

Abstract
Acetylation of key Lysine residues characterizes aggregates of the microtubule-associated protein tau constituting the neuropathological hallmark of many neurodegenerative diseases, such as Alzheimer’s disease (AD) and Progressive Supranuclear Palsy (PSP). This has led to the idea that acetylation influences tau aggregation. Using a HEK293 cell-based aggregation assay, we tested whether acetylation-mimicking substitutions (K→Q) on five AD-associated acetyl-modified sites (AcK-311, 353, 369, 370, 375) influenced its propensity to aggregate when exposed to tau seeds derived from two clinically distinctive diseases – AD and PSP. In combination, the presence of 5K→Q sites ablated tau aggregation induced by seeds from both AD and PSP patients, indicating that acetylation within the filament core domain of tau could have an inhibitory effect on seed-mediated aggregation. We had previously identified that a phosphorylation-mimetic on Ser305 (S→E) abrogated tau aggregation by seeds from AD patients, without affecting seeding by PSP patients. Combining the S305→E to the 5K→Q acetyl-modified sites, we found that this tau could now be seeded only by PSP patients, but not by AD patients, confirming Ser305 as a critical determinant of strain-specific tau seeding. On the other hand, acetylation-nullifying substitutions (K→R or K→A) on these same Lys sites did not alter tau seeding abilities compared to the parental tau construct. Notably, the combined acetylation-nullifying Alanine substitutions on these 5 Lys sites resulted in spontaneous self-aggregation, with the filaments resembling amorphous deposits. All together, we demonstrate that cooperative acetyl-occupancy in the tau filament core influences seeded propagation of misfolded tau as well as drives self-aggregation.
Keywords: post-translational modification, seeding, aggregation, Alzheimer’s disease, Progressive Supranuclear Palsy
Introduction
Tau is a microtubule binding and stabilizing protein that regulates axonal transport as well as participates in neuroplasticity [1, 2]. Aggregated tau is a characteristic hallmark of a broad category of neurodegenerative dementias, known as tauopathies, some examples of which are Alzheimer’s disease (AD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and primary age-related tauopathies [3]. Aggregated tau pathology in the brain is directly correlated with a decline in mental acuity and neurodegeneration, underpinning its pathological role in tauopathies and necessitating therapeutic targeting of pathological species of tau [4].
Neuropathological and biochemical analysis of tau isolated from tauopathy patients shows that it undergoes extensive post-translational modifications (PTMs) [5]. Both tau that remains detergent-soluble, as well as detergent-insoluble tau within neurofibrillary tangle (NFT), are extensively modified by phosphorylation, acetylation, methylation, ubiquitination and glycosylation [6–8]. While phosphorylation is the major PTM that has been extensively studied, recently acetylation of Lys (K) residues has been revealed to mechanistically modify tau function and structure [9–14]. There are 44 lysine residues on tau that can be potentially acetylated. Previously, acetylation of residues 174 and 280/281 has been shown experimentally to induce tau aggregation [11, 12, 15, 16], albeit through different mechanisms. These sites are found to be robustly acetylated in multiple types of tauopathies [11, 17]. However, acetylation on other residues (AcK259, AcK290, AcK298, AcK321, AcK353) can suppress tau fibrillization [10, 14]. Acetylation could also prevent the availability of the Lys to other PTMs, such as methylation, glycation, ubiquitination and SUMOylation [18], thus altering the structure-function profile of tau [19]. In addition, specific pseudo-acetylation of K321 in the microtubule-binding repeat domain 3 (MTBR-R3) has been shown to prevent neighboring phosphorylation [20]. Thus, acetylation could not only be associated with tau aggregation, but also influence the homeostatic function of tau as well as tau clearance. Overall, there seems to be a complex series of biological outcomes when tau is acetylated leading to changes in ‘pathological’ phosphorylation patterns that are found in neurodegenerative dementias.
In this study, we specifically focused on five acetylation sites (K311, K353, K369, K370, K375) identified in late-stage AD patients with high frequency [6]. As these sites were found in late AD and are within the core domain of tau filaments identified in AD [21], we reasoned that these sites could play a key role in tau misfolding and aggregation. As 0N/4R tau was enriched in the insoluble tau fraction in AD patients, we constructed recombinant 0N/4R P301L tau with acetyl-mimetics (K→Q) and acetyl-nullifying substitutions (K→R or K→A) on these five sites. Using a previously described transfected HEK293 cell model [22], we examined the aggregation of these variants after seeding with detergent-insoluble tau seeds derived from AD or PSP patients. We observed that tau harboring the K→Q acetyl-mimicking substitutions on all 5 sites combined was resistant to seeded aggregation induced by tau seeds from AD or PSP brains. Acetyl-nullifying K→R and K→A substituted tau showed equivalent levels of seeded aggregation relative to the parent P301L construct. Interestingly, K→A substituted tau forms robust aggregated tau even in the absence of any brain-derived tau seeds. Our results suggest that hyperacetylation of tau on select epitopes within the filament core domain may be a defense mechanism to slow the aggregation or propagation of misfolded tau.
Results
Pseudo-acetylated tau shows differential aggregation in the presence of tau seeds
A proteomic analysis of detergent-insoluble tau extracted from a cohort of 49 AD patients identified major phosphorylation and acetylation sites present on tau across various stages of the disease [6]. This study predicted that while phosphorylation signatures were present across all stages of the disease, acetylation modifications occurred at late Braak stages (V/VI) and exclusively along the MTBR and C-terminus of tau that overlaps with the AD-tau filament core domain. To understand how acetylation within the AD-tau filament core domain affected the pathological cascade of tau aggregation, we selected 5 lysine sites (K311, K353, K369, K370, and K375) that were identified in this study as associated with tau pathology. We hypothesized that acetylation could modulate the propagation of misfolded tau or the formation of aggregates. Using a robust HEK293T cell seeding model established in our lab [22], we determined whether AD-tau seeds and PSP-tau seeds extracted from human postmortem brains could propagate misfolded conformations to tau containing acetyl-mimicking or acetyl-nullifying residues on these selected sites.
In the first study, all 5 lysine residues were mutated combinatorically to K→Q (acetyl-mimicking or acetyl-plus, 5K-Q), K→R (acetyl-null, 5K-R), or K→A (acetyl-null, 5K-A) on the 0N/4R P301L tau backbone to investigate changes in the propagation of brain-derived seeds (Fig. 1a). We used two different acetyl-nullifying mimetics, Lys→Arg and Lys→Ala, because substitutions to Arg conserves charge, although this change may also render tau susceptible to methylation [5]. We assessed the biophysical properties of these different acetyl-variants in the context of the AD-tau filament core (PDB:7QL4; [23]) or PSP-tau filament core (PDB:7P65; [24]) (Fig. 1a). The proportion of hydrophobic residues was highest in the 5K-A variant of the AD-tau (37.3%) and PSP-tau (33.6%) compared to corresponding wild type tau (30.7% for AD-tau and 29.1% for PSP-tau) (Fig. 1a). The isoelectric pH (pI) of 5K-Q and 5K-A were shifted towards neutral pH in the context of the AD-tau filament whereas the pI of the acetyl-modified variants in the PSP-tau filament were similar to the wild type tau (Fig. 1a).
Figure 1: Comparative effect of combinatorial acetyl variants in the tau core domain on seed-induced P301L tau aggregation.

