

Abstract
Increased fructose intake from sugar-sweetened beverages and highly processed sweets is a well-recognized risk factor for the development of obesity and its complications. Fructose strongly supports lipogenesis on a normal chow diet by providing both, a substrate for lipid synthesis and activation of lipogenic transcription factors. However, the negative health consequences of dietary sugar are best observed with the concomitant intake of a HFD. Indeed, the most commonly used obesogenic research diets, such as “Western diet”, contain both fructose and a high amount of fat. In spite of its common use, how the combined intake of fructose and fat synergistically supports development of metabolic complications is not fully elucidated. Here we present the preponderance of evidence that fructose consumption decreases oxidation of dietary fat in human and animal studies. We provide a detailed review of the mitochondrial β-oxidation pathway. Fructose affects hepatic activation of fatty acyl-CoAs, decreases acylcarnitine production and impairs the carnitine shuttle. Mechanistically, fructose suppresses transcriptional activity of PPARα and its target CPT1α, the rate limiting enzyme of acylcarnitine production. These effects of fructose may be, in part, mediated by protein acetylation. Acetylation of PGC1α, a co-activator of PPARα and acetylation of CPT1α, in part, account for fructose-impaired acylcarnitine production. Interestingly, metabolic effects of fructose in the liver can be largely overcome by carnitine supplementation. In summary, fructose decreases oxidation of dietary fat in the liver, in part, by impairing acylcarnitine production, offering one explanation for the synergistic effects of these nutrients on the development of metabolic complications, such as NAFLD.
Keywords: Sugar, Fructose, Fatty acid oxidation (FAO), Western diet, Non-alcoholic fatty liver disease (NAFLD)
1. Introduction
Consumption of a high-fat diet (HFD) was initially considered to be the primary driver of the worldwide epidemic of obesity and its associated metabolic complications [1]. Thus, over the last three decades a strong emphasis has been placed on reducing total fat intake. Despite successfully lowering the percentage of energy intake from dietary fat on a population level [2], the incidence of obesity, heart disease, and non-alcoholic fatty liver disease (NAFLD) has not been tamed. Recently, there has been a paradigm shift and more attention has been focused on the detrimental effects of sugar, primarily fructose, as a contributor to the obesity epidemic [3]. The most sugar in our diet is consumed as sucrose (glucose and fructose disaccharide) or high-fructose corn syrup (55% fructose and 45% glucose monosaccharide). Indeed, the combined intake of fructose and sucrose robustly supports weight gain, whereas consumption of fructose alone does not always result in increased weight gain [4]. However, even weight neutral fructose intake may worsen metabolic complications [5,6].
Several metabolic properties of fructose make it an ideal candidate to support the development of obesity and its complications. First, fructose consumption contributes to an increased caloric load. For example, desserts are consumed after a meal when the subject is no longer hungry; and sugar sweetened beverages are consumed to quench thirst, not hunger, resulting in excess caloric intake. On the other hand, even isocaloric fructose restriction may improve weight gain and metabolic dysfunction [7–9]. Next, fructose is considered a highly lipogenic nutrient and serves as both a substrate for lipogenesis and a key inducer of the lipogenic pathway. Additionally, fructose metabolism is exceedingly fast compared to glucose metabolism leading to depletion of adenosine triphosphate (ATP) and uric acid production. Uric acid can lead to lower AMP-activated protein kinase (AMPK) activity [10] and thus decreased fatty acid oxidation. The interaction between fructose and fat metabolism may be the most interesting aspect of fructose-induced effects since these nutrients are commonly consumed together in the obesogenic “Western diet.” In spite of the strong evidence showing that the most detrimental effects of fructose are observed in the setting of a HFD intake, the effects of fructose on fat oxidation are poorly defined. Therefore, the aim of this review is to summarize the available evidence and propose a mechanistic understanding of how fructose intake decreases fatty acid oxidation.
2. Is high fat diet or high sugar intake causing obesity? The truth lies in between
A positive energy balance is required for the development of obesity and its complications. Due to its high caloric density, diets high in fat have indisputably been found to strongly induce obesity. Some studies, in mice, even suggest that dietary fat is the necessary and sufficient nutrient to induce obesity. A comparison of 29 different diets with varying amounts of protein, fat, and sugar found that only increasing the fat content up to 60% of the total calories results in greater obesity, whereas increasing sugar or protein content from 5 to 30% of the calories had no effect on weight gain [11]. This study did not examine the effects of sugar free diets and interestingly, increasing the percent of fat above 60% of the total calories, at an expense of reducing carbohydrate intake, resulted in lower weight gain [11]. This part of the study is in agreement with a well-known paradigm that high-fat, sugar-free keto genic diets promote decreased weight gain [12]. Indeed, high-fat, ketogenic diets reduce adiposity, as long as they are free of sugar, since even a small amount of sugar precludes ketosis. Thus, reducing sugar intake from an obesogenic high-fat diet, even at the expense of further increasing dietary fat, results in reduced weight gain [13], diminished liver lipid accumulation [14–16], decreased insulin resistance [17,18], and an improved lipid profile [19,20]. In addition to being sugar free, ketogenic diets are also low in total carbohydrate load. A proposed mechanism for metabolic improvements is that sugar-free ketogenic diets stimulate a fasting-like state [21] and rely on fat oxidation to sustain metabolic functions [22]. In agreement with this, we published that abrogating fructose metabolism in the livers of mice on a most commonly utilized obesogenic HFD (Research diets, D12492, 60% calories from fat), which also contains 6.7% of sugar, leads to increased mitochondrial fatty acid oxidation and improved metabolic health [23]. Thus, while HFDs strongly induce obesity and metabolic dysfunction, a relatively small amount of dietary sugar is a required co-ingredient to turn off fatty acid oxidation and allow for the full manifestation of metabolic derangements to become evident.
Sugar intake is clearly associated with weight gain and metabolic dysfunction in human [6,24–27] and animal studies [28–30]. Studies showing a positive correlation between sugar intake and obesity generally use a supra-physiologic amount of sugar, such as greater than 60% fructose in solid diet [31–35], or provide sugar-sweetened beverages (SSBs) on top of a standard diet [27–29,36], inducing a hypercaloric state. Therefore, the prevailing opinion is that sugar is a vehicle for increased caloric intake and that the caloric load principally drives the negative health consequences associated with sugar intake [37]. This hypothesis decreases the enthusiasm for avoiding sugar intake, as a greater decrease in caloric load can be achieved by reducing fat, which contains a higher caloric load. However, emerging data shows that isocaloric fructose intake [5,7,38] or fructose, as compared to equicaloric glucose intake [6,39,40], supports the development of metabolic complications independent of total caloric load.
In our studies, we find that 30% fructose-sweetened water consumption does result in increased weight gain in mice on standard chow diet (LabDiet, 9F 5020, 21.6% calories from fat), but this does not lead to severe metabolic complications after 10 weeks on the diet [41]. Interestingly, we observed that 30% fructose supplementation of mice on a low-fat diet (Research Diets, D12450K, 10% calories from fat) over the same time period does not result in weight gain (unpublished observation). Others have also shown that fructose consumption on a normal diet (e.g., 13% calories from fat) does not induce weight gain [42], suggesting that a higher percent of fat in a diet is required to observe the obesogenic properties of fructose. This hypothesis is in line with the well-established fact that fructose intake from fruits and vegetables, as a part of well-balanced diet, does not lead to obesity and metabolic complications [43,44]. On the other hand, we showed that 30% fructose supplementation of mice on a HFD worsens weight gain and metabolic dysfunction as compared to 30% glucose supplementation [41]. Others have also observed that the most severe metabolic derangements are induced by the combined intake of fructose and a HFD [45–50]. Therefore, a Western diet, which is high in both fat and sugar has become a diet of choice in animal studies to induce obesity [51]. Indeed, a recent systemic review of 3,920 rodent models of NAFLD found that the livers of rodents fed a high-fat, high-fructose diet most closely resemble the pattern of severe liver injury observed in human subjects with NAFLD [52]. Taken together, these studies suggest that the effects of fructose on metabolic dysfunction are dependent on higher dietary fat intake, and that a HFD must contain some sugar to fully support development of obesity-associated complications. While the combined intake of fructose and fat in our Western diet is necessary for the optimal obesogenic phenotype, the mechanism that governs these synergistic effects remains elusive.
3. Fructose supports de novo lipogenesis
Most studies in the literature, including our early work, report on a high lipogenic potential of fructose. Indeed, fructose stimulates hepatic de novo lipogenesis (DNL) to a greater extent than glucose [36,53,54], starch [5,55,56], or HFD [14,57,58]. Mechanistically (Fig. 1), fructose uptake across the plasma membrane is largely facilitated by glucose transporters (GLUT2, GLUT5), and the sodium glucose cotransporter 5 (SGLT5) [59,60]. Tissue-specific distribution of these transporters accounts for highly compartmentalized fructose metabolism in the selected organs, such as in the liver (GLUT2), intestine (GLUT5), and kidney (GLUT5/SGLT5) [60]. Phosphorylation and subsequent metabolism of fructose by the rate-limiting enzyme ketohexokinase (KHK) and downstream aldolase B (ALDOB) provides the triose-phosphate intermediates, dihydroxyacetone phosphate (DHAP) and glyceraldehyde (GA), while triose kinase/FMN Cyclase (TKFC) produces glyceraldehyde 3-phosphate (GA3P) from GA. These three carbon metabolites are also intermediates of glycolysis and serve as precursors for lipid synthesis. Aside from providing substrate for lipogenesis, fructose metabolism activates lipogenic transcription factors that govern the expression of lipogenic enzymes.
Fig. 1.
