, Pereda E, Maestú F. Hypersynchronization in mild cognitive impairment: the ‘X’ model. Brain. 2019;142:3936–50. doi: 10.1093/brain/awz320. [DOI] [PubMed] [Google Scholar]
- 158.Roy DS, Arons A, Mitchell TI, Pignatelli M, Ryan TJ, Tonegawa S. Memory retrieval by activating engram cells in mouse models of early Alzheimer’s disease. Nature. 2016;531:508–12. doi: 10.1038/nature17172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hijazi S, Heistek TS, van der Loo R, Mansvelder HD, Smit AB, van Kesteren RE. Hyperexcitable parvalbumin interneurons render hippocampal circuitry vulnerable to amyloid beta. IScience. 2020;23:101271. doi: 10.1016/j.isci.2020.101271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rice HC, Marcassa G, Chrysidou I, Horre K, Young-Pearse TL, Muller UC, et al. Contribution of GABAergic interneurons to amyloid-beta plaque pathology in an APP knock-in mouse model. Mol Neurodegener. 2020;15:3. doi: 10.1186/s13024-019-0356-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dávila-Bouziguet E, Targa-Fabra G, Ávila J, Soriano E, Pascual M. Differential accumulation of Tau phosphorylated at residues Thr231, Ser262 and Thr205 in hippocampal interneurons and its modulation by Tau mutations (VLW) and amyloid-β peptide. Neurobiol Dis. 2019;125:232–44. doi: 10.1016/j.nbd.2018.12.006. [DOI] [PubMed] [Google Scholar]
- 162.Höfling C, Shehabi E, Kuhn P-H, Lichtenthaler SF, Hartlage-Rübsamen M, Roßner S. Cell type-specific human APP transgene expression by hippocampal interneurons in the Tg2576 mouse model of Alzheimer’s Disease. Front Neurosci. 2019;13:137. doi: 10.3389/fnins.2019.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kim DY, Gersbacher MT, Inquimbert P, Kovacs DM. Reduced sodium channel Na(v)1.1 levels in BACE1-null mice. J Biol Chem. 2011;286:8106–16. doi: 10.1074/jbc.M110.134692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Park A, Jacob AD, Walters BJ, Park S, Rashid AJ, Jung JH, et al. A time-dependent role for the transcription factor CREB in neuronal allocation to an engram underlying a fear memory revealed using a novel in vivo optogenetic tool to modulate CREB function. Neuropsychopharmacology. 2020;45:916–24. doi: 10.1038/s41386-019-0588-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ren SQ, Yao W, Yan JZ, Jin C, Yin JJ, Yuan J, et al. Amyloid beta causes excitation/inhibition imbalance through dopamine receptor 1-dependent disruption of fast-spiking GABAergic input in anterior cingulate cortex. Sci Rep. 2018;8:302. doi: 10.1038/s41598-017-18729-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fu H, Rodriguez GA, Herman M, Emrani S, Nahmani E, Barrett G, et al. Tau pathology induces excitatory neuron loss, grid cell dysfunction, and spatial memory deficits reminiscent of early Alzheimer’s Disease. Neuron. 2017;93:533–41.e5. doi: 10.1016/j.neuron.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Levenga J, Krishnamurthy P, Rajamohamedsait H, Wong H, Franke TF, Cain P, et al. Tau pathology induces loss of GABAergic interneurons leading to altered synaptic plasticity and behavioral impairments. Acta Neuropathol Commun. 2013;1:34. doi: 10.1186/2051-5960-1-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pooler AM, Polydoro M, Maury EA, Nicholls SB, Reddy SM, Wegmann S, et al. Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer’s disease. Acta Neuropathol Commun. 2015;3:14. doi: 10.1186/s40478-015-0199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pontecorvo MJ, Devous MD, Navitsky M, Lu M, Salloway S, Schaerf FW, et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain. 2017;140:748–63. doi: 10.1093/brain/aww334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wang L, Benzinger TL, Su Y, Christensen J, Friedrichsen K, Aldea P, et al. Evaluation of tau imaging in staging alzheimer disease and revealing interactions between beta-amyloid and tauopathy. JAMA Neurol. 2016;73:1070–7. doi: 10.1001/jamaneurol.2016.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Busche MA, Wegmann S, Dujardin S, Commins C, Schiantarelli J, Klickstein N, et al. Tau impairs neural circuits, dominating amyloid-beta effects, in Alzheimer models in vivo. Nat Neurosci. 2019;22:57–64. doi: 10.1038/s41593-018-0289-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Alibhai JD, Diack AB, Manson JC. Unravelling the glial response in the pathogenesis of Alzheimer’s disease. FASEB J. 2018;32:5766–77. doi: 10.1096/fj.201801360R. [DOI] [PubMed] [Google Scholar]