a. Schematic depiction of 0N/4R P301L tau variants where AcK sites contained in the core domain (K311, K353, K369, K370, K375) were mutated from Lys→Gln (Pseudo-Acetyl: 5K-Q), Lys→Arg (Acetyl-Null: 5K-R), or Lys→Ala (Acetyl-Null: 5K-A). Depiction not to scale. All numbers correspond to 2N4R tau. Representation of AD-tau core or PSP-tau core containing acetyl substitutions, showing isoelectric pH and amino acid composition (bottom panel). b-d. HEK293T cell seeding assay using the acetyl variants seeded with AD-tau seeds or PSP-tau seeds. Samples were fractionated into detergent-soluble and detergent-insoluble lysates and probed for total tau (t-tau) and p-tau (AT8). Representative immunoblot and % Aggregation of acetyl-tau variants seeded with AD:Patient A tau seeds or PSP:Patient A tau seeds. e-g. Representative immunoblot and AT8 insolubility index (%AT8) of tau acetyl variants seeded with AD:Patient A tau seeds or PSP:Patient A tau seeds. %AT8 calculated as a ratio of insoluble to soluble AT8 signal. Relative molecular masses (kDa) are indicated on the left of each blot. N=3 for each experimental replicate. 2-way ANOVA with Dunnett’s multiple comparisons test, with single pooled variance. *p<0.05, ****p<0.0001. Blots showing seeding results from AD:Patient B tau seeds or PSP:Patient B tau seeds are in Fig. S1.
To investigate potential patient heterogeneity in seeding, we used seed preparations from two individuals with AD and two individuals with PSP (referred to as Patients A and B for each diagnosis: AD-A, AD-B, PSP-A, PSP-B). All patients used for this study were neuropathologically confirmed and their seeding profiles were characterized in an earlier study (Table S1) [22]. As wild-type 0N4R tau is not templated efficiently in cellular models, we used human 0N/4R P301L tau as the recipient tau with the pseudo-acetylation residues [22]. 0.5 μg of AD-tau and PSP-tau from Patient A (Fig. 1) or Patient B (Fig. S1) were used to seed HEK293T cells expressing each acetyl-variant construct. After 2 days, cell lysates were fractionated, separated by SDS-PAGE, and probed with a total tau antibody (t-tau) and phosphorylation-specific tau antibody (AT8 - pSer202/Thr205) to reveal the amount of tau that became misfolded for each construct, represented by %Aggregation in the detergent-insoluble fraction. We used AT8 as this antibody is widely found in NFT and is generally absent in normal brain tissue [25]. First, we confirmed that the expression levels and AT8 patterns of the soluble acetyl-variant tau forms were equivalent (Fig. 1b, e; Fig. S1a, d). We also performed an AlamarBlue cell viability assay to confirm that none of these acetyl-variant tau causes cytotoxicity (Fig. S1g). Examining tau levels in the insoluble fraction following seeding, we found that neither AD-tau nor PSP-tau seeds from two different patients could efficiently propagate 5K-Q tau (p<0.0001 relative to parent P301L tau) (Fig. 1c, d, f, g; Fig. S1b, c, e, f). By contrast, both the acetylation-null mimetics, 5K-R tau and 5K-A tau, showed similar levels of induced aggregation (detected by total tau and AT8-tau antibodies) relative to parent P301L tau, when these were exposed to AD-tau or PSP-tau seeds (Fig. 1d, g; Fig. S1c, f). Additionally, we observed that 5K-A tau spontaneously formed AT8-positive aggregates in the absence of any brain-derived tau seeds (unseeded: Fig. 1c, d, f, g; Fig. S1b, c, e, f), indicating that substitution of these Lys residues with Ala renders tau unstable. AT8 reactivity was not observed in the insoluble cellular fraction of unseeded HEK293T cells expressing 5K-Q, 5K-R and P301L tau (Fig. 1c, d, f, g; Fig. S1c, d, f, g).
To complement the biochemical data, we conducted immunofluorescence for aggregated tau following seeding of the pseudo-acetylation mimetics, 5K-Q, 5K-R and parental P301L tau (Fig. S1h, i). Representative images for each condition are displayed, with the amyloid-specific stain ThioS (green) and total tau (red). We normalized ThioS positivity to total tau signal, finding that the patterns of Thioflavin S staining reflected our biochemical data for 5K-Q tau showing reduced aggregation in the presence of AD-tau seeds and PSP-tau seeds when compared to P301L tau (Fig. S1i).
Tau aggregation levels of individual pseudo-acetyl tau seeded by AD-tau and PSP-tau seeds
Previous research has shown that acetylation on individual Lys residues influence tau function and structure [26]. Since we observed that combinatorial K→Q substitutions resulted in a dramatic loss of seeding, we next investigated whether any of these substituted sites, by themselves, play a significant role in tau seeding (Fig. 2a). 0.5 μg of AD-tau from Patient A (Fig. 2b–g) or Patient B (Fig. S2a–f) were used to seed HEK293T cells expressing individual pseudo-acetyl tau at K311, K353, K369, K370, and K375. After 2 days, cell lysates were fractionated, separated by SDS-PAGE, and probed with a total tau (t-tau) and phosphorylation-specific tau antibody (AT8 - pSer202/Thr205) to reveal %Aggregation and %AT8 insolubility index respectively. The individual pseudo-acetyl tau expressed equally and showed comparable AT8 levels in the soluble fraction (Fig. 2 b, e; Fig. S2a, d). Although we found that seeds from AD Patient A were less effective in propagating tau aggregation in cells expressing the K369Q and K375Q tau variants (p<0.05; Fig. 2d), these differences were not seen in experiments with seeds from AD Patient B (Fig. S2c, f), indicating inter-patient heterogeneity. The levels of AT8 reactive insoluble tau were variable but none of the AD-tau seeded pseudo-acetyl tau showed differential AT8 phosphorylation relative to each other or parent P301L tau (Fig. 2g; Suppl. Fig. S2f).
Figure 2: Individual pseudo-acetyl variants in the tau filament core domain show variable seeding propensity.

a. Linearized depiction of individual 0N/4R P301L tau mimetic variants where the sites contained (K311, K353, K369, K370, K375) were mutated to Lys→Gln (Acetyl-Plus). b-d. Representative total tau immunoblot of individual acetyl-mimetic P301L tau seeded with AD:Patient A tau seeds from detergent-soluble and insoluble fractions. Quantification of % aggregation for each tau variant are shown (d). e-g. Representative AT8 immunoblot of individual acetyl-mimetic P301L tau seeded with AD:Patient A tau seeds. h-j. Representative total tau immunoblot of individual acetyl-mimetic P301L tau seeded with PSP:Patient A tau seeds from detergent-soluble and insoluble fractions. Quantification of % aggregation for each variant are shown (j). k-m. Representative AT8 immunoblot of individual acetyl-mimetic P301L tau seeded with PSP:Patient A tau seeds. Relative molecular masses (kDa) are indicated on the left of each blot. N=3 for each experimental replicate. 1-way ANOVA with Sidak’s multiple comparisons test, with single pooled variance. *p<0.05, **p<0.01, ***p<0.001. Blots showing seeding results from AD:Patient B tau seeds or PSP:Patient B tau seeds are in Fig. S2.
We next tested how PSP-tau seeds from two different patients affected seeding efficiency of the individual pseudo-acetyl tau (Fig 2h–m; Fig. S2g–l). We found inter-patient variability in seeding efficiency in this case also. Specifically, PSP-tau seeds from Patient A resulted in lower tau seeding efficiency for K311Q (p<0.001), K353Q (p<0.05), K369Q (p<0.01) and K375Q (p<0.05), but not K370Q tau relative to parent P301L tau (Fig. 2i–j). However, AT8 levels were mostly comparable among the pseudo-acetyl tau variants and P301L tau, except for K353Q which showed higher AT8 occupancy than P301L tau, when seeded with PSP-tau (Fig. 2m; p<0.001). For PSP-tau seeds derived from Patient B, we did not find any differences in seeding efficiency for any of the pseudo-acetyl tau (Fig. S2g–l). Interestingly, similar to Patient A, PSP-tau from Patient B also induced higher levels of AT8-tau in K353Q tau relative to P301L tau (Fig. S2l; p<0.05). None of the unseeded pseudo-acetyl tau variants displayed detectable AT8 or total tau in the insoluble cellular fraction (Fig. 2i, l; Suppl. Fig. S2h, k). Overall, individual pseudo-acetyl tau showed diverse tau aggregation propensity when seeded with seeds derived from different patients.