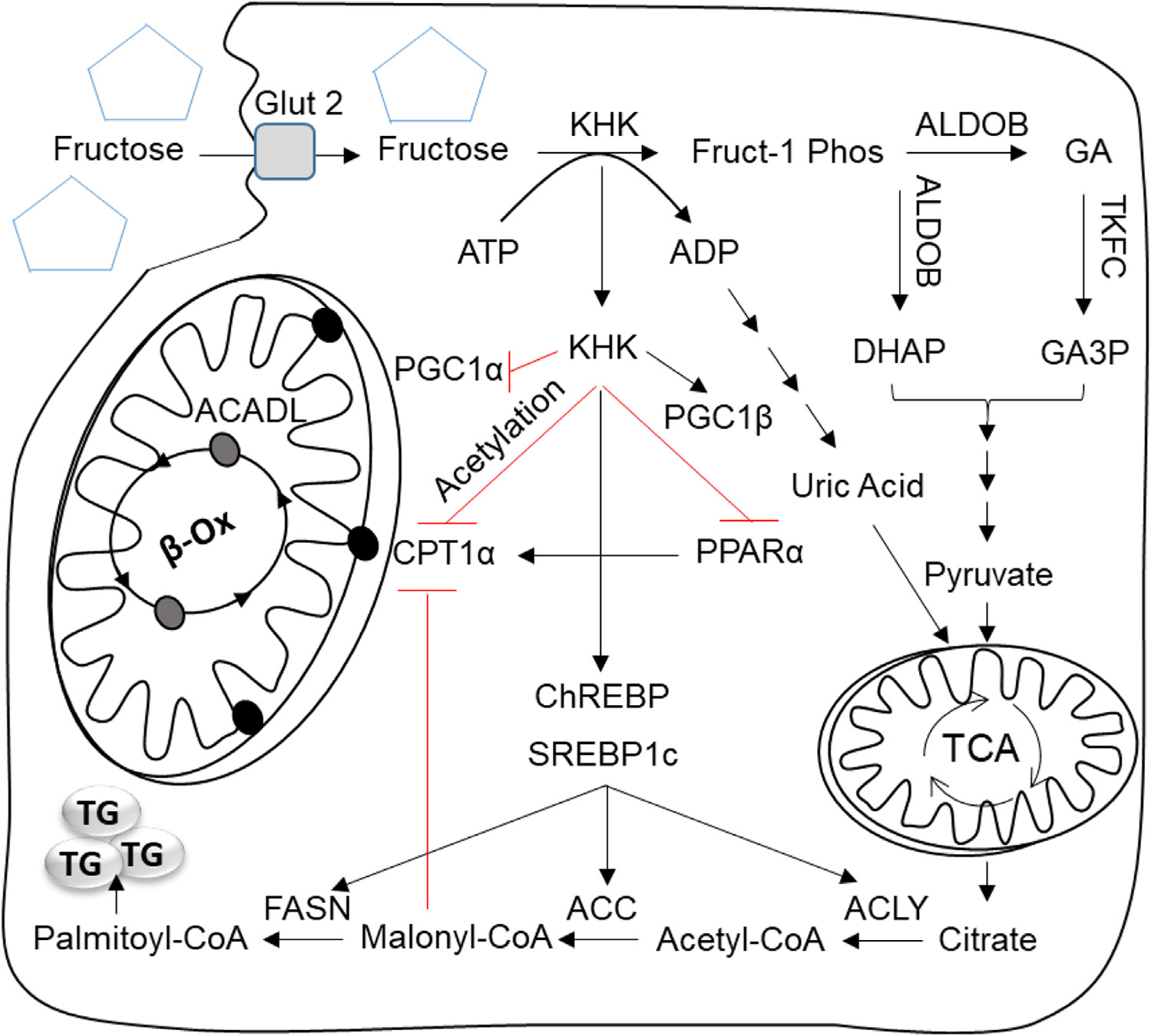
The effect of fructose on lipogenesis and mitochondrial oxidation. In the liver fructose is taken up by solute carrier family 2, facilitated glucose transporter, member 2 (SLC2A2 aka GLUT2) and phosphorylated by ketohexokinase (KHK) to fructose-1 phosphate (Fruct-1 Phos). KHK mediated fructose phosphorylation is rapid leading to depletion of adenosine triphosphate (ATP), accumulation of adenosine diphosphate (ADP) and eventually production of uric acid. Fruct-1 Phos is further metabolized by aldolase b (ALDOB) into dihydroxyacetone phosphate (DHAP) and glyceraldehyde (GA). GA is then phosphorylated by triokinase and FMN cyclase (TKFC) into glyceraldehyde-3 phosphate (GA3P). DHAP and GA3P are also intermediates of glycolysis pathway and they are metabolized into pyruvate. Pyruvate enters the tricarboxylic acid cycle (TCA) to produce electrons for energy production. When the cellular energy stores are plentiful citrate is transported out of mitochondria into the cytosol to serve as a substrate for de novo lipogenesis (DNL). Uric acid produced by fructose metabolism inhibits aconites, an enzyme in TCA cycle, to further increase citrate production. In the cytosol, citrate is converted to acetyl-CoA by the enzyme ATP citrate lyase (ACLY). Acetyl-CoA carboxylase (ACC1), then catalyzes the production of malonyl-CoA and fatty acid synthase (FASN) extends the growing fatty acid chain to synthesize palmitoyl-CoA, a major building block of triglyceride (TG) formation. Malonyl-CoA, an intermediate in DNL also inhibits carnitine palmitoyltransferase 1 alpha (CPT1α) the rate limiting enzyme of mitochondrial fatty acid oxidation (FAO). In addition to providing substrate for DNL fructose through KHK affects the function of transcription factors mediating DNL and FAO. KHK and carbohydrate responsive element binding protein (ChREBP) regulate each other via a bidirectional loop, and upregulated ChREBP increases the expression of genes involved in DNL. Fructose through KHK also upregulates another lipogenic transcription factor, sterol regulatory element-binding protein 1c (SREBP1c), either directly via PPARG coactivator 1 beta (PGC1β) or indirectly by inducing selective insulin resistance and hyperinsulinemia. We present additional evidence that fructose through KHK decreases FAO. These effects are mediated, in part, via lower peroxisome proliferator activated receptor alpha (PPARα), a transcription factor that regulates expression of FAO enzymes, leading to decreased CTP1α protein. Additionally, fructose decreases PGC1α, a cofactor required for optimal PPARα activity via increasing PGC1α acetylation. Lastly, our research indicates that fructose through KHK increases CPT1α acetylation and decreases its protein stability.
In human [61] and animal [62] studies, fructose robustly upregulates carbohydrate response element-binding protein (ChREBP) transcription factor, as well as its binding to DNA [63]. ChREBP then upregulates the transcription of enzymes involved in fatty acid synthesis including ATP citrate lyase (Acly), acetyl-CoA carboxylase (Acc), fatty acid synthase (Fasn), and stearoyl-CoA desaturase-1 (Scd1). This then enables lipid synthesis associated with fructose intake. At the same time, ChREBP also upregulates gluconeogenesis, as well as fructolytic enzymes Khk, AldoB, Tkfc, and Glut5 in a feed forward cycle [64]. Moreover, ChREBP knockout in the liver leads to decreased expression of KHK and other fructolytic enzymes [65]. A bidirectional relationship between fructose metabolism and ChREBP is further highlighted by the studies utilizing a small molecule KHK inhibitor reporting that decreased fructose metabolism leads to ChREBP inactivation [66]. In spite of the strong evidence that ChREBP mediates some aspects of fructose metabolism in the liver, its role in fructose-induced metabolic dysfunction is complex. First, it has been reported that ChREBP dissociates hepatic lipid accumulation from the development of insulin resistance [67]. Next, hepatic ChREBP is required to protect the liver from fructose-induced hepatotoxicity, as fructose-fed liver-specific ChREBP knockout mice exhibit dysregulated glycogen metabolism, cholesterol synthesis, and ATP homeostasis [33,35]. While ChREBP functions in the liver to mediate some fructose-induced metabolic effects, in the intestine ChREBP regulates sugar absorption and taste preference for sweets. Therefore, ChREBP knockout in the intestine leads to fructose malabsorption, bacterial proliferation, and symptoms consistent with irritable bowel syndrome [65,68,69]. In summary, fructose intake induces ChREBP, which in turn further increases fructolysis, but ChREBP upregulation appears to protect the liver from the toxic effects induced by this dietary sugar.
Fructose can also induce DNL via upregulation of sterol regulatory element-binding protein 1c (SREBP1c) transcription factor. SREBP1c upregulates the expression of largely overlapping DNL genes as does ChREBP. Unlike its direct action on ChREBP, fructose effects on SREBP1c are mainly thought to be mediated via its strong propensity to induce insulin resistance. We have recently reviewed the data and mechanistic details of how fructose induces hepatic insulin resistance [70]. Insulin resistance leads to hyperinsulinemia, and insulin is a strong mediator of the SREBP1c-dependent signaling machinery. Insulin stimulates all aspects of SREBP1c control including transcription, proteolytic cleavage into its active form, and nuclear translocation [71]. However, SREBP1c nuclear translocation, as well as concomitant increases in lipogenic genes, can be induced by fructose feedings even in mice with liver-specific knockout of insulin receptor, indicating that fructose can stimulate SREBP1c action in the setting of a complete lack of insulin signaling [72]. Peroxisome proliferator-activated receptor-gamma coactivator 1 beta (PGC1β), a transcriptional coactivator for SREBP1, may mediate these effects, since a knockout of PGC1β reduces SREBP1c expression and downstream lipogenic genes in the liver [73]. Therefore, upregulation of SREBP1c by fructose likely occurs via insulin dependent and independent mechanisms and contributes to transcriptional activation of hepatic lipogenesis.
Lastly, fructose metabolism leads to depletion of ATP, thereby resulting in uric acid production [62,74]. Elevated uric acid can promote lipogenesis by activating ChREBP [62] and SREBP1c [75]. In addition, uric acid inhibits aconitase, an enzyme in the tricarboxylic acid cycle (TCA) cycle i.e., responsible for catabolizing citrate, further supporting the dual action of fructose to increase transcriptional DNL machinery and to increase the substrate availability for lipogenesis.
While fructose strongly supports lipogenesis in rodent models on a normal chow diet, we find that the effects of fructose on lipid synthesis are decreased in the presence of a HFD [41]. This observation is intuitive, as a HFD provides ample supply of free fatty acids (FFA) and obviates the need for ongoing DNL. Although lipogenesis is reduced with the combined intake of fructose and a HFD, the metabolic complications on a Western diet are more evident than on either a HFD or fructose diet alone. Therefore, fructose-induced lipogenesis does not fully account for the metabolic effects of fructose. Rather, the combined intake of fructose and a HFD synergistically supports the development of severe metabolic dysregulation even in the setting of reduced lipogenesis.