- 173.Dzamba D, Harantova L, Butenko O, Anderova M. Glial cells - the key elements of Alzheimer’s Disease. Curr Alzheimer Res. 2016;13:894–911. doi: 10.2174/1567205013666160129095924. [DOI] [PubMed] [Google Scholar]
- 174.Perez-Nievas BG, Serrano-Pozo A. Editorial: the role of glia in Alzheimer’s disease. Front Neurol. 2018;9:1161. doi: 10.3389/fneur.2018.01161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mederos S, Perea G. GABAergic-astrocyte signaling: a refinement of inhibitory brain networks. Glia. 2019;67:1842–51. doi: 10.1002/glia.23644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–31. doi: 10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Araque A, Navarrete M. Glial cells in neuronal network function. Philos Trans R Soc Lond B Biol Sci. 2010;365:2375–81. doi: 10.1098/rstb.2009.0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Badia-Soteras A, Heistek TS, Kater MSJ, Mak A, Negrean A, van den Oever MC, et al. Retraction of astrocyte leaflets from the synapse enhances fear memory. Biol Psychiatry. 2022:S000632232201705X. 10.1016/j.biopsych.2022.10.013. Online ahead of print. [DOI] [PubMed]
- 179.Huang AY, Woo J, Sardar D, Lozzi B, Bosquez Huerta NA, Lin CJ, et al. Region-specific transcriptional control of astrocyte function oversees local circuit activities. Neuron. 2020;10:2020. doi: 10.1016/j.neuron.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, et al. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–92. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Majumdar A, Chung H, Dolios G, Wang R, Asamoah N, Lobel P, et al. Degradation of fibrillar forms of Alzheimer’s amyloid beta-peptide by macrophages. Neurobiol Aging. 2008;29:707–15. doi: 10.1016/j.neurobiolaging.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yang CN, Shiao YJ, Shie FS, Guo BS, Chen PH, Cho CY, et al. Mechanism mediating oligomeric Abeta clearance by naive primary microglia. Neurobiol Dis. 2011;42:221–30. doi: 10.1016/j.nbd.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 183.Zhao R, Hu W, Tsai J, Li W, Gan WB. Microglia limit the expansion of beta-amyloid plaques in a mouse model of Alzheimer’s disease. Mol Neurodegener. 2017;12:47. doi: 10.1186/s13024-017-0188-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Boutajangout A, Wisniewski T. The innate immune system in Alzheimer’s disease. Int J Cell Biol. 2013;2013:576383. doi: 10.1155/2013/576383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–82. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 186.Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol. 1999;154:1673–84. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet. 2022;54:412–36. doi: 10.1038/s41588-022-01024-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bellenguez C, Charbonnier C, Grenier-Boley B, Quenez O, Le Guennec K, Nicolas G, et al. Contribution to Alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol Aging. 2017;59:220.e1–220.e9. doi: 10.1016/j.neurobiolaging.2017.07.001. [DOI] [PubMed] [Google Scholar]
- 189.Perez SE, Nadeem M, He B, Miguel JC, Malek-Ahmadi MH, Chen K, et al. Neocortical and hippocampal TREM2 protein levels during the progression of Alzheimer’s disease. Neurobiol Aging. 2017;54:133–43. doi: 10.1016/j.neurobiolaging.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Yeh FL, Hansen DV, Sheng M. TREM2, microglia, and neurodegenerative diseases. Trends Mol Med. 2017;23:512–33. doi: 10.1016/j.molmed.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 191.Audoy-Remus J, Bozoyan L, Dumas A, Filali M, Lecours C, Lacroix S, et al. GPR84 deficiency reduces microgliosis, but accelerates dendritic degeneration and cognitive decline in a mouse model of Alzheimer’s disease. Brain Behav Immun. 2015;46:112–20. doi: 10.1016/j.bbi.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 192.Bie B, Wu J, Foss JF, Naguib M. Activation of mGluR1 mediates C1q-dependent microglial phagocytosis of glutamatergic synapses in Alzheimer’s rodent models. Mol Neurobiol. 2019;56:5568–85. doi: 10.1007/s12035-019-1467-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–6. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Bowser DN, Khakh BS. ATP excites interneurons and astrocytes to increase synaptic inhibition in neuronal networks. J Neurosci. 2004;24:8606–20. doi: 10.1523/JNEUROSCI.2660-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Egawa K, Yamada J, Furukawa T, Yanagawa Y, Fukuda A. Cl(-) homeodynamics in gap junction-coupled astrocytic networks on activation of GABAergic synapses. J Physiol. 2013;591:3901–17. doi: 10.1113/jphysiol.2013.257162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Henstridge CM, Tzioras M, Paolicelli RC. Glial contribution to excitatory and inhibitory synapse loss in neurodegeneration. Front Cell Neurosci. 2019;13:63. doi: 10.3389/fncel.2019.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kersante F, Rowley SC, Pavlov I, Gutierrez-Mecinas M, Semyanov A, Reul JM, et al. A functional role for both -aminobutyric acid (GABA) transporter-1 and GABA transporter-3 in the modulation of extracellular GABA and GABAergic tonic conductances in the rat hippocampus. J Physiol. 2013;591:2429–41. doi: 10.1113/jphysiol.2012.246298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kozlov AS, Angulo MC, Audinat E, Charpak S. Target cell-specific modulation of neuronal activity by astrocytes. Proc Natl Acad Sci USA. 2006;103:10058–63. doi: 10.1073/pnas.0603741103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Li K, Li J, Zheng J, Qin S. Reactive astrocytes in neurodegenerative diseases. Aging Dis. 2019;10:664–75. doi: 10.14336/AD.2018.0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Shigetomi E, Tong X, Kwan KY, Corey DP, Khakh BS. TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat Neurosci. 2011;15:70–80. doi: 10.1038/nn.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med. 2014;20:886–96. doi: 10.1038/nm.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wu Z, Guo Z, Gearing M, Chen G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s [corrected] disease model. Nat Commun. 2014;5:4159. doi: 10.1038/ncomms5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yarishkin O, Lee J, Jo S, Hwang EM, Lee CJ. Disinhibitory action of astrocytic GABA at the perforant path to dentate gyrus granule neuron synapse reverses to inhibitory in Alzheimer’s Disease model. Exp Neurobiol. 2015;24:211–8. doi: 10.5607/en.2015.24.3.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Assefa BT, Gebre AK, Altaye BM. Reactive astrocytes as drug target in Alzheimer’s Disease. Biomed Res Int. 2018;2018:4160247. doi: 10.1155/2018/4160247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Frost GR, Li YM. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017;7:170228. doi: 10.1098/rsob.170228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Papuć E, Rejdak K. The role of myelin damage in Alzheimer’s disease pathology. Arch Med Sci. 2018;16:345–51. doi: 10.5114/aoms.2018.76863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chen J-F, Liu K, Hu B, Li R-R, Xin W, Chen H, et al. Enhancing myelin renewal reverses cognitive dysfunction in a murine model of Alzheimer’s disease. Neuron. 2021;109:2292–.e5. doi: 10.1016/j.neuron.2021.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Knowles JK, Xu H, Soane C, Batra A, Saucedo T, Frost E, et al. Maladaptive myelination promotes generalized epilepsy progression. Nat Neurosci. 2022;25:596–606. doi: 10.1038/s41593-022-01052-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kole K, Voesenek BJB, Brinia ME, Petersen N, Kole MHP. Parvalbumin basket cell myelination accumulates axonal mitochondria to internodes. Nat Commun. 2022;13:7598. doi: 10.1038/s41467-022-35350-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hu H, Roth FC, Vandael D, Jonas P. Complementary tuning of Na(+) and K(+) channel gating underlies fast and energy-efficient action potentials in GABAergic interneuron axons. Neuron. 2018;98:156–65.e6. doi: 10.1016/j.neuron.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Mondragon-Rodriguez S, Gu N, Manseau F, Williams S. Alzheimer’s transgenic model is characterized by very early brain network alterations and beta-CTF fragment accumulation: reversal by beta-secretase inhibition. Front Cell Neurosci. 2018;12:121. doi: 10.3389/fncel.2018.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhang H, Zhang L, Zhou D, He X, Wang D, Pan H, et al. Ablating ErbB4 in PV neurons attenuates synaptic and cognitive deficits in an animal model of Alzheimer’s disease. Neurobiol Dis. 2017;106:171–80. doi: 10.1016/j.nbd.2017.07.001. [DOI] [PubMed] [Google Scholar]