Inclusion of phospho-mimicking Ser305Glu substitution enables pseudo-acetylated tau to show disease-specific seeding characteristic
Previous studies have indicated that acetylation on specific residues is related to phosphorylation patterns and these together regulate tau polymerization [20]. This prompted us to consider whether a phosphorylation site (Ser305Glu [22]) that we had previously characterized as underlying disease-strain specific seeding would have any deterministic role in seeding of pseudo-acetylated tau. Specifically, we showed that P301L tau containing S305E can be seeded by PSP-tau but not by AD-tau seeds [22]. We introduced the S305E substitution into 5K-Q tau, which showed the least induction of tau aggregates following seeding by both AD-tau and PSP-tau seeds (Fig. 3a). We first confirmed that the expression levels of the different tau variants, including the S305E containing 5K-Q, were equivalent to the other tau variants being tested (Fig. 3b). We observed ablation of seeding when S305E was combined with 5K-Q tau in the presence of AD-tau seeds, similar to the inhibition noted in parental P301L tau carrying the S305E substitution (Fig. 3c–d). Interestingly, this new phospho-acetyl mimetic (5K-Q/S305E) retained limited seeding responsiveness to PSP-tau, when compared to the parent 5K-Q tau (Fig. 3c–d). We also confirmed that parent P301L with the S305E substitution can be seeded by PSP-tau, as shown earlier [22]. None of the constructs showed any insoluble tau when unseeded. These findings indicate that the phosphorylation status at Ser305 determines AD/PSP strain-specific seeding, even in the presence of acetylation mimicking modifications in the filament core domain of tau.
Figure 3: Presence of phospho-mimicking Ser305Glu modifies the ability of pseudo-acetyl 5K-Q tau to be differentially seeded by AD-tau and PSP-tau seeds.

a. Linearized depiction of pseudo-acetyl 5K-Q tau carrying the phosphorylation-mimetic substitution on Ser305 (Ser→Glu) generated on human 0N/4R P301L mutant tau. b-d. Seeded HEK293T cells were fractionated into detergent-soluble and detergent-insoluble lysates and probed for total tau (t-tau) and p-tau (AT8). Representative immunoblots and % tau aggregation are shown. N=3 for each experimental replicate. Relative molecular masses (kDa) are indicated on the left of each blot. 2-way ANOVA with Dunnett’s multiple comparisons test, with single pooled variance. *p<0.05, **p<0.01.
Phosphorylation profile of AD-tau and PSP-tau seeded acetyl tau variants
To further investigate the cooperative relationship between acetylation and phosphorylation, we probed for the presence of different phosphorylation epitopes that are associated generally with advanced stages of tauopathies. Using AD-tau and PSP-tau seeded insoluble tau isolated from cells expressing 5K-Q, 5K-R, 5K-Q/S305E, and P301L tau, we examined the relative level of PHF1 (pSer396/404), AT8 (pSer202/pThr205), AT270 (pThr181), and AT180 (pThr231) signals (Fig. 4a). Using total tau antibody, we confirmed our earlier observation that seeding is ablated on 5K-Q tau but not on 5K-R tau, when compared to parent P301L tau. Presence of the different phosphorylation epitopes was consistent with this observation showing that while 5K-Q tau seeded with AD-tau and PSP-tau seeds showed minimal phosphorylation at these epitopes while 5K-R tau showed higher phospho-occupancy (Fig. 4b). Of note, when 5K-Q/S305E tau was seeded with AD-tau seeds, there was low to undetectable PHF1, AT8, AT270 and AT180 epitopes. However, in the presence of PSP-tau seeds, 5K-Q/S305E tau regained PHF1 and AT270 epitope phosphorylation, though both AT8 and AT180 remained undetectable in the seeded 5K-Q/S305E tau (Fig. 4b).
Figure 4: Phosphorylation profile of seeded acetyl-substituted tau variants.
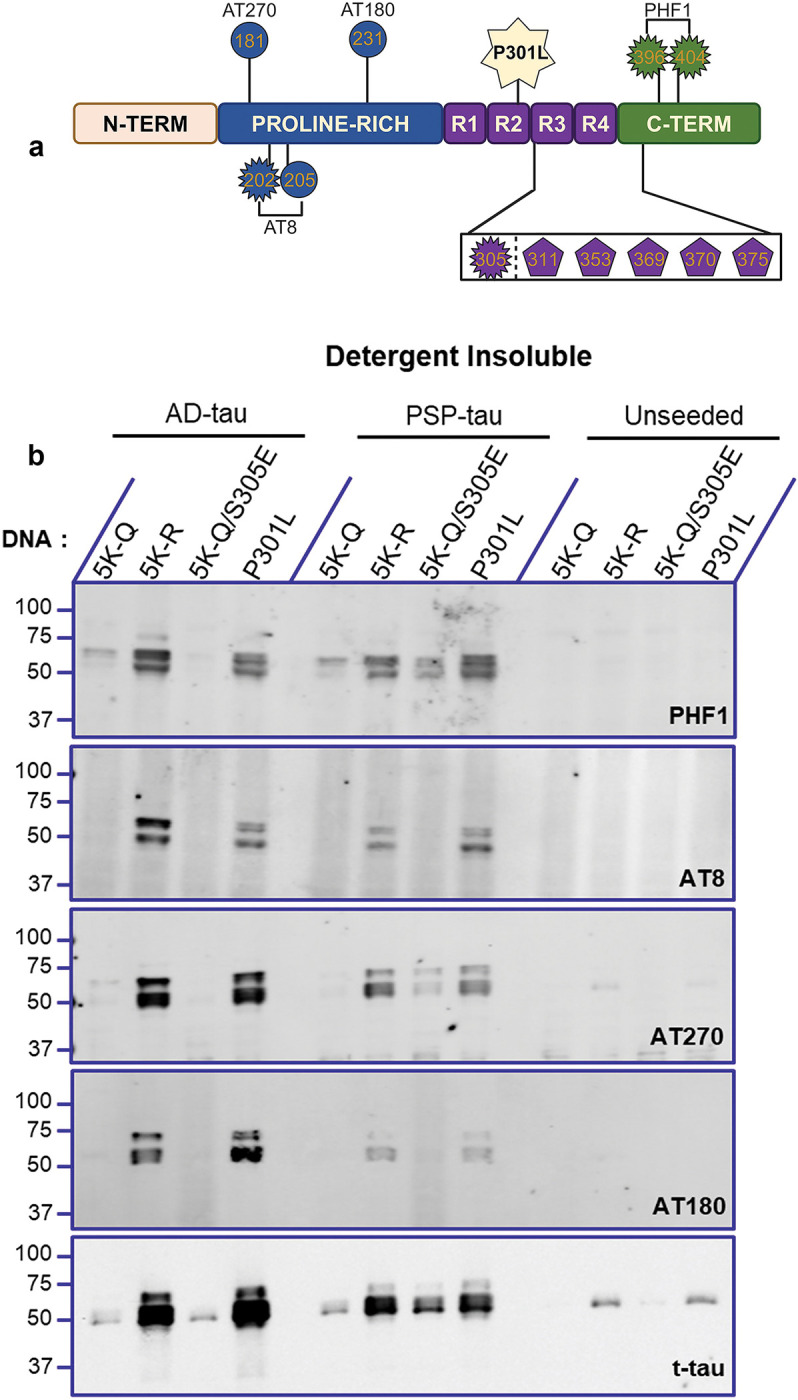
a. Schematic depiction of acetyl-mimetic 0N/4R P301L tau variants with antibody epitopes depicted. Depiction not to scale. b. The detergent-insoluble fractions of 5K-Q, 5K-R, 5K-Q/S305E and parent P301L tau variants seeded with AD:Patient A tau seeds or PSP:Patient A tau seeds, or unseeded were immunoblotted and probed with different p-tau epitopes PHF1 (pSer396/404), AT8 (pSer202/pThr205), AT270 (pThr181), and AT180 (Thr231). Representative blot from n=2 replicates.