4. Fructose impairs fatty acid oxidation in human and animal studies
Based on the cumulative negative effects when fructose and fat are co-ingested, it is reasonable to assume that their concomitant metabolism acts synergistically. Indeed, numerous studies document that fructose decreases the oxidation of dietary fat (Table 1). This hypothesis is supported by clinical studies in human subjects. In the 1980’s, Tappy and colleagues showed that 4h after ingesting a single 75g dose of fructose, seventeen healthy volunteers experienced a significantly greater decrease in lipid oxidation vs. subjects ingesting the same amount of glucose [76]. Similarly, Chong et al., gave fructose or glucose test meals labeled with (2H2)Palmitate and (13C)D-fructose or (13C)D-glucose to fourteen subjects after an overnight fast. The subjects who consumed fructose had a higher respiratory exchange ratio (RER) and more carbons from fructose recovered in breath CO2 indicative of greater carbohydrate utilization. Conversely, net fat oxidation and beta-hydroxybutyrate (BHB) were significantly lower after fructose intake. The authors concluded that fructose contribution to DNL is small, but its effect on altering the partitioning of fatty acids toward esterification may be considerable when fructose and fat are co-ingested [77]. These single dose studies are in agreement with a short-term study of 6 d fructose overfeeding in eight male subjects showing decreased lipid oxidation (0.28±0.11 mg/kg/min) when compared to the basal reading (0.54±0.11 mg/kg/min) on a diet containing 35% of calories from fat [78]. Lastly, in a seminal paper, Cox et al., showed that long-term consumption of fructose- but not glucose-sweetened beverages for 10 weeks reduced net postprandial fat oxidation and energy expenditure in overweight/obese men and women [40].
Table 1.
Studies documenting fructose-induced decrease in fatty acid oxidation.
| Model | Diet comparison | Dose & duration | Fatty acid oxidation | Mechanism | Refs |
|---|---|---|---|---|---|
|
| |||||
| Humans | Fructose or Glucose, 75g | A single drink, 4h metabolic monitoring | -Higher RER | N/A | Tappy, 1986 [76] |
| Humans | Fructose or Glucose in a drink containing fat | A single drink containing 0.75 g sugar/kg body weight and 0.5g/kg of oil | -Higher RER -Higher 13 CO2 from labeled fructose -lower BHB |
N/A | Chong, 2007 [77] |
| Humans | Fructose drink in addition to regular diet vs. regular diet alone | 25% additional calories from fructose (3.5mg/kg dose) for 6 d | -Decreased lipid oxidation mg/kg/min and -BHB only in male subjects | N/A | Couchepin, 2008 [78] |
| Humans | Fructose or Glucose-drinks on ad libitum diet | 25% of energy requirement on normal diet containing 30% fat for 10-wk | -Decreased postprandial fat oxidation and -increased carbohydrate oxidation | N/A | Cox, 2012 [40] |
| Perfused Rat Liver | Fructose | 25 and 45 mg fructose/100 mL of blood | Decreased 14 C incorporation into CO2 and lower ketone bodies. | N/A | Topping 1972 [81] |
| Rat Liver and serum | Fructose, Glucose, Glyceraldehyde, Sorbitol injection in fasted rats | 1 mL of 30% fructose, glucose, glyceraldehyde, or sorbitol were injected intramuscular | Decreased in ketone bodies | N/A | Rawat 1975 [79] |
| Rat Liver | Fructose addition to mitochondria-supernatant system | 5.56 mM fructose treatment along with 14 C labeled palmitate | Decreased conversion of 14 Cpalmitate to 14 CO2 | N/A | Prager 1976 [80] |
| Rat Liver Isolated rat hepatocytes | High fructose diet 67% carbohydrate (98%fructose) | −8 wk of high fructose diet -Isolated hepatocytes in 25mM fructose | Decreased expression of FAO genes | -Reduced PPARα protein and activity -Decreased CPT1α |
Nagai 2002 [87] |
| Rat Liver | Fructose drink on chow diet | 10% fructose in water for 2 wk | Decreased β-oxidation activity nmol/min/mg | Decreased PPARα and target gene CPT1α | Roglans 2002 [91] |
| Rat Liver | Fructose or glucose drink on normal diet | 10% fructose or glucose in water for 14 d | Decreased β-oxidation activity nmol/min/mg | Decreased PPARα and CPT1α protein & mRNA | Roglans 2007 [88] |
| Rat Liver & human hepatocytes | Sucrose in rats/fructose in hepatocytes | 40% sucrose diet for 10 wk/ 5mM fructose in vitro |
Decreased BHB | AMPD2 mediated decrease in AMPK activity | Lanaspa 2012 [132] |
| Rat Liver Rat hepatoma cells | Fructose drink on of regular diet | −10% fructose drink for 14 d; | Decreased β-oxidation activity | -Decreased PPARα and Sirt1 | Rebollo 2014 [93] |
| Human hepatocytes | In vitro fructose, glucose and mannitol | −25 mM fructose for in vitro experiments | nmol/min/mg | -Increased acetylation of PGC1α | |
| Rat Liver | Fructose supplementation of regular diet | 20% fructose solution for 14 wk | Decreased FAO gene expression | DNA methylation at PPARa and CPT1A promoter regions | Ohashi 2015 [90] |
| Mouse liver and Hepatocytes | 30% Fructose or Glucose drinks on chow and HFD | In vitro 25 mM fructose vs. glucose for 24 hr | Decreased β-oxidation (OCR pmol/min) with 25 mM fructose | Acetylation of metabolic enzymes dependent on KHK | Softic 2019 [23] |
| Mouse liver | 15% or 30% fructose in water on chow diet | Wild type and KHK A/C KO mice treated for 25 wk | Decreased BHB with fructose | KHK KO restored BHB | Ishimoto 2012 [28] |
Decreased fat oxidation with fructose feeding has also been reported in rodent studies since the 1970’s [79–82], providing a model to investigate the underlying mechanism. Since then, several hypotheses have been proposed to mediate the fructose-induced decrease in oxidation of dietary fat. Given the high propensity of fructose to support de novo lipogenesis, the first mechanism explored the role of malonyl-CoA. Malonyl-CoA is produced from acetyl-CoA by the action of ACC1 and is the first committed step in fatty acid synthesis. Subsequently, FASN sequentially adds malonyl-CoA to extend the growing fatty acid chain by two carbons to form saturated fatty acids, such as palmitoyl-CoA. Besides supporting lipogenesis, malonyl-CoA also acts as a allosteric inhibitor of CPT1α [83], the rate limiting enzyme in mitochondrial fatty acid oxidation. By inhibiting CTP1α, malonyl-CoA prevents the two competing processes, fatty acid synthesis and fatty acid oxidation, to occur at the same time. However, regulation of liver CPT1α isoform is much less sensitive to malonyl-CoA inhibition than its counterpart CPT1β found in the muscle [84]. Moreover, malonyl-CoA levels are regulated, in part, by the nutrient sensor AMPK. When AMP and/or ADP levels increase relative to ATP, such as during fasting and fructose catabolism, AMPK is activated leading to phosphorylation and inactivation of its substrate ACC1 and decreases conversion of acetyl-CoA to malonyl-CoA. Consistent with this finding, we have found that hepatic levels of malonyl-CoA are not increased by fructose feeding on a HFD [23]. Therefore, despite the strong propensity of fructose to actively drive lipogenesis, there is no evidence that fructose metabolism on a HFD leads to accumulation of malonyl-CoA in the liver to explain the decrease in FAO observed with fructose feeding.
Another mode of regulation by which fructose can impair FAO is through disruption of a gene regulatory network. The nuclear receptor peroxisome proliferator-activated receptor α (PPARα) controls the transcriptional regulation of genes involved in FAO including Cpt1α [85]. While stimulation of PPARα drives fatty acid utilization, PPARα deficiency leads to hepatic lipid accumulation and inflammation in response to a HFD-feeding [86]. Numerous reports have demonstrated that fructose feeding lowers the expression and activity of PPARα [87–91] and leads to a reduction in target gene expression and ultimately lower FAO. For example, Nagai et al., demonstrated that eight weeks of high-fructose feeding in rats decreased PPARα mRNA and CPT1α protein levels, which can be partially restored by treatment with the PPARα agonist fenofibrate [87]. However, unraveling the mechanisms by which fructose alters PPARα signaling warrants further investigation.
In addition to transcriptional regulation, fructose may also impede FAO by altering the post-translational modification (PTM) of proteins involved in FAO and leading to their decreased enzymatic activity and/or protein stability. Acetylation is the most common nutrient dependent PTM of mitochondrial proteins. We have published that fructose or glucose supplementation uniquely alters acetylation of mitochondrial proteins in mice on either chow or HFD [92]. Further, it has been shown that fructose reduces Sirt1, a major protein deacetylase, and leads to acetylation and inactivation of peroxisome proliferator activated receptor gamma coactivator 1α (PGC1α), resulting in reduced FAO [93]. In our studies of the combined intake of fructose and fat, we find that fructose increases the acetylation of mitochondrial proteins, specifically ACADL and CPT1α, which mediate FAO. These effects are dependent on KHK, as a knockdown of KHK via siRNA lowers ACADL acetylation and increases CPT1α protein [23,92]. Thus, fructose-induced acetylation of mitochondrial proteins contributes to the decreased FAO observed with dietary fructose intake.
Here we present a large amount of data that fructose decreases FAO in human and rodent studies. The effects of fructose are mediated, in part, via faster fructose metabolism producing greater carbohydrate oxidation, high lipogenic potential of fructose stimulating hepatic malonyl-CoA, lower PPARα transcriptional activity and fructose-induced acetylation of mitochondrial proteins (Fig. 1). Based on these studies, we propose that the fructose component of dietary sugar decreases mitochondrial fatty acid oxidation and consequently augments HFD-induced lipotoxicity. This hypothesis explains why fructose intake on a HFD, but not on low-fat diet, leads to cumulative detrimental effects in terms of obesity and the development of metabolic complications.