- 213.Ali F, Baringer SL, Neal A, Choi EY, Kwan AC. Parvalbumin-positive neuron loss and amyloid-beta deposits in the frontal cortex of Alzheimer’s Disease-related mice. J Alzheimers Dis. 2019;72:1323–39. doi: 10.3233/JAD-181190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Giesers NK, Wirths O. Loss of hippocampal calretinin and parvalbumin interneurons in the 5XFAD mouse model of Alzheimer’s Disease. ASN Neuro. 2020;12:1759091420925356. doi: 10.1177/1759091420925356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Scott L, Feng J, Kiss T, Needle E, Atchison K, Kawabe TT, et al. Age-dependent disruption in hippocampal theta oscillation in amyloid-beta overproducing transgenic mice. Neurobiol Aging. 2012;33:1481.e13–23.. doi: 10.1016/j.neurobiolaging.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 216.Zhang Z, Jing Y, Ma Y, Duan D, Li B, Holscher C, et al. Driving GABAergic neurons optogenetically improves learning, reduces amyloid load and enhances autophagy in a mouse model of Alzheimer’s disease. Biochem Biophys Res Commun. 2020;12:2020. doi: 10.1016/j.bbrc.2020.03.004. [DOI] [PubMed] [Google Scholar]
- 217.Klein AS, Donoso JR, Kempter R, Schmitz D, Beed P. Early cortical changes in gamma oscillations in Alzheimer’s Disease. Front Syst Neurosci. 2016;10:83. doi: 10.3389/fnsys.2016.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Zhang Q, Yang C, Liu T, Liu L, Li F, Cai Y, et al. Citalopram restores short-term memory deficit and non-cognitive behaviors in APP/PS1 mice while halting the advance of Alzheimer’s disease-like pathology. Neuropharmacology. 2018;131:475–86. doi: 10.1016/j.neuropharm.2017.12.021. [DOI] [PubMed] [Google Scholar]
- 219.Hamm V, Heraud C, Bott JB, Herbeaux K, Strittmatter C, Mathis C, et al. Differential contribution of APP metabolites to early cognitive deficits in a TgCRND8 mouse model of Alzheimer’s disease. Sci Adv. 2017;3:e1601068. doi: 10.1126/sciadv.1601068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Goutagny R, Gu N, Cavanagh C, Jackson J, Chabot JG, Quirion R, et al. Alterations in hippocampal network oscillations and theta-gamma coupling arise before Abeta overproduction in a mouse model of Alzheimer’s disease. Eur J Neurosci. 2013;37:1896–902. doi: 10.1111/ejn.12233. [DOI] [PubMed] [Google Scholar]
- 221.Ahnaou A, Moechars D, Raeymaekers L, Biermans R, Manyakov NV, Bottelbergs A, et al. Emergence of early alterations in network oscillations and functional connectivity in a tau seeding mouse model of Alzheimer’s disease pathology. Sci Rep. 2017;7:14189. doi: 10.1038/s41598-017-13839-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Adaikkan C, Middleton SJ, Marco A, Pao PC, Mathys H, Kim DN, et al. Gamma entrainment binds higher-order brain regions and offers neuroprotection. Neuron. 2019;102:929–43.e8. doi: 10.1016/j.neuron.2019.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Martorell AJ, Paulson AL, Suk HJ, Abdurrob F, Drummond GT, Guan W, et al. Multi-sensory gamma stimulation ameliorates Alzheimer’s-associated pathology and improves cognition. Cell. 2019;177:256–71.e22. doi: 10.1016/j.cell.2019.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kam K, Duffy ÁM, Moretto J, LaFrancois JJ, Scharfman HE. Interictal spikes during sleep are an early defect in the Tg2576 mouse model of β-amyloid neuropathology. Sci Rep. 2016;6:20119. doi: 10.1038/srep20119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Corbett BF, Leiser SC, Ling H-P, Nagy R, Breysse N, Zhang X, et al. Sodium channel cleavage is associated with aberrant neuronal activity and cognitive deficits in a mouse model of Alzheimer’s disease. J Neurosci. 2013;33:7020–6. doi: 10.1523/JNEUROSCI.2325-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Nakazono T, Lam TN, Patel AY, Kitazawa M, Saito T, Saido TC, et al. Impaired in vivo gamma oscillations in the medial entorhinal cortex of knock-in Alzheimer model. Front Syst Neurosci. 2017;11:48. doi: 10.3389/fnsys.2017.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Witton J, Staniaszek LE, Bartsch U, Randall AD, Jones MW, Brown JT. Disrupted hippocampal sharp-wave ripple-associated spike dynamics in a transgenic mouse model of dementia. J Physiol. 