Differential microtubule binding patterns of pseudo-acetyl tau variants
Tau plays a key role in assembly of tubulin protein and further in maintaining structural stability of axonal microtubules [27]. To investigate whether acetylation of key residues within the tau filament core domain affects microtubule binding, we performed a microtubule binding assay in the presence or absence of the microtubule-stabilizing compound Paclitaxel (Fig. 5, Fig. S3). HEK293T cells expressing the pseudo-acetyl tau were harvested with high salt buffer supplemented with Paclitaxel and GTP, followed by ultracentrifugation to reveal tau unbound (S) or bound to microtubules (P), and each of these fractions was probed for total tau (t-tau) and β-tubulin (β-tub) (Fig. 5). First, we confirmed that there was significantly more tubulin present in the pellet fraction of HEK293T cells expressing the pseudo-acetyl tau in the presence of paclitaxel compared to conditions in the absence of paclitaxel (Fig. S3a), indicating that paclitaxel was able to stabilize microtubules as expected. In the presence of paclitaxel, the 5K-Q tau remained largely unbound to the microtubule (p<0.05) while 5K-R tau had similar microtubule binding affinity as P301L tau (Fig. 5a,b). The 5K-A tau showed a significantly higher proportion of tau in the pellet fraction of paclitaxel-treated (Fig. 5a,b; p<0.0001) or paclitaxel-untreated reactions (Fig. S3b,c; p<0.0001), indicating that it has an inherent strong affinity for microtubules. We also investigated the relative microtubule-binding affinities of the individual pseudo-acetyl variants (Fig. 5c, d). All of these pseudo-acetyl variants shows equivalent levels of microtubule binding relative to each other and P301L tau (Fig. 5c,d), except for K370Q and K375Q tau that showed increased presence in the pellet fractions (Fig. 5c,d; p=0.0271 for K370Q; p=0.1413 for K375Q). When the S305E was paired with 5K-Q tau, we found increased tau bound to the microtubules in presence or absence of paclitaxel, though the levels were lower than P301L tau (Fig. 5e,f, p<0.0001; Fig. S3f,g, p=0.0014). Neither GFP expression nor naïve HEK293T cells showed detectable tau in this assay (Fig. 5e; Fig. S3f).
Figure 5: Comparative microtubule binding properties of acetyl-substituted tau.

HEK293T cell-based microtubule binding assay was performed in the presence of Paclitaxel with cells transfected to express different P301L tau with acetyl-substituted epitopes, with GFP and no-DNA control. Antibody specific for β-tubulin was used to assess the microtubule polymerization under the same conditions. a-b. Quantification of microtubule-binding efficiency of 5K-Q, 5K-R, 5K-A compared to parent P301L tau. c-d. Quantification of microtubule-binding efficiency of K311Q, K353Q, K369Q, K370Q, K375Q compared to parent P301L tau. e-f. Quantification of microtubule-binding efficiency of 5K-Q/S305E mimetic compared to 5K-Q and parent P301L tau. The relative molecular masses of protein markers are indicated on the left. N=3 for each experimental replicate. 1-way ANOVA with Dunnett’s multiple comparisons test, with single pooled variance. *p<0.05, ****p<0.0001. S= supernatant fraction; P= pellet fraction. Data from microtubule binding assay without Paclitaxel treatment are shown in Fig. S3.
Acetyl-null substitutions in the tau filament core display unique seeding properties
To further investigate the properties of the 5K-A tau which has spontaneous self-aggregating properties, HEK293T cells expressing 5K-A tau were exposed to AD-tau, PSP-tau or left unseeded to generate aggregated tau (Fig. 6a; ‘primary passage’). The detergent-insoluble tau aggregates from primary passage were then used as tau seeds on HEK293T cells expressing wild type tau, P301L tau or cells were left untreated (Fig. 6a; ‘secondary passage’). We used the secondary passaging strategy commonly used in the prion field to assess if 5K-A tau has the unique strain properties depending on the original seeds that it was exposed to [28].
Figure 6: 5K-A tau displays unique self-aggregating properties.

a. Schematic description of secondary passaging paradigm. Briefly, HEK293T cells expressing 5K-A tau constructs were either seeded with AD-tau, PSP-tau, or left unseeded. The detergent-insoluble fraction was harvested and used again (‘secondary 5K-A seeds’) for seeding of WT tau or P301L tau. b. Representative immuno-EM images of total tau antibody-stained detergent-insoluble 5K-A tau seeded with either AD-tau, PSP-tau, or unseeded. All filaments were stained with 10nm gold conjugated secondary antibody followed by negative staining with 1% uranyl acetate. Arrows depict 10nm gold particles. Additional EM images for each condition are shown in Fig. S4. c-d. Representative total tau and AT8-tau blots depicting outcomes of secondary passaging of 5K-A seeds on WT tau and P301L tau. e. Characterization of the phosphorylation profile for primary passaged 5K-A tau seeds (top row) and secondary passaged 5K-A seeds on P301L tau (bottom row). Representative blots from n=2 replicates.
We first investigated the micro-structure of 5K-A tau that was exposed to AD-tau seeds, PSP-tau seeds or was allowed to self-aggregate from primary passage experiment. We performed immunogold electron microscopy (immuno-EM) using anti total tau CP27 antibody on detergent insoluble 5K-A tau primed with either AD-tau, PSP-tau or unseeded (Fig. 6b, Fig. S4). We observed amorphous deposits in all of these preparations that were negatively stained with uranyl acetate and were simultaneously labeled with 10nm immunogold particles (Fig. 6b). We confirmed that in the absence of primary antibody, there was no immunogold labeling (Fig. S4d).
In the secondary passaging experiment, we wanted to determine if the 5K-A tau aggregates were capable of seeding both P301L tau or WT tau. For these experiments, we extracted detergent-insoluble fraction from cells expressing 5K-A tau that were seeded with AD-tau or PSP-tau, and incubated these extracts on cells expressing WT tau, P301L tau or no vector condition (Fig. 6c). For these secondary passaging experiments, the 5K-A seeds (AD-tau primed, PSP-tau primed or Native) were diluted 1:10 (~50ng), as the undiluted samples caused cells to lift off within 24 hours of application. We found that all forms of 5K-A seeds (AD-tau primed or PSP-tau primed or native) were able to secondarily seed P301L tau and induce AT8 positivity (Fig. 6c, d). Cells expressing WT tau did not develop aggregates after seeding with any of these seeds (Fig. 6d). We further examined the phosphorylation patterns of 5K-A seeds from primary passage seeding and secondary passaging (Fig. 6e). In the primary passage, detergent-insoluble 5K-A showed phosphorylation on PHF1, AT180, AT8, AT270, and CP13 epitopes, irrespective of whether 5K-A tau was seeded with AD-tau, PSP-tau or left unseeded. Notably, unseeded 5K-A showed lower levels of the 65kDa hyperphosphorylated tau than AD-tau and PSP-tau seeded 5K-A. When these different 5K-A seeds were exposed to P301L tau recipient, the resulting detergent-insoluble tau showed phosphorylation on PHF1, AT8, AT270, and CP13 epitopes that is comparable to the primary passage-generated tau. Notably, the secondary passage generated tau seeds completely lacked phosphorylation of the AT180 epitope (Fig. 6e).
Discussion
Specific acetylation patterns on tau are associated with disease stage-specific tau aggregation in tauopathies and thus are broadly thought to underlie the progressive neurodegenerative cascade in tauopathies [6, 7]. Previous research has demonstrated the pathological role of tau acetylated in the proline-rich region and MTBR R1-R2 domains [29]. Recently, specific acetylation events residing on MTBR R3-R4 domains spanning the tau filament core domain (K311, K353, K369, K370 and K375) were identified at high frequency in AD patients [6], leading us to examine whether presence of acetylation on these sites influence templated tau aggregation. Utilizing a biochemical approach with acetyl-mimetic or acetyl-nullifying residues on 0N/4R P301L tau, we show that: (1) combinatorial acetyl-mimetic construct is not templated efficiently by tau seeds derived from AD and PSP patients; (2) combinatorial acetyl-null constructs does not affect the seeding ability of human brain-derived tau seeds; (3) an acetyl-null construct with alanine substitution results in self-aggregation in the absence of any brain-derived tau seeds, and (4) strain-specific features of the S305E phospho-variant were preserved when combined with the 5K-Q hyper-acetyl tau variant. Thus, our results demonstrate that hyperacetylation within the tau filament core could prevent seeding-induced tau aggregation and that acetylation and phosphorylation can act cooperatively to determine strain-specific seeding.