5. A review of the mitochondrial β-oxidation pathway
Next, we will provide a detailed review of fatty acid oxidation pathway in the liver (Fig. 2). Adipose tissue lipolysis provides the major source of fatty acids delivered to the liver [94]. Fatty acids bound to albumin circulate in the blood and are taken up into the liver via plasma membrane-associated proteins. Several transporters mediate fatty acid uptake in the liver, such as fatty acid translocase (FAT, aka CD36), liver-fatty acid binding protein (L-FABP), caveolins, and fatty acid transport proteins (FATPs). CD36 plays a major role in fatty acid uptake in heart and skeletal muscle. Its expression is relatively low in the liver, but it is highly inducible by lipid overload [95]. Moreover, increased hepatic expression of CD36 in mice contributes to the dyslipidemia associated with diet-induced obesity [96]. Expression of L-FABP is very high in the liver and it may represent 2–5% of cytosolic protein [97]. L-FABP levels are elevated in NAFLD patients [98], whereas silencing of L-FABP ameliorates hepatic steatosis in mice [99]. Caveolins (types 1–3) were initially characterized as cholesterol transporters and later implicated in fatty acid transport [100]. Mice deficient in caveolin-1 show reduced fat accumulation in the hepatocytes. Together, CD36, L-FABP Caveolin-1, and calcium-independent membrane phospholipase A2 are thought to form a heterotetrameric protein complex within the hepatocyte plasma membrane to promote fatty acid uptake [101]. Another pathway that facilitates fatty acid uptake across cellular plasma membrane is via FATPs. There are six members of this family, but only FATP2 and FATP5 contribute to lipid transport in the hepatocytes [102]. FATP5 is localized on the plasma membrane, while FATP2 is found in the endoplasmic reticulum [103]. These proteins possess translocase activity, but also esterify fatty acids into fatty acyl-CoAs [104]. Fatty acids activated into their acyl-CoA thioesters are unable to passively diffuse out of the cell and are committed to utilization within the cell. This step is of paramount importance to the subsequent partitioning of fatty acids into unique metabolic processes so that FATPs have been renamed for their acyl-CoA synthesis activity, such that FATP2 is also known as very long-chain acyl-CoA synthetase 1 (ACSVL1), and FATP5 as ACSVL6. There are 26 enzymes that possess acyl-CoA synthetase activity and this is highly regulated process dependent on the nutrient status. Once synthesized, acyl-CoAs support a variety of cellular processes such as the synthesis of complex lipids including triglycerides, phospholipids, and cholesterol esters. On the other hand, they can be converted to acyl-carnitines, which is a process that commits them to oxidation and energy production.
Fig. 2.
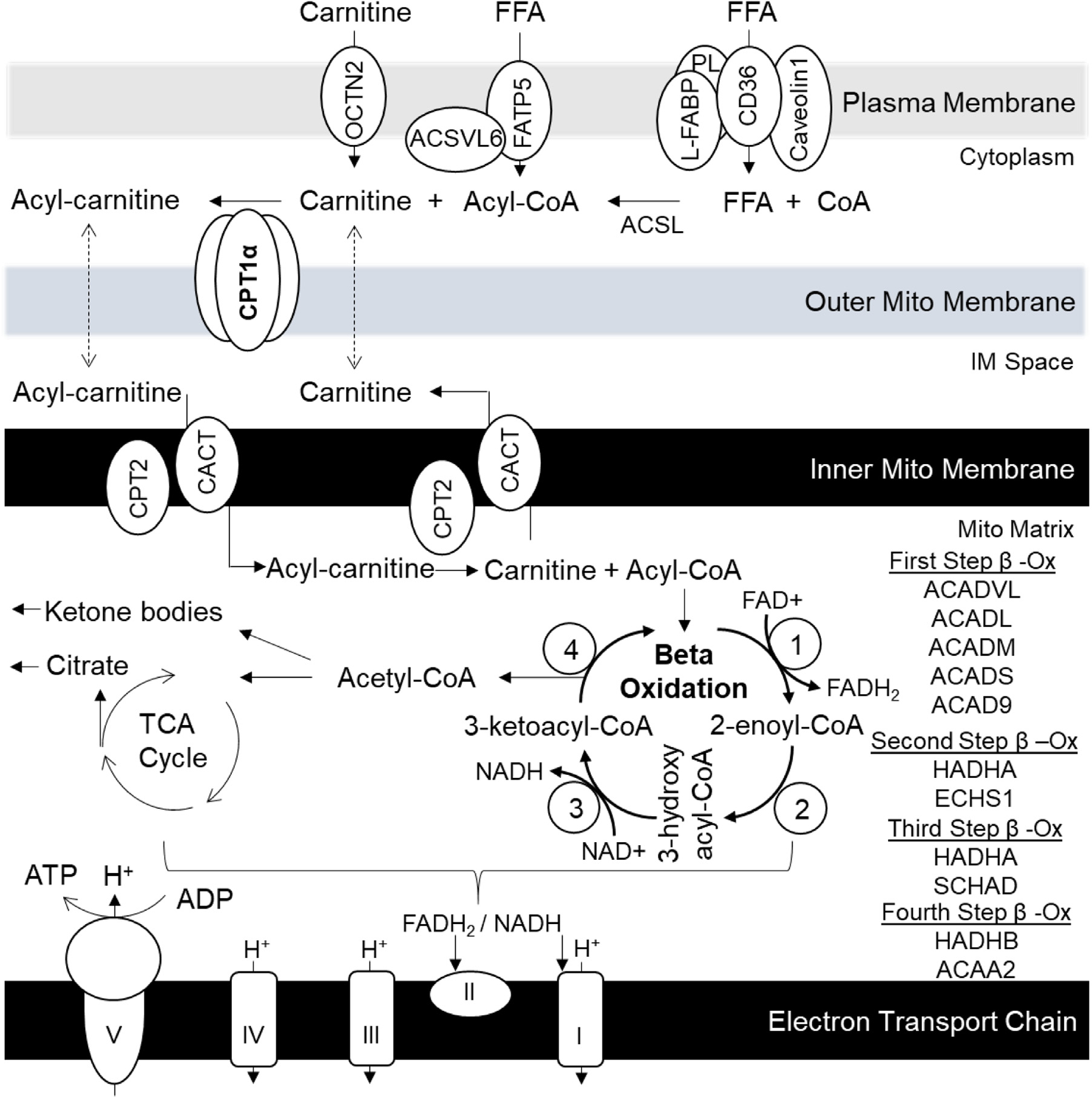
The Pathway of Mitochondrial Fatty Acid Oxidation in the Liver. Long-chain fatty acids are transported into the liver via a complex of proteins consisting of fatty acid translocase (FAT, aka CD36), liver-fatty acid binding protein (L-FABP), caveolins and phospholipase A2 (PL). In the cytosol fatty acids are activated into fatty acyl-CoA by the actions of long-chain acyl-CoA synthetases (ACSL). Acyl-CoAs are unable to diffuse out of the cell and are committed to different cellular processes such as lipid synthesis or oxidation. The metabolic fate of acyl-CoAs is likely mediated by unique ACSL isoforms. Another pathway that mediates fatty acid import into the hepatocytes is via fatty acid transport protein 5 (FATP5). This enzyme complex additionally contains very long-chain acyl-CoA synthetase 6 (ACSVL6) activity to generate Acyl-CoA. Acyl-CoAs designated for energy production are conjugated to carnitine by carnitine palmitoyltransferase 1 alpha (CPA1a). This is the rate limiting enzyme of fatty acid oxidation accounting for 80% of the control over the pathway. Carnitine is transported into the hepatocytes by OCTN2. Acyl-carnitines can cross outer mitochondrial membrane and are transported across the inner mitochondrial membrane by carnitine-acylcarnitine translocase (CACT) in an exchange for free carnitine transported from mitochondrial matrix into inter membrane space. Inside the mitochondrial matrix carnitine palmitoyltransferase 2 (CPT2) converts acyl-carnitines into free carnitine and acyl-CoA. Beta oxidation involves a four-step process where long chain acyl-CoA are progressively shortened by two carbons to generate acetyl-CoA and two electrons. The first step dehydrogenates acyl-CoA into 2-enoly-CoA, and is mediated by acyl-CoA dehydrogenases (ACADs) of different chain lengths. This dehydrogenation step yields FADH2, which donates electrons to complex II of the mitochondrial electron transport (ETC) chain for ATP production. The next three steps are mediated by mitochondrial trifunctional protein (MTP). The second step is hydration step and it produces 3-hydroxyacyl-CoA by the action of 2-enoyl-CoA hydratases (ECH). The third step is another dehydrogenation step mediated by 3-hydroxyacyl-CoA dehydrogenase (HADH) to produce 3-ketoacyl-CoA. This dehydrogenation step yields NADH, which donates electrons to complex I of the mitochondrial ETC chain for ATP production. The fourth step is mediated by acetyl-CoA acyltransferase 2 (ACAA2) to produce shortened acyl-CoA chain and acetyl-CoA. Acetyl-CoA can be further reduced in tricarboxylic acid (TCA) cycle and it yields citrate when energy stores are plentifully. During fasting, acetyl-CoA may be converted to ketone bodies that are used during fasting as energy source in other tissue.
Indeed, conversion of acyl-CoAs into acyl-carnitines is the rate-limiting step of fatty acid oxidation. Mitochondrial β-oxidation is the primary site in the liver for oxidation of short, medium, and long-chain fatty acids. Carnitine palmitoyltransferase 1 (CPT1) catalyzes the conversion of long chain (12–18 carbons) acyl-CoAs into acyl-carnitines. CPT1 is localized on the outer mitochondrial membrane and is the rate-limiting enzyme of mitochondrial FAO. There are three CPT1 isoforms encoded by different genes. CPT1α is mainly found in the liver, CPT1β localizes to the muscle and adipose tissue, while CPT1c is present in the brain and testes. Due to its paramount importance, CPT1α is regulated at many levels. As previously mentioned, CPT1α is transcriptionally governed by PPARα and post-translationally regulated by malonyl-CoA and insulin. Its activity is also dependent on dimer and tetramer assemble [105], as well as acetylation of its lysine residues [23]. Long chain acyl-carnitines made by CPT1α are transported across the mitochondrial membrane by carnitine-acylcarnitine translocase (CACT). Inside the mitochondrial matrix acyl groups are transferred back to CoA, and free carnitine is generated by the enzyme CPT2. CPT2 in the mitochondrial matrix catalyzes the reverse reaction of CPT1 in the cytosol. Although CPT1 accounts for 80% of control over the pathway flux [106], recycling of carnitine via the carnitine shuttle is crucial for FAO to work properly. Additionally, carnitine is shuttled back to the mitochondrial inter-membrane space by CACT and then passively diffuses across the outer mitochondrial membrane into the cytosol, so that the cycle can begin anew. Cytoplasmic carnitine levels are also mediated by organic cation/carnitine transporter 2 (OCTN2), which transports carnitine from the plasma across the hepatocyte cytoplasmic membrane. Plasma carnitine levels are mainly a reflection of dietary L-carnitine intake.