2016;594:4615–30. doi: 10.1113/jphysiol.2014.282889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Booth CA, Ridler T, Murray TK, Ward MA, de Groot E, Goodfellow M, et al. Electrical and network neuronal properties are preferentially disrupted in dorsal, but not ventral, medial entorhinal cortex in a mouse model of tauopathy. J Neurosci. 2016;36:312–24. doi: 10.1523/JNEUROSCI.2845-14.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Ciupek SM, Cheng J, Ali YO, Lu H-C, Ji D. Progressive functional impairments of hippocampal neurons in a tauopathy mouse model. J Neurosci. 2015;35:8118–31. doi: 10.1523/JNEUROSCI.3130-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Driver JE, Racca C, Cunningham MO, Towers SK, Davies CH, Whittington MA, et al. Impairment of hippocampal gamma-frequency oscillations in vitro in mice overexpressing human amyloid precursor protein (APP) Eur J Neurosci. 2007;26:1280–8. doi: 10.1111/j.1460-9568.2007.05705.x. [DOI] [PubMed] [Google Scholar]
- 231.Zallo F, Gardenal E, Verkhratsky A, Rodríguez JJ. Loss of calretinin and parvalbumin positive interneurones in the hippocampal CA1 of aged Alzheimer’s disease mice. Neurosci Lett. 2018;681:19–25. doi: 10.1016/j.neulet.2018.05.027. [DOI] [PubMed] [Google Scholar]
- 232.Soler H, Dorca-Arevalo J, Gonzalez M, Rubio SE, Avila J, Soriano E, et al. The GABAergic septohippocampal connection is impaired in a mouse model of tauopathy. Neurobiol Aging. 2017;49:40–51. doi: 10.1016/j.neurobiolaging.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 233.Loreth D, Ozmen L, Revel FG, Knoflach F, Wetzel P, Frotscher M, et al. Selective degeneration of septal and hippocampal GABAergic neurons in a mouse model of amyloidosis and tauopathy. Neurobiol Dis. 2012;47:1–12. doi: 10.1016/j.nbd.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 234.Aso E, Andres-Benito P, Ferrer I. Genetic deletion of CB1 cannabinoid receptors exacerbates the Alzheimer-like symptoms in a transgenic animal model. Biochem Pharm. 2018;157:210–6. doi: 10.1016/j.bcp.2018.08.007. [DOI] [PubMed] [Google Scholar]
- 235.Xiao MF, Xu D, Craig MT, Pelkey KA, Chien CC, Shi Y, et al. NPTX2 and cognitive dysfunction in Alzheimer’s Disease. Elife. 2017;6:e23798. doi: 10.7554/eLife.23798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Villette V, Poindessous-Jazat F, Simon A, Lena C, Roullot E, Bellessort B, et al. Decreased rhythmic GABAergic septal activity and memory-associated theta oscillations after hippocampal amyloid-beta pathology in the rat. J Neurosci. 2010;30:10991–1003. doi: 10.1523/JNEUROSCI.6284-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kurudenkandy FR, Zilberter M, Biverstal H, Presto J, Honcharenko D, Stromberg R, et al. Amyloid-beta-induced action potential desynchronization and degradation of hippocampal gamma oscillations is prevented by interference with peptide conformation change and aggregation. J Neurosci. 2014;34:11416–25. doi: 10.1523/JNEUROSCI.1195-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Nerelius C, Sandegren A, Sargsyan H, Raunak R, Leijonmarck H, Chatterjee U, et al. Alpha-helix targeting reduces amyloid-beta peptide toxicity. Proc Natl Acad Sci USA. 2009;106:9191–6. doi: 10.1073/pnas.0810364106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Pena-Ortega F. Amyloid beta-protein and neural network dysfunction. J Neurodegener Dis. 2013;2013:657470. doi: 10.1155/2013/657470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Gutierrez-Lerma AI, Ordaz B, Pena-Ortega F. Amyloid Beta peptides differentially affect hippocampal theta rhythms in vitro. Int J Pept. 2013;2013:328140. doi: 10.1155/2013/328140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Leao RN, Colom LV, Borgius L, Kiehn O, Fisahn A. Medial septal dysfunction by Abeta-induced KCNQ channel-block in glutamatergic neurons. Neurobiol Aging. 2012;33:2046–61. doi: 10.1016/j.neurobiolaging.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 242.Watremez W, Jackson J, Almari B, McLean SL, Grayson B, Neill JC, et al. Stabilized low-n amyloid-beta oligomers induce robust novel object recognition deficits associated with inflammatory, synaptic, and GABAergic dysfunction in the rat. J Alzheimers Dis. 2018;62:213–26. doi: 10.3233/JAD-170489. [DOI] [PubMed] [Google Scholar]