

Abstract
Type 1 diabetes (T1D) is characterized by the destruction of pancreatic β-cells. Several observations have renewed the interest in β-cell RNA sensors and editors. Here, we report that N6-methyladenosine (m6A) is an adaptive β-cell safeguard mechanism that controls the amplitude and duration of the antiviral innate immune response at T1D onset. m6A writer methyltransferase 3 (METTL3) levels increase drastically in β-cells at T1D onset but rapidly decline with disease progression. m6A sequencing revealed the m6A hyper methylation of several key innate immune mediators, including OAS1, OAS2, OAS3 and ADAR1 in human islets and EndoC-βH1 cells at T1D onset. METTL3 silencing enhanced 2′-5′-oligoadenylate synthetase levels by increasing its mRNA stability. Consistently, in vivo gene therapy to prolong Mettl3 overexpression specifically in β-cells delayed diabetes progression in the non-obese diabetic mouse model of T1 D. Mechanistically, the accumulation of reactive oxygen species blocked upregulation of METTL3 in response to cytokines, while physiological levels of nitric oxide enhanced METTL3 levels and activity. Furthermore, we report that the cysteines in position C276 and C326 in the zinc finger domains of the METTL3 protein are sensitive to S-nitrosylation and are important to the METTL3-mediated regulation of oligoadenylate synthase mRNA stability in human β-cells. Collectively, we report that m6A regulates the innate immune response at the β-cell level during the onset of T1D in humans.
The ability to distinguish self from non-self DNA or RNA is a fundamental function of the innate immune system1. Consequently, several autoimmune diseases are triggered by the over-activation of the innate immune system2. Furthermore, substantial evidence points to the activation of multiple genes that mediate the innate immune system before the onset of type 1 diabetes (T1D)3,4. This gains significance since nucleic acid sensors involved in the innate immune response are upregulated in insulitic islets at T1D onset5,6. Among the upregulated genes include the 2′-5′-oligoadenylate synthetase (OAS) family, a class of nucleotidyltransferases that, once activated, either act independently or produce 2′–5′-linked oligoadenylates to activate RNase L (ref. 7). Notably, polymorphisms in the OAS gene cluster have been associated with susceptibility to T1D8–10. Intriguingly, β-cells are unique among pancreatic islet cells in possessing an ability to upregulate OAS expression in response to interferon (IFN)-α or poly(I:C) (a double-stranded RNA (dsRNA) mimetic)11,12. OAS overexpression in β-cells leads to proliferation arrest and apoptosis12,13, while mice deficient in RNase L are protected from diabetes in a dsRNA-induced mouse model of T1D, consistent with the notion that over-activation of the OAS–RNase L pathway leads to β-cell death and T1D14.
N6-methyladenosine (m6A) is the most abundant modification in messenger RNA15–17. Adenosine methylation levels are regulated by ‘writer’ proteins such as methyltransferase 3 (METTL3) and 14 (METTL14)15. Several RNA-binding proteins, ‘readers,’ including YT521-B homology family proteins (for example, YTHDF), recognize methylated adenosines and regulate several aspects of mRNA biology including mRNA decay18–20. METTL3 is the only enzyme in the m6A writer complex that presents catalytic activity21. Recent work has demonstrated that METTL3 activity can be regulated by SUMOylation22 and phosphorylation23. However, the role of cysteine oxidative modifications such as S-nitrosylation (SNO) has not been explored.
Recent discoveries have led to the suggestion that m6A machinery regulates the innate immune response by accelerating the turnover of type I IFN genes in fibroblasts24, via promoting adenosine-to-inosine (A-to-I) RNA editing through regulation of ADAR1 (ref. 25) or by blocking the synthesis of endogenous aberrant dsRNAs26,27. However, the biological roles of m6A in T1D and, more specifically, their contribution towards mediating β-cell innate immune responses are unknown.
In this Article, we show that METTL3 levels are increased at the onset of T1D followed by a rapid decline. Furthermore, we identified m6A hypermethylation of OAS genes and demonstrated that METTL3 downregulation in both human pseudoislets and EndoC-βH1 cells leads to the upregulation of OAS proteins. We also observed that m6A accelerates the mRNA decay of OAS via SNO of the cysteine residues (C276 and C326) in the redox-sensitive zinc finger domains of METTL3. The ability of a sustained overexpression of Mettl3 in β-cells to limit the upregulation of Oas and protect the non-obese diabetic (NOD) mouse model of T1D28 from developing diabetes supports the translational significance of these findings.
Together, our studies identify m6A as an adaptive β-cell safeguard mechanism that controls the innate antiviral immune response at the onset of T1D.
Results
m6A writer (METTL3) levels peak at the onset and decrease drastically with progression of T1D
We have previously reported that a decrease in m6A levels leads to the downregulation of genes essential for β-cell function, identity and survival29. Upon further analyses of the upregulated genes (Extended Data Fig. 1a), we were intrigued to find a significant enrichment in pathways related to the immune response (Extended Data Fig. 1b). This interesting observation provided us with the rationale to determine if the m6A modulators, and in particular the major m6A writers, were impacted in T1D.
To begin, we isolated islets from NOD and an NOD congenic insulitis and diabetes-resistant model (NOR)30. We focused on mice that were 4 or 8 weeks of age, since at 4 weeks there is an initial phase of myeloid cell infiltration and a surge in the type I IFN signature (innate immune response), while the 8-week age marks the intense infiltration of major leukocyte subsets and T cell activation (adaptive immune response) in the NOD mouse model31.
Body weights and glucose levels did not differ between groups (Extended Data Fig. 1c,d). Mettl3 and Mettl14 were upregulated in islets from NOD compared with NOR mice at 4 weeks of age (Fig. 1a), followed by their downregulation at age 8 weeks (Fig. 1a). To confirm these changes, we subjected sorted β- and non-β-cells32 that were negatively selected for CD45 (Extended Data Fig. 1e) to RNA sequencing (RNA-seq). The CD45-negative β-cell and non-β-cell populations were enriched for insulin and glucagon, respectively (Extended Data Fig. 1f), and segregated by group (Extended Data Fig. 1g), with the β-cell fractions enriched for β-cell identity genes (Extended Data Fig. 1h). Consistently, β-cells from 8-week-old NOD mice presented upregulation of several immune genes involved in T1D (Extended Data Fig. 1j). Consistently, Mettl14 and Wtap were downregulated specifically in β-cell fractions in the 8-week-old versus 4-week-old mice, as well as a numerical decrease in Mettl3 (Fig. 1b).
Fig. 1 |. m6A writer METTL3 levels peak at T1D onset but decrease drastically with disease progression.
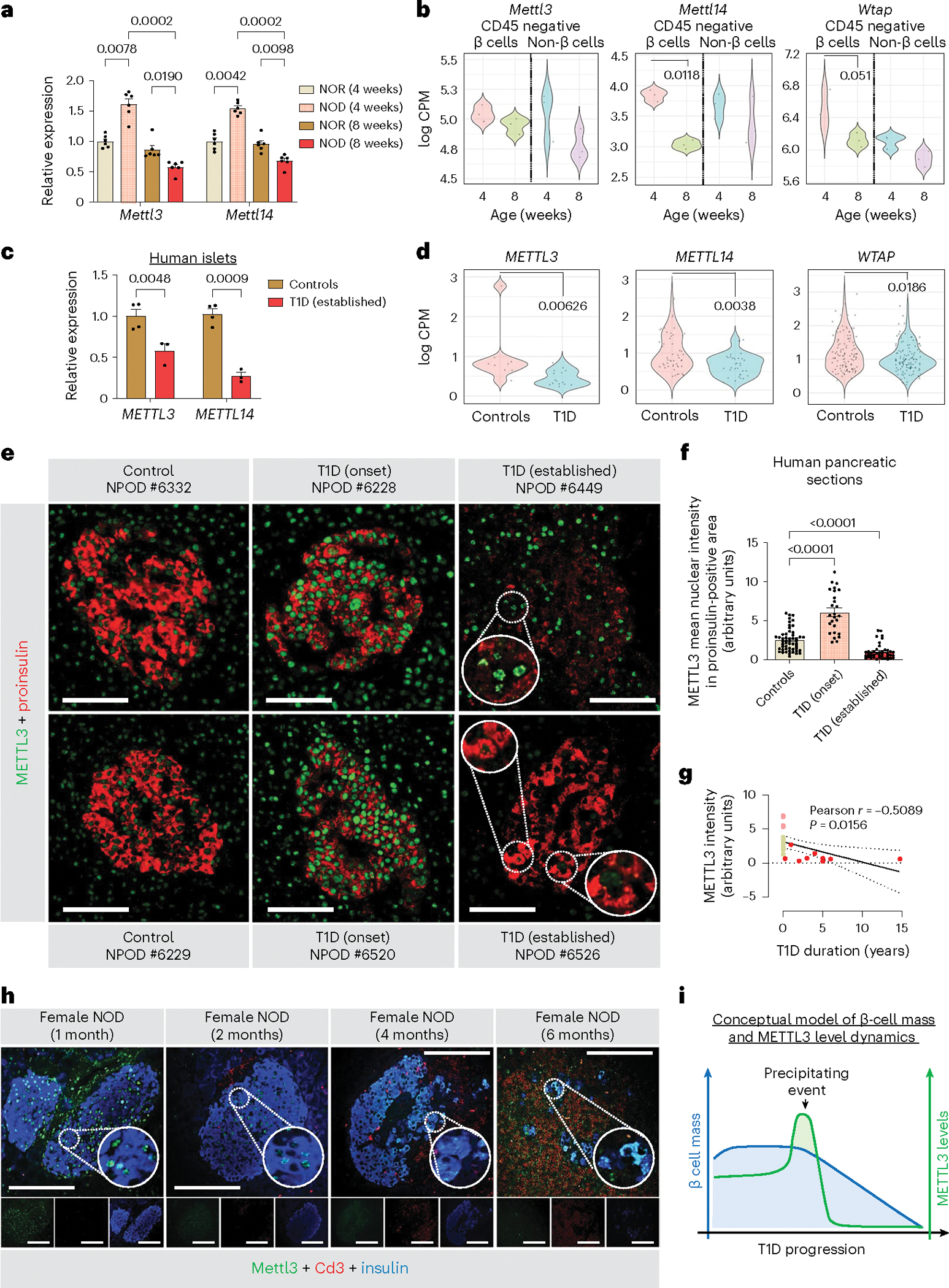
a, Quantitative reverse transcription (qRT)–PCR analyses of the m6A writer genes in whole islets isolated from 4-week-old or 8-week-old NOR or NOD mice (n = 6 mice per group). b, Violin-plots representation of the distribution of gene expression of m6A writers in CD45-negative sorted β- and non-β-cells in 4- or 8-week-old NOD mice (n = 3 pools of 3 mice per group). c, qRT–PCR analyses of METTL3 and METTL14 in whole islets isolated from human non-diabetic controls (n = 4), and human patients with established T1D (n = 3 samples). d, Violin-plots representation of the distribution of gene expression of m6A writers in single β-cells (high insulin gene expression) from control (n = 12 human patients) and established T1D (n = 4 human patients) (GSE121863). e, Representative pictures of immunofluorescence staining of proinsulin+METTL3 in pancreatic sections collected from non-diabetic (control) (n = 9), T1D onset (n = 4) and established T1D (n = 7) humans (scale bar, 100 μm; insert, 3× magnification; insert in nPOD #6526, 1.7× top and 3× bottom). f, Quantification of METTL3 intensity in proinsulin-positive area in pancreatic sections collected from non-diabetic (control) (n = 52 histological fields from 9 patients), T1D onset (n = 27 histological fields from 4 patients) and established T1D (n = 41 histological fields from 7 patients) humans. g, Pearson correlation of METTL3 nuclear intensity levels with T1D duration (control, green; T1D onset, pink; T1D established, red). h, Representative pictures of immunofluorescence staining of METTL3 in pancreatic sections collected from NOD female mice with 1, 2, 4 and 6months of age (n = 3 mice per group) (scale bar, 50 μm; insert, 3× magnification). i, Conceptual schematic representation of METTL3 and β-cell mass dynamics in the progression of human T1D. All samples in each panel are biologically independent. Data are expressed as mean ± standard error of the mean. Statistical analysis was performed by two-way analysis of variance (ANOVA) with Tukey multiple comparison test in a; Benjamini–Hochberg procedure in b and d; two-way ANOVA with Holm–Šídák’s multiple comparisons test in c and f. Numerical source data are available in Source data.
Next, we obtained freshly isolated islets from patients with established T1D (patient information in Supplementary Table 1). METTL3 and METTL14 were downregulated in established T1D islets (Fig. 1c). To confirm this, we downloaded and re-analysed a single-cell (sc) RNA-seq dataset performed in islets of patients with established T1D and non-diabetic controls33. Pancreatic β-cells were identified by insulin (INS) gene expression (Extended Data Fig. 2a) and easily segregated from other islet cell types including α-cells (Extended Data Fig. 2b). The expression of the m6A writers METTL3, METTL14 and WTAP were downregulated in β-cells from patients with established T1D (Fig. 1d).
METTL3 is considered an attractive therapeutic target as it is the only subunit of the m6A writer complex possessing enzymatic activity21,34. We took advantage of the abundance of METTL3 in human β-cells29 to perform immunofluorescence staining in pancreas sections (Network for Pancreatic Organ Donors with Diabetes (nPOD) patient information in Supplementary Table 1). Using a pipeline on ImageJ35 to measure unbiased METTL3 nuclear intensity in the proinsulin + area (Extended Data Fig. 2c), we detected an upregulation of METTL3 at T1D onset, followed by downregulation in established T1D (Fig. 1e,f). This downregulation correlated with the disease duration (Fig. 1g). METTL3 upregulation was also noticeable in the peri-islet area at T1D onset (Fig. 1e). The ease of identification of human islets from T1D-onset patients even without proinsulin co-staining reflected the robust upregulation of METTL3 predominantly in islets and specifically in β-cells (Extended Data Fig. 2d).
Next, to further dissect the METTL3 dynamics in T1D progression, we performed immunostaining for Mettl3, insulin and Cd3 (a T cell marker) in pancreas sections from NOD mice that were extensively phenotyped previously36 (Fig. 1h and Extended Data Fig. 2e). Consistent with human data, protein levels of Mettl3 were low in female NODs at late stages of T1D (Fig. 1h), while being unaltered in age-matched males (Extended Data Fig. 2e).
Altogether T1D onset is characterized by a significant upregulation of METTL3 levels in β-cells followed by its downregulation that spans established T1D (Fig. 1i) and coincides with the notion of a ‘precipitating event’ as postulated in the Eisenbarth model of T1D37.
Stimulation of human β-cells with IL-1β and IFN-α recapitulates the upregulation of METTL3
Next, we aimed to create an in vitro tool to recapitulate and study the METTL3 upregulation seen at T1D onset in human β-cells. Cytokine treatment of human islets, and in particular β-cells, has been reported to recapitulate several pathophysiological aspects of the molecular landscape of T1D38,39.
First, we challenged human islets or a human β-cell line (EndoC-βH1)40 with interleukin (IL)-1β, IFN-α, IFN-γ or various combinations for 48 h (Fig. 2a). We then measured total m6A levels in mRNA by liquid chromatography (LC)–tandem mass spectrometry (MS/MS) and analysed METTL3 and METTL14 protein levels (Fig. 2a). m6A levels were upregulated by IFN-α and even more robustly by the combination of IL-1β+IFN-α (Fig. 2b,c). Overall, METTL3 and METTL14 protein levels increased significantly in human islets exposed to cytokines (Fig. 2d). Similar results were observed in EndoC-βH1 cells (Fig. 2e).
Fig. 2 |. Co-treatment of human β-cells with IL-1β and IFN-α recapitulates the METTL3 upregulation seen at human T1D onset.
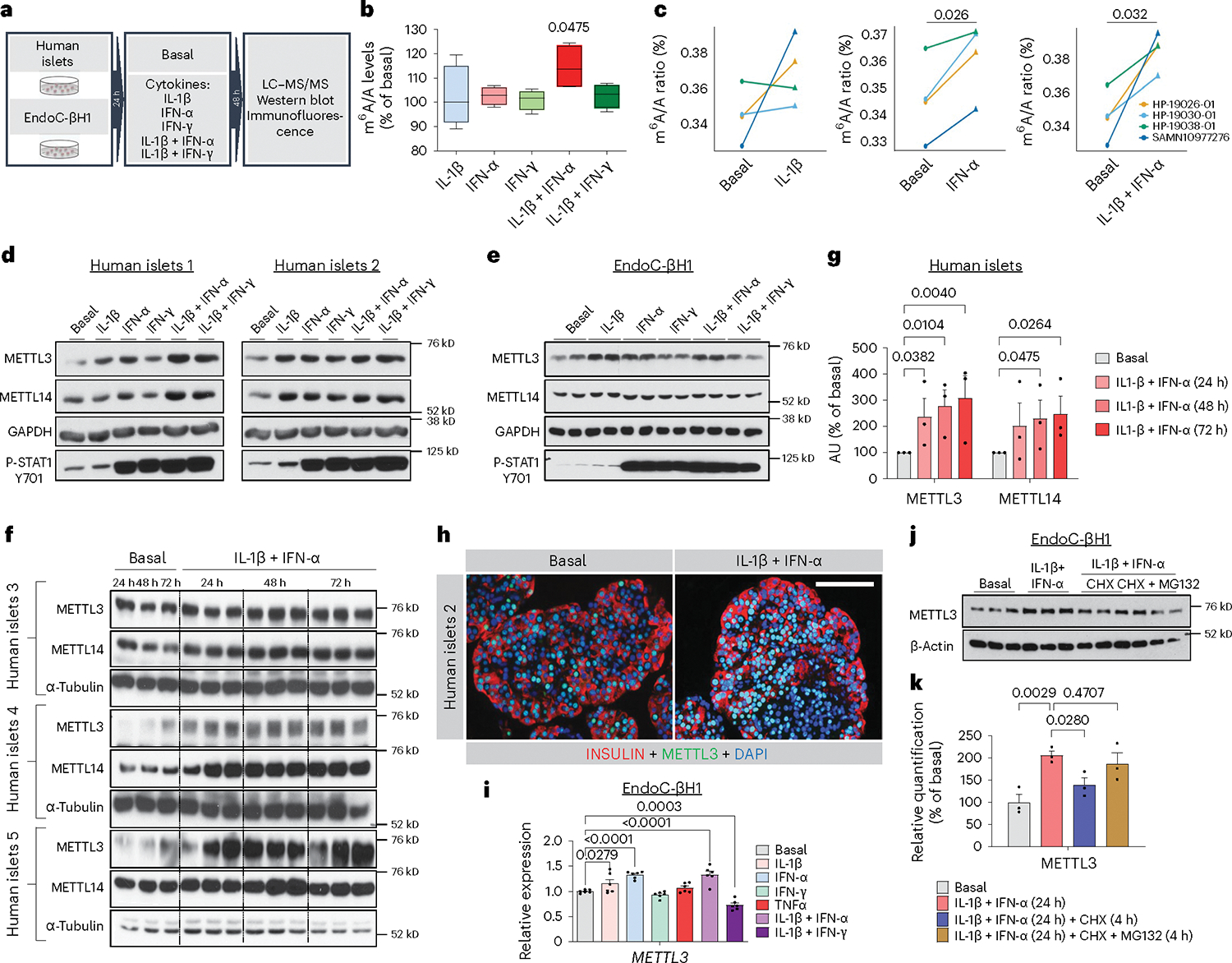
a, Summary scheme of the experimental plan. b, m6A/A levels measured by LC–MS/MS in human islets treated with IL-1β, IFN-α, IFN-γ, a combination of IL-1β plus IFN-α, or IL-1β plus IFN-γ for 48 h compared with PBS-treated (n = 4 independent biological samples). c, m6A levels measured by LC–MS/MS in individual human islet donors treated with IL-1β, IFN-α or IL-1β plus IFN-α compared with basal (PBS-treated) (n = 4 independent biological samples). Box plot shows the median, box edges show first and third quartiles, and whiskers show the minimum and maximum. d, Western-blot analyses of indicated proteins in human islets treated with the represented cytokines or cytokine combinations for 48 h (n = 2 independent biological samples). e, Western-blot analyses of indicated proteins in EndoC-βH1 cells treated with the represented cytokines or cytokine combinations for 48 h (n = 2 independent biological samples). f, Western-blot analyses of indicated proteins in human islets treated with IL-1β plus IFN-α or PBS (basal) for 24, 48 or 72h (n = 3 independent biological samples). g, Protein quantification of f. AU, arbitrary units. h, Representative pictures of immunofluorescence staining analyses of METTL3 in agar-embedded islets collected from human islets treated with IL-1β plus IFN-α or PBS (basal) for 48 h (n = 2 independent biological samples) (scale bar, 100 μm). i, Reverse transcription PCR analyses of METTL3 proteins in EndoC-βH1 cells treated with the represented cytokines or cytokine combinations for 48 h (n = 6 independent experiments/group). j, Western-blot analyses of indicated proteins in EndoC-βH1 cells treated with PBS and DMSO (basal) for 24 h, IL-1β+IFN-α for 24 h, Il-1β+IFN-α for 24 h and 10 μM cyclohexamide (CHX) for 4 h, or Il-1β+IFN-α for 24 h and 10 μM cyclohexamide+5 μM MG132 for 4 h (n = 3 independent experiments/group). k, Protein quantification of j. All samples in each panel are biologically independent. Data are expressed as mean ± standard error of the mean. Statistical analysis was performed by one-way analysis of variance (ANOVA) with Holm–Šídák’s multiple comparisons test in b; two-tailed paired t-test in c; two-way ANOVA with Fisher’s least significant difference test in g, i and k. Source numerical data and unprocessed gels are available in Source data.