Many studies have established the pathogenic properties of acetylated tau in the context of tauopathies. Of the >30 potential AcK epitopes, several studies have shown that AcK174, AcK274, AcK280 and AcK281 are found in NFT from patient brains and underlie toxicity in mice [11, 15, 16, 30]. These AcK sites are located in the proline-rich region and MTBR R1-R2, regions that are outside of the canonical tau filament core [21]. The mechanisms proposed to explain the role of acetylation in tau aggregation vary from disruption of tau-microtubule interaction, impaired turnover and clearance, synaptic dysregulation and increasing the rate of tau fibrillization [9, 15, 30, 31]. Our study is based on selected AcK sites that have been identified within the filament core domain of tau from AD patients using sensitive mass spectrometry techniques [6]. Using these sites, we have asked the question whether acetylation of tau within the canonical tau filament core influences its templating activity.
Previous studies have provided insights into how AcK residues regulate tau aggregation. In a recent study using 4R tau, Cohen and colleagues demonstrated that P301L tau protein that has an acetyl-nullifying K280→R substitution showed delayed aggregation kinetics whereas the pseudo-acetyl K280→Q showed modestly increased aggregation [12]. Another recent study showed that acetylation of 4R tau strongly inhibits in vitro fibrillization kinetics [14], consistent with our cellular seeding data using the five combinatorial AcK sites around MTBR domain 4. Notably, cooperative hyperacetylation on the KXGS motifs spanning the MTBR regions, specifically, K259/290/321/353Q substitution inhibits tau aggregation [10], though individually K353Q did not influence tau fibrillization in vitro [20]. In addition, single site substitutions on AcK311 and AcK369 also did not substantially alter tau fibrillization [20], suggesting that inter-AcK cooperativity is critical. Our data would also be consistent with the theory that cooperative hyperacetylation on specific residues within the filament core domain (AcK311, AcK353, AcK369, AcK370 and AcK375) prevents seeding-induced tau misfolding, irrespective of the disease-type signature present in the tau seeds. Additionally, we also found that single site pseudo-acetylated tau could also influence seeded aggregation, but to various levels showing inter-patient heterogeneity.
Since not much is known specifically about the cooperativity between these AcK sites, we performed in silico modeling analysis on the known filament core domains from AD and PSP [21, 24] by substituting these residues with Q residues on K311, K353, K369, K370 and K375 (Fig. 7). We predicted how the K to Q substitutions could potentially influence intramolecular interactions on tau. In AD-tau, the Q substitutions were predicted to cause subtle changes in the positioning of the amino acid side chains and affect interactions with neighboring residues, which is reflected in altered inter-molecular distances in K311Q and K370Q (Fig. 7a, d). Notably, there is a potential loss of salt bridge interaction between K370 and D314 which could impact the folding (Fig. 7d). In some cases, contact with phospho-epitopes were also modified, which could potentially affect the availability of these Ser/Thr residues to PTM on AD-tau. For example, K353Q moves closer to Ser356 (6.0Å 4.3Å) and K375Q moves away from Thr373 (3.8Å 5.5Å) (Fig. 7b, e). Comparing the corresponding pseudo-acetyl residues on the PSP-tau PDB framework (Fig. 7f–j), we found similarly that the K to Q substitutions showed subtle changes in interactions between neighboring residues, especially K311Q, K353Q and K369Q (Fig. 7f–h). In the in-silico models of contact prediction involving pseudo-acetylated PSP-tau, we did not observe any potential altered interactions with Ser/Thr residues. Overall, these structural predictions, in combination with functional and molecular-level structural data in the future, could provide insights into how hyperacetylation profoundly inhibits seeded tau aggregation.
Figure 7: In silico predictive modeling of 5K-Q tau contained in the tau core domains found in AD and PSP.

In silico predictions of WT tau and 5K-Q tau generated on the AD-tau core (a-e) (7QL4, [23]) and PSP- tau core (f-j) (7P65 [24]) using ChimeraX.
Biochemical and neuropathologic data suggests that the predominant form of misfolded tau that is found in different tauopathies have unique structures [32]. This finding has led to the strain hypothesis of tau, whereby tau adopts specific conformations that determines how it is templated and propagated along specific pathways and affecting specific cell types. This strain hypothesis could explain the intra-individual phenotypic, neuropathological and clinical diversity within each tauopathy as well as between different tauopathies [33]. Indeed, rodent modeling studies [34–38] is now supported by elegant molecular-level cryo-EM data [24, 39] which show that native tau transforms into specific filamentous patterns that are unique to the clinical delineation of the tauopathy. While cellular and molecular studies establish how phosphorylation determines the seeding propensity of tau [22, 35], less is known about acetylation as a determinant factor. Recent data shows that acetylation on K298/K294 (MTBR-R2 domain) and K311 (MTBR-R3 domain) could result in differential disease-specific tau strains based on tau isoforms [14, 40]. Specifically, acetylation on K298/294 delays 4R tau aggregation whereas AcK311 favors 3R tau aggregation. Another study showed that while acetylated forms of recombinant P301L tau seeds did not influence aggregation propensity, enhancing cellular acetylation environment by overexpressing CBP/p300 acetyltransferase promoted tau oligomerization, seeding and aggregation [12]. Our data now provides additional evidence on how cooperative and individual acetylation of AD-associated AcK sites within the tau filament core could determine the availability of 0N/4R tau to be seeded.
Proteomic and neuropathologic data from human patient samples suggest that cooperativity between different PTM sites determine tau misfolding [6, 8]. Acetylation-mediated downstream activity (such as those occurring in histones) generally follow phosphorylation signals, which act as initiating cellular signaling stimuli in normal homeostatic conditions in the nucleus. Whether such a directional modality exists in the life cycle of tau as posited by Wesseling and colleagues [6] is unknown. In addition, Lysine residues are modified not only by acetylation, but also by methylation, ubiquitination, sumoylation and neddylation in a mutually exclusive manner, thus increasing the complexity of the PTM code that could underlie AD-specific PTM code. Thus, with regard to tau, this is a complex relationship as specific PTM could alter tau aggregation in both positive and negative manner and that one type of PTM could preclude another type, thus complicating the outcome [5]. Some molecular evidence shows such PTM-site cooperativity in the context of tau. For example, pseudo-acetyl K259/290/321/353Q tau reduces tau fibrillization in vitro and further modifies the phosphorylation on specific Ser epitopes in its vicinity [10, 20], suggesting cooperativity between phosphorylation and acetylation status. We provide in silico modeling data to suggest that hyperacetylation in the AD-tau filament core could potentially affect phosphorylation on K353 and K375. We were also able to demonstrate the importance of Ser305Glu as a determinant of PSP strain-specific tau seeding, even in the presence of pseudo-acetyl motifs. This would suggest that phosphorylation on specific epitopes could impact strain-like characteristics to tau, even in the presence of other PTMs such as acetylation.
Limitations of our study
One limitation of our study is that we used the FTD-associated P301L tau to examine tau seeding. We have previously shown that WT tau is not receptive to templated aggregation [22] in these cellular assays which is supported by similar assays from other research groups using P301L or P301S backbone [37, 41]. Another limitation is that our analysis is done using 0N/4R tau and given the newly emerging data on the differential toxicity of 3R and 4R tau in different tauopathies [14, 40, 42], future studies will need to consider the relative seeding propensity of acetylated 3R and 4R isoform tau.
Conclusions
In conclusion, our study shows that hyperacetylation in the tau filament core prevents the templating propensity of soluble tau when exposed to human brain-derived tau seeds and further there is cooperative cross-talk between phosphorylation and acetylation leading up to seed-induced tau aggregation.