Inside the mitochondrial matrix, newly transported acyl-CoAs undergo progressive shortening by oxidative removal of two carbon (acetyl) units via a four-step process. This process is termed mitochondrial beta oxidation. The first step is mediated by acyl-CoA dehydrogenases (ACAD). Dependent on acyl-CoA chain length, short, medium, long, and very long chain acyl-CoA dehydrogenases catalyze the production of 2-enoyl-CoA. ACAD9 has been more recently recognized as acyl-CoA dehydrogenase i.e., active in brain tissue. This dehydrogenation step utilizes FAD + to make FADH2, which subsequently donates electrons to complex II of the mitochondrial electron transport chain. The second step is mediated by 2-enoyl-CoA hydratases (ECH) and produces 3-hydroxyacyl-CoA. There are two ECH enzymes: short-chain (ECHS1) and long-chain, which is a part of trifunctional protein (HADHA). This hydration step reduces a double bond between carbons two and three of the acyl-CoA chain. The third step is another dehydrogenation reaction mediated by 3-hydroxyacyl-CoA dehydrogenase (HADH) to produce 3-ketoacyl-CoA. It also reduces NAD + to NADH which donates electrons to complex I to support ATP synthesis. Similar to ECH, there are short- (SCHAD) and long-chain HADHs (HADHA). The fourth and final step of the beta cycle is mediated by acetyl-CoA acyltransferase 2 (ACAA2) to produce acetyl-CoA and shortened acyl-CoA chain. There are short-, medium- (ACAA2) and long-chain (HADHB) enzymes that possess thiolase function, although the short-chain may not play a role in beta oxidation. The last three reactions, which metabolize longer chain acyl-CoAs, are catalyzed by the multi-domain mitochondrial trifunctional protein (MTP). MTP is made of eight subunits. Four alpha subunits are encoded by the HADHA gene, and four beta subunits are produced from the HADHB gene. The alpha subunits harbor long-chain enoyl-CoA hydratase and long-chain 3-hydroxyacyl-CoA dehydrogenase enzymes. The beta subunits contain long-chain acetyl-CoA acyltransferase enzyme. In summary, once cycle of beta oxidation yields a fatty acyl-CoA shortened by two carbons, one acetyl-CoA molecule, and two electrons. The cycle is repeated until two acetyl-CoA molecules remain. Acetyl-CoA can be further reduced in the TCA cycle to generate citrate and additional electrons to support ATP production. In the liver, acetyl-CoA may be also be converted to ketone bodies that are used during fasting as energy source in other tissue.
6. The effects of fructose on import, activation, transport and oxidation of FFA
First, we will examine the effects of fructose on fat transporters. CD36 knockout mice show improved glucose uptake in the muscle on normal chow diet; however, as expected, they develop glucose intolerance and insulin resistance on a HFD. As previously stated, CD36 is not abundant in the liver as compared to the muscle, so whole body CD36 KO results in increased hepatic fatty acid uptake. Interestingly, CD36 KO mice develop worse glucose intolerance and insulin resistance on a high-fructose diet as compared to a HFD, suggesting that fructose decreases fatty acid oxidation in these mice [107]. On the other hand, L-FABP KO mice are protected from high-fat and high-fructose diet-induced NAFLD and liver fibrosis. This provides further evidence that fructose intake is insufficient to induce NAFLD without adequate fat supply to the liver [108]. While there is no direct evidence that dietary fructose impairs hepatic fat transporters, the perturbations that result in increased or decreased fat transport in the liver dictate the metabolic outcomes of a high-fructose diet.
There is direct evidence that fructose downregulates activation of fatty acids to their corresponding acyl-CoAs in the liver. Dong et al., showed that a high-fructose diet markedly reduces the protein and mRNA expression of ACSL3 in hamster livers [109]. Pharmacologic activation of ACSL3 in these mice reduced hepatic triglyceride accumulation. Others have also show that the expression of several ACSL isoforms is differentially controlled by fasting followed by refeeding a 69% sucrose diet [110]. For example, fasting increased ACSL1 and ACSL4 mRNA abundance in the liver, while refeeding fructose decreased their expression. Conversely, ACSL3 and ACSL5 are reduced in fasted mice, and their mRNA is restored by feeding a high-fructose diet [110]. Based on these studies it can be concluded that fructose decreases activation of specific ACSL, which likely function to shunt fatty acyl-CoA towards mitochondrial oxidation.
We have already presented multiple studies documenting strong downregulation of CPT1α by dietary fructose (Table 1). We [23] and others [28] have shown that these effects are dependent on KHK mediated fructose metabolism [111]. We could not find evidence that fructose directly affects other enzymes in the carnitine shuttle, such as CACT and OCTN2. However, treating fructosefed rats with carnitine is sufficient to reverse fructose-induced metabolic derangements [112]. Similarly, L-Carnitine supplementation in mice attenuates fructose-mediated lipid accumulation, counteracts mitochondrial damage, and decrease the production of reactive oxygen species [113]. Together this evidence shows the strong propensity of fructose to lower fatty acid oxidation by decreasing acyl-carnitine production, an effect that can be reversed by carnitine supplementation.
The reports documenting the direct effects of fructose on enzymes mediating the four steps of mitochondrial β-oxidation are lacking. In summary, we can conclude that fructose inhibits FAO by decreasing CPT1α-mediated acylcarnitine production and activation of fatty acids into specific acyl-CoAs, while the data is lacking that fructose directly alters fatty acid uptake and the enzymes mediating mitochondrial β-oxidation. Thus, the profound effects of fructose to decrease FAO can be explained by decreased acylcarnitine production to account for the additive effects of fructose and a HFD on NAFLD pathogenesis.
7. Is FAO impaired in patients with NAFLD?
Obesity is a major risk factors for development of NAFLD [114,115]. We propose that a mechanism by which a Western diet induces NAFLD is mediated, in part, via a fructose-induced decrease in fat oxidation. Dietary fructose clearly increases the risk of developing a more severe form of NAFLD [116,117], but is there evidence that FAO is impaired in patients with NAFLD? This question is challenging to answer due to difficulty in measuring FAO, thus leading to conflicting reports. Naguib et al., reported that FAO of orally-delivered 13C-labeled palmitate, as a part of mixed meal test, is decreased in NAFLD patients compared to healthy controls [118]. The authors suggest that this is likely due to decreased mitochondrial β-oxidation and propose the use of a 13C-palmitate breath test to noninvasively assess FAO. Another study also found reduced hepatic fat oxidation in patients with NAFLD [119]. Conversely, Kotronen et al., published that whole-body lipid oxidation is increased because of peripheral insulin resistance, but that hepatic lipid oxidation is unchanged in NAFLD patients [120]. They used labeled glucose, indirect calorimetry, and a euglycemic hyperinsulinemic clamp to measure metabolic flux in patients with NAFLD. However, some studies have even found that fatty acid oxidation is higher in patients with non-alcoholic steatohepatitis (NASH) [121–123]. In spite of the lack of clear evidence from tracer studies that FAO is impaired in NAFLD, it has been reported that PPARα is downregulated in patients with advanced fatty liver disease compared to those with simple steatosis [124], and that PPARα expression negatively correlates with severity of NAFLD. Similarly CPT1α has been documented to be decreased in patients with NAFLD [125]. Therefore, approaches to increase FAO are currently being investigated as treatment options for NAFLD [126,127].
Elafibranor, is a dual PPARα and δ agonist that was clinically tested for treatment of NAFLD. It showed promising results in a phase 2b clinical study [128]. However, in a phase 3 clinical trial in patients with NASH, elafibranor did not meet the predefined primary endpoint and has been discontinued. Next in line, the new generation pan-PPAR agonist, lanifibranor, has been subsequently shown to lower steatosis, inflammation, and fibrosis in humans and mice [129,130]. Lanifibranor is currently in stage 3 clinical trials studying its effectiveness in improving NASH and fibrosis. Similarly, gene therapy to increase CPT1α activity and hepatic fatty acid oxidation has proved effective to decrease hepatic steatosis in mice [131]. Together, these studies suggest that restoring fructose-induced decrease in hepatic fatty acid oxidation may be a potentially promising strategy to treat NAFLD.
8. Conclusions
Fructose is a highly lipogenic nutrient that has been implicated in the development of obesity and its complications, such as NAFLD. Here we present a large body of evidence demonstrating that fructose also decreases dietary fat oxidation. In spite of the strength and abundance of the evidence, this aspect of fructose metabolism has been largely understudied and often overlooked. However, the enormous relevance of these findings to those affected by consumption of a Western diet warrant further investigation. Future studies are needed to elucidate the mechanism of how dietary fructose decreases PPARα and CPT1α function. These effects are not likely to be mediated by malonyl-CoA in the liver, but could be secondary to protein acetylation leading to decreased PPARα transcriptional activity and lower CPT1α protein levels. A deeper understanding of this not so simple sugar metabolism may offer new treatment options for management of obesity-associated complications.
Funding
This work was also supported by NASPGHAN Foundation Young Investigator Award, Pediatric Scientist Development Program Award (HD0 0 0850) and COCVD Pilot and Feasibility Grant (GM127211) awarded to SS. National Institutes of Health grants K01DK128022 and NIH National Center for Advancing Translational Sciences through grant number UL1TR001998 awarded to RNH.
Footnotes
Declaration of Competing Interests
The authors declare that there are no conflicts of interest.