Next, we explored METTL3 upregulation dynamics (Fig. 2f). METTL3 and METTL14 showed a time-dependent increase in response to stimulation with IL-1β and IFN-α (Fig. 2g). We visually confirmed METTL3 upregulation upon IL-1β and IFN-α stimulation by immunostaining (Fig. 2h). Finally, to study the mechanism(s) underlying METTL3 upregulation, we first examined the transcriptional regulation of METTL3 in EndoC-βH1 cells (Fig. 2i). METTL3 expression did not increase more than 30% upon IL-1β, IFN-α or IL-1β+IFN-α compared with basal (Fig. 2i). Second, to gain further insight into the stability of the METTL3 protein, we performed chase experiments (Fig. 2i,j). Interestingly, METTL3 upregulation was blocked with cyclohexamide, and this was rescued with MG132 (Fig. 2i,j). These findings suggest that the upregulation of METTL3 is primarily mediated by increased protein synthesis concomitantly with a modest transcriptional upregulation. Overall, these results demonstrate that the stimulation of human islets and β-cells with IL-1β and IFN-α in vitro accurately reproduces the upregulation of METTL3 observed during the onset of human T1D.
m6A landscape analyses reveal hypermethylation of antiviral innate immune genes with cytokines
To directly evaluate the m6A landscape during T1D onset, we employed RNA-seq and m6A-seq of islets from 15 human donors (Supplementary Table 1) stimulated with cytokines (IL-1β+IFN-α) for 48 h (Fig. 3a). The transcriptome (Fig. 3b) and m6A methylome (Fig. 3c) of cytokine-treated islets showed segregation from phosphate-buffered saline (PBS)-treated samples. However, the m6A response was heterogeneous among donors (Fig. 3c). Transcriptomic changes induced by cytokines were characterized by the downregulation of 5,181 genes and upregulation of 5,005 genes (Fig. 3d). Analyses of the combined differentially regulated gene sets revealed enrichment in pathways involved in nonsense-mediated decay and innate immune pathways (Fig. 3e).
Fig. 3 |. m6A landscape analyses of human islets treated with IL-1β and IFN-α reveal hypermethylation of 2′,5′-OAS genes.
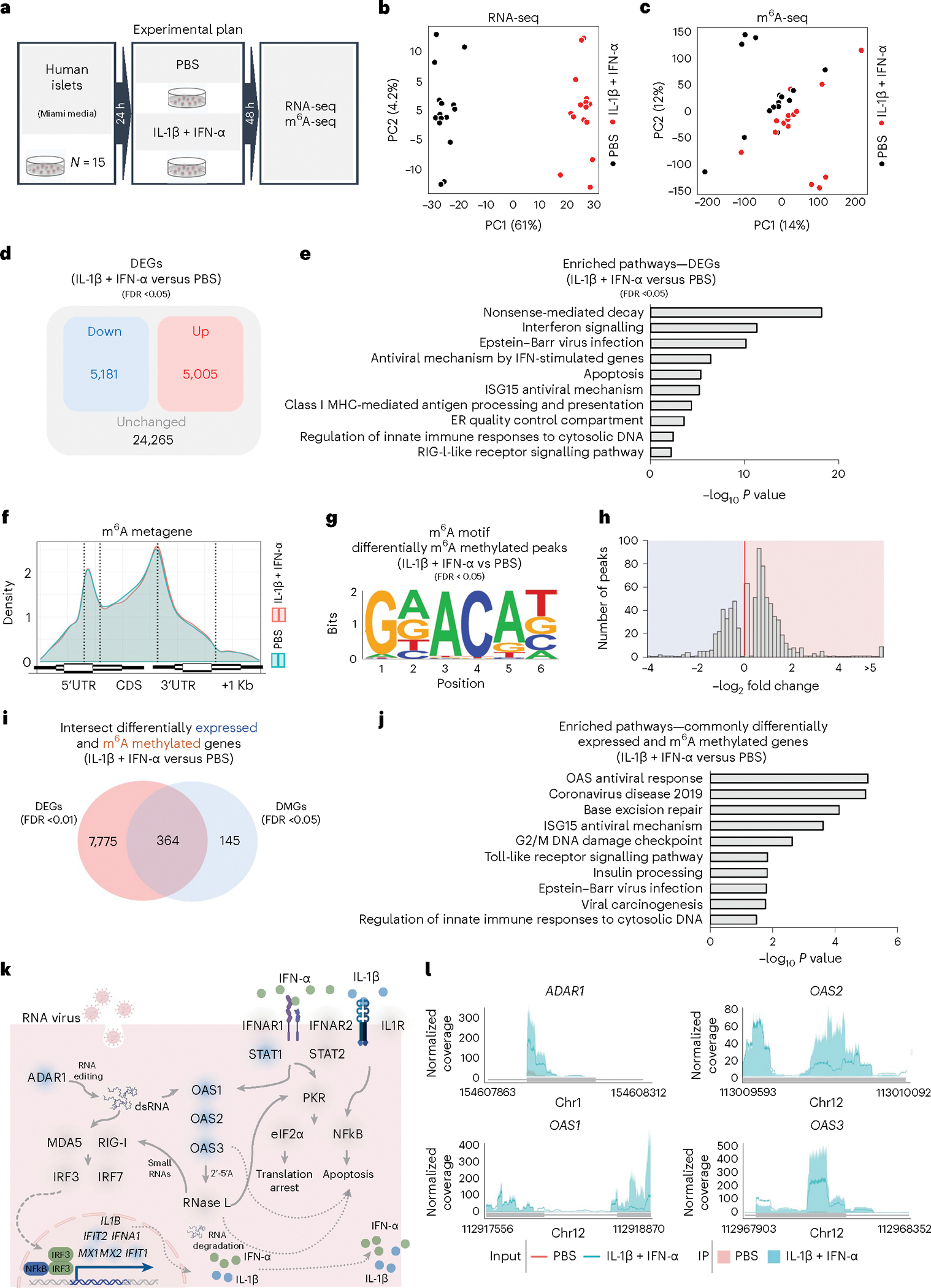
a, Summary scheme of the experimental plan. b, PCA plot of RNA-seq in human islets treated with PBS (black dots) or IL-1β and IFN-α (red dots) (n = 15 human patients). PC, principal component. c, PCA plot of m6A-seq in human islets treated with PBS (black dots) or IL-1β and IFN-α (red dots) (n = 15 human patients). d, Diagram representation of the upregulated (red), downregulated (blue) and unchanged genes (black) in human islets treated with IL-1β and IFN-α compared with PBS. Statistical analyses were performed using the Benjamini–Hochberg procedure and genes were filtered for FDR <0.05. e, Pathway enrichment analyses of upregulated and downregulated genes in human islets treated with IL-1β and IFN-α compared with PBS. f, Metagene of m6A enriched peaks in PBS- (blue) and IL-1β plus IFN-α-treated (red) human islets. CDS, coding sequence; UTR, untranslated region. g, Enrichment for known m6A consensus motif RRACH. h, Histogram of log2-fold change showing the distribution of differential m6A loci fold changes from IL-1β plus IFN-α-treated versus PBS. i, Venn diagram representation of the intersection between differentially methylated and expressed genes in human islets treated with IL-1β and IFN-α compared with PBS (n = 15 human patients). Statistical analyses were performed using the Benjamini–Hochberg procedure and DEGs were filtered for FDR <0.01 and m6A-methylated genes for FDR <0.05. j, Pathway enrichment analyses of intersected genes in i. k, Representation of antiviral innate immune pathway based on Kyoto encyclopedia of genes and genomes (KEGG) and Wikipathway annotations depicting several m6A hypermethylated genes (blue shade) and unchanged genes (grey shade) in human islets treated with IL-1β and IFN-α compared with PBS-treated (genes filtered for FDR <0.05). l, Coverage plots of m6A peaks in ADAR1, OAS1, OAS2 and OAS3 genes in human Islets treated with L-1β and IFN-α (blue) or PBS (red). Plotted coverages are the median of the n replicates presented. All samples in each panel are biologically independent. Pvalues of pathway enrichment analysis were calculated according to the hypergeometric test based on the number of physical entities present in both the pre-defined set and user-specified list of physical entities. Source numerical data are available in Source data.
Next, analyses of the m6A methylome showed changes that were consistent with patterns from previous studies16,17 in that the m6A peaks were enriched at the start and stop codons (Fig. 3f), and characterized by the canonical GGACU motif (Fig. 3g). m6A sequencing (m6A-seq) revealed 800 differently methylated sites in 509 genes with a higher number of hypermethylated m6A sites in cytokine-treated compared with PBS-treated islets (Fig. 3h). Since IL-1β and IFN-α stimulation of human islets led to extensive transcriptomic remodelling, we hypothesized that some of these pathways are modulated by m6A at the mRNA level.
To test this hypothesis, we intersected the differentially expressed genes (DEGs) and differentially m6A-methylated genes (DMGs) (Fig. 3i and Extended Data Fig. 3a). Pathway enrichment analyses on these common 364 intersected genes (Fig. 3i) revealed interconnected pathways involving the antiviral innate immune system (Fig. 3j). Specifically, several dsRNA sensors such as OAS and the RNA editor ADAR1 were highly hypermethylated and upregulated in human islets in response to IL-1β and IFN-α (Fig. 3k,l and Extended Data Fig. 3b). Other hypermethylated and upregulated genes included several IFN-induced genes such as IFN-induced protein with tetratricopeptide (IFIT1 and IFIT2), and MX dynamin-like GTPase (MX1 and MX2) (Fig. 3k). Conversely, genes displaying hypermethylation and subsequent downregulation exerted influences on pathways linked to insulin secretion (Extended Data Fig. 3c). Additionally, a set of hypomethylated genes exhibited enrichment in antiviral defence pathways (Extended Data Fig. 3d,e), including multiple MHC class I genes responsible for antigen presentation (Extended Data Fig. 3f,g), such as endoplasmic reticulum aminopeptidase 1 (ERAP1; Extended Data Fig. 3h–j). This enzyme is known to play a critical role in trimming peptides for antigen presentation41, including the preproinsulin signal peptide antigen42. Overall, these results indicate a dynamic transcriptomic remodelling in human islets upon IL-1β and IFN-α stimulation and show that genes that are differentially expressed and m6A-decorated are mainly involved in the antiviral innate immune response.
OAS genes are upregulated and m6A decorated in human β-cells treated with IL-1β and IFN-α
To validate the m6A regulation of OAS genes specifically in human β-cells, we challenged EndoC-βH1 cells with cytokines (IL-1β+IFN-α) or PBS (Fig. 4a). The transcriptome (Fig. 4b) and the m6A methylome (Fig. 4c) of cytokine-treated cells showed clear segregation between groups. Cytokine stimulation resulted in the downregulation of 3,166 genes and the upregulation of 3,203 genes compared with PBS-treated EndoC-βH1 cells (Extended Data Fig. 4a). Pathway enrichment analyses of the combined upregulated and downregulated genes identified nonsense-mediated decay and antiviral innate immune pathways (Extended Data Fig. 4b).
Fig. 4 |. m6A controls the mRNA stability of 2′–5′-OAS genes.
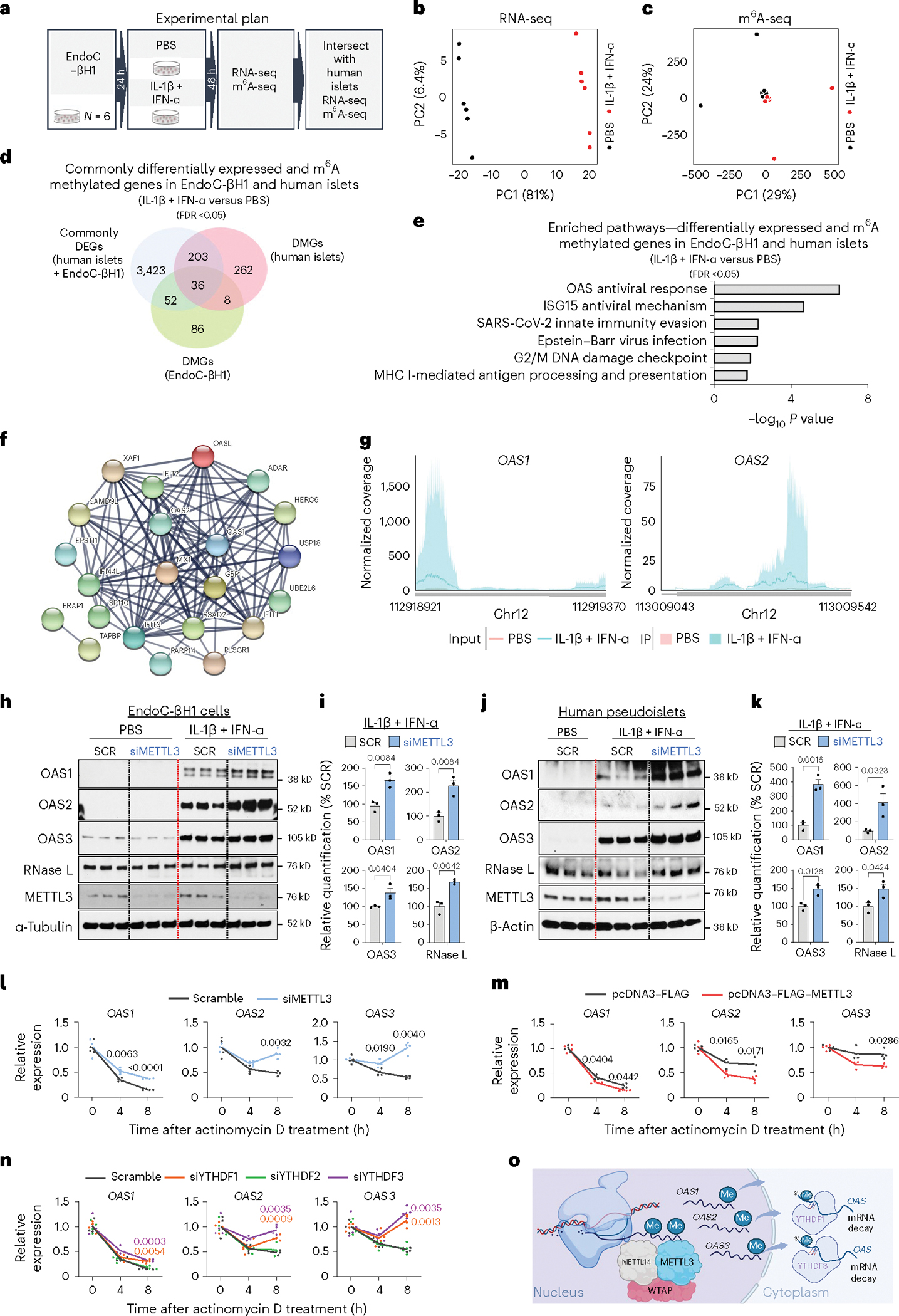
a, Summary scheme of experimental plan. b, PCA plot of RNA-seq in EndoC-βH1 cells treated with PBS (black dots) or IL-1β and IFN-α (red dots) (n = 6 independent experiments per group). PC, principal component. c, PCA plot of m6A-seq in EndoC-βH1 treated with PBS (black dots) or IL-1β and IFN-α (red dots) (n = 6 independent experiments per group). d, Venn diagram representation of the intersection between DEGs in human islets and EndoC-βH1 treated with IL-1β and IFN-α compared with PBS-treated with DMGs in human islets and EndoC-βH1 treated with IL-1β and IFN-α compared with PBS-treated. Statistical analyses were performed using the Benjamini–Hochberg procedure and genes were filtered for FDR <0.05. e, Pathway enrichment analyses of intersected genes in d. f, STRING functional protein–protein interaction network of 36 intersected genes, showing differential expression and m6A methylation in human islets and Endoc-βH1 cells treated with IL-1β and IFN-α compared with PBS. g, Coverage plots of m6A peaks in OAS1 and OAS2 genes in EndoC-βH1 cells treated with L-1β and IFN-α or PBS. Plotted coverages are the median of the n replicates presented. h, Western-blot analyses of indicated proteins after IL-1β plus IFN-α or PBS stimulation in EndoC-βH1 cells harbouring METTL3 KD or scramble (SCR) (n = 3 independent experiments per group). Same experiment of Extended Data Fig. 5h, with same loading control. i, Protein quantification of indicated protein in h. j, Western-blot analyses of indicated proteins after IL-1β plus IFN-α or PBS stimulation in human pseudoislets METTL3 KD or SCR (n = 3 biological independent samples). k, Protein quantification of indicated protein in j. l Quantitative reverse transcription (qRT)–PCR analyses of OAS genes after IL-1β plus IFN-α stimulation of METTL3 KD or scramble EndoC-βH1 cells after a time-course treatment with actinomycin D (ActD) (n = 4 independent experiments per group). m, qRT–PCR analyses of OAS genes after IL-1β plus IFN-α stimulation in METTL3-overexpressing (OE) and control (FLAG) EndoC-βH1 cells after a time-course treatment with ActD (n = 4 independent experiments per group). n, qRT–PCR analyses of OAS genes after IL-1β plus IFN-α stimulation in scramble, YTHDF1, YTHDF2 or YTHDF3 KD EndoC-βH1 cells after a time-course treatment with ActD (n = 4 independent experiments per group). o, Model depicting the role of METTL3-mediated m6A hypermethylation of OAS genes in response to IL-1β and IFN-α or at T1D onset that leads to OAS nuclear export to the β-cell cytoplasm and recognition by m6A readers YTHDF1 and YTHDF3 leading to accelerated mRNA decay. All samples in each panel are biologically independent. Data are expressed as mean ± standard error of the mean. Statistical analysis was performed by two-tailed unpaired t-test in i and k; two-way analysis of variance with Holm–Šídák’s multiple comparisons test in l–n; or as otherwise stated above. P values of pathway enrichment analysis were calculated according to the hypergeometric test based on the number of physical entities present in both the pre-defined set and user-specified list of physical entities. Source unprocessed gel images and numerical data are available in Source data.
Since human islets are composed of several cell types, we focused on the DEGs in human islets that overlapped with EndoC-βH1 cells to ensure β-cell specificity. Intersection of the DEGs in human islets and EndoC-βH1 cells revealed 1,577 and 1,547 commonly upregulated and downregulated genes, respectively (Extended Data Fig. 4c). Enrichment pathway analyses on the intersected genes again revealed nonsense-mediated decay and the antiviral innate immune response (Extended Data Fig. 4d). The gene expression similarities between human islets (Extended Data Fig. 4e) and EndoC-βH1 cells (Extended Data Fig. 4f) in response to cytokines was remarkable. Indeed, virtually all innate immune sensor genes were consistently upregulated in both models (Extended Data Fig. 4e,f). The translational relevance of these findings is supported by the observation that numerous innate immune genes that were commonly upregulated in both human islets and EndoC-βH1 cells treated with IL-1β and IFN-α overlap with those upregulated in the human insulitic islets from T1D-onset patients, and notably include the OAS1, OAS2 and OAS3 genes8.