Experimental Procedures
Generation of tau acetyl-variant constructs
Acetyl-variant tau were generated in the human 0N4R P301L tau backbone under contract with Genscript, carrying hybrid chicken β-actin promoter, Woodchuck promoter regulatory element and bovine poly A sequence. The pI of individual acetyl constructs on the AD-tau filament core and PSP-tau filament core was calculated from Vector NTI and the biophysical properties were imputed from https://www.peptide2.com/N_peptide_hydrophobicity_hydrophilicity.php.
Purification of insoluble Tau from AD and PSP brains.
Human brain tissues from two AD cases, two PSP cases (all cases from the UF Brain Bank brain bank) were selected for this study (Table S1). All cases were diagnosed based on accepted neuropathology criteria. Purification of pathological, insoluble tau from the temporal cortex of AD and non-demented control cases, as well as lentiform nucleus of PSP cases were performed as previously described [22]. Briefly, for the purification of AD-tau and PSP-tau, 50–100 mg of temporal cortical gray matter or lentiform nucleus was homogenized in nine volumes (v/w) of high-salt buffer (10 mm Tris with 0.8 m NaCl, pH7.4) with 0.1% sarkosyl and 10% sucrose added, and centrifuged at 10,000 × g for 10 min at 4°C. Pellets were re-extracted twice using the same high-salt buffer and the supernatants from all three extractions were filtered and pooled. Additional sarkosyl was added to the pooled supernatants to reach 1% and the samples were nutated for 1 h at 37°C. The samples were centrifuged at 150,000 × g for 60 min at 15°C and the resulted 1% sarkosyl-insoluble pellets containing pathological tau were resuspended in PBS. The resuspended sarkosyl-insoluble pellets were further purified by a brief sonication using a handheld probe (Qsonica), followed by centrifugation at 100,000 × g for 30 min at 4°C. The pellets were resuspended in PBS at 1/2 to 1/5 of the pre-centrifugation volume, sonicated, and spun at 10,000 × g for 30 min at 4°C to remove large debris. The final purified supernatants contained insoluble, pathological tau, and are identified as AD-tau and PSP-tau in subsequent experiments. The final fraction was analyzed by Western blotting and sandwich ELISA for tau (Invitrogen #KHB0041) as previously characterized in [22].
Cell Culture and Transfection
HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin/100 μg/ml streptomycin at 37°C and 5% CO2. For transfections, cells were plated on 24-well polystyrene plate with 1 mL of media. Once cells reached ~60% confluency, 0.4 μg of plasmid DNA expressing 0N4R tau was combined with Lipofectamine 3000 (Thermo Fisher Scientific). This mixture was incubated at room temperature for 10–15 minutes before adding dropwise to the media in each well and placed in the incubator for 30 minutes. For seeding, 0.5 μg of AD-tau or PSP-tau was incubated with Lipofectamine reagent for 20 minutes and added to the cells. Cells were harvested 48 hours after transfection and fractionated.
Biochemical Cellular Fractionation of HEK cells
Cells were harvested in 50 μL of High Salt Buffer (50 mM Tris-HCl, pH 7.4, 250 mM NaCl, 2 mM EDTA, 1% Triton X-100, 20 mM NaF) and a cocktail of protease and phosphatase inhibitors (Pierce #A32959). Samples were sedimented at 150,000 × g for 30 minutes at 4°C and the supernatants collected, washed and sedimented again. Supernatants were removed and the pellets were resuspended in 50 μL of High salt buffer. SDS sample buffer (final concentration of 250 mM Tris-Cl, pH 6.8, 5% β-Mercaptoethanol, 0.02% Orange G, 10% SDS, 30% glycerol) was added to the collected supernatants and resuspended pellets, referred to as the detergent-soluble and detergent-insoluble fractions respectively. To resuspend the pellet, detergent-insoluble samples were probe-sonicated. These samples were then heated at 90°C for 10 minutes.
Preparation of Secondary Seeds and Passaging of 5K-A tau
Briefly, HEK293T cells transfected with 5K-A tau were seeded with 0.5 μg of AD-tau or PSP-tau or left unseeded and harvested after 48 hours. Cellular material was then fractionated and the detergent-insoluble fraction was used for secondary passaging experiments. Detergent-insoluble seeds were then characterized via immunoblotting and ImmunoEM, followed by secondary passaging. The secondary seeds are labeled as 5KA-AD, 5KA-PSP and 5KA-U to denote that the seeds were derived from HEK293T cells expressing 5K-A and seeded with AD-tau or PSP-tau or unseeded. For secondary passaging experiments, HEK293T cells transfected with WT tau or P301L tau were seeded with 50ng of these secondary seeds and harvested after 48 hours. Cellular material was further fractionated into detergent soluble and insoluble fractions, followed by characterization of seeding via immunoblot.
Microtubule Binding Assay
Cells were lysed in 100 μl of PEM buffer (80 mM PIPES, pH 6.8, 1 mM EGTA, 1 mM MgCl2) supplemented with 0.1% Triton X-100, 2 mM GTP, 20 μM paclitaxel, and a mix of protease inhibitors as described previously [22]. For the “without paclitaxel” condition, cells were lysed in PEM buffer supplemented with 0.1% Triton X-100 and a mix of protease and phosphatase inhibitors. Cell lysates were incubated in a 37 °C water bath for 30 min and then centrifuged at 100,000 × g for 30 min to pellet microtubules (MT). Supernatant was transferred to a new tube, and the pellet (MT fraction with bound proteins) was resuspended in PEM buffer. The pellet fraction was briefly sonicated, and SDS gel loading buffer was added to both fractions. Equivalent amounts of supernatant and pellet were loaded on SDS-polyacrylamide gels for Western blot analysis. Percent MT bound tau was calculated as pellet/(supernatant + pellet) × 100. Tubulin polymerization was calculated as a ratio of β-tubulin in the Pellet (P):Supernatant (S) fraction.
Western blotting
8 μl of detergent-soluble and 15 μl of detergent-insoluble samples were loaded on 4–20% Tris-Glycine gels (Invitrogen #XP04205) and separated for 1.5hrs at 100V. Blots were then transferred to 0.45 μm PVDF membranes (Millipore #IPFL85R) for 2hrs at 300 mAmps before being blocked in 0.5% Casein for 1 hr. Blots were incubated in appropriate primary antibodies overnight as indicated in each figure (Table S2), and then incubated with IRDye-labeled secondary antibodies and scanned using an ODY-2536 Imager.
Immunocytochemistry
HEK293T cells were washed 3x with PBS and fixed with 10% formalin for 15 min at room temperature. Staining was performed with 0.0125% (wt/vol) ThioS in 50% ethanol for 3 minutes. ThioS was differentiated in 50% ethanol in PBS for 30 seconds. Coverslips were mounted using ProLong Glass Antifade Mountant with NucBlue Stain (Invitrogen #P36981) to label cell nuclei. A Keyence microscope was used to acquire immunofluorescence images. For the quantification of ThioS positivity, at least 10 of 20X frames were taken at random along the coverslip and percent Thioflavin reactivity was calculated from a ratio of Thioflavin positive to tau positive cells.
Cell Viability Assay
Confluent HEK cells in a 24-well plate were used to test viability of cells via alamarBlue (Invitrogen #DAL1025). Briefly, each well received alamarBlue reagent and a well with only media was used as a blank control. Cells were incubated for different time points to track viability in which 100 μl of media was drawn from each condition and stored in a clear, round-bottom 96-well plate and sealed until all time points were collected. Fluorescence excitation/emission was detected at 540–570/580–610 nm and absorbance at 570 nm.
Immuno-gold, negative-stain electron microscopy
Tau filaments from detergent-insoluble HEK293 cell lysates were deposited on 300 mesh carbon film-coated copper grids (Ted Pella 01843-F) for 10 minutes, blocked for 15 min with PBS + 2% BSA, and incubated with anti-tau CP27 antibody (1:500) in PBS + 1% BSA overnight. Grids were washed with blocking buffer and incubated with 10 nm gold-conjugated anti-mouse IgG (Sigma G7652) diluted 1:50 in PBS + 1% BSA for 2 hrs. The grids were then washed with water and stained with 1% uranyl acetate for 60 s. Images were acquired using a Tecnai G2 Spirit at 120 kV (University of Florida ICBR Electron Microscopy Core Facility, RRID:SCR_019146)
In Silico Predictions
Bioinformatic modeling of tau acetyl-mimetics were carried out using the ChimeraX program [43]. Using the previously published tau core domains for AD (V306-F378) and PSP (G272-N381) [21, 24], individual amino acid residues corresponding to 5K-Q were in silico substituted and the intramolecular environmental distances were measured against adjacent residues.