References
- [1].Bray GA, Popkin BM. Dietary fat intake does affect obesity!. Am J Clin Nutr 1998;68:1157–73. [DOI] [PubMed] [Google Scholar]
- [2].Austin GL, Ogden LG, Hill JO. Trends in carbohydrate, fat, and protein intakes and association with energy intake in normal-weight, overweight, and obese individuals: 1971–2006. Am J Clin Nutr 2011;93:836–43. [DOI] [PubMed] [Google Scholar]
- [3].Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr 2004;79:537–43. [DOI] [PubMed] [Google Scholar]
- [4].Sievenpiper JL, de Souza RJ, Mirrahimi A, Yu ME, Carleton AJ, Beyene J, et al. Effect of fructose on body weight in controlled feeding trials: a systematic review and meta-analysis. Ann Intern Med 2012;156:291–304. [DOI] [PubMed] [Google Scholar]
- [5].Schwarz JM, Noworolski SM, Wen MJ, Dyachenko A, Prior JL, Weinberg ME, et al. Effect of a high-fructose weight-maintaining diet on lipogenesis and liver fat. J Clin Endocrinol Metab 2015;100:2434–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Geidl-Flueck B, Hochuli M, Nemeth A, Eberl A, Derron N, Kofeler HC, et al. Fructose- and sucrose- but not glucose-sweetened beverages promote hepatic de novo lipogenesis: a randomized controlled trial. J Hepatol 2021;75:46–54. [DOI] [PubMed] [Google Scholar]
- [7].Lustig RH, Mulligan K, Noworolski SM, Tai VW, Wen MJ, Erkin-Cakmak A, et al. Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obesity (Silver Spring) 2016;24:453–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Schwarz JM, Noworolski SM, Erkin-Cakmak A, Korn NJ, Wen MJ, Tai VW, et al. Effects of dietary fructose restriction on liver fat, de novo lipogenesis, and insulin kinetics in children with obesity. Gastroenterology 2017;153:743–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Radulescu A, Killian M, Kang Q, Yuan Q, Softic S. Dietary counseling aimed at reducing sugar intake yields the greatest improvement in management of weight and metabolic dysfunction in children with obesity. Nutrients 2022;14(7):500. doi: 10.3390/nu14071500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cicerchi C, Li N, Kratzer J, Garcia G, Roncal-Jimenez CA, Tanabe K, et al. Uric acid-dependent inhibition of AMP kinase induces hepatic glucose production in diabetes and starvation: evolutionary implications of the uricase loss in hominids. FASEB J 2014;28:3339–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hu S, Wang L, Yang D, Li L, Togo J, Wu Y, et al. Dietary fat, but not protein or carbohydrate, regulates energy intake and causes adiposity in mice. Cell Metab 2018;28:415–31. [DOI] [PubMed] [Google Scholar]
- [12].Bueno NB, de Melo IS, de Oliveira SL, da Rocha Ataide T. Very-low-carbohydrate ketogenic diet v. low-fat diet for long-term weight loss: a meta-analysis of randomised controlled trials. Br J Nutr 2013;110:1178–87. [DOI] [PubMed] [Google Scholar]
- [13].Foster GD, Wyatt HR, Hill JO, McGuckin BG, Brill C, Mohammed BS, et al. A randomized trial of a low-carbohydrate diet for obesity. N Engl J Med 2003;348:2082–90. [DOI] [PubMed] [Google Scholar]
- [14].Kennedy AR, Pissios P, Otu H, Roberson R, Xue B, Asakura K, et al. Maratos-Flier E: a high-fat, ketogenic diet induces a unique metabolic state in mice. Am J Physiol Endocrinol Metab 2007;292:E1724–39. [DOI] [PubMed] [Google Scholar]
- [15].D’Abbondanza M, Ministrini S, Pucci G, Nulli Migliola E, Martorelli EE, Gandolfo V, et al. Very Low-carbohydrate ketogenic diet for the treatment of severe obesity and associated non-alcoholic fatty liver disease: the role of sex differences. Nutrients 2020;12(9):2748 doi: 10.3390/nu12092748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Luukkonen PK, Dufour S, Lyu K, Zhang XM, Hakkarainen A, Lehtimaki TE, et al. Yki-Jarvinen H: Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 2020;117:7347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Al-Khalifa A, Mathew TC, Al-Zaid NS, Mathew E, Dashti HM. Therapeutic role of low-carbohydrate ketogenic diet in diabetes. Nutrition 2009;25:1177–85. [DOI] [PubMed] [Google Scholar]
- [18].Accurso A, Bernstein RK, Dahlqvist A, Draznin B, Feinman RD, Fine EJ, et al. Dietary carbohydrate restriction in type 2 diabetes mellitus and metabolic syndrome: time for a critical appraisal. Nutr Metab (Lond) 2008;5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dashti HM, Mathew TC, Khadada M, Al-Mousawi M, Talib H, Asfar SK, et al. Beneficial effects of ketogenic diet in obese diabetic subjects. Mol Cell Biochem 2007;302:249–56. [DOI] [PubMed] [Google Scholar]
- [20].Sharman MJ, Kraemer WJ, Love DM, Avery NG, Gomez AL, Scheett TP, et al. A ketogenic diet favorably affects serum biomarkers for cardiovascular disease in normal-weight men. J Nutr 2002;132:1879–85. [DOI] [PubMed] [Google Scholar]
- [21].Paoli A, Bianco A, Damiani E, Bosco G. Ketogenic diet in neuromuscular and neurodegenerative diseases. Biomed Res Int 2014;2014:474296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Paoli A Ketogenic diet for obesity: friend or foe? Int J Environ Res Public Health 2014;11:2092–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Softic S, Meyer JG, Wang GX, Gupta MK, Batista TM, Lauritzen H, et al. Dietary sugars alter hepatic fatty acid oxidation via transcriptional and post-translational modifications of mitochondrial proteins. Cell Metab 2019;30:735–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dhingra R, Sullivan L, Jacques PF, Wang TJ, Fox CS, Meigs JB, et al. Soft drink consumption and risk of developing cardiometabolic risk factors and the metabolic syndrome in middle-aged adults in the community. Circulation 2007;116:480–8. [DOI] [PubMed] [Google Scholar]
- [25].Taskinen MR, Soderlund S, Bogl LH, Hakkarainen A, Matikainen N, Pietilainen KH, et al. Adverse effects of fructose on cardiometabolic risk factors and hepatic lipid metabolism in subjects with abdominal obesity. J Intern Med 2017;282:187–201. [DOI] [PubMed] [Google Scholar]
- [26].Le KA, Ith M, Kreis R, Faeh D, Bortolotti M, Tran C, et al. Fructose overconsumption causes dyslipidemia and ectopic lipid deposition in healthy subjects with and without a family history of type 2 diabetes. Am J Clin Nutr 2009;89:1760–5. [DOI] [PubMed] [Google Scholar]
- [27].Faeh D, Minehira K, Schwarz JM, Periasamy R, Park S, Tappy L. Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes 2005;54:1907–13. [DOI] [PubMed] [Google Scholar]
- [28].Ishimoto T, Lanaspa MA, Le MT, Garcia GE, Diggle CP, Maclean PS, et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc Natl Acad Sci U S A 2012;109:4320–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009;50:1094–104. [DOI] [PubMed] [Google Scholar]
- [30].Andres-Hernando A, Orlicky DJ, Kuwabara M, Ishimoto T, Nakagawa T, Johnson RJ, et al. Deletion of Fructokinase in the liver or in the intestine reveals differential effects on sugar-induced metabolic dysfunction. Cell Metab 2020;32:117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shapiro A, Mu W, Roncal C, Cheng KY, Johnson RJ, Scarpace PJ. Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. Am J Physiol Regul Integr Comp Physiol 2008;295:R1370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Miyazaki M, Dobrzyn A, Man WC, Chu K, Sampath H, Kim HJ, et al. Stearoyl-CoA desaturase 1 gene expression is necessary for fructose-mediated induction of lipogenic gene expression by sterol regulatory element-binding protein-1c-dependent and -independent mechanisms. J Biol Chem 2004;279:25164–71. [DOI] [PubMed] [Google Scholar]
- [33].Shi JH, Lu JY, Chen HY, Wei CC, Xu X, Li H, et al. Liver ChREBP protects against fructose-induced glycogenic hepatotoxicity by regulating l-type pyruvate kinase. Diabetes 2020;69:591–602. [DOI] [PubMed] [Google Scholar]
- [34].Todoric J, Di Caro G, Reibe S, Henstridge DC, Green CR, Vrbanac A, et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat Metab 2020;2:1034–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhang D, Tong X, VanDommelen K, Gupta N, Stamper K, Brady GF, et al. Lipogenic transcription factor ChREBP mediates fructose-induced metabolic adaptations to prevent hepatotoxicity. J Clin Invest 2017;127:2855–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest 2009;119:1322–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].van Buul VJ, Tappy L, Brouns FJ. Misconceptions about fructose-containing sugars and their role in the obesity epidemic. Nutr Res Rev 2014;27:119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Erkin-Cakmak A, Bains Y, Caccavello R, Noworolski SM, Schwarz JM, Mulligan K, et al. Isocaloric fructose restriction reduces serum d-lactate concentration in children with obesity and metabolic syndrome. J Clin Endocrinol Metab 2019;104:3003–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Silbernagel G, Machann J, Unmuth S, Schick F, Stefan N, Haring HU, et al. Effects of 4-week very-high-fructose/glucose diets on insulin sensitivity, visceral fat and intrahepatic lipids: an exploratory trial. Br J Nutr 2011;106:79–86. [DOI] [PubMed] [Google Scholar]
- [40].Cox CL, Stanhope KL, Schwarz JM, Graham JL, Hatcher B, Griffen SC, et al. Consumption of fructose-sweetened beverages for 10 weeks reduces net fat oxidation and energy expenditure in overweight/obese men and women. Eur J Clin Nutr 2012;66:201–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Softic S, Gupta MK, Wang GX, Fujisaka S, O’Neill BT, Rao TN, et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest 2017;127:4059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Glendinning JI, Breinager L, Kyrillou E, Lacuna K, Rocha R, Sclafani A. Differential effects of sucrose and fructose on dietary obesity in four mouse strains. Physiol Behav 2010;101:331–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tajima R, Kimura T, Enomoto A, Saito A, Kobayashi S, Masuda K, et al. No association between fruits or vegetables and non-alcoholic fatty liver disease in middle-aged men and women. Nutrition 2019;61:119–24. [DOI] [PubMed] [Google Scholar]