Examination of the m6A methylome revealed m6A peaks enriched at the start and stop codons (Extended Data Fig. 5a) and characterized by the canonical GGACU motif (Extended Data Fig. 5b). Analysis of m6A-seq revealed 301 differently methylated sites in 182 genes and a higher number of sites with increased levels of m6A methylation in IL-1β and IFN-α-treated compared with PBS (Extended Data Fig. 5c).
To further explore the m6A regulation of the OAS innate immune sensors in β-cells, we intersected the commonly expressed and m6A-methylated genes in human islets or EndoC-βH1 cells (Fig. 4d). This analysis identified 36 commonly expressed and m6A-regulated genes mainly involved in the innate immune pathway including OAS and ADAR1 (Fig. 4e,f). Protein–protein interaction analyses on the common m6A-regulated and expressed genes revealed a close relationship among OAS1, OAS2 and OAS3 proteins with other innate immune mediators (Fig. 4f). Importantly, we were able to validate the m6A hypermethylation of OAS1 and OAS2 in EndoC-βH1 cells by m6A-seq (Fig. 4g), and OAS1, OAS2 and OAS3 by m6A immunoprecipitation (m6A-IP)–quantitative polymerase chain reaction (qPCR) in EndoC-βH1 cells (Extended Data Fig. 5d).
Finally, the observation that different pancreatic islet cell types are capable of mounting divergent antiviral responses12 prompted us to dissect the OAS response in other pancreatic cells. To this end, we challenged EndoC-βH1, 266-6 (mouse acinar-cell line; Extended Data Fig. 5e), PANC-1 (human ductcell line; Extended Data Fig. 5f) and α-TC6 (mouse α-cell line; Extended Data Fig. 5g) with IL-1β and IFN-α or PBS in parallel for 48 h. These data showed that METTL3 and OAS upregulation in response to cytokines was prominent in human β-cells.
m6A mRNA methylation promotes OAS mRNA decay in human β-cells
Next, to test the hypothesis that m6A impacts the upregulation of OAS protein levels we challenged EndoC-βH1 cells harbouring METTL3 knockdown (KD) or control scramble with cytokines (IL-1β+IFN-α) or PBS for 16 h (Fig. 4h). OAS1, OAS2 and OAS3 were upregulated in scramble cells stimulated with cytokines compared with PBS (Fig. 4h). Furthermore, METTL3 silencing led to an even greater increase in OAS protein levels in response to cytokines (Fig. 4h,i). A similar approach in human pseudoislets (Fig. 4j) with METTL3 silencing exhibited a greater upregulation of OAS proteins than scramble controls (Fig. 4j,k). Furthermore, we noted alterations in the expression of other RNA sensors and editors (Extended Data Fig. 5h). For example, METTL3 silencing in EndoC-βH1 cells led to downregulation of isoform p150 of ADAR1 (Extended Data Fig. 5i), in contrast to the upregulation of OAS, suggesting that m6A regulates RNA editing in β-cells. Consistently, ADAR1 has recently been shown to be induced by cytokines and to increase the A-to-I RNA editing of Alu-containing mRNAs in human β cells43 and ADAR1p150 has been reported to prevent fatal auto-inflammation44.
Next, we explored whether METTL3 controls the innate immune response in β-cells by accelerating the mRNA decay of OAS genes. To this end, we challenged EndoC-βH1 cells harbouring either METTL3 KD or scramble KD with actinomycin D (a transcription inhibitor) or dimethyl sulfoxide (DMSO) in the presence of cytokines (Fig. 4l). We observed that METTL3 silencing increases the mRNA stability of OAS genes (Fig. 4l). To validate these findings, we asked whether the converse, that is upregulating METTL3, accelerates decay of OAS genes. Indeed, overexpression of METTL3 in EndoC-βH1 cells accelerated the decay of OAS genes in response to cytokines (Fig. 4m).
To validate the m6A regulation of OAS expression and identify the putative m6A reader proteins controlling OAS mRNA decay, we independently silenced the readers, YTHDF1, YTHDF2 or YTHDF3, in EndoC-βH1 cells (Fig. 4n and Extended Data Fig. 5j). YTHDF1 or YTHDF3 downregulation increased OAS mRNA stability, suggesting that m6A hypermethylation of OAS transcripts accelerates the decay of their mRNAs mediated by YTHDF1 and YTHDF3 (Fig. 4n). In summary, these data reveal that METTL3 upregulation and consequent m6A hypermethylation of OAS1, OAS2 and OAS3 control the innate immune response in human β-cells by promoting their mRNA decay via YTHDF1 and YTHDF3 (Fig. 4o).
OAS upregulation leads to an extensive downregulation of metallothioneins in β-cells
To gain insight into the transcriptomic alterations induced by OAS in human β-cells, we individually overexpressed OAS1, OAS2 or OAS3 in EndoC-βH1 cells in the basal cellular state (Fig. 5a).
Fig. 5 |. OAS upregulation leads to an extensive downregulation of metallothioneins in β-cells.
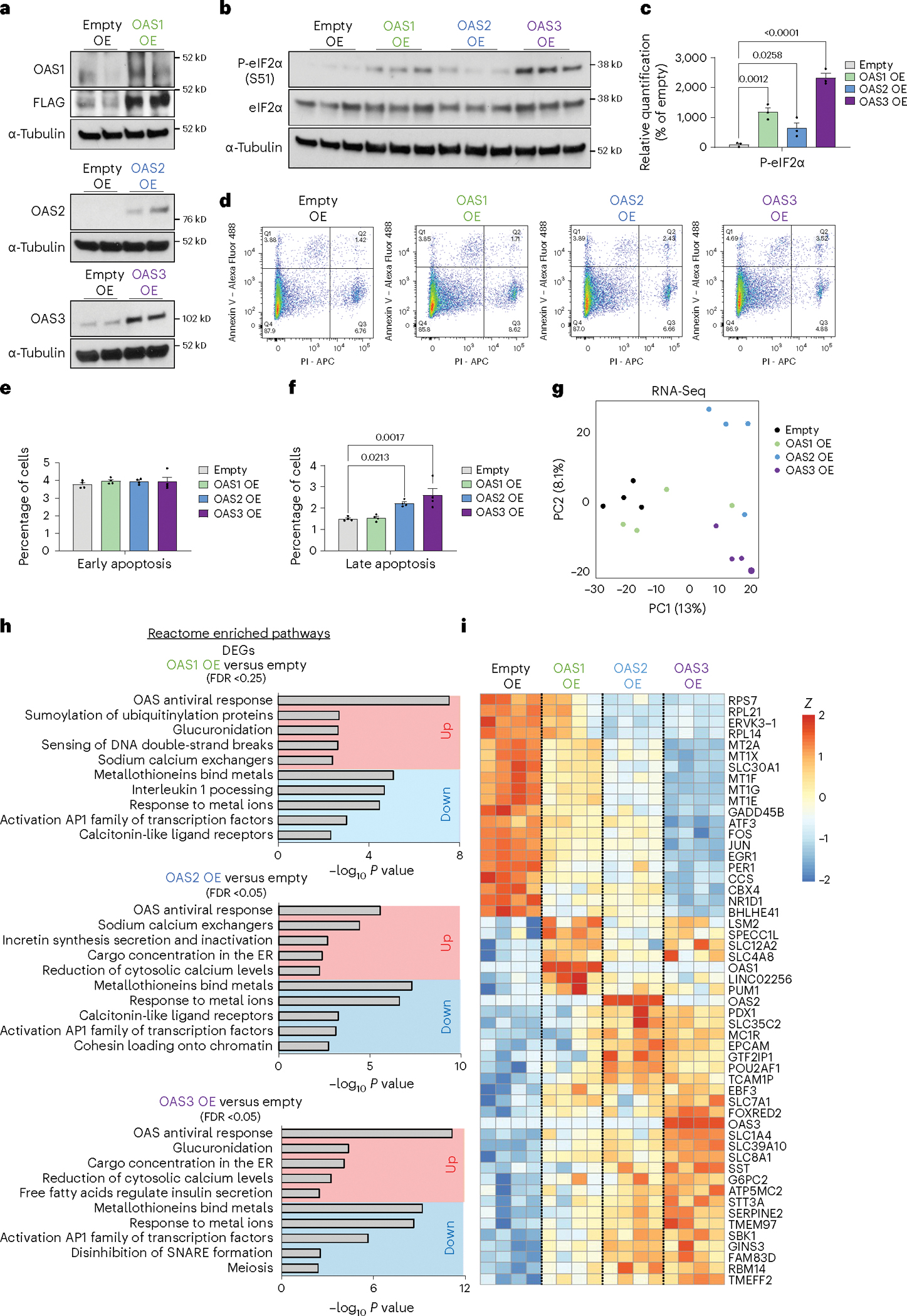
a, Western-blot validation of OAS1, OAS2 or OAS3 protein overexpression (OE) in EndoC-βH1 cells (n = 2 experiments). b, Western-blot analyses of indicated proteins in EndoC-βH1 cells overexpressing OAS1, OAS2, OAS3 or a stuffer control plasmid for 72h (n = 3 independent experiments per group). c, Protein quantification of b. d, Flow cytometry analyses of PI and Annexin V in EndoC-βH1 cells overexpressing OAS1, OAS2, OAS3 or a stuffer control plasmid (n = 4 independent experiments per group). e,f, Quantification of d, showing early (e) and late (f) apoptosis rates. g, PCA plot of RNA-seq in EndoC-βH1 cells overexpressing OAS1 (green), OAS2 (blue), OAS3 (purple) or a stuffer control (black) plasmid for 72h (n = 4 independent experiments per group). PC, principal component. h, Pathway enrichment analyses of DEGs. i, Heat-map representation of top DEGs. All samples in each panel are biologically independent. Heat map represents clipped Z-scored log CPM. Data are expressed as mean ± standard error of the mean. Statistical analysis was performed by Benjamini–Hochberg procedure or one-way analysis of variance with Holm–Šídák’s multiple comparisons test in c and f. P values of pathway enrichment analysis were calculated according to the hypergeometric test based on the number of physical entities present in both the pre-defined set and user-specified list of physical entities. Source unprocessed gel images and numerical data are available in Source data.
Notably, the overexpression of OAS proteins induced the phosphorylation of eukaryotic initiation factor-2α (eIF2α) at serine 51, even in the absence of IFN stimulation, indicative of translation inhibition (Fig. 5b,c). Moreover, OAS2 and OAS3 overexpression, but not OAS1, resulted in elevated β-cell apoptosis rates (Fig. 5d–f). The impact of OAS upregulation in β-cells remains relatively unexplored, and to further clarify this phenomenon, we conducted RNA-seq analyses in EndoC-βH1 cells overexpressing OAS1, OAS2 or OAS3 individually (Fig. 5g). Our analyses revealed clear distinctions, with OAS2 and OAS3 overexpression producing more profound changes in the transcriptomic profiles as compared with OAS1 (Fig. 5g). Pathway enrichment analyses using the Rotation Gene Set Test (Roast) method (Methods) showed upregulation of pathways associated with the OAS response and endoplasmic reticulum (ER) function. In stark contrast, OAS upregulation resulted in a consistent and widespread downregulation of metallothionein (MT) genes, including MT1E, MT1X and MT2A (Fig. 5i). MTs are low-molecular-weight, cysteine-rich proteins known for their high-affinity binding to heavy metals45,46. They play pivotal roles in metal metabolism and detoxification and, owing to their numerous cysteine residues, function as potent antioxidants45,46. Notably, the overexpression of MTs in β-cells has been demonstrated to confer broad resistance to oxidative stress45. Thus, our findings indicate that OAS overexpression triggers a widespread downregulation of MTs, compromising the antioxidant capacity of the β-cells and ultimately rendering them more susceptible to apoptosis.
m6A landscape of established T1D is enriched in β-cell identity and function genes
To explore the m6A landscape of established T1D, we performed RNA-seq and m6A-seq in human islets from patients with established T1D or non-diabetic controls (Fig. 6a). The transcriptome (Fig. 6b) and the m6A methylome (Fig. 6c) exhibited clear segregation in the principal component analysis (PCA). T1D islets presented downregulation of 5,441 genes and an upregulation of 4,913 genes compared with controls (Fig. 6d). Pathway enrichment analyses of the combined upregulated and downregulated genes (Fig. 6d) revealed pathways involved in translation initiation, nonsense-mediated decay, insulin secretion and maturity-onset of diabetes of the young (MODY; Fig. 6e).
Fig. 6 |. m6A landscape of established T1D is enriched in β-cell identity and function genes.
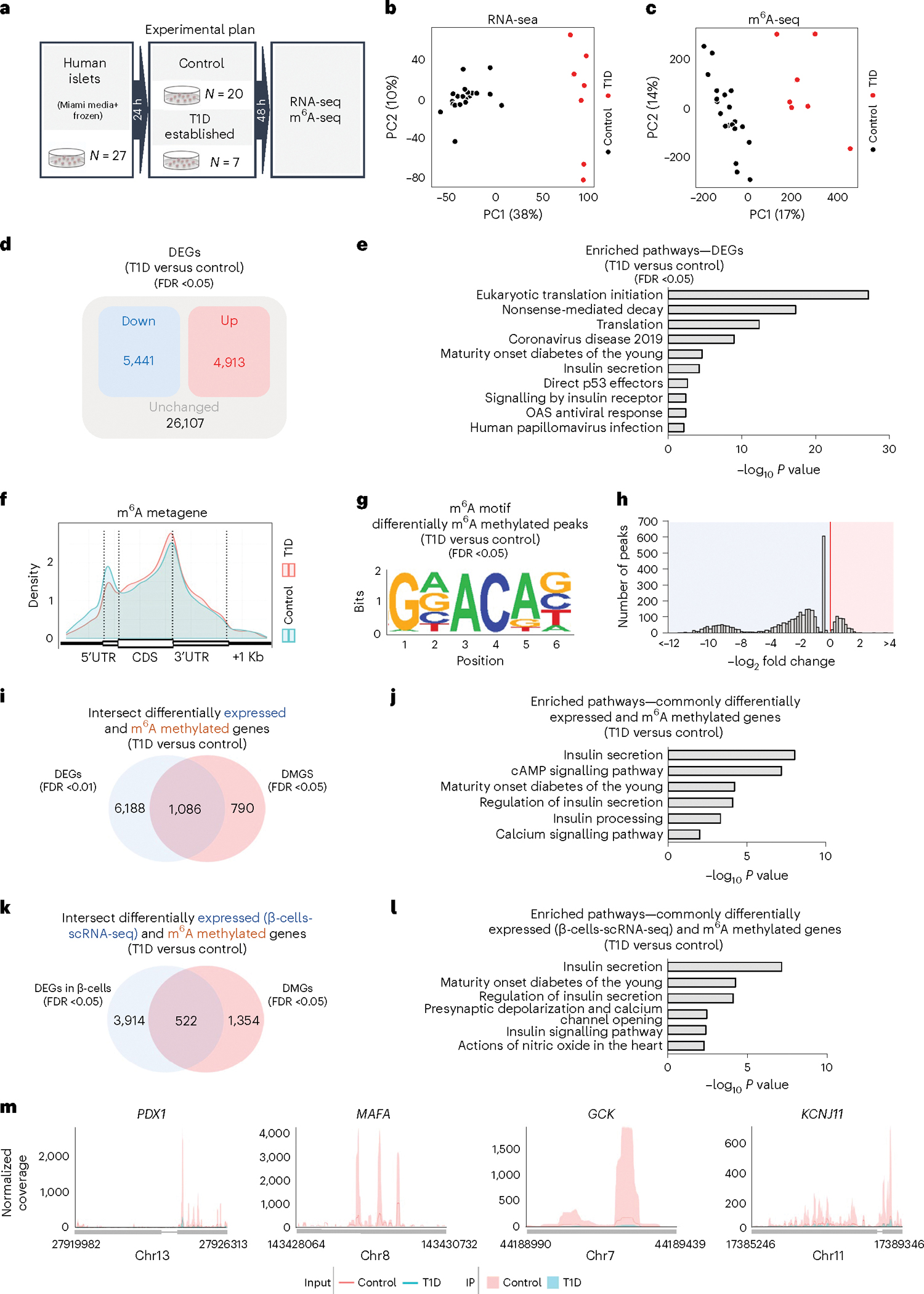
a, Summary scheme of the experimental plan. b, PCA plot of RNA-seq in human islets from control (black dots) (n = 20 biological independent samples) or established T1D (red dots) (n = 7 biological independent samples). PC, principal component. c, PCA plot of m6A-seq in human islets from control (black dots) (n = 20 biological independent samples) or established T1D (red dots) (n = 7 biological independent samples). d, Diagram representation of the upregulated (red), downregulated (blue) and unchanged genes (black) in human islets from control or T1D. e, Pathway enrichment analyses of upregulated and downregulated genes in human islets from T1D compared with controls. f, Metagene of m6A enriched peaks in control (blue) and T1D (red) human islets. UTR, untranslated region; CDS, coding sequence. g, Enrichment for known m6A consensus motif RRACH. h, Histogram showing the distribution of differential m6A loci log2 fold changes from T1D versus control human islets. i, Intersection of differentially expressed and m6A-methylated genes in T1D compared with control human islets. j, Pathway enrichment analyses of intersected genes in i. k, Intersection of DEGs in T1D β-cells compared with control from a published scRNA-seq dataset33 and our m6A dataset comparing the DMGs in established T1D with control human islets. l, Pathway enrichment analyses of intersected genes in k. m, Coverage plots of m6A peaks in β-cell identity genes in human islets from established T1D compared with controls. Plotted coverages are the median of the n replicates presented. Human islets: controls n = 20 and T1D n = 7 biologically independent samples. EndoC-βH1 cells: n = 6 biologically independent samples. Statistical analyses were performed using the Benjamini–Hochberg procedure. P values of pathway enrichment analysis were calculated according to the hypergeometric test based on the number of physical entities present in both the pre-defined set and user-specified list of physical entities. Source numerical data are available in Source data.
m6A-seq analysis confirmed the enrichment of m6A peaks at the start and stop codons (Fig. 6f) and was characterized by the canonical GGACU motif (Fig. 6g). There were 3,485 differently methylated sites in 1,876 genes and a higher number of sites presenting m6A hypomethylation in established T1D compared with controls (Fig. 6h) consistent with our findings of downregulation of METTL3 with T1D progression (Fig. 1e,f).
To dissect the m6A regulation of gene expression in established T1D, we intersected and performed pathway enrichment analysis on the differentially expressed and m6A-methylated genes in human islets from patients with established T1D (Fig. 6i). This analysis identified enrichment for pathways associated with β-cell function and identity (Fig. 6j). Next, to overcome the limitations of the number of insulin-positive β-cells being drastically decreased in established T1D, we intersected the differentially m6A-decorated genes with the β-cell transcriptome from a scRNA-seq dataset performed in islets from patients with established T1D and non-diabetic controls (GSE121863; Fig. 6k and Extended Data Fig. 6a). Pathway analyses on the intersected differentially expressed and m6A-methylated genes revealed enrichment for insulin secretion and MODY (Fig. 6l and Extended Data Fig. 6b–e). Several master regulators of β-cell function and identity, such as PDX1, MAFA, GCK and KCNJ11 presented m6A hypomethylation and were downregulated in established T1D (Fig. 6m and Extended Data Fig. 6e). Together, these results identify distinct m6A landscapes at the onset and in established T1D specifically in human β-cells and point to m6A regulation of β-cell function and identity genes in established disease.
Sustained overexpression of Mettl3 in β-cells delays diabetes progression in the NOD mouse
To test whether prolonged upregulation of Mettl3 in the NOD mouse β-cells would lead to a faster turnover and/or decrease in expression of Oas and delay T1D progression, we designed two different adeno-associated virus serotype 8 (AAV8) driving enhanced green fluorescent protein (eGFP) or Mettl3 under the control of the rat insulin promoter II (RIP2)47 (Fig. 7a and Extended Data Fig. 7a). We chose to infuse PBS or AAV8 into 4-week-old animals (Fig. 7b) because our data showed that Mettl3 levels start to decline after this age in NOD females (Fig. 1a,h). We confirmed Mettl3 protein overexpression and specificity at 12 weeks of age (Fig. 7c and Extended Data Fig. 7b) and followed these mice for up to 25 weeks of age, similar to previous studies48–50. Body weight trajectories did not change until 17 weeks of age, when mice that received AAV8 driving eGFP overexpression (AAV8-eGFP) or PBS started to lose weight compared with mice that received AAV8 driving Mettl3 overexpression (AAV8-Mettl3) (Fig. 7d). Random-fed blood glucose levels increased with age in AAV8-eGFP and PBS groups, in contrast to the AAV8-Mettl3 group (Fig. 7e) indicating a delayed progression of T1D (Fig. 7f).