Statistical Analysis
Western blot signals were quantified based on densitometric analysis using ImageJ. For statistical comparisons, we performed both one-way and two-way analysis of variance (ANOVA) with either a Dunnett’s or Sidak’s multiple comparisons test, to compare each group to the control.
Supplementary Material
Figure S1: Comparative effect of combinatorial acetyl variants in the tau core domain on seed-induced P301L tau aggregation. a-f. HEK293T cell seeding assay using the acetyl variants seeded with AD-tau seeds or PSP-tau seeds. Samples were fractionated into detergent-soluble and detergent-insoluble lysates and probed for total tau (t-tau) and p-tau (AT8). Representative immunoblot and % Aggregation of acetyl-tau variants seeded with AD:Patient B tau seeds or PSP:Patient B tau seeds. N=2 for each experimental replicate. g. AlamarBlue cell viability was conducted on cells transfected with different plasmids. Measurements were taken at 4 different time points. h-i. Immunofluorescence of HEK293T expressing 5K-Q, 5K-R, and P301L tau, seeded with either AD-tau, PSP-tau, or unseeded were probed with ThioS (green) and total tau (red) and DAPI (blue) for nuclei. Quantification of immunofluorescent images as a ratio of ThioS-positive cells to tau-positive cells calculated from 10 individual fields of view for each group (i). 2-way ANOVA with Dunnett’s multiple comparisons test, with single pooled variance. ****p<0.0001.
Figure S2: Comparative profiles of individual pseudo-acetyl tau variants seeded with AD-tau and PSP-tau seeds. a-f. Representative immunoblots and quantitation depicting % tau aggregation (a-c) and AT8 insolubility index (d-f) of individual acetyl-mimetic tau seeded with AD:Patient B tau seeds. g-l. Representative immunoblots and quantitation depicting % tau aggregation (g-i) and AT8 insolubility index (j-l) of individual acetyl-mimetic tau seeded with PSP:Patient B tau seeds. Relative molecular masses (kDa) are indicated on the left of each blot. N=2 for each experimental replicate. 1-way ANOVA with Sidak’s multiple comparisons test, with single pooled variance. *p<0.05.
Figure S3: Microtubule binding of acetyl-substituted tau variants in the absence of paclitaxel. HEK293T cells were transfected with acetyl-substituted tau variants as indicated and cell lysates used for microtubule binding assay. a. Quantification of the tubulin polymerization efficiency for conditions treated with and without Paclitaxel. b-c. Quantification of microtubule-binding of 5K-Q, 5K-R, 5K-A compared to parent P301L tau. d-e. Quantification of microtubule-binding of K311Q, K353Q, K369Q, K370Q, K375Q compared to parent P301L tau. f-g. Quantification of microtubule-binding for 5K-Q/S305E, 5K-Q and P301L tau. 1-way ANOVA with Dunnett’s multiple comparisons test, with single pooled variance. **p<0.01, ***p<0.001, ****p<0.0001. S= supernatant fraction; P= pellet fraction.
Figure S4: Additional immuno-EM images for 5K-A tau filaments. a-c. Representative immuno-EM images of detergent-insoluble 5K-A filaments from HEK293T cells that were seeded with AD-tau (a), PSP-tau (b) or left unseeded (c). d. Representative Immuno-EM images of 5K-A filaments exposed to secondary antibody only. All filaments were stained with 10nm gold conjugated secondary antibody followed by negative staining with 1% uranyl acetate.
Supplementary Table S1. Demographics and diagnoses of the AD and PSP brain donors.
Individually characterized brains in the University of Florida Neuromedicine Human Brain and Tissue Bank were used in the study. Experimental Group denotes the designation for this study. NP Dx1, primary diagnosis based on neuropathology; NP Dx2 and NP Dx3, secondary diagnoses based on neuropathology; Thal phase, burden of immunostained amyloid deposits in cortical and subcortical area; Braak stage, CERAD score, neuritic plaque frequency; AD, Alzheimer’s disease; ARTAG, Aging-related tau astrogliopathy; CAA, cerebral amyloid angiopathy; CVD, cardiovascular disease; HC, healthy control with no dementia; LATE, limbic-predominant age-related TDP-43 encephalopathy; PSP, Progressive supranuclear Palsy. N/A: Not applicable; ND, not done.
Supplementary Table S2. Antibodies used in this study.
Acknowledgements.
We thank the University of Florida Neuromedicine Human Brain and Tissue Bank for sharing human brain samples (funded in part by NIA P30AG066506).
Funding and Additional Information.
The work was supported partially by NIA R01 AG078734 (PC, DRB) and NIH T32-NS082128 (EDS).
Abbreviations:
- AcK
acetylated Lysine
- AD
Alzheimer’s disease
- AD-tau
detergent-insoluble tau filaments isolated from AD patients
- 5K-Q, 5K→Q
Gln (Q) substitution on 5 Lysine (K) sites in 0N/4R P301L tau
- 5K-R, 5K→R
Arg (R) substitution on 5 Lysine (K) sites in 0N/4R P301L tau
- 5K-A, 5K→Q
Ala (A) substitution on 5 Lysine (K) sites in 0N/4R P301L tau
- CBD
corticobasal degeneration
- Immuno-EM
immunogold electron microscopy
- pI
isoelectric pH
- MT
microtubules
- MTBR
microtubule binding repeat domains
- NFT
neurofibrillary tangle
- PSP
progressive supranuclear palsy
- PSP-tau
detergent-insoluble tau filaments isolated from PSP patients
- PTM
post-translational modification
Footnotes
Conflict of interest. The authors declare that they have no competing interests.
Supporting Information
This article contains supporting information.
Data Availability
All data generated or analyzed during this study are included in this published article (and its supporting information files).