- [44].Sharma SP, Chung HJ, Kim HJ, Hong ST. Paradoxical effects of fruit on obesity. Nutrients 2016;8(10):633. doi: 10.3390/nu8100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ferrere G, Leroux A, Wrzosek L, Puchois V, Gaudin F, Ciocan D, et al. Activation of Kupffer cells is associated with a specific dysbiosis induced by fructose or high fat diet in mice. PLoS One 2016;11:e0146177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Charlton M, Krishnan A, Viker K, Sanderson S, Cazanave S, McConico A, et al. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol 2011;301:G825–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ishimoto T, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, Orlicky DJ, Cicerchi C, et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013;58:1632–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, et al. High--fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010;52:934–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sellmann C, Priebs J, Landmann M, Degen C, Engstler AJ, Jin CJ, et al. Diets rich in fructose, fat or fructose and fat alter intestinal barrier function and lead to the development of nonalcoholic fatty liver disease over time. J Nutr Biochem 2015;26:1183–92. [DOI] [PubMed] [Google Scholar]
- [50].Volynets V, Louis S, Pretz D, Lang L, Ostaff MJ, Wehkamp J, et al. Intestinal barrier function and the gut microbiome are differentially affected in mice fed a western-style diet or drinking water supplemented with fructose. J Nutr 2017;147:770–80. [DOI] [PubMed] [Google Scholar]
- [51].Hintze KJ, Benninghoff AD, Cho CE, Ward RE. Modeling the western diet for preclinical investigations. Adv Nutr 2018;9:263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Im YR, Hunter H, de Gracia Hahn D, Duret A, Cheah Q, Dong J, et al. A systematic review of animal models of NAFLD finds high-fat, high-fructose diets most closely resemble human NAFLD. Hepatology 2021;74:1884–901. [DOI] [PubMed] [Google Scholar]
- [53].Lecoultre V, Egli L, Carrel G, Theytaz F, Kreis R, Schneiter P, et al. Effects of fructose and glucose overfeeding on hepatic insulin sensitivity and intrahepatic lipids in healthy humans. Obesity (Silver Spring) 2013;21:782–5. [DOI] [PubMed] [Google Scholar]
- [54].Parks EJ, Skokan LE, Timlin MT, Dingfelder CS. Dietary sugars stimulate fatty acid synthesis in adults. J Nutr 2008;138:1039–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Laube H, Klor HU, Fussganger R, Pfeiffer EF. The effect of starch, sucrose, glucose and fructose on lipid metabolism in rats. Nutr Metab 1973;15:273–80. [DOI] [PubMed] [Google Scholar]
- [56].Timlin MT, Parks EJ. Temporal pattern of de novo lipogenesis in the postprandial state in healthy men. Am J Clin Nutr 2005;81:35–42. [DOI] [PubMed] [Google Scholar]
- [57].Softic S, Cohen DE, Kahn CR. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig Dis Sci 2016;61:1282–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Samuel VT. Fructose induced lipogenesis: from sugar to fat to insulin resistance. Trends Endocrinol Metab 2011;22:60–5. [DOI] [PubMed] [Google Scholar]
- [59].Hannou SA, Haslam DE, McKeown NM, Herman MA. Fructose metabolism and metabolic disease. J Clin Invest 2018;128:545–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Helsley RN, Moreau F, Gupta MK, Radulescu A, DeBosch B, Softic S. Tissue-specific fructose metabolism in obesity and diabetes. Curr Diab Rep 2020;20:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Fisher FM, Kim M, Doridot L, Cunniff JC, Parker TS, Levine DM, et al. A critical role for ChREBP-mediated FGF21 secretion in hepatic fructose metabolism. Mol Metab 2017;6:14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lanaspa MA, Sanchez-Lozada LG, Cicerchi C, Li N, Roncal-Jimenez CA, Ishimoto T, et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS One 2012;7:e47948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Koo HY, Miyashita M, Cho BH, Nakamura MT. Replacing dietary glucose with fructose increases ChREBP activity and SREBP-1 protein in rat liver nucleus. Biochem Biophys Res Commun 2009;390:285–9. [DOI] [PubMed] [Google Scholar]
- [64].Lee HJ, Cha JY. Recent insights into the role of ChREBP in intestinal fructose absorption and metabolism. BMB Rep 2018;51:429–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kim M, Astapova II, Flier SN, Hannou SA, Doridot L, Sargsyan A, et al. Intestinal, but not hepatic, ChREBP is required for fructose tolerance. JCI Insight 2017;2(24):e96703. doi: 10.1172/jci.insight.96703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Gutierrez JA, Liu W, Perez S, Xing G, Sonnenberg G, Kou K, et al. Pharmacologic inhibition of ketohexokinase prevents fructose-induced metabolic dysfunction. Mol Metab 2021;48:101196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Benhamed F, Denechaud PD, Lemoine M, Robichon C, Moldes M, Bertrand-Michel J, et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest 2012;122:2176–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Oh AR, Sohn S, Lee J, Park JM, Nam KT, Hahm KB, et al. ChREBP deficiency leads to diarrhea-predominant irritable bowel syndrome. Metabolism 2018;85:286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kato T, Iizuka K, Takao K, Horikawa Y, Kitamura T, Takeda J. ChREBP-Knockout mice show sucrose intolerance and fructose malabsorption. Nutrients 2018;10(3):340. doi: 10.3390/nu10030340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Softic S, Stanhope KL, Boucher J, Divanovic S, Lanaspa MA, et al. Fructose and hepatic insulin resistance. Crit Rev Clin Lab Sci 2020;57(5):308–22. doi: 10.1080/10408363.2019.1711360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Shimomura I, Bashmakov Y, Ikemoto S, Horton JD, Brown MS, Goldstein JL. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc Natl Acad Sci U S A 1999;96:13656–13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Haas JT, Miao J, Chanda D, Wang Y, Zhao E, Haas ME, et al. Hepatic insulin signaling is required for obesity-dependent expression of SREBP-1c mRNA but not for feeding-dependent expression. Cell Metab 2012;15:873–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Nagai Y, Yonemitsu S, Erion DM, Iwasaki T, Stark R, Weismann D, et al. The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metab 2009;9:252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, Roncal-Jimenez CA, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem 2012;287:40732–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Choi YJ, Shin HS, Choi HS, Park JW, Jo I, Oh ES, et al. Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP-1c activation in hepatocytes. Lab Invest 2014;94:1114–25. [DOI] [PubMed] [Google Scholar]
- [76].Tappy L, Randin JP, Felber JP, Chiolero R, Simonson DC, Jequier E, et al. Comparison of thermogenic effect of fructose and glucose in normal humans. Am J Physiol 1986;250:E718–24. [DOI] [PubMed] [Google Scholar]
- [77].Chong MF, Fielding BA, Frayn KN. Mechanisms for the acute effect of fructose on postprandial lipemia. Am J Clin Nutr 2007;85:1511–20. [DOI] [PubMed] [Google Scholar]
- [78].Couchepin C, Le KA, Bortolotti M, da Encarnacao JA, Oboni JB, Tran C, et al. Markedly blunted metabolic effects of fructose in healthy young female subjects compared with male subjects. Diabetes Care 2008;31:1254–6. [DOI] [PubMed] [Google Scholar]
- [79].Rawat AK, Menahan LA. Antiketogenic action of fructose, glyceraldehyde, and sorbitol in the rat in vivo. Diabetes 1975;24:926–32. [DOI] [PubMed] [Google Scholar]
- [80].Prager GN, Ontko JA. Direct effects of fructose metabolism on fatty acid oxidation in a recombined rat liver mitochondria-hish speed supernatant system. Biochim Biophys Acta 1976;424:386–95. [DOI] [PubMed] [Google Scholar]
- [81].Topping DL, Mayes PA. The immediate effects of insulin and fructose on the metabolism of the perfused liver. Changes in lipoprotein secretion, fatty acid oxidation and esterification, lipogenesis and carbohydrate metabolism. Biochem J 1972;126:295–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Ontko JA. Metabolism of free fatty acids in isolated liver cells. Factors affecting the partition between esterification and oxidation. J Biol Chem 1972;247:1788–800. [PubMed] [Google Scholar]
- [83].McGarry JD, Mannaerts GP, Foster DW. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J Clin Invest 1977;60:265–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Lopez-Vinas E, Bentebibel A, Gurunathan C, Morillas M, de Arriaga D, Serra D, et al. Gomez-Puertas P: Definition by functional and structural analysis of two malonyl-CoA sites in carnitine palmitoyltransferase 1A. J Biol Chem 2007;282:18212–24. [DOI] [PubMed] [Google Scholar]
- [85].Song S, Attia RR, Connaughton S, Niesen MI, Ness GC, Elam MB, et al. Peroxisome proliferator activated receptor alpha (PPARalpha) and PPAR gamma coactivator (PGC-1alpha) induce carnitine palmitoyltransferase IA (CPT-1A) via independent gene elements. Mol Cell Endocrinol 2010;325:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Regnier M, Polizzi A, Smati S, Lukowicz C, Fougerat A, Lippi Y, et al. Hepatocyte-specific deletion of Pparalpha promotes NAFLD in the context of obesity. Sci Rep 2020;10:6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Nagai Y, Nishio Y, Nakamura T, Maegawa H, Kikkawa R, Kashiwagi A. Amelioration of high fructose-induced metabolic derangements by activation of PPARalpha. Am J Physiol Endocrinol Metab 2002;282:E1180–90. [DOI] [PubMed] [Google Scholar]
- [88].Roglans N, Vila L, Farre M, Alegret M, Sanchez RM, Vazquez-Carrera M, et al. Impairment of hepatic Stat-3 activation and reduction of PPARalpha activity in fructose-fed rats. Hepatology 2007;45:778–88. [DOI] [PubMed] [Google Scholar]