Fig. 7 |. In vivo AAV8-mediated overexpression of Mettl3 in NOD mouse β-cells delays T1D progression.
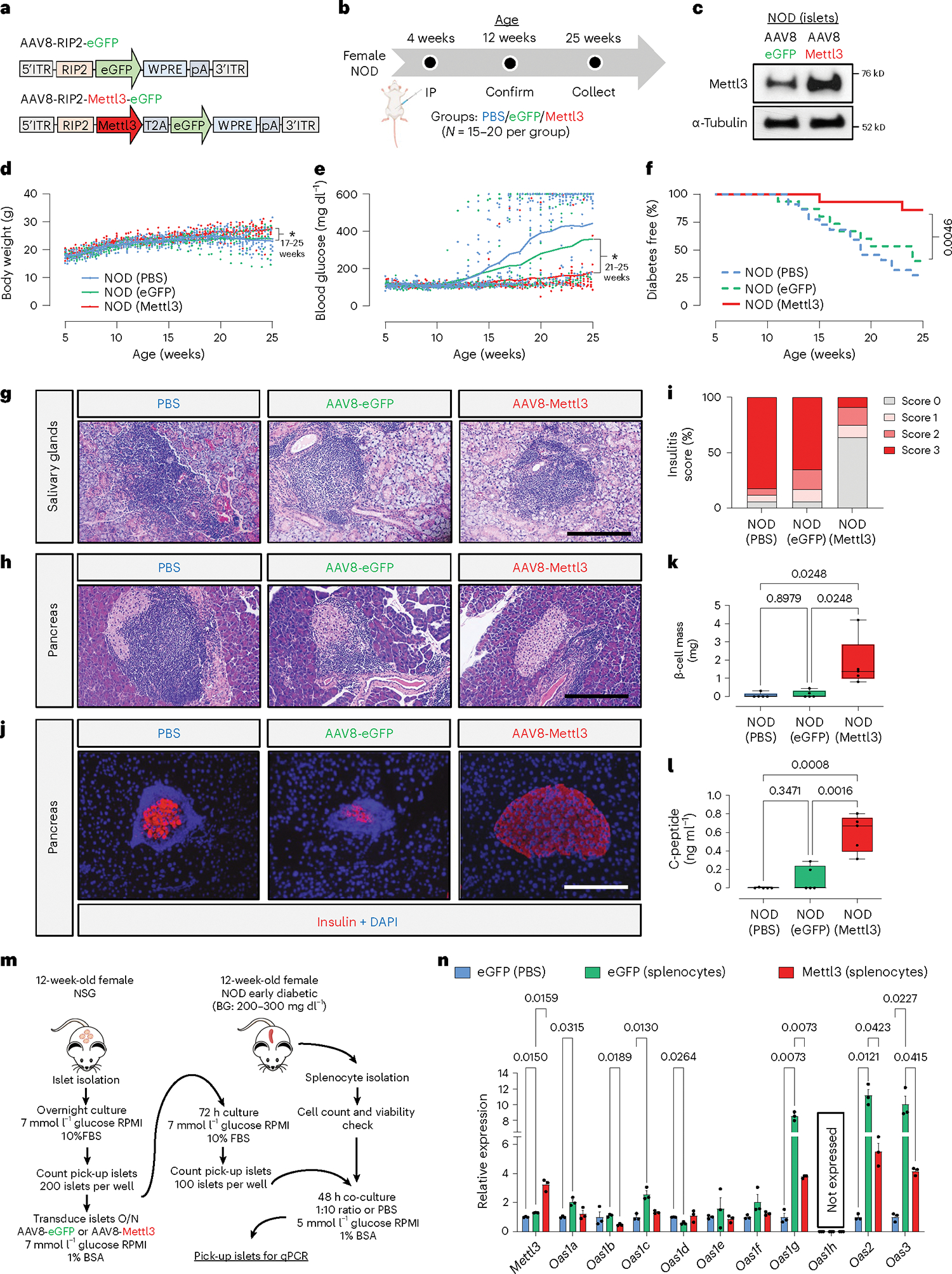
a, Schematic diagram showing the construction of AAV8 driving eGFP or Mettl3 under the control of a RIP2. b, Scheme of experimental approach depicting NOD mice receiving PBS (blue), AAV8 overexpressing eGFP (green) or AAV8 overexpressing Mettl3 (red). N = 20 mice in NOD (PBS), and n = 15 mice in NOD (eGFP) or NOD (Mettl3). c, Western-blot validation of Mettl3 overexpression in isolated NOD female islets after 8 weeks of in vivo transduction (n = 3 independent experiments). d, Body weight trajectories of NOD (PBS; blue dots/line), NOD (eGFP; green dots/line) and NOD (Mettl3; red dots/line) (P = 0.0215, 17 weeks of age; P = 0.0034, 25 weeks of age, NOD-PBS versus NOD-Mettl3). e, Blood glucose trajectories of NOD (PBS; blue dots/line), NOD (eGFP; green dots/line) and NOD (Mettl3; red dots/line) (P = 0.0239, 21 weeks of age; P = 0.0028, 25 weeks of age, NOD-PBS versus NOD-Mettl3). f, Percentage diabetes-free NOD (PBS), NOD (eGFP) and NOD (Mettl3) at the end of the 25 weeks of age. g, Representative haematoxylin and eosin (H&E) staining showing immune cell infiltration in salivary glands in NOD (PBS), NOD (eGFP) or NOD (Mettl3) (scale bar, 200 μm) (n = 5 mice per group). h, Representative H&E staining showing immune cell infiltration in pancreatic islets in NOD (PBS), NOD (eGFP) or NOD (Mettl3) (scale bar, 200 μm) (n = 5 mice per group). i, Quantification of insulitis score of pancreatic sections from h (n = 5 per group). j, Representative immunofluorescence images showing insulin (red) and DAPI (blue) in pancreatic sections from NOD (PBS), NOD (eGFP) or NOD (Mettl3) (scale bar, 200 μm). k, β-cell mass estimations of NOD (PBS), NOD (eGFP) and NOD (Mettl3) at 25 weeks of age (n = 5 mice per group). l, Serum C-peptide levels in NOD (PBS), NOD (eGFP) and NOD (Mettl3) at 25 weeks of age (n = 5 mice per group). m, Schematic representation of the co-culture experimental plan. BG, blood glucose. n, Quantitative reverse transcription PCR analyses of Oas genes in NSG islets transduced with eGFP and co-cultured with PBS (blue bars) or NOD diabetogenic splenocytes (green bars), or NSG islets transduced with AAV8 overexpressing Mettl3 and co-cultured with NOD diabetogenic splenocytes (red bars) (n = 3 per group; islets from 3 pools of 5 mice each pool). All samples in each panel are biologically independent. Data are expressed as mean ± standard error of the mean. Statistical analysis was performed by mixed-effects analysis with Dunnet’s multiple comparison test in d and Šídák’s multiple comparison test in e; log-rank (Mantel–Cox) test in f; two-way analysis of variance with Holm–Šídák’s multiple comparisons test in k, l and n. Source numerical data and unprocessed gel images are available in Source data.
NOD mice present immune cell infiltration in salivary glands as well as pancreatic islets during progression of T1D51. All groups presented salivary gland infiltration (Fig. 7g), but only the AAV8-Mettl3-treated mice showed decreased immune infiltration in pancreatic islets (Fig. 7h) and consequently decreased insulitis scores compared with AAV8-eGFP- or PBS-treated groups (Fig. 7i). Furthermore, an extensive immune cell profiling analysis of pancreatic lymph nodes (Extended Data Fig. 8a–n) revealed a decrease in the populations of CD4, CD8 and Th1 cells in AAV8-Mettl3-treated mice compared with AAV8-eGFP. Conversely, no notable changes in immune cell subpopulations were detected in splenocytes (Extended Data Fig. 9). These findings were further corroborated by an increase in β-cell mass observed in AAV8-Mettl3-treated mice (Fig. 5j,k), along with elevated serum C-peptide levels compared with mice treated with AAV8-eGFP or PBS (Fig. 6l). Collectively, these results demonstrate that the prolonged upregulation of Mettl3 protects β-cells and effectively delays the onset of T1D in the NOD mouse model.
Mettl3 overexpression hampers Oas immune response in NOD islets
To confirm the hypothesis that Mettl3 overexpression in mouse β-cells limits Oas upregulation in response to a T1D immune insult, we employed co-culture experiments (Fig. 7m). For this, we first transduced islets from 12-week-old female immunodeficient NOD SCID gamma (NSG) mice with AAV8-eGFP or AAV8-Mettl3, and then co-cultured them with either PBS or diabetogenic splenocytes from 12-week-old diabetic NOD females (Fig. 7m).
Co-culture of islets from AAV8-eGFP-transduced NSG mice with diabetogenic splenocytes increased Mettl3 expression compared with AAV8-eGFP islets treated with PBS (Fig. 7n). In addition, AAV8-Mettl3 islets presented a greater Mettl3 upregulation compared with AAV-eGFP islets when also challenged with splenocytes (Fig. 7n). Mouse Oas1 exhibits eight paralogue genes that have been described to differ in their antiviral activity52. Co-culture of diabetogenic splenocytes with islets transduced with AAV8-eGFP induced the upregulation of Oas1a, Oas1c, Oas1g, Oas2and Oas3 compared with islets transduced with AAV8-eGFP that were challenged with PBS (Fig. 7n). On the other hand, overexpression of Mettl3 (AAV8-Mettl3) in β-cells before co-culture with diabetogenic splenocytes blunted the upregulation of Oas genes compared with β-cells transduced with AAV8-eGFP and co-cultured with diabetogenic splenocytes (Fig. 7n). These data provide strong evidence for the existence of a conserved METTL3 regulation of the OAS innate immune response in both mouse and human β-cells.
ROS-induced oxidative stress and ER stress constrain METTL3 upregulation
To examine the mechanisms involved in the dynamic regulation of METTL3, we considered recent reports describing the involvement of mitochondrial dysfunction, reactive oxygen species (ROS) and ER stress as contributors to the development of T1D53–55.
We began by challenging human islets with treatment of PBS, ROS (hydrogen peroxide alone) or H2O2 in combination with a ROS scavenger (N-acetyl cysteine, ‘NAC’). METTL3 protein levels were significantly downregulated by H2O2, and this downregulation was rescued in the presence of NAC (Fig. 8a,b).
Fig. 8 |. OAS mRNA stability is regulated by the SNO of METTL3 in human β-cells.
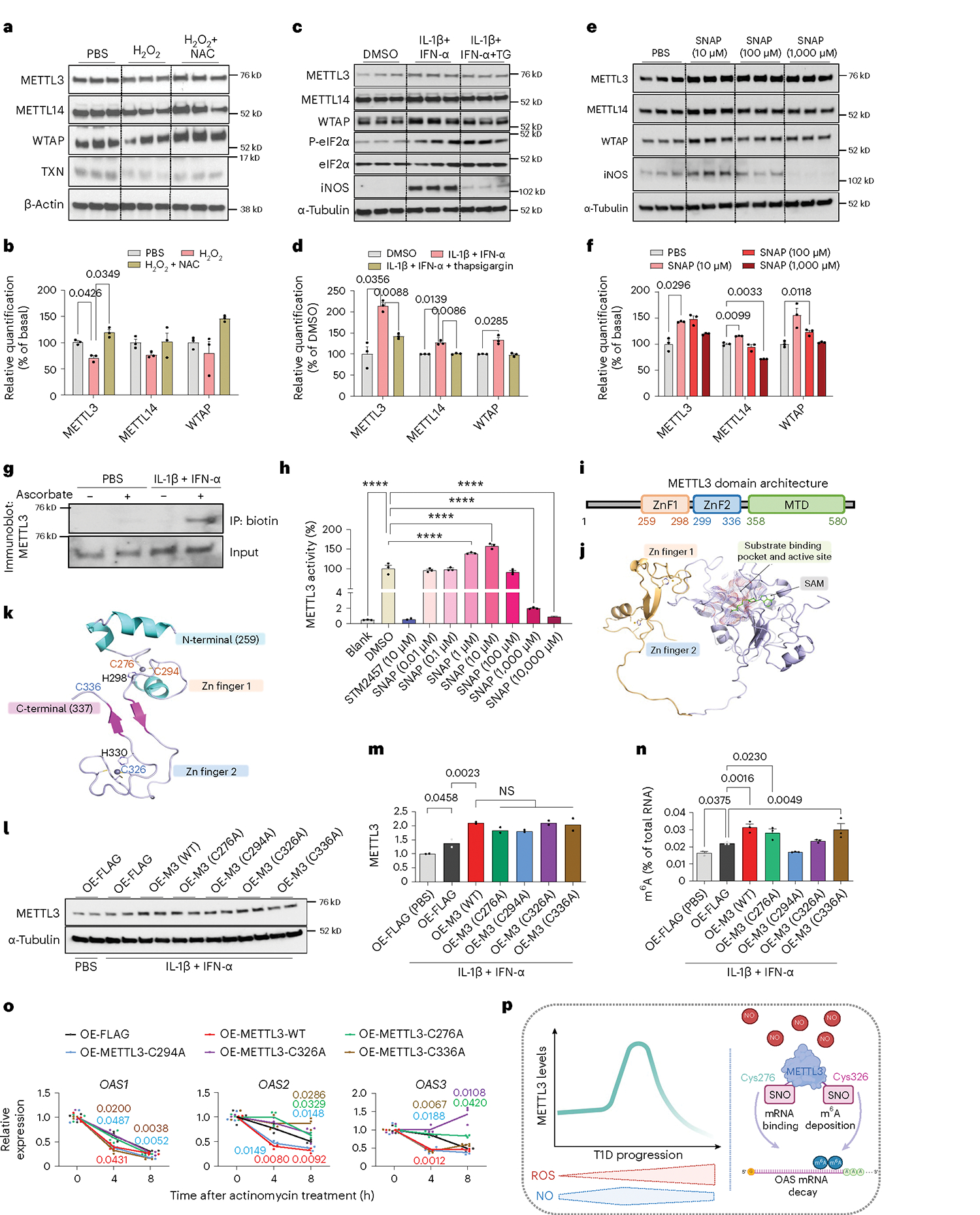
a, Western-blot analyses of indicated proteins in human islet cells treated with H2O2 or H2O2 plus NAC for 24 h (n = 3 independent biological samples per group). b, Protein quantification of indicated proteins related to a. c, Western-blot analyses of indicated proteins in human islets treated with DMSO, IL-1β plus IFN-α pre-treated with thapsigargin plus IL-1β and IFN-α for a total of 24 h (n = 3 independent biological samples per group). d, Protein quantification of indicated proteins related to c. e, Western-blot analyses of indicated proteins in human islets treated with PBS or represented doses of SNAP for 24 h (n = 3 independent biological samples per group). f, Protein quantification of indicated proteins related to e. g, Western-blot analyses of biotin-switch assay on METTL3 in EndoC-βH1 cells treated with PBS or IL-1β plus IFN-α (n = 3 independent experiments per group). h, METTL3:METTL14 complex methyltransferase activity with DMSO, STM2457 (a METTL3 inhibitor) or different concentrations of SNAP (n = 3 independent experiments per group; n = 2 independent experiments/STM2457). **** P < 0.001. i,j, METTL3 protein domains representing zinc finger domains (ZnF) and MTD. k, Structure of METTL3 zinc finger domains depicting the identified cysteines sensitive to SNO. l, Western-blot analyses of METTL3 in EndoC-βH1 cells (n = 2 independent experiments per group). m, Protein quantification of METTL3 related to l. n, m6A levels measured by a colourimetric ELISA kit of total RNA isolated from EndoC-βH1 cells overexpressing the represented plasmids and treated with PBS or IL-1β plus IFN-α for 48 h (n = 3 independent experiments per group). NS, not significant. o, Quantitative reverse transcription PCR analyses of OAS genes after IL-1β plus IFN-α stimulation in EndoC-βH1 overexpressing the represented plasmids after a time-course treatment with actinomycin D (ActD) (n = 3 independent experiments per group). p, Model depicting the role of SNO in controlling METTL3 function and OAS mRNA decay. All samples in each panel are biologically independent. Data are expressed as mean ± standard error of the mean. Statistical analysis was performed by two-way analysis of variance (ANOVA) with Fisher’s least significant difference test or one-way with ANOVA Holm–Šídák’s test in h and n. Source numerical data and unprocessed gel images are available in Source data.
We then examined the impact of ER stress on METTL3 regulation. First, we treated human islets with thapsigargin or tunicamycin (Extended Data Fig. 10a). ER stress alone had a minor impact on m6A writers (Extended Data Fig. 10b). To analyse the impact of ER stress in the context of T1D, we incubated human islets with thapsigargin before treatment with cytokines (Fig. 8c). Cytokine treatment led to the upregulation of all three components of the m6A writer complex (Fig. 8c,d). However, induction of ER stress by prior incubation with thapsigargin blunted upregulation of the m6A writers (Fig. 8c,d).
To study whether the downregulation of METTL3 was mediated by proteasomal degradation, we exposed EndoC-βH1 cells to H2O2, either alone or in conjunction with MG132 (Extended Data Fig. 10c). As anticipated, H2O2 downregulated METTL3, a response that was effectively reversed with MG132 (Extended Data Fig. 10d).
Next, we considered the mechanism(s) for the converse, that is, METTL3 upregulation during the early stages of T1D. In this context, it is notable that, while physiological levels of nitric oxide (NO) are protective, the persistent accumulation of ROS and NO to pathological levels may trigger β-cell apoptosis53. To examine the involvement of NO, we challenged human islets with different concentrations of an NO donor (S-nitroso-N-acetyl-dl-penicillamine, ‘SNAP’). Physiological levels of SNAP increased METTL3 and iNOS protein abundance, while this response was lost in islets treated with high doses of SNAP (Fig. 8e,f). Altogether, these data support the concept that at early stages of T1D an initial increase in NO induces upregulation of METTL3. However, as the disease progresses, the accumulation of NO and ROS in β-cells coupled with exacerbation of ER stress downregulates METTL3 and activates a persistent OAS response.
Regulation of OAS mRNA decay in β-cells is dependent on SNO of METTL3
To define the mechanism(s) involved in the regulation of METTL3 by NO, we first confirmed SNO of METTL3 in EndoC-βH1 cells (Fig. 8g). Next, we assessed METTL3 enzymatic activity. While low levels of NO increased METTL3 activity, this effect was reversed with high levels of NO (Fig. 8h). To identify the covalently modified Cys residues that are susceptible to SNO, we optimized a mass spectrometry-based protocol on human recombinant METTL3 protein treated with SNAP (Extended Data Fig. 10e). These analyses identified the SNO of four METTL3 Cys residues (Cys276, Cys294, Cys326 and Cys336) (Extended Data Fig. 10f). The domain architecture of human METTTL3 is characterized by the existence of two zinc finger domains (CCCH) and a methyltransferase domain (MTD)21 (Fig. 8i,j and Extended Data Fig. 10g). Interestingly, the METTL3 Cys residues sensitive to SNO were evolutionarily conserved (Extended Data Fig. 10h) and located in the zinc fingers (Fig. 8k).
We next tested the hypothesis that SNO of the identified cysteine residues were fundamental for the METTL3 regulation by performing site-directed mutagenesis and overexpressed either wild-type (WT) METTL3 constructs or mutant Cys METTL3 constructs in EndoC-βH1 cells. Cells overexpressing a FLAG empty plasmid (OE-FLAG) treated with cytokines presented upregulation of METTL3 compared with OE-FLAG cells treated with PBS (Fig. 8l,m). Overexpression of WT METTL3 in EndoC-βH1 cells was successful and importantly, all Cys mutants upregulated METTL3 similarly to WT METTL3 compared with a FLAG empty plasmid (Fig. 8l,m). However, while overexpression of WT METTL3 in EndoC-βH1 cells increased m6A levels in total RNA, this increase was blocked by mutating Cys294 and Cys326 (Fig. 8n). These data show that SNO at Cys294 and Cys326 residues in METTL3 is important for m6A deposition.