References
- 1.Wang Y. and Mandelkow E.. (2016) Tau physiology and pathology. Nat Rev Neurosci. 17, 5–21. [DOI] [PubMed] [Google Scholar]
- 2.Koller E.J., Chakrabarty P.. (2020) Tau-Mediated Dysregulation of Neuroplasticity and Glial Plasticity. Front Mol Neurosci. 13, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gotz J., Halliday G., Nisbet R.M. (2019) Molecular Pathogenesis of the Tauopathies. Annu Rev Pathol. 14, 239–261. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y., et al. (2022) Tauopathies: new perspectives and challenges. Mol Neurodegener. 17, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alquezar C., Arya S., Kao A.W. (2020) Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front Neurol. 11, 595532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wesseling H., et al. (2020) Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer’s Disease. Cell. 183, 1699–1713 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arakhamia T., et al. (2021) Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell. 184, 6207–6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kyalu Ngoie Zola N., et al. (2023) Specific post-translational modifications of soluble tau protein distinguishes Alzheimer’s disease and primary tauopathies. Nat Commun. 14, 3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Min S.W., et al. (2010) Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 67, 953–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook C., et al. (2014) Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum Mol Genet. 23, 104–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Min S.W., et al. (2015) Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med. 21, 1154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tseng J.H., et al. (2021) Tau seeds are subject to aberrant modifications resulting in distinct signatures. Cell Rep. 35, 109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shin M.K., et al. (2021) Reducing acetylated tau is neuroprotective in brain injury. Cell. 184, 2715–2732 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chakraborty P., et al. (2023) Acetylation discriminates disease-specific tau deposition. Nat Commun. 14, 5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen T.J., et al. (2011) The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irwin D.J., et al. (2012) Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain. 135, 807–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irwin D.J., Lee V.M., Trojanowski J.Q. (2013) Parkinson’s disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci. 14, 626–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang X.J., Seto E. (2008) Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell. 31, 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas S.N., et al. (2012) Dual modification of Alzheimer’s disease PHF-tau protein by lysine methylation and ubiquitylation: a mass spectrometry approach. Acta Neuropathol. 123, 105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carlomagno Y., et al. (2017) An acetylation-phosphorylation switch that regulates tau aggregation propensity and function. J Biol Chem. 292, 15277–15286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fitzpatrick A.W.P., et al. (2017) Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith E.D., et al. (2023) Human tauopathy strains defined by phosphorylation in R1-R2 repeat domains of tau. Acta Neuropathol Commun. 11, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lovestam S., et al. (2022) Assembly of recombinant tau into filaments identical to those of Alzheimer’s disease and chronic traumatic encephalopathy. Elife. 11, e76494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi Y., et al. (2021) Structure-based classification of tauopathies. Nature. 598, 359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mercken M., et al. (1992) Monoclonal antibodies with selective specificity for Alzheimer Tau are directed against phosphatase-sensitive epitopes. Acta Neuropathol. 84, 265–72. [DOI] [PubMed] [Google Scholar]
- 26.Limorenko G., Lashuel H.A. (2022) Revisiting the grammar of Tau aggregation and pathology formation: how new insights from brain pathology are shaping how we study and target Tauopathies. Chem Soc Rev. 51, 513–565. [DOI] [PubMed] [Google Scholar]
- 27.Cook C., et al. (2014) Acetylation: a new key to unlock tau’s role in neurodegeneration. Alzheimers Res Ther. 6, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makarava N., et al. (2020) Posttranslational modifications define course of prion strain adaptation and disease phenotype. J Clin Invest. 130, 4382–4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kontaxi C., Piccardo P., Gill A.C. (2017) Lysine-Directed Post-translational Modifications of Tau Protein in Alzheimer’s Disease and Related Tauopathies. Front Mol Biosci. 4, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tracy T.E., et al. (2016) Acetylated Tau Obstructs KIBRA-Mediated Signaling in Synaptic Plasticity and Promotes Tauopathy-Related Memory Loss. Neuron. 90, 245–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haj-Yahya M., Lashuel H.A. (2018) Protein Semisynthesis Provides Access to Tau Disease-Associated Post-translational Modifications (PTMs) and Paves the Way to Deciphering the Tau PTM Code in Health and Diseased States. J Am Chem Soc. 140, 6611–6621. [DOI] [PubMed] [Google Scholar]
- 32.Chung D.C., et al. (2021) Cellular and pathological heterogeneity of primary tauopathies. Mol Neurodegener. 16, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vaquer-Alicea J., Diamond M.I., Joachimiak L.A. (2021) Tau strains shape disease. Acta Neuropathol. 142, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar M., et al. (2024) Alzheimer proteopathic tau seeds are biochemically a forme fruste of mature paired helical filaments. Brain. 147, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dujardin S., et al. (2020) Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat Med. 26, 1256–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaufman S.K., et al. (2016) Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron. 92, 796–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holmes B.B., Diamond M.I. (2014) Prion-like properties of Tau protein: the importance of extracellular Tau as a therapeutic target. J Biol Chem. 289, 19855–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Narasimhan S., et al. (2017) Pathological Tau Strains from Human Brains Recapitulate the Diversity of Tauopathies in Nontransgenic Mouse Brain. J Neurosci. 37, 11406–11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scheres S.H., et al. (2020) Cryo-EM structures of tau filaments. Curr Opin Struct Biol. 64, 17–25. [DOI] [PubMed] [Google Scholar]
- 40.Trzeciakiewicz H., et al. (2020) An HDAC6-dependent surveillance mechanism suppresses tau-mediated neurodegeneration and cognitive decline. Nat Commun. 11, 5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lathuiliere A., et al. (2023) Specific detection of tau seeding activity in Alzheimer’s disease using rationally designed biosensor cells. Mol Neurodegener. 18, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schoch K.M., et al. (2016) Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model. Neuron. 90, 941–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meng E.C., et al. (2023) UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 32, e4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Comparative effect of combinatorial acetyl variants in the tau core domain on seed-induced P301L tau aggregation. a-f. HEK293T cell seeding assay using the acetyl variants seeded with AD-tau seeds or PSP-tau seeds. Samples were fractionated into detergent-soluble and detergent-insoluble lysates and probed for total tau (t-tau) and p-tau (AT8). Representative immunoblot and % Aggregation of acetyl-tau variants seeded with AD:Patient B tau seeds or PSP:Patient B tau seeds. N=2 for each experimental replicate. g. AlamarBlue cell viability was conducted on cells transfected with different plasmids. Measurements were taken at 4 different time points. h-i. Immunofluorescence of HEK293T expressing 5K-Q, 5K-R, and P301L tau, seeded with either AD-tau, PSP-tau, or unseeded were probed with ThioS (green) and total tau (red) and DAPI (blue) for nuclei. Quantification of immunofluorescent images as a ratio of ThioS-positive cells to tau-positive cells calculated from 10 individual fields of view for each group (i). 2-way ANOVA with Dunnett’s multiple comparisons test, with single pooled variance. ****p<0.0001.
Figure S2: Comparative profiles of individual pseudo-acetyl tau variants seeded with AD-tau and PSP-tau seeds. a-f. Representative immunoblots and quantitation depicting % tau aggregation (a-c) and AT8 insolubility index (d-f) of individual acetyl-mimetic tau seeded with AD:Patient B tau seeds. g-l. Representative immunoblots and quantitation depicting % tau aggregation (g-i) and AT8 insolubility index (j-l) of individual acetyl-mimetic tau seeded with PSP:Patient B tau seeds. Relative molecular masses (kDa) are indicated on the left of each blot. N=2 for each experimental replicate. 1-way ANOVA with Sidak’s multiple comparisons test, with single pooled variance. *p<0.05.
Figure S3: Microtubule binding of acetyl-substituted tau variants in the absence of paclitaxel. HEK293T cells were transfected with acetyl-substituted tau variants as indicated and cell lysates used for microtubule binding assay. a. Quantification of the tubulin polymerization efficiency for conditions treated with and without Paclitaxel. b-c. Quantification of microtubule-binding of 5K-Q, 5K-R, 5K-A compared to parent P301L tau. d-e. Quantification of microtubule-binding of K311Q, K353Q, K369Q, K370Q, K375Q compared to parent P301L tau. f-g. Quantification of microtubule-binding for 5K-Q/S305E, 5K-Q and P301L tau. 1-way ANOVA with Dunnett’s multiple comparisons test, with single pooled variance. **p<0.01, ***p<0.001, ****p<0.0001. S= supernatant fraction; P= pellet fraction.
Figure S4: Additional immuno-EM images for 5K-A tau filaments. a-c. Representative immuno-EM images of detergent-insoluble 5K-A filaments from HEK293T cells that were seeded with AD-tau (a), PSP-tau (b) or left unseeded (c). d. Representative Immuno-EM images of 5K-A filaments exposed to secondary antibody only. All filaments were stained with 10nm gold conjugated secondary antibody followed by negative staining with 1% uranyl acetate.
Supplementary Table S1. Demographics and diagnoses of the AD and PSP brain donors.
Individually characterized brains in the University of Florida Neuromedicine Human Brain and Tissue Bank were used in the study. Experimental Group denotes the designation for this study. NP Dx1, primary diagnosis based on neuropathology; NP Dx2 and NP Dx3, secondary diagnoses based on neuropathology; Thal phase, burden of immunostained amyloid deposits in cortical and subcortical area; Braak stage, CERAD score, neuritic plaque frequency; AD, Alzheimer’s disease; ARTAG, Aging-related tau astrogliopathy; CAA, cerebral amyloid angiopathy; CVD, cardiovascular disease; HC, healthy control with no dementia; LATE, limbic-predominant age-related TDP-43 encephalopathy; PSP, Progressive supranuclear Palsy. N/A: Not applicable; ND, not done.
Supplementary Table S2. Antibodies used in this study.
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its supporting information files).