- [89].Rebollo A, Roglans N, Baena M, Padrosa A, Sanchez RM, Merlos M, et al. Liquid fructose down-regulates liver insulin receptor substrate 2 and gluconeogenic enzymes by modifying nutrient sensing factors in rats. J Nutr Biochem 2014;25:250–8. [DOI] [PubMed] [Google Scholar]
- [90].Ohashi K, Munetsuna E, Yamada H, Ando Y, Yamazaki M, Taromaru N, et al. High fructose consumption induces DNA methylation at PPARalpha and CPT1A promoter regions in the rat liver. Biochem Biophys Res Commun 2015;468:185–9. [DOI] [PubMed] [Google Scholar]
- [91].Roglans N, Sanguino E, Peris C, Alegret M, Vazquez M, Adzet T, et al. Atorvastatin treatment induced peroxisome proliferator-activated receptor alpha expression and decreased plasma nonesterified fatty acids and liver triglyceride in fructose-fed rats. J Pharmacol Exp Ther 2002;302:232–9. [DOI] [PubMed] [Google Scholar]
- [92].Meyer JG, Softic S, Basisty N, Rardin MJ, Verdin E, Gibson BW, et al. Temporal dynamics of liver mitochondrial protein acetylation and succinylation and metabolites due to high fat diet and/or excess glucose or fructose. PLoS One 2018;13:e0208973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Rebollo A, Roglans N, Baena M, Sanchez RM, Merlos M, Alegret M, et al. Liquid fructose downregulates Sirt1 expression and activity and impairs the oxidation of fatty acids in rat and human liver cells. Biochim Biophys Acta 2014;1841:514–24. [DOI] [PubMed] [Google Scholar]
- [94].Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115:1343–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Inoue M, Ohtake T, Motomura W, Takahashi N, Hosoki Y, Miyoshi S, et al. Increased expression of PPARgamma in high fat diet-induced liver steatosis in mice. Biochem Biophys Res Commun 2005;336:215–22. [DOI] [PubMed] [Google Scholar]
- [96].Koonen DP, Jacobs RL, Febbraio M, Young ME, Soltys CL, Ong H, et al. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes 2007;56:2863–71. [DOI] [PubMed] [Google Scholar]
- [97].Smathers RL, Petersen DR. The human fatty acid-binding protein family: evolutionary divergences and functions. Hum Genomics 2011;5:170–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Akbal E, Kocak E, Akyurek O, Koklu S, Batgi H, Senes M. Liver fatty acid-binding protein as a diagnostic marker for non-alcoholic fatty liver disease. Wien Klin Wochenschr 2016;128:48–52. [DOI] [PubMed] [Google Scholar]
- [99].Mukai T, Egawa M, Takeuchi T, Yamashita H, Kusudo T. Silencing of FABP1 ameliorates hepatic steatosis, inflammation, and oxidative stress in mice with nonalcoholic fatty liver disease. FEBS Open Bio 2017;7:1009–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Pohl J, Ring A, Stremmel W. Uptake of long-chain fatty acids in HepG2 cells involves caveolae: analysis of a novel pathway. J Lipid Res 2002;43:1390–1399. [DOI] [PubMed] [Google Scholar]
- [101].Stremmel W, Staffer S, Wannhoff A, Pathil A, Chamulitrat W. Plasma membrane phospholipase A2 controls hepatocellular fatty acid uptake and is responsive to pharmacological modulation: implications for nonalcoholic steatohepatitis. FASEB J 2014;28:3159–70. [DOI] [PubMed] [Google Scholar]
- [102].Softic S, Kirby M, Berger NG, Shroyer NF, Woods SC, Kohli R. Insulin concentration modulates hepatic lipid accumulation in mice in part via transcriptional regulation of fatty acid transport proteins. PLoS One 2012;7:e38952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Alves-Bezerra M, Cohen DE. Triglyceride Metabolism in the Liver. Compr Physiol 2017;8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Gimeno RE. Fatty acid transport proteins. Curr Opin Lipidol 2007;18:271–6. [DOI] [PubMed] [Google Scholar]
- [105].Faye A, Esnous C, Price NT, Onfray MA, Girard J, Prip-Buus C. Rat liver carnitine palmitoyltransferase 1 forms an oligomeric complex within the outer mitochondrial membrane. J Biol Chem 2007;282:26908–16. [DOI] [PubMed] [Google Scholar]
- [106].Bartlett K, Eaton S. Mitochondrial beta-oxidation. Eur J Biochem 2004;271:462–9. [DOI] [PubMed] [Google Scholar]
- [107].Hajri T, Han XX, Bonen A, Abumrad NA. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J Clin Invest 2002;109:1381–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Chen A, Tang Y, Davis V, Hsu FF, Kennedy SM, Song H, et al. Liver fatty acid binding protein (L-Fabp) modulates murine stellate cell activation and diet-induced nonalcoholic fatty liver disease. Hepatology 2013;57:2202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Dong B, Kan CF, Singh AB, Liu J. High-fructose diet downregulates long-chain acyl-CoA synthetase 3 expression in liver of hamsters via impairing LXR/RXR signaling pathway. J Lipid Res 2013;54:1241–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Mashek DG, Li LO, Coleman RA. Rat long-chain acyl-CoA synthetase mRNA, protein, and activity vary in tissue distribution and in response to diet. J Lipid Res 20 06;47:20 04–10. [DOI] [PubMed] [Google Scholar]
- [111].Park SH, Helsley RN, Noetzli L, Tu HC, Wallenius K, O’Mahony G, et al. A luminescence-based protocol for assessing fructose metabolism via quantification of ketohexokinase enzymatic activity in mouse or human hepatocytes. STAR Protoc 2021;2:100731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Rajasekar P, Anuradha CV. Fructose-induced hepatic gluconeogenesis: effect of L-carnitine. Life Sci 2007;80:1176–83. [DOI] [PubMed] [Google Scholar]
- [113].Montesano A, Senesi P, Vacante F, Mollica G, Benedini S, Mariotti M, et al. L–Carnitine counteracts in vitro fructose-induced hepatic steatosis through targeting oxidative stress markers. J Endocrinol Invest 2020;43:493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Radulescu A, Dugan AJ, Killian M, Attia SL, Mouzaki M, Fuchs GJ, et al. Softic S: Stratification by obesity class, rather than age, can identify a higher percent of children at risk for non-alcoholic fatty liver disease and metabolic dysfunction. Pediatr Obes 2021;17(3):e12862. doi: 10.1111/ijpo.12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Softic S, Kahn CR. Fatty liver disease: is it nonalcoholic fatty liver disease or obesity-associated fatty liver disease? Eur J Gastroenterol Hepatol 2019;31:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Abdelmalek MF, Suzuki A, Guy C, Unalp-Arida A, Colvin R, Johnson RJ, et al. Nonalcoholic steatohepatitis clinical research n: increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010;51:1961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Ouyang X, Cirillo P, Sautin Y, McCall S, Bruchette JL, Diehl AM, et al. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol 2008;48:993–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Naguib G, Morris N, Yang S, Fryzek N, Haynes-Williams V, Huang WA, et al. Dietary fatty acid oxidation is decreased in non-alcoholic fatty liver disease: a palmitate breath test study. Liver Int 2020;40:590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Croci I, Byrne NM, Choquette S, Hills AP, Chachay VS, Clouston AD, et al. Whole-body substrate metabolism is associated with disease severity in patients with non-alcoholic fatty liver disease. Gut 2013;62:1625–33. [DOI] [PubMed] [Google Scholar]
- [120].Kotronen A, Seppala-Lindroos A, Vehkavaara S, Bergholm R, Frayn KN, Fielding BA, et al. Liver fat and lipid oxidation in humans. Liver Int 2009;29:1439–46. [DOI] [PubMed] [Google Scholar]
- [121].Dasarathy S, Kasumov T, Edmison JM, Gruca LL, Bennett C, Duenas C, et al. Glycine and urea kinetics in nonalcoholic steatohepatitis in human: effect of intralipid infusion. Am J Physiol Gastrointest Liver Physiol 2009;297:G567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Dasarathy S, Yang Y, McCullough AJ, Marczewski S, Bennett C, Kalhan SC. Elevated hepatic fatty acid oxidation, high plasma fibroblast growth factor 21, and fasting bile acids in nonalcoholic steatohepatitis. Eur J Gastroenterol Hepatol 2011;23:382–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Bugianesi E, Gastaldelli A, Vanni E, Gambino R, Cassader M, Baldi S, et al. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: sites and mechanisms. Diabetologia 2005;48:634–42. [DOI] [PubMed] [Google Scholar]
- [124].Fujita K, Nozaki Y, Wada K, Yoneda M, Fujimoto Y, Fujitake M, et al. Dysfunctional very-low-density lipoprotein synthesis and release is a key factor in nonalcoholic steatohepatitis pathogenesis. Hepatology 2009;50:772–80. [DOI] [PubMed] [Google Scholar]
- [125].Kohjima M, Enjoji M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med 2007;20:351–8. [PubMed] [Google Scholar]
- [126].Attia SL, Softic S, Mouzaki M. Evolving role for pharmacotherapy in NAFLD/NASH. Clin Transl Sci 2021;14:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Softic S, Kohli R. Pediatric NASH therapies: a speedbump on the road to success. Hepatology 2022;76(2):292–4. doi: 10.1002/hep.32322. [DOI] [PubMed] [Google Scholar]
- [128].Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Group G-IS: elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and -delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016;150:1147–59. [DOI] [PubMed] [Google Scholar]
- [129].Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. Group NS: a randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N Engl J Med 2021;385:1547–58. [DOI] [PubMed] [Google Scholar]
- [130].Wettstein G, Luccarini JM, Poekes L, Faye P, Kupkowski F, Adarbes V, et al. The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol Commun 2017;1:524–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Weber M, Mera P, Casas J, Salvador J, Rodriguez A, Alonso S, et al. Liver CPT1A gene therapy reduces diet-induced hepatic steatosis in mice and highlights potential lipid biomarkers for human NAFLD. FASEB J 2020;34:11816–37. [DOI] [PubMed] [Google Scholar]
- [132].Lanaspa MA, Cicerchi C, Garcia G, Li N, Roncal-Jimenez CA, Rivard CJ, et al. Counteracting roles of AMP deaminase and AMP kinase in the development of fatty liver. PLoS One 2012;7:e48801. [DOI] [PMC free article] [PubMed] [Google Scholar]