Finally, to determine which of the four cysteines directly influences METTL3 control OAS mRNA decay, we overexpressed FLAG, WT METTL3 or Cys mutant constructs independently in EndoC-βH1 cells. METTL3 overexpression accelerated the mRNA decay of OAS1, OAS2 and OAS3 (Fig. 8o). Overall, mutant C294A overexpression cells behaved similarly to WT METTL3 (Fig. 8o), suggesting that SNO at this Cys residue does not control the mRNA decay of OAS despite impacting global m6A deposition. Overexpression of C336A accelerated the mRNA decay of OAS1 and OAS3 similarly to WT METTL3. However, SNO at this specific Cys was necessary for the mRNA decay of OAS2 (Fig. 8o). On the other hand, overexpression of the mutants C276A and C336A behaved similarly to FLAG overexpressing cells and did not accelerate OAS mRNA decay as seen in the WT METTL3-overexpressing cells (Fig. 8o). Together, these data suggest that SNO regulates METTL3 and OAS mRNA decay by two different mechanisms: while SNO at Cys326 impacts METTL3 global m6A deposition capacity, Cys276 is probably important for the OAS RNA binding to METTL3 and does not disturb METTL3 m6A deposition in total RNA (Fig. 8p).
Discussion
There is growing evidence for a central role of the β-cell in triggering autoimmunity in T1D56. Here we report that METTL3 is dynamically regulated and that m6A mRNA methylation provides key negative feedback on the antiviral innate immune response at the onset of T1D, preventing excessive and deleterious local inflammation. This protective effect is, however, dependent on the β-cell redox state, and we describe SNO as a post-translational regulatory mechanism for METTL3 function and OAS mRNA decay.
We present several lines of evidence to validate the ability of METTL3 to control the mRNA decay of OAS genes via the m6A pathway. First, silencing METTL3 increased OAS protein levels. Second, EndoC-βH1 cells deficient in METTL3 exhibited increased stability of OAS mRNA, while overexpression of METTL3 accelerated OAS mRNA decay. Third, we demonstrated that the mRNA decay of OAS genes is mediated by the m6A readers YTHDF1 and YTHDF3. Finally, we report that the β-cell overexpression of Mettl3 in mice ex vivo dampened Oas upregulation in response to diabetogenic splenocytes. Although individual YTHDF proteins exhibited different behaviours20, it is worth mentioning that YTHDF2 directly participates in regulating mRNA decay57. Notably, we observed that YTHDF2 KD did not impact Oas mRNA decay in β-cells. This observation may be explained by the possibility that YTHDF2 is not a critical m6A reader in β-cells. Further work is necessary to examine the β cell-specific role of YTHDF2.
In the context of the pathophysiology of T1D, repeated interactions with viruses may have resulted in the development of temporal safeguard mechanisms in host cells. For example, there may be development of activation thresholds for innate immune sensors before they can trigger elimination and/or prevent immuno-stimulatory endogenous nucleic acids. We report m6A as a safeguard mechanism to control the innate antiviral immune response, thus establishing a link between antiviral innate immunity and T1D. Recent reports support this concept. For example, Gao and colleagues show that loss of Mettl3 in the foetal liver promotes the formation of deleterious dsRNAs and activation of innate immune pathways in the absence of a viral infection26. On the other hand, deletion of METTL3 (ref. 24) or METTL14 (ref. 58) in fibroblasts increases the mRNA stability of IFN-β upon viral infection. Further studies are warranted to elucidate if m6A levels impact the β-cell susceptibility to viral infection and/or if downregulation of m6A levels in β-cells can trigger the formation of aberrant endogenous immune-stimulatory dsRNAs.
The m6A landscape of β-cells at T1D onset seemed to be distinct from that observed in established T1D. This argues for distinct features between the transcriptome and m6A landscape of β-cells at T1D onset—such as that observed in human islets and EndoC-βH1 cells treated with cytokines—compared with β-cells from patients with established T1D. This suggests that, as T1D progresses, m6A decorates and controls genes important for β-cell function and identity. We have demonstrated in previous studies that silencing METTL3 or METTL14 in β cells leads to cell-cycle arrest and apoptosis29. Therefore, it is reasonable to hypothesize that the downregulation of METTL3 during T1D progression accelerates the decline in β-cell mass, while its overexpression potentially limits this decline. Additional experiments are needed to show the direct link between METLL3 overexpression and β-cell mass preservation. We utilized input RNA-seq data to normalize m6A-seq in established T1D. Furthermore, we intersected the m6A-seq data in established T1D with a published resource on scRNA-seq in T1D β-cells to control for potential alterations in gene expression due to islet cell composition changes. Nevertheless, additional experiments are necessary to inform that changes in m6A are specific to β-cells in established T1D. Overall, these results point to m6A as a mechanism involved in the loss of β-cell identity and functional mass in the latter stages of T1D.
To begin to dissect the mechanism(s) involved in the dynamic regulation of METTL3 during T1D progression, we considered recent findings reporting increased accumulation of intracellular ROS before the development of T1D53,59 and the upregulation of ER-stress markers in human T1D β-cells60,61. We propose that during the early stages of T1D when the initial immune attack and cytokine release occurs, the rising levels of NO are within the physiological range and promote upregulation of METTL3. However, as the disease advances with progressive accumulation of NO and ROS, the heightened redox sensitivity of METTL3 triggers its downregulation and decreased enzymatic activity.
The upregulation of OAS genes at the onset of T1D despite an increase in METTL3 could be due to stressed β-cells failing to increase expression of the m6A writer in a timely manner to counterbalance the rapid rise of OAS. The fact that human islets pre-exposed to ER stress by thapsigargin treatment do not exhibit upregulation of METTL3 similar to control islets supports this contention. Furthermore, boosting Mettl3 overexpression in β-cells in vitro co-culture experiments was sufficient to limit the upregulation of Oas in islets from immunodeficient NSG mice in response to diabetogenic splenocytes.
FInally, other mechanisms could regulate the METTL3 m6A deposition independently of its total protein levels. For example, SUMOylation of METTL3 does not alter its stability, localization, or interaction with other m6A writers, but significantly represses its m6A methyltransferase activity22. SNO has been reported to be essential for diverse aspects of β-cell function62. We identified the four cysteines that are modified by SNO to be in the zinc finger domains of METTL3. This gains significance considering SNO of zinc fingers disrupts their structures and modulates enzymatic activity impacting the binding capacities of RNA-binding proteins63,64. A recent study has also reported that cysteines 294 and 326 in the zinc finger domains of METTL3 are essential for its enzymatic activity65. We observed that the SNO of the METTL3 Cys276 and Cys326 is needed for the mRNA decay of OAS in response to cytokines. Interestingly, while the mutation of Cys326 impacted m6A deposition, mutation of Cys276 did not alter m6A levels. This suggests that, while SNO of Cys326 controls METTL3 enzymatic activity, SNO of Cys276 might regulate METTL3 structural binding to OAS mRNA. Overall, these results demonstrate that redox signalling controls METTL3 and OAS mRNA stability in β-cells at T1D onset.
In conclusion, we provide evidence that m6A methylation acts as a β-cell protective mechanism that controls the OAS innate immune response at the onset of T1D in mice and humans (Extended Data Fig. 10i). Our data suggest that increased m6A promotes accelerated mRNA decay of OAS genes in β-cells. Importantly, we observed that SNO represents a previously unidentified mechanism with the capacity to modulate METTL3 protein function and the potential mRNA binding affinity to OAS mRNA. Based on these results, we propose that therapeutic targeting of METTL3 before seroconversion or at T1D onset has the potential to promote β-cell survival and improve secretory function during disease progression.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41556-024-01368-0.
Methods
Study approval
All animal experiments were conducted following the Association for Assessment and Accreditation of Laboratory Animal Care. All protocols were approved by the Institutional Animal Care and Use Committee of the Joslin Diabetes Center following National Institutes of Health (NIH) guidelines. All human studies and protocols used were approved by the Joslin Diabetes Center’s Committee on Human Studies (CHS#5-05). Formal consent from human islet donors was not required because samples were discarded islets from de-identified humans.
Human islet isolation and processing
Human islets were obtained from the Integrated Islet Distribution Program, Prodo Laboratories, ADI isletcore, and provided by Alvin C. Powers MD (Vanderbilt University). Freshly isolated islets were cultured overnight (16 h) in Miami Media #1A (Cellgro) upon arrival. Islets were then handpicked and seeded on ultra low-attachment six-well plates (Corning) (200 islets per well) for experiments. Snap-frozen control and T1D islets from ADI isletcore were immediately lysed in TRIzol (ThermoFisher) upon arrival and stored at −80 °C for RNA isolation.
EndoC-βH1 cell culture
The EndoC-βH1 cell line (EndoC-βH1, Human Cell Design) was cultured and passaged as previously described66. Briefly, culture plates were coated with Dulbecco’s modified Eagle medium (DMEM, glucose 4.5gl−1; Gibco) containing penicillin and streptomycin (PS) (1%; Gibco), fibronectin (2 μgml−1; Sigma), and matrigel (1% vol/vol; Sigma) and incubated for at least 1 h in 5% CO2 at 37 °C before the cells were seeded. EndoC-βH1 cells were grown on Matrigel/fibronectin-coated (Sigma) culture plates containing DMEM (glucose 1gl−1; Gibco), bovine serum albumin (BSA) fraction V (2% wt/vol; Roche Diagnostics), 2-mercaptoethanol (50 μM; Gibco), nicotinamide (10mM; Sigma), transferrin (5.5 μgml−1; Sigma), sodium selenite (6.7ngml−1; Sigma) and PS (1%; Gibco)66.
266-6 cell culture
Briefly, 10-cm culture plates were coated with DMEM (glucose 4.5gl−1; Gibco) containing PS (1%; Gibco), fibronectin (2 μgml−1; Sigma) and matrigel (1% vol/vol; Sigma) and incubated for at least 1 h in 5% CO2 at 37 °C before the cells were seeded. The 266-6 cells (CRL-2151, ATCC) were grown in 10-cm culture plates containing DMEM (glucose 4.5gl−1; Gibco), foetal bovine serum (FBS, 10%; Gibco) and PS (1%; Gibco).
PANC-1 cell culture
The PANC-1 cells (CRL-1469, ATCC) were grown in 10-cm culture plates containing DMEM (glucose 4.5gl−1; Gibco), FBS (10%; Gibco) and PS (1%; Gibco).
αTC-6 cell culture
The αTC-6 cells (CRL-2934, ATCC) were grown in 10-cm culture plates containing DMEM (glucose 1gl−1; Gibco), FBS (15%; Gibco), BSA fraction V (0.1% wt/vol; Roche Diagnostics), non-essential amino acids (1%; Gibco) and PS (1%; Gibco).
Human islets and cell treatments
Cytokines treatments.
Overnight cultured human islets or EndoC-βH1 cells were challenged with vehicle (PBS), IL-1β (50Uml−1; R&D Systems), IFN-α (2,000Uml−1; PBL Assay Science), IFN-γ (1,000Uml−1; Peprotech) or a combination of IL-1β+IFN-α, or IL-1β+IFN-γ in respective culture media. After treatments, islets were then handpicked, washed twice with ice-cold Dulbecco’s phosphate-buffered saline (DPBS; Gibco) by self-sedimentation, and immediately lysed in TRIzol for RNA isolation, RIPA buffer (ThermoFisher) for protein isolation, or fixed and embedded in agar for immunofluorescence staining as previously described67.
Thapsigargin treatments.
Overnight cultured human islets were treated with thapsigargin (1 μM; Selleckchem) or vehicle (DMSO) in Miami Media #1A for 16 h and collected for protein isolation.
H2O2 and NAC treatments.
Overnight cultured human islets were treated with 25 μM of H2O2 (MiliporeSigma), or H2O2 plus 1mM of NAC (Cayman Chemical), or vehicle (PBS) in Miami Media #1A for 24 h and collected for protein isolation.
SNAP treatments.
Overnight cultured human islets were treated with 10, 100 or 1,000nM of SNAP (Cayman Chemical), or vehicle (DMSO) in Miami Media #1A for 24 h and collected for protein isolation.
Actinomycin D treatments.
EndoC-βH1 cells were cultured as described above, and at 48 h post-seeding/KD or overexpression were challenged with IL-1β+IFN-α as described above. At 72h post-seeding/KD or overexpression, cells were treated with 10 μgμl−1 actinomycin D (ThermoFisher) or DMSO for 0, 4 or 8h.
Cyclohexamide treatments.
EndoC-βH1 cells were cultured as described above, and at 48 h post-seeding were challenged with IL-1β+IFN-α or PBS as described above for 24 h. Cells were treated with 10 μM of cyclohexamide (Cell Signaling) or DMSO for 4 h after 20 h of culture with cytokines and collected for protein isolation.
MG132 treatments.
EndoC-βH1 cells were cultured as described above, and at 48 h post-seeding were challenged with IL-1β+IFN-α or PBS as described above for 24 h. Cells were treated with 5 μM of MG132 (Cell Signaling) or DMSO for 4 h after 20 h of culture with cytokines and collected for protein isolation.
Transfections
KD experiments.
Reverse transfections were performed as previously described29. Briefly, EndoC-βH1 cells or dispersed human islet cells were mixed with Lipofectamine RNAiMAX Reagent (Life Technologies) and small interfering RNA (siRNA) complexes (Dharmacon) at a final concentration of 15nmoll−1 siRNA according to manufacturer instructions. EndoC-βH1 cells were seeded at a density of 6 × 104 cells cm−2 in Matrigel/fibronectin-coated (MiliporeSigma) culture plates. Human dispersed islets were seeded at a density of 5 × 104 cells cm−2 on ultra low-attachment plates (ThermoFIsher) and allowed to form spontaneous pseudoislets. EndoC-βH1 cells and human pseudoislets were collected 96h post-transfection. siRNAs included ON-TARGETplus Non-Targeting Control Pool D-001810-10-05, ON-TARGETplus METTL3 siRNA L-005170-02-0005, ON-TARGETplus Human YTHDF1 siRNA L-018095-02-0005, ON-TARGETplus Human YTHDF2 siRNA L-021009-02-0005, ON-TARGETplus Human YTHDF3 siRNA L-017080-01-0005 (Dharmacon).
Overexpression experiments.
EndoC-βH1 cells (1 × 106 cells) were seeded in Matrigel/fibronectin-coated (MiliporeSigma) six-well culture plates. After 24 h, medium was changed and cells were forward-transfected with 2 μg of plasmid. c-FLAG pcDNA3 (Addgene #20011) and pcDNA3/Flag-METTL3 (Addgene #53739) were obtained from Addgene. FLAG-tagged OAS1 (NM_016816.4), OAS2 (NM_002535.3) and OAS3 (NM_006187.4) plasmids were generated by VectorBuilder. Transfections were performed using Lipofectamine 3000 (Invitrogen) and Opti-MEM (Invitrogen) according to manufacturer protocols. Medium was exchanged after 16 h of transfection, and at 48 h cells were further used for experiments including cytokine treatments and actinomycin D/MG132 treatments.
Mouse studies
Female NOD/shiLtJ (‘NOD’; Jackson Laboratories #001976), NOR/LtJ (‘NOR’; Jackson Laboratories #002050) and NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (‘NOD NSG’; Jackson Laboratories #005557) mice were used. Mice were housed on a 12-h light/12-h dark cycle with water and food ad libitum. Mice were weaned and maintained on a chow diet (PicoLab mouse diet 20-5058). Female mice were used for all experiments throughout the study as male NOD mice do not develop T1D as consistently or within the same timeframe. Body weight and blood glucose were measured weekly for follow-up studies, and mice were considered diabetic when two consecutive measurements of blood glucose exceeded 250mg dl−1. Serum C-peptide levels were measured using enzyme-linked immunosorbent assay (ELISA) kits (Crystal Chem) according to manufacturer guidelines. All mice were kept in a specific pathogen-free facility in the Animal Facility at Joslin Diabetes Center, and animal protocols were approved by the Institutional Animal Care and Use Committee. Sample sizes for animal experiments were chosen on the basis of experience in previous in-house studies of metabolic phenotypes and to balance the ability to detect significant differences with minimization of the number of animals used following NIH guidelines.
Mouse islet isolations.
Islets were isolated from female NSG mice as previously described68. In brief, 3-month-old mice were anaesthetized and their pancreas was infused with liberase (Roche). Following incubation at 37 °C for 17 min the digested pancreases were washed, filtered through a 400-μm filter and run on a Histopaque (Sigma) gradient. The purified islets were handpicked, counted and cultured overnight in 7mM glucose RPMI medium (containing 10% FBS and 1% PS; Gibco).
β-cell sorting by fluorescence-activated cell sorting.
Overnight cultured mouse islets were dispersed with a solution of 1mgml−1 trypsin and 30 μgml−1 DNase followed by incubation for 15 min at 37 °C. During the digestion, the islets were vortexed every 5 min for 10s. Cold medium including serum was added to stop the digestion, and the cells were washed two times in DPBS containing 1% fatty-acid-free BSA. Before sorting islet cells were filtered through a 35-μm filter and sorted using MoFlo Cytometer (Dako), where cells were gated according to forward scatter and then sorted on the basis of endogenous fluorescence32 and CD45 staining (BioLegend # QA17A26).
Co-culture of splenocytes and islets.
Total splenocytes were purified as previously described36. Briefly, freshly collected spleens of 12–13-week-old female NOD mice with early diabetes were filtered through a nylon mesh by followed lysis of the red blood cells with ACK Lysing buffer (Lonza). After starvation, 100 size-matched islets from NOD NSG mice were co-cultured with NOD splenocytes in 5 mmoll−1 glucose RPMI at a ratio of 1:10 as previously described69. At 48 h islets were handpicked, washed in ice-cold DPBS and lysed in TRIzol for RNA isolation.
NOD immune cell profiling by fluorescence-activated cell sorting.
Splenocytes and pancreatic lymph nodes were collected and stained with a viability dye Fvd (APC-Cy7, #6508614, eBioscience). Subsequently, various antibodies were applied at a 1:300 dilution, as follows: CD4 (Alexa Fluor-700, #2081383, eBioscience), CD8a (PerCP-Cy5.5, #2151510, eBioscience), CD25 (Pacific Blue, #102022, BioLegend), CD69 (PE-Cy5, #104510, BioLegend), CCR6 (PE-Cy7, #129816, BioLegend), CD19 (BV785, #115543, BioLegend), CD11C (BV711, #117349, BioLegend), CD27 (BV650, #124233, BioLegend), CD103 (BV605, #121433, BioLegend), CD86 (PE/Dazzle594, #105042, BioLegend), RorT (APC, #2193857, eBioscience), FOXP3 (FITC, #2290357, eBioscience) and Tbet (PE, #644810, BioLegend). Isotype controls were employed to mitigate the effects of nonspecific binding and to ensure proper gating. All incubations were conducted on ice and protected from light. A minimum of 100,000 cells were counted using a Fortessa flow cytometer (BD Biosciences) and subsequently analysed using FlowJo software.
In vivo Mettl3 overexpression.
AAV8 overexpressing Mettl3 (NM_019721.2) or eGFP under the control of rat insulin II promoter (Addgene #15029) with a WPRE element were synthesized by VectorBuilder. Briefly, 4-week-old NOD female mice received an intraperitoneal injection of 200 μl PBS+0.01% Pluronic F-68 (Sigma) containing 1 × 1011 gene copies per mouse of AAV8 overexpressing eGFP or Mettl3 and were followed for 20 weeks.
RNA isolation and reverse transcription PCRs
Total RNA was isolated as previously described70. In brief, high-quality total RNA (>200nt) was extracted using standard TRIzol reagent (Invitrogen) according to manufacturer instructions and the resultant aqueous phase was mixed (1:1) with 70% RNA-free ethanol and added to Qiagen RNeasy mini kit columns (Qiagen) and the kit protocol was followed. RNA quality and quantity were analysed using Nanodrop 1000 and used for reverse transcription using the high-capacity cDNA synthesis kit (Applied Biosciences). Complementary DNA was analysed using the ABI 7900HT system (Applied Biosciences), and gene expression was calculated using the ΔΔCt method. Data were normalized to GADPH.
m6A-IP–qPCR
EndoC-βH1 cells were grown as described above in 15-cm Matrigel/fibronectin-coated plates and treated with IL-1β+IFN-α or PBS for 48 h. Total RNA was isolated as described above, and 50 μg was fractionated using NEBNext Magnesium RNA Fragmentation Module (NEB, #E6150S). Eighteen microlitres of total RNA were used for m6A-IP using the EpiMark N6-methyladenosine enrichment kit (NEB, #E1610S) according to the kit protocol, and 2 μl was kept as input. The resulting IP and input RNA were cleaned up using the Monarch RNA Cleanup Kit (NEB #T2030), and 200ng was used for reverse transcription using the high-capacity cDNA synthesis kit (Applied Biosciences). cDNA was analysed using the ABI 7900HT system (Applied Biosciences), and gene expression was calculated using the ΔΔCt method.
Protein isolation and western blotting
Total proteins were collected from cell lines using M-PER (Thermo Fisher), and tissue (for example, islets) using RIPA protein extraction reagent (Thermo Fisher) supplemented with proteinase and phosphatase inhibitors (Sigma) according to standard protocol. Protein concentrations were determined using the BCA standard protocol followed by the standard western immunoblotting protocol of proteins. Primary antibodies used for western blotting with a 1:1,000 dilution included METTL3 (#195352, Abcam), METTL14 (#HPA038002, Sigma), WTAP (#60188-1-Ig, Proteintech), OAS1 (#14955-1-AP, Proteintech), OAS2 (#19279-1-AP, Proteintech), OAS3 (#21915-1-AP, Proteintech), RNASEL (#22577-1-AP, Proteintech), ADAR1 (#14175, Cell Signaling), phospho-STAT1 (#9167, Cell Signaling), STAT3 (#12640, Cell Signaling), iNOS (#ab178945, Abcam), phospho-eIF2a (#3398, Cell Signaling), eIF2a (#5324, Cell Signaling), Thioredoxin 1 (#2429, Cell Signaling), β-actin (#4970, Cell Signaling), α-tubulin (#7291, Abcam) and GAPDH (#5174, Cell Signaling). The blots were developed using chemiluminescent substrate ECL (ThermoFisher) and quantified using Image studio Lite Ver. 5.2 software (LICOR).
Pancreas immunostaining and analyses
Mouse pancreas was collected and fixed in 4% formaldehyde at 4 °C overnight, followed by paraffin embedding. Five-micrometre-thick slides were cut and subjected to immunostaining. Slides were heated in 10mM sodium citrate, followed by blocking with donkey serum, and incubated with various primary antibodies: proinsulin (DSHB, #GS-9A8, dilution 1:2,500), insulin (Abcam, #ab7842, dilution 1:500), glucagon (Sigma, #G2654, dilution 1:8,000), somatostatin (Abcam, #ab30788, dilution 1:10,000) and METTL3 (Abcam, #195352, dilution 1:5,000). Specific signals were detected by using fluorescence-conjugated secondary antibodies (Jackson Immunoresearch, Alexa 488, Alexa 594 and AMCA). Images were captured using Zeiss AXIO Imager A2 upright fluorescence microscope. Insulitis was evaluated as reported previously36. Quantification of β-cell mass was performed as previously described67.
Measuring total m6A levels
Total m6A levels were measured by employing LC–MS/MS or a quantitative colourimetric ELISA.
LC–MS/MS quantification of m6A.
Total m6A levels among all adenosines were measured by triple-quad LC–MS/MS. We first purified mRNA from human islet’s total RNA by two rounds of polyA selection using polyA beads. Fifty nanograms of purified mRNA was subject to digestion by 1U of nuclease P1 (Sigma #N8630-1VL) in 25 μl of buffer containing 20mM of NH4Ac at 42 °C for 2h followed by phosphatase treatment using 1 μl of FastAP Thermosensitive Alkaline Phosphatase (ThermoFisher #EF0651) at 37 °C for 1 h. The digested nucleotides were filtered by a 0.22-μm syringe filter (Millipore) and then analysed by a C18 reverse phase column on HPLC (Agilent) followed by triple-quad MS/MS quantification (Sciex). The concentration of each type of nucleotide was calibrated by standard curves measured from pure nucleoside standards in each experiment. The m6A/A ratio was computed using the estimated m6A and A concentrations.
Colorimetric quantification of m6A.
EpiQuik m6A RNA Methylation Quantification Kit (EpigenTek) was used to measure the percentage of m6A methylation level in total RNA. EndoC-βH1 cells harbouring WT or mutant METTL3 overexpression were used according to the protocols of the manufacturer using the kit provided negative control, and positive control, and our samples consisting of 200 μg of total RNA from EndoC-βH1 cells. The m6A percentage in total RNA was calculated using the following formula: m6A%=(sample OD−NC OD)/S)/(PC OD−NC OD)/p) × 100%, where NC is the negative control, PC is the positive control, S is the amount of input sample RNA and p is the amount of input positive control. Equal amounts of RNA samples were used.
m6A immunoprecipitation and sequencing
For patient islet samples, polyA-selected mRNA was adjusted to 15ng μl−1 in 100 μl and fragmented using a Bioruptor ultrasonicator (Diagenode) with 30s on/off for 30 cycles. m6A-IP was performed using the monoclonal m6A antibody from the EpiMark N6-methyladenosine enrichment kit (NEB, #E1610S). Input and eluted total RNA from m6A-IP were used to prepare libraries with Takara Pico-Input Strand-Specific Total RNA-seq for Illumina v2 (Takara). Sequencing was performed on Illumina Nova-seq according to the manufacturer’s instructions. Approximately 30 million paired-end 150-bp reads were generated for each sample.
Differential methylation analysis for m6A-seq
Human genome sequences and gene annotations were downloaded from the UCSC golden path version hg38. We generated genome indexes using the genomeGenerate module of the STAR aligner71 with sjdbOverhang as 52 for 53-bp reads and 59 for 60-bp reads. The reads were trimmed for adapters and poly(A/T) tails, and then filtered by sequencing Phred quality (≥Q15) using fastp72. We aligned the adapter-trimmed reads to the genome using STAR with the two-pass option and indexed the BAM files with samtools73. Using the R package RADAR74, we counted the mapped reads in 50-bp consecutive bins of each gene for each pair of input and m6A-IP samples. Counts were normalized for library size and IP counts were adjusted for expression level by the gene-level read counts of input libraries. Bins with average IP-adjusted counts lower than 10 in both CTRL and CASE groups were removed. Then bins that were not enriched in IP were also filtered out. To construct PCA plots, we used the removeBatchEffect function in the limma package75 to remove the subject effect for the human islets; the clone effect for the EndoC-βH1 cells; the batch, gender, age and body mass index effects for the T1D islet study. We performed differential methylation analysis of count data using the R package DESeq2 (ref. 76). To consider the pairing of m6A IP and input, we used the normalized, expression-level (that is, input)-adjusted, and low-read-count-filtered IP counts. Using the Wald test, we tested for significant effects of cytokine treatment or T1D on the m6A enrichment/depletion. To discover cytokine treatment effects in human islets or EndoC-βH1 cells, we performed paired tests so that each cytokine-treated sample was compared with its own paired baseline sample. To discover T1D effects in human islets using all controls, we adjusted for batch, gender, age and body mass index. We then merged the neighbouring significant bins. P values of these bins were combined by Fisher’s method77. We adjusted for multiple testing using the Benjamini–Hochberg false discovery rate (FDR) controlling procedure.
Differential expression analysis of m6A-seq input samples
The input libraries of m6A-seq are essentially mRNA-seq libraries, so we performed gene-level differential expression analysis on them. After STAR alignment, alignments were assigned to genomic features (for example, the exons for spliced RNAs) using featureCounts78. Multi-mapping reads were counted as fractions. R package DESeq2 (ref. 76) was used to test for differential expression where sequencing batch, gender and age were included as covariates. We adjusted for multiple testing using the Benjamini–Hochberg FDR procedure.
RNA-seq analyses of sorted NOD mouse β-cells
Reversely stranded 100-bp single-end reads were trimmed for adapters and filtered by sequencing Phred quality (≥Q15) using fastp72. Reads were aligned to the mouse transcriptome (Ensembl version 98) using kallisto79, and transcript counts were converted to gene counts using tximport80. To filter out low-expressing genes, we only kept genes that had counts per million (CPM) more than 1 in at least three samples. We then normalized counts by weighted trimmed mean of M-values81. To use linear models in the following analysis, we transformed counts into logCPM with Voom82. To discover the differential genes, we used the linear regression modelling R package limma75, which applied moderated t-tests to detect genes that are differentially expressed between groups. We adjusted for multiple testing using the Benjamini–Hochberg FDR procedure.
RNA-seq of EndoC-βH1 cells overexpressing OAS
Sequencing was performed by BGI. Briefly, RNA samples were denatured at an appropriate temperature to disrupt their secondary structure, and mRNA was enriched using magnetic beads attached with oligo (dT). A reaction system was configured, and RNAs were fragmented after a set duration at an optimal temperature. The first-strand synthesis reaction system was added to the fragmented mRNA to synthesize the first-strand cDNA. Next, the second-strand synthesis reaction system (including dUTP) was prepared, and the second-strand cDNA was synthesized. Following the configuration and setup of reaction systems and programs, double-stranded cDNA fragments underwent end repair, and a single ‘A’ nucleotide was added to the 3′ ends of the blunt fragments. Adaptors were then ligated to the cDNAs, with corresponding reaction systems and programs. PCR reactions were set up to amplify the cDNAs. Library quality control protocols were chosen on the basis of specific product requirements. Denaturation resulted in the production of single-stranded PCR products. The reaction system and program for circularization were subsequently configured and set up. This process generated single-stranded circularized products, while linear DNA molecules that did not cyclize were digested. Single-stranded circular DNA molecules were replicated through rolling cycle amplification, producing DNA nanoballs containing multiple copies of DNA. High-quality DNA nanoballs were loaded onto patterned nanoarrays using high-intensity DNA nanochip techniques and sequenced using the combinatorial probe-anchor synthesis method. RNA-seq analyses were performed as described above for NOD β-cells, with the difference that, to filter out low-expressing genes, we kept genes that have CPM more than 0.49 in at least four samples.
Re-analyses of scRNA-seq dataset
Data were obtained from Gene Expression Omnibus under the accession number GSE121863 (ref. 33). First, we computed some quality control metrics with R package scater83. We removed outlier cells whose library size and number of expressed genes were too low or whose proportion of counts assigned to mitochondrial genes was too high using thresholds of 2,000, 1,000 and 10%. We removed genes that have average counts of 0. We clustered similar cells together using a graph-based clustering algorithm and using genes that have average counts of more than 0.1. The algorithm normalizes the cells in each cluster using the deconvolution method84. Finally, it performs scaling to ensure that size factors of cells in different clusters are comparable. Next, we estimated the technical noise by assuming the noise follows a Poisson distribution. We used the 1,000 genes that have the largest biological variations and performed PCA. We selected the first 100 principal components for the following analysis. We constructed t-distributed stochastic neighbour embedding plots from the principal components where each point represented a cell and was coloured according to the variable diabetes. The insulin expression levels had four modes. We then used the Gaussian finite mixture model to identify cells in the third and fourth mode, that is, those with the highest insulin expression85. To perform differential gene expression in β-cells, we selected β-cells and analysed genes that are expressed in at least ten cells using limma75, which applied moderated t-test to detect genes that are differentially expressed between the established T1D and non-T1D. We adjusted for multiple testing using the Benjamini–Hochberg FDR procedure.
Pathway enrichment analysis
Pathway enrichment analyses were performed using ConsensusPathDB86 or Metascape87 using default settings. Gene Ontology terms tree were constructed using Cytoscape88. Protein–protein functional networks were constructed using STRING using default settings89.
Biotin switch assay
We used the biotin switch method that converts SNO into biotinylated groups using a detection kit (Cayman Chemical), to detect SNO of METTL3 in EndoC-βH1 cells treated with IFN-α and IL-1β according to the protocols of the manufacturer.
LC–MS/MS analysis on METTL3 SNO
Ten micrograms recombinant human METTL3 was incubated with 1mM dithiothreitol for 30 min at room temperature, and buffer was exchanged into 30 μl of 50mM HEPES (pH 7.4) containing 1mM ethylenediaminetetraacetic acid, 0.1mM neocuproine and 0.05% sodium dodecyl sulfate using 30k spin columns. METTL3 was incubated with and without 200 μM SNAP in dark at room temperature for 1 h. Excessive SNAP was removed by buffer exchange with 50mM HEPES (pH 7.4). Free thiols in all samples were blocked by 20mM N-ethylmaleimide (NEM) at room temperature for 30min. NEM was removed by washing with 50mM NH4HCO3(pH 8) containing 1mM ethylenediaminetetraacetic acid, 0.1mM neocuproine and 8M urea. SNO modifications in proteins were reduced with 20mM sodium ascorbate and alkylated by 20mM iodoacetamide at room temperature for 1 h. Proteins were then digested by trypsin (enzyme-to-protein ratio 1:20) overnight. Peptides were eluted by centrifugation in 50mM NH4HCO, and the concentration of each sample was adjusted to 0.05 μgμl−1 for LC–MS/MS. LC–MS/MS analysis was conducted using a nanoAcquity UPLC system (Waters) coupled to a Q-Exactive mass spectrometer as previously described90. MS/MS raw data were searched against the Uniprot FASTA file of Homo Sapiens using MS-GF+ algorithm. Dynamic modifications included the oxidation of methionine (15.9949Da), NEM on cysteine (125.0477Da) and iodoacetamide on cysteine (57.0215Da).
Site-directed mutagenesis
Site-directed mutagenesis was performed by GenScript. Briefly, vector pcDNA3/Flag-METTL3 (Addgene #53739) was transformed and the plasmid was extracted with Axygen kit (Corning). Next, sequence verification and enzyme digestion were performed for vector verification. Plasmid was linearized by digestion with HindIII and XboI to obtain a ~5.2kb vector backbone. Gene fragments were prepared by using two specific pairs of primers with overlap for each construct. Gene fragment amplification was performed by two rounds of PCR to generate the target gene fragment with the designed point mutation using the WT as a template. The PCR product was purified and cloned, and ligated with a linearized vector with T4 ligase. Gene products were transformed into competent cells, and single colonies were picked for screening and sequence verification.
METTL3 protein structure modelling
Structural figures of METTL3 were generated using Pymol (Delano) and coordinates from AlphaFold accession number AF-Q86U44-F1-model_v4 highlighting the zinc finger cluster and catalytic domain inter-domain interactions (also shown in the predicted aligned error grid in Extended Data Fig. 10g)91. The zinc finger cluster is shown in light orange, the catalytic SET domain in light-blue ribbon format and the SAM cofactor in stick format. The majority of the active site, including the cofactor and substrate binding pockets, is shown in dotted surface format. The solution structure of the zinc finger cluster from PDB 5YZ9 (ref. 65) is also shown in greater detail in ribbon format including the zinc atoms and coordinating cysteine residues implicated in SNO.
METTL3/METTL14 methyltransferase activity
METTL3/METTL14 activity was analysed using the METTL3/METTL14 Complex Chemiluminescent Assay Kit (BPS Biosciences, #79614) in the presence of DMSO, SNAP (Cayman Chemical) or STM2457 (Selleckchem) according to the kit protocol.
Statistics and reproducibility
The sample size was chosen on the basis of having an 80% power in detecting a difference at a significant level of α = 0.05. Sample sizes for animal experiments were chosen based on experience in previous in-house studies of metabolic phenotypes and to balance the ability to detect significant differences with minimization of the number of animals used following NIH guidelines. Replication attempts were successful. All replication experiments were included in the study. All experiments were performed using a minimum of three biological replicates unless specified in the legends. All cell line experiments were performed in three independent experiments. Animals were randomly assigned to experimental groups and matched for age and gender. β-cell mass analyses and insulitis scores were performed blindly. In vitro experiments with genetic manipulation such as KD experiments were performed with awareness of groups to distinguish scramble from KD/overexpressing cells. Blinding was not relevant or possible in other experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. T1D pathways are upregulated in Mettl14 deficient mouse β-cells and pre-diabetic NOD β-cells show enrichment in pathways associated with Type 1 diabetes (related to Fig. 1).
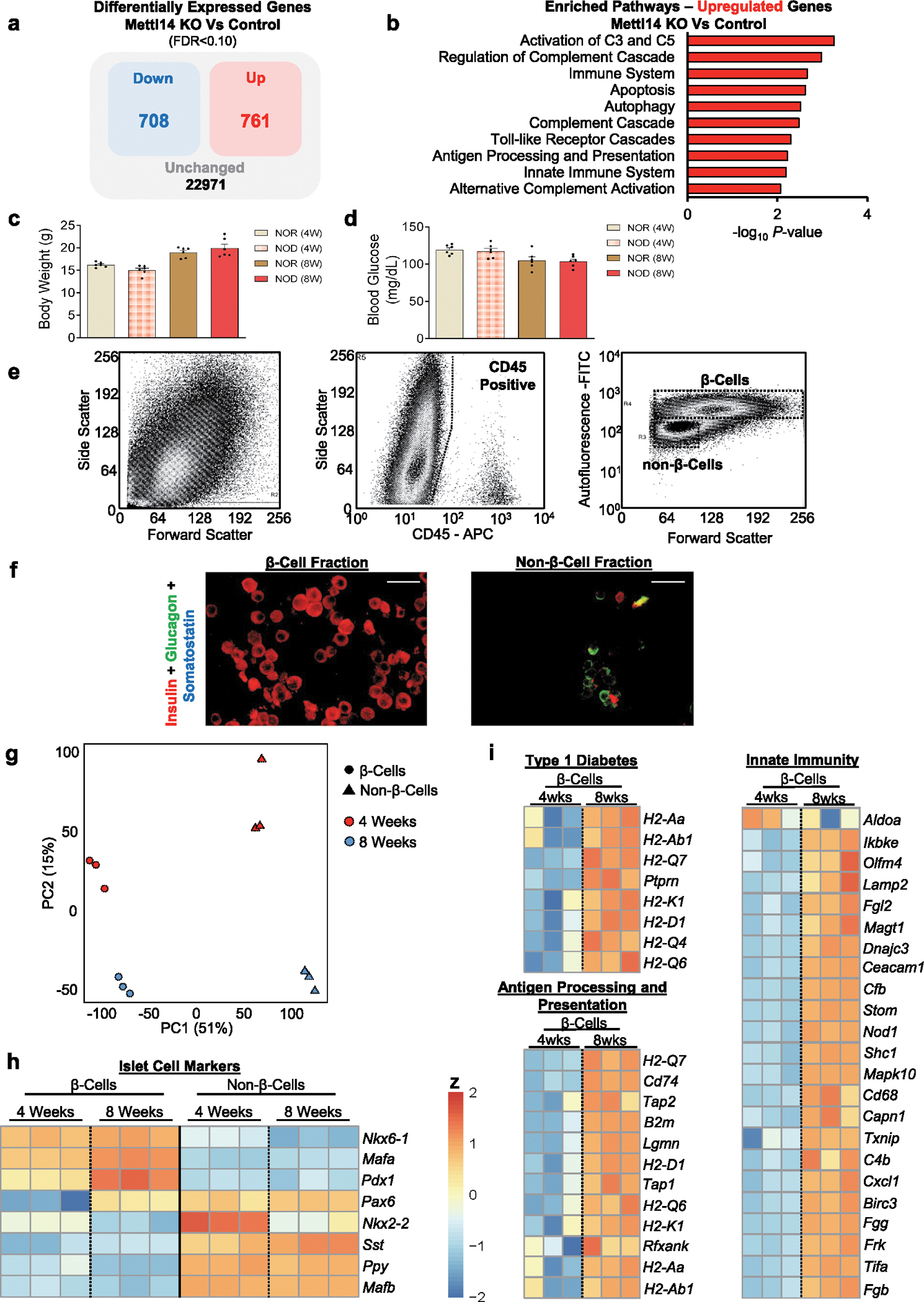
a, Diagram of differentially expressed genes in Mettl14 KO β-cells compared to controls. b, Pathway enrichment analyses of genes upregulated in Mettl14 KO β-cells compared to controls (Controls, n = 4 pools, 2 animals/pool; M14KO, n = 4 pools, 4 animals/pool). c, Body weight of NOR and NOD females at 4- or 8-weeks of age (n = 6 mice/group). d, Random-fed serum glucose levels of NOR and NOD females at 4- or 8-weeks of age (n = 6 mice/group). e, Representative FACS sorting strategy to deplete CD45 positive cells, and obtain an enriched β-cell population based on size, granularity, and autofluorescence (FITC) from NOD female mice. f, Immunofluorescence staining of sorted β- and non-β-cell fractions showing insulin staining in red, glucagon in green, and somatostatin in blue (n = 3 independent experiments/group). Scale bar = 20 μM. g, PCA plot of RNA-seq samples of 4- or 8-week-old FACS sorted β-cells or non-β-cells (n = 3 pools of 3 mice per pool). h, Heat-map representation of islet identity genes, showing enrichment for β cell identity genes in the β-cell fraction compared to non-β-cells. i, Heat-map of represented genes associated with Type 1 diabetes, innate immunity, and antigen processing and presentation and upregulated in pre-diabetic 8-week-old β-cells compared to 4-week-old. All samples in each panel are biologically independent. Data were expressed as means ± SEM. Heat maps represent clipped Z-scored log CPM. Statistical analyses were performed using the Benjamini-Hochberg procedure and genes were filtered for FDR<0.05 or 0.10 in ‘a’. P values of pathway enrichment analysis were calculated according to the hypergeometric test based on the number of physical entities present in both the predefined set and user-specified list of physical entities. Data in ‘a’ and ‘b’ were downloaded and reanalyzed from dataset GSE132306 (ref. 35). Source numerical data are available in source data.
Extended Data Fig. 2 |. scRNA-seq re-analyses of established T1D islets, upregulation of METTL3 in human β-cells at T1D onset, and Mettl3 downregulation in β-cells with T1D progression in female NOD mice (related to Fig. 1).
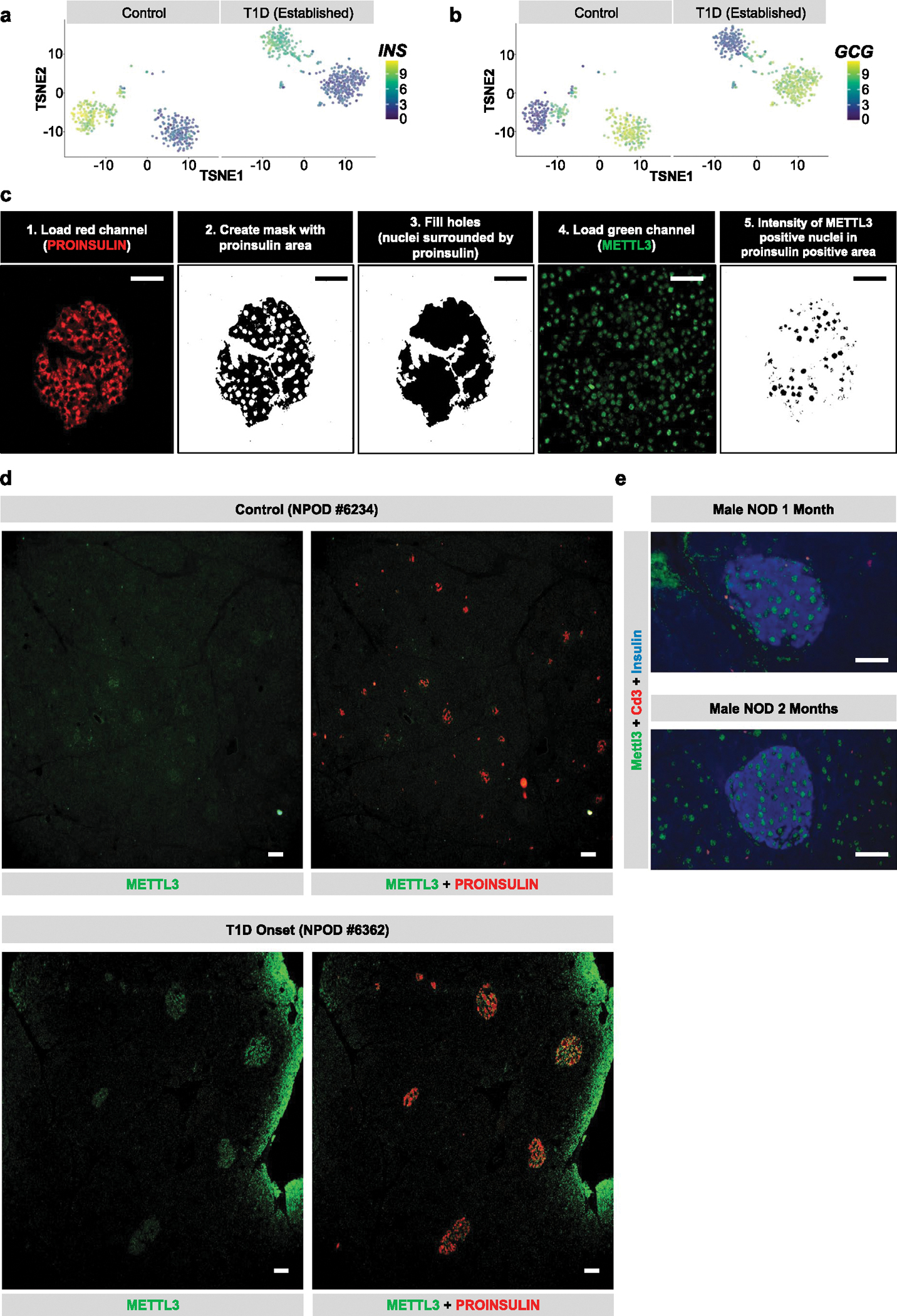
a-b, t-SNE representation of β-cells (a) (high insulin expression) or α-cells (b) (high glucagon expression) in control and established Type 1 diabetes (T1D) (GSE121863)40. c, Schematic representation of the Image J pipeline used to quantify the METTL3 nuclear intensity in proinsulin positive area in human pancreatic sections from the Network for Pancreatic Organ Donors with Diabetes (nPOD). d, Representative low magnification pictures of immunofluorescence staining of METTL3 (green) and Proinsulin (red) in pancreatic sections from human control and T1D onset showing a robust islet enriched upregulation of METTL3 at T1D onset. (Control, n = 9 patients; T1D Onset, n = 4 patients; established T1D, n = 7 patients). e, Representative pictures of immunofluorescence staining of Mettl3 (green), Insulin (blue), and Cd3 (red) in pancreatic sections from NOD male mice without any histological pancreatic immune cell infiltration patterns (n = 3 mice/group). Scale bar = 100 μM. Source numerical data are available in source data.
Extended Data Fig. 3 |. m6A landscape analyses of human islets treated with IL-1β and IFN-α reveal differential methylation of class I MHC-mediated antigen processing and presentation genes (related to Fig. 3).
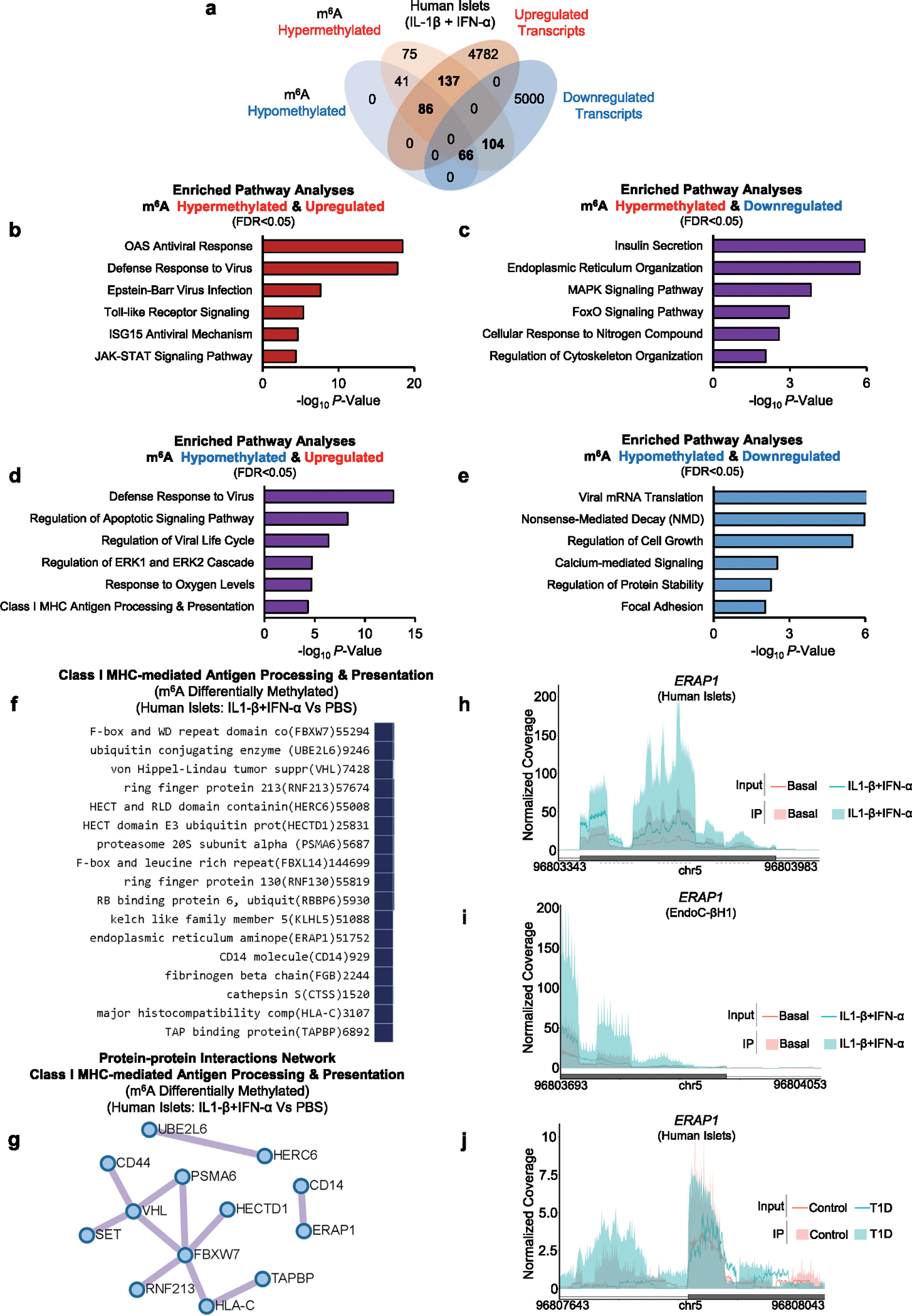
a, Venn diagram representation of the m6A hypermethylated, m6A hypomethylated, upregulated, or downregulated genes in human islets treated with IL-1β and IFN-α compared to PBS. Statistical analyses were performed using the Benjamini-Hochberg procedure and genes were filtered for FDR<0.05. b-e, Pathway enrichment analyses of m6A hypermethylated and upregulated (a), m6A hypermethylated and downregulated (c), m6A hypomethylated and upregulated (d), or m6A hypomethylated and downregulated genes (e) in human islets treated with IL-1β and IFN-α compared to PBS. P values were calculated according to the hypergeometric test based on the number of physical entities present in both the predefined set and user-specified list of physical entities. f, MHC class I differentially m6A methylated genes in human islets treated with IL-1β and IFN-α compared to PBS. g, Protein-protein interactions network of class I MHC-mediated antigen processing and presentation differentially m6A methylated in human islets treated with IL-1β and IFN-α compared to PBS. h-j, Coverage plots of m6A peaks ERAP1in human islets treated with IL-1β and IFN-α or PBS (h) (n = 15 biological independent samples/group), EndoC-βH1 cells treated with IL-1β and IFN-α or PBS (i) (n = 6 independent experiments/group), or human T1D and Control islets (j) (Controls, n = 20; T1D, n = 7 biological independent samples). Plotted coverages are the median of the n replicates presented. All samples in each panel are biologically independent. P values of pathway enrichment analysis were calculated according to the hypergeometric test based on the number of physical entities present in both the predefined set and user-specified list of physical entities. Source numerical data are available in source data.
Extended Data Fig. 4 |. Human islets and EndoC-βH1 cells present an extensive overlap in the innate immune response to IL-1β and IFN-α (related to Figs. 3 and 4).
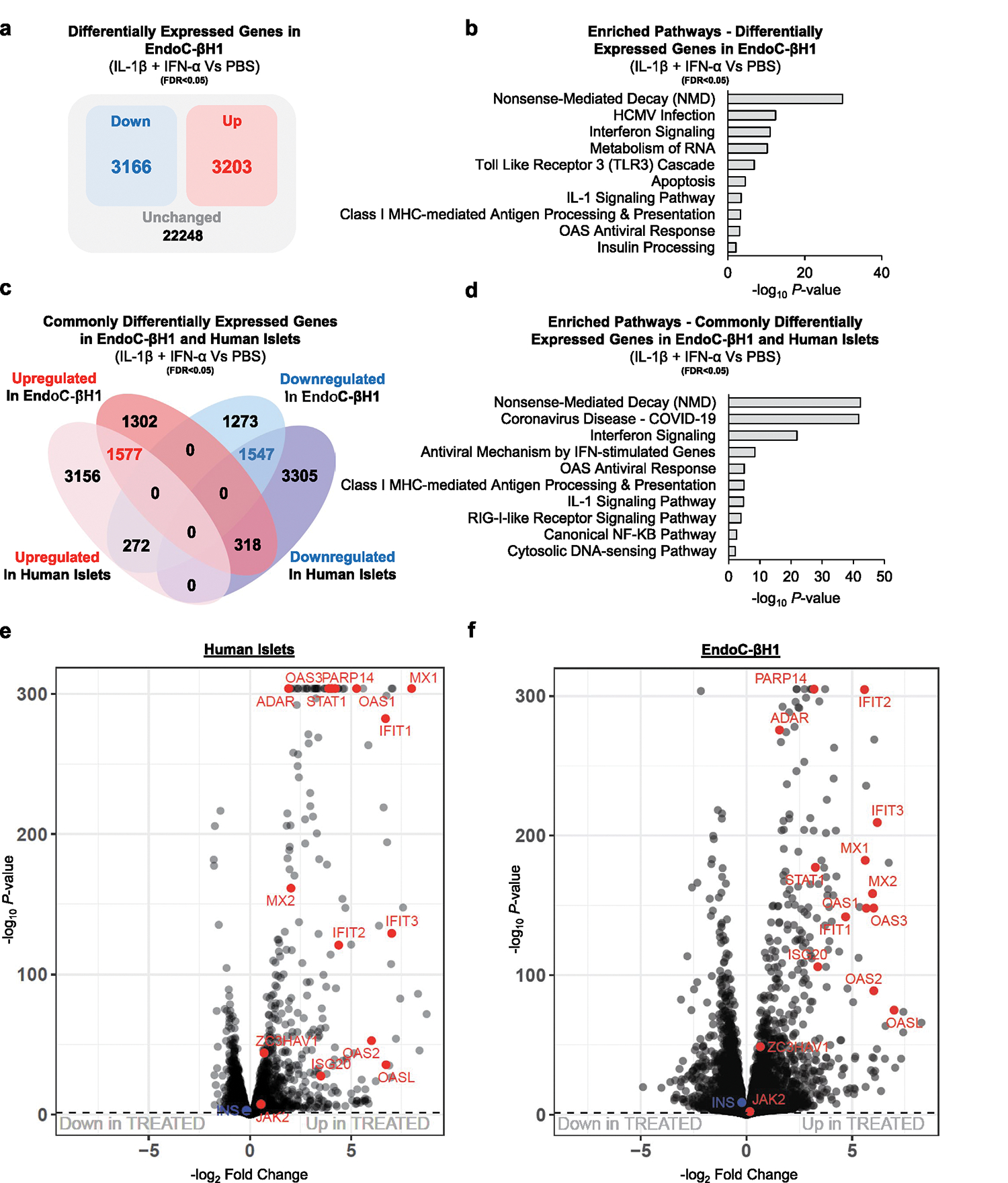
a, Diagram representation of the upregulated (red), downregulated (blue), and unchanged genes (black) in EndoC-βH1 cells treated with IL-1β and IFN-α compared to PBS. Statistical analyses were performed using the Benjamini-Hochberg procedure and genes were filtered for FDR<0.05. b, Pathway enrichment analyses of upregulated and downregulated genes in EndoC-βH1 cells treated with IL-1β and IFN-α compared to PBS. c, Venn diagram representation of the commonly upregulated (red), downregulated (blue), and unchanged genes (black) of the intersected genes in EndoC-βH1 and human islets cells treated with IL-1β and IFN-α compared to PBS. Statistical analyses were performed using the Benjamini-Hochberg procedure and genes were filtered for FDR<0.05. d, Pathway enrichment analyses of commonly upregulated and downregulated genes in human islets and EndoC-βH1 cells treated with IL-1β and IFN-α compared to PBS. e-f, Volcano-plot representation of differentially expressed genes in human islets (e) and EndoC-βH1 cells (f) treated with IL-1β and IFN-α compared to PBS. Innate immune genes are depicted in red and show a near absolute overlap between human islets and EndoC-βH1 cells. Human islets: n = 15 biologically independent samples. EndoC-βH1 cells: n = 6 independent experiments/group. Statistical analyses were performed using the Benjamini-Hochberg procedure. P values of pathway enrichment analysis were calculated according to the hypergeometric test based on the number of physical entities present in both the predefined set and user-specified list of physical entities. Source numerical data are available in source data.
Extended Data Fig. 5 |. OAS upregulation is more prominent in β-cells, and METTL3 silencing leads to the downregulation of ADAR1 p150 isoform in β-cells (related to Fig. 4).
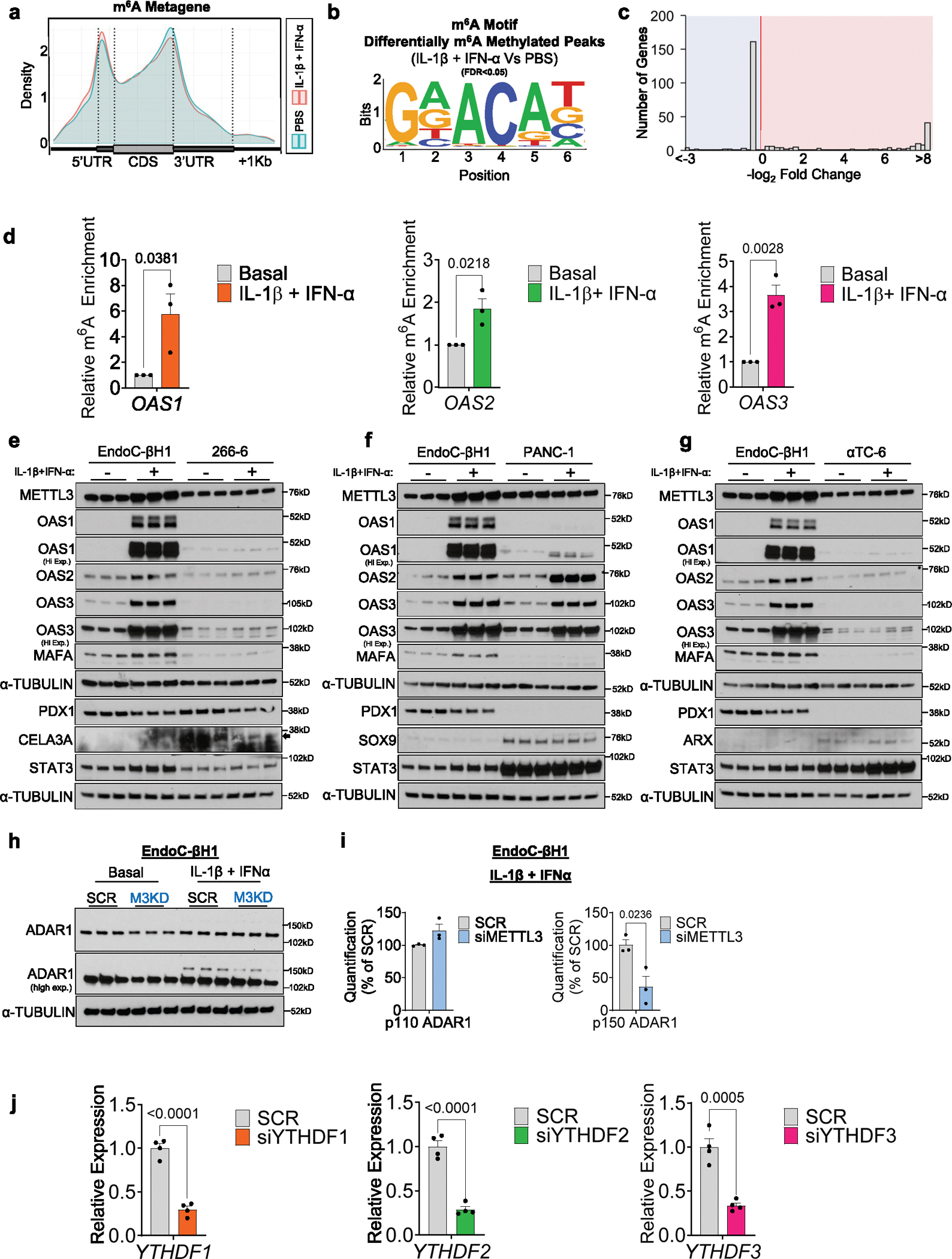
a, Metagene of m6A enriched peaks in PBS (blue) or IL-1β + IFN-α-treated (red) EndoC-βH1 cells. b, Enrichment for known m6A consensus motif RRACH. c, Histogram of the distribution of differential m6A loci log2 fold changes from IL-1β plus IFN-α-treated versus PBS in EndoC-βH1 cells. d, m6A-IP-qPCR showing increased m6A in OAS1, OAS2, and OAS3 in IL-1β + IFN-α-treated EndoC-βH1 cells compared to PBS (Basal) (n = 3 independent experiments/group). e-g, Western-blot analyses of indicated proteins in EndoC-βH1 cells and 266-6 cells (e), or PANC-1 (f), or αTC-6 (g) treated with PBS or IL1-β plus IFN-α for 24h (n = 3 independent experiments/group). h, Western-blot analyses of indicated proteins EndoC-βH1 cells harboring METTL3 silencing or Scramble and treated with PBS or IL1-β plus IFN-α (n = 3 independent experiments/group). Same experiment of Fig. 4h, with same loading control. i, Protein quantification of indicated proteins related to (h). j, qRT-PCR analyses of YTHDF genes after IL-1β plus IFN-α stimulation in scramble, YTHDF1, YTHDF2, or YTHDF3 KD EndoC-βH1 cells (n = 4 independent experiments/group). All samples in each panel are biologically independent. Data were expressed as means ± SEM. Statistical analysis was performed by two-tailed unpaired t-test. Source numerical data and unprocessed gel images are available in source data.
Extended Data Fig. 6 |. m6A landscape analyses of established T1D reveal differential methylation of master regulators of β-cell identity and function (related to Fig. 6).
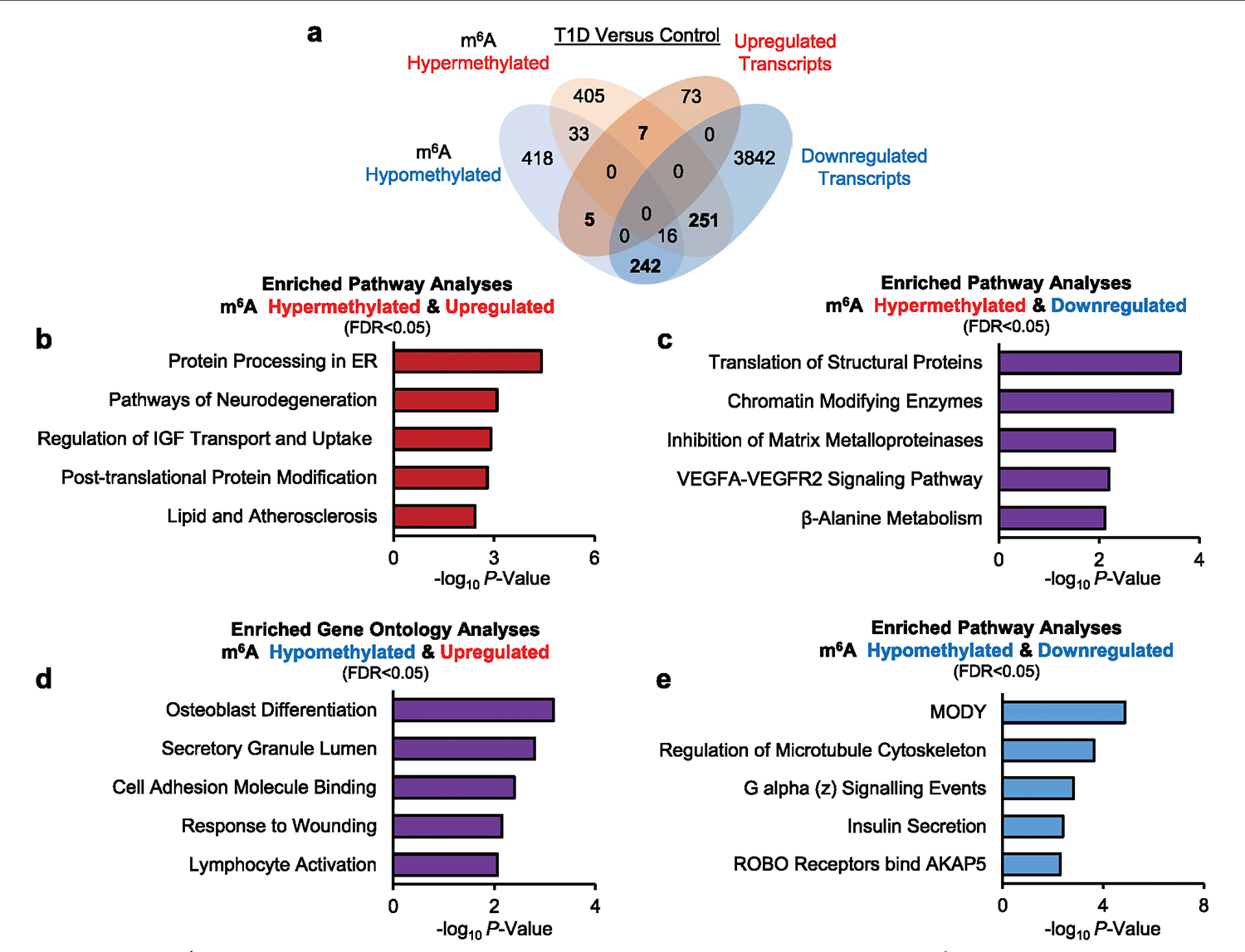
a, Venn diagram representation of the m6A hypermethylated, m6A hypomethylated, upregulated, or downregulated genes in human islets from patients with established T1D or non-diabetic Controls. Statistical analyses were performed using the Benjamini-Hochberg procedure and genes were filtered for FDR<0.05. Human islets: Controls n = 20 and T1D n = 7 biologically independent samples. b-e, Pathway enrichment analyses of m6A hypermethylated and upregulated (a), m6A hypermethylated and downregulated (c), m6A hypomethylated and upregulated (d), or m6A hypomethylated and downregulated genes (e) in human islets from established T1D compared to Controls. P values of pathway enrichment analysis were calculated according to the hypergeometric test based on the number of physical entities present in both the predefined set and user-specified list of physical entities. Source numerical data are available in source data.
Extended Data Fig. 7 |. Mettl3 overexpression in NOD β-cells (related to Fig. 7).
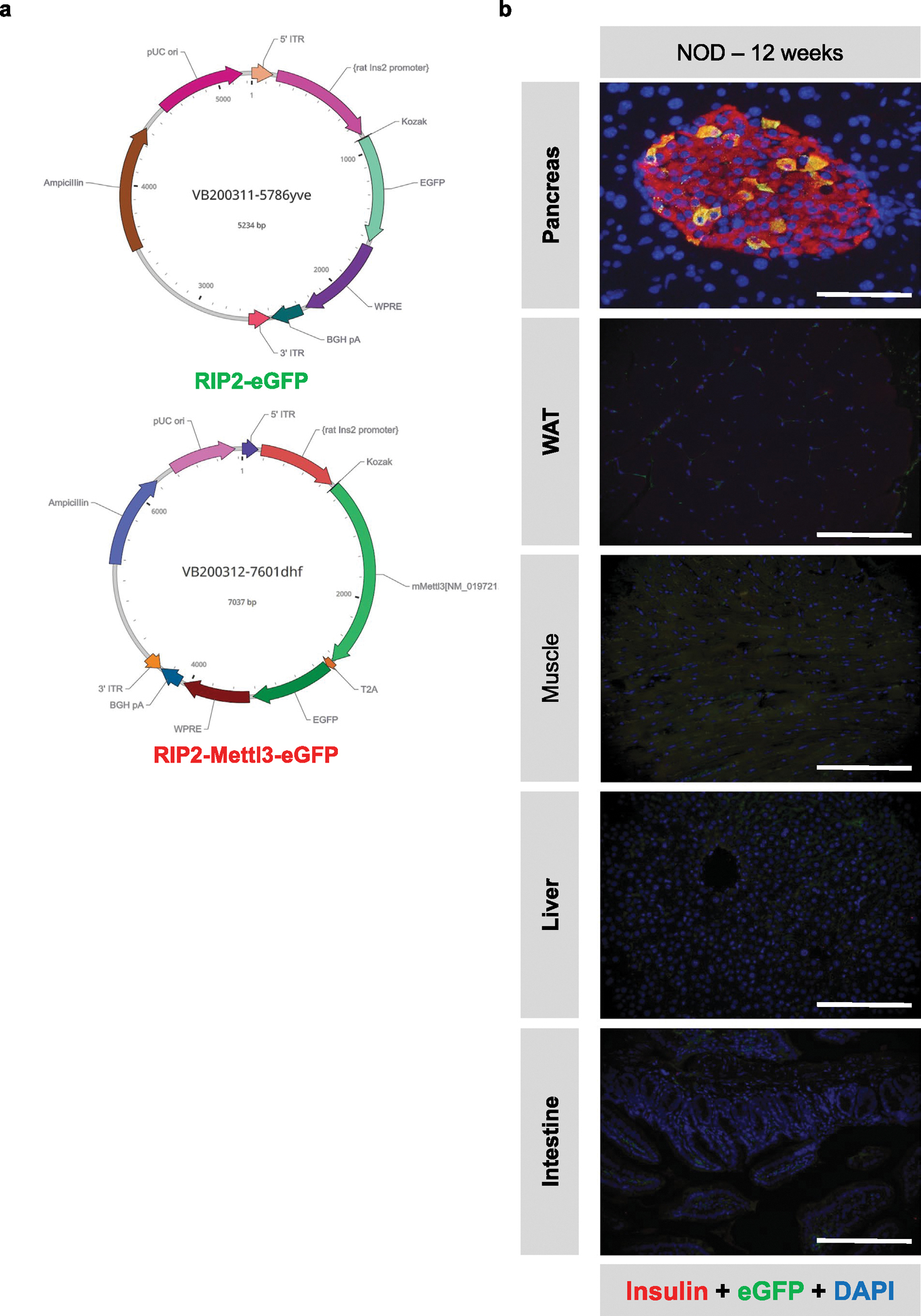
a, Schematic diagram showing the vector design of AAV8 driving eGFP or Mettl3 under the control of a rat insulin promoter II. b, Representative immunofluorescence images of pancreas, white adipose tissue (WAT), muscle, liver, and intestine showing insulin (red), eGFP (green), and DAPI (blue) (n = 4 mice/group). Scale bar = 100 μM.
Extended Data Fig. 8 |. Immune cell profiling of NOD pancreatic lymph nodes (related to Fig. 7).
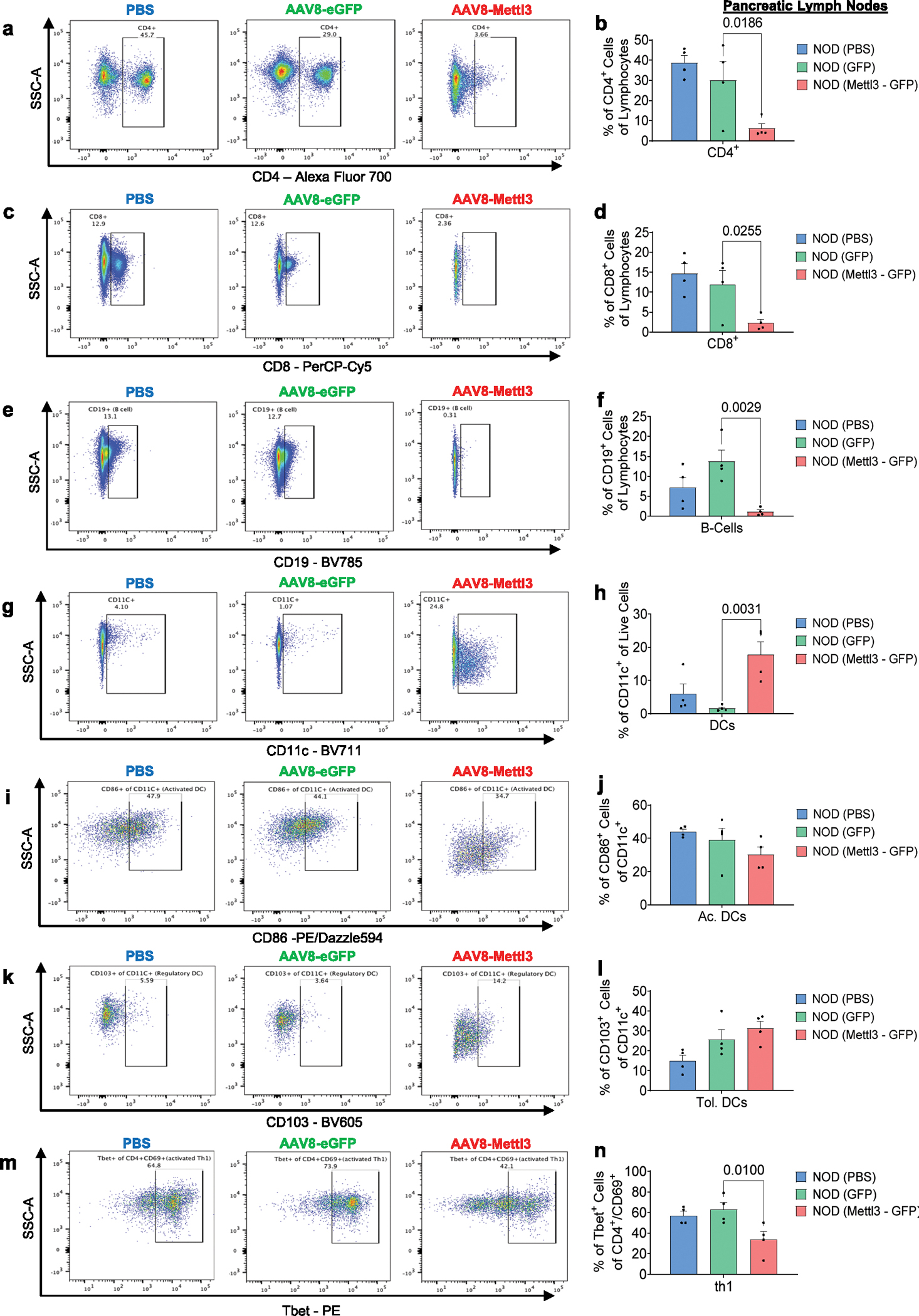
a-n, Flow cytometry analyses of CD4 (a,b), CD8 (c,d), B-cells (e,f), dendritic cells (g,h), activated dendritic cells (i,j), tolerogenic dendritic cells (k,l), or th1 cells (m,n) isolated from pancreatic lymph nodes of NOD mice receiving PBS, eGFP, or Mettl3 overexpression (n = 4 mice/group). All samples in each panel are biologically independent. Data were expressed as meansx ± SEM. Statistical analysis was performed by One-Way ANOVA with Holm-Sidak test. Source numerical data are available in source data.
Extended Data Fig. 9 |. Immune cell profiling of NOD splenocytes (related to Fig. 7).
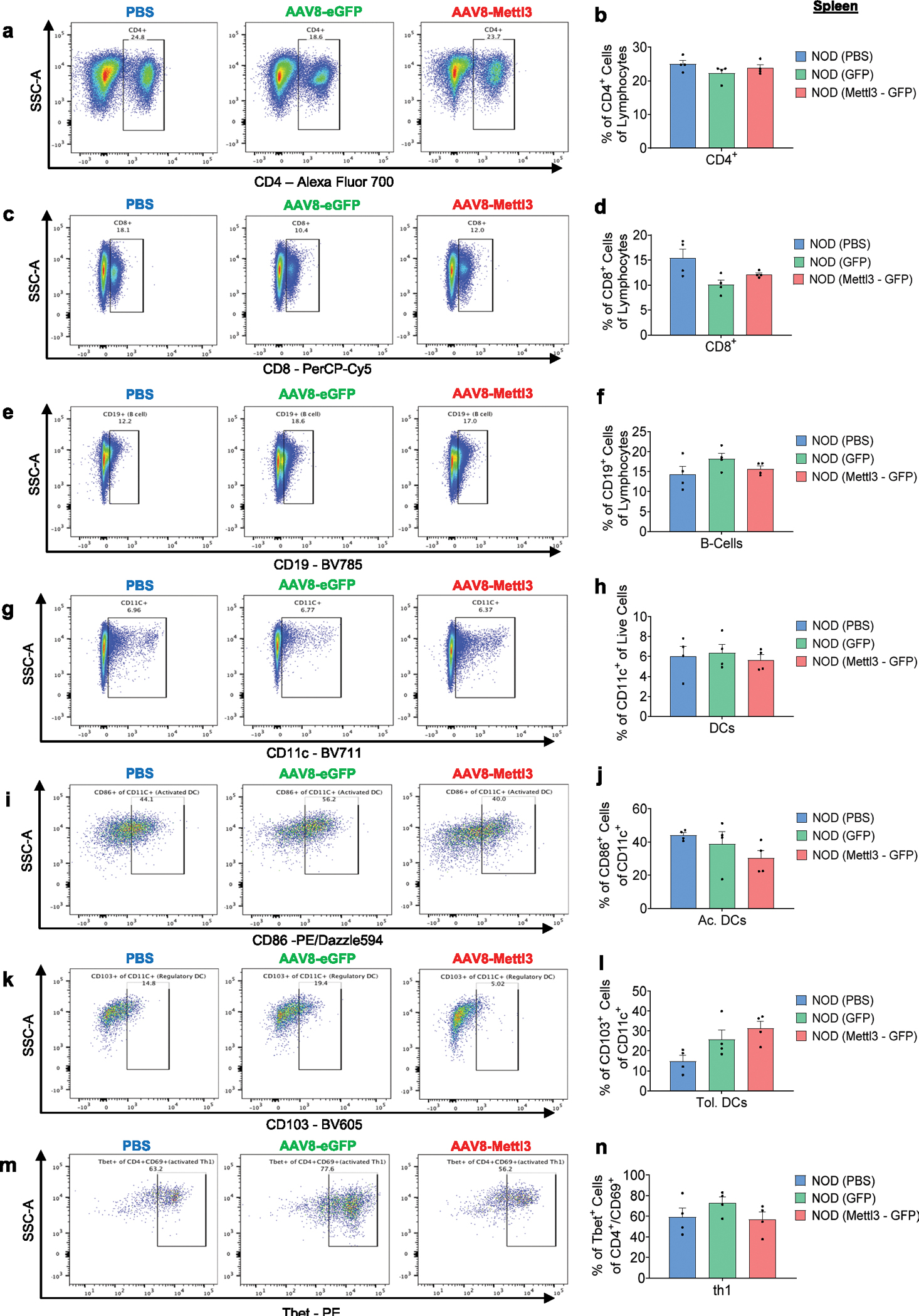
a-n, Flow cytometry analyses of CD4 (a,b), CD8 (c,d), B-cells (e,f), dendritic cells (g,h), activated dendritic cells (i,j), tolerogenic dendritic cells (k,l), or th1 cells (m,n) isolated from spleens of NOD mice receiving PBS, eGFP, or Mettl3 overexpression (n = 4 mice/group). All samples in each panel are biologically independent. Data were expressed as means ± SEM. Statistical analysis was performed by One-Way ANOVA with Holm-Sidak test. Source numerical data are available in source data.
Extended Data Fig. 10 |. Summary schemes of experimental approaches and METTL3 protein structural modeling (related to Fig. 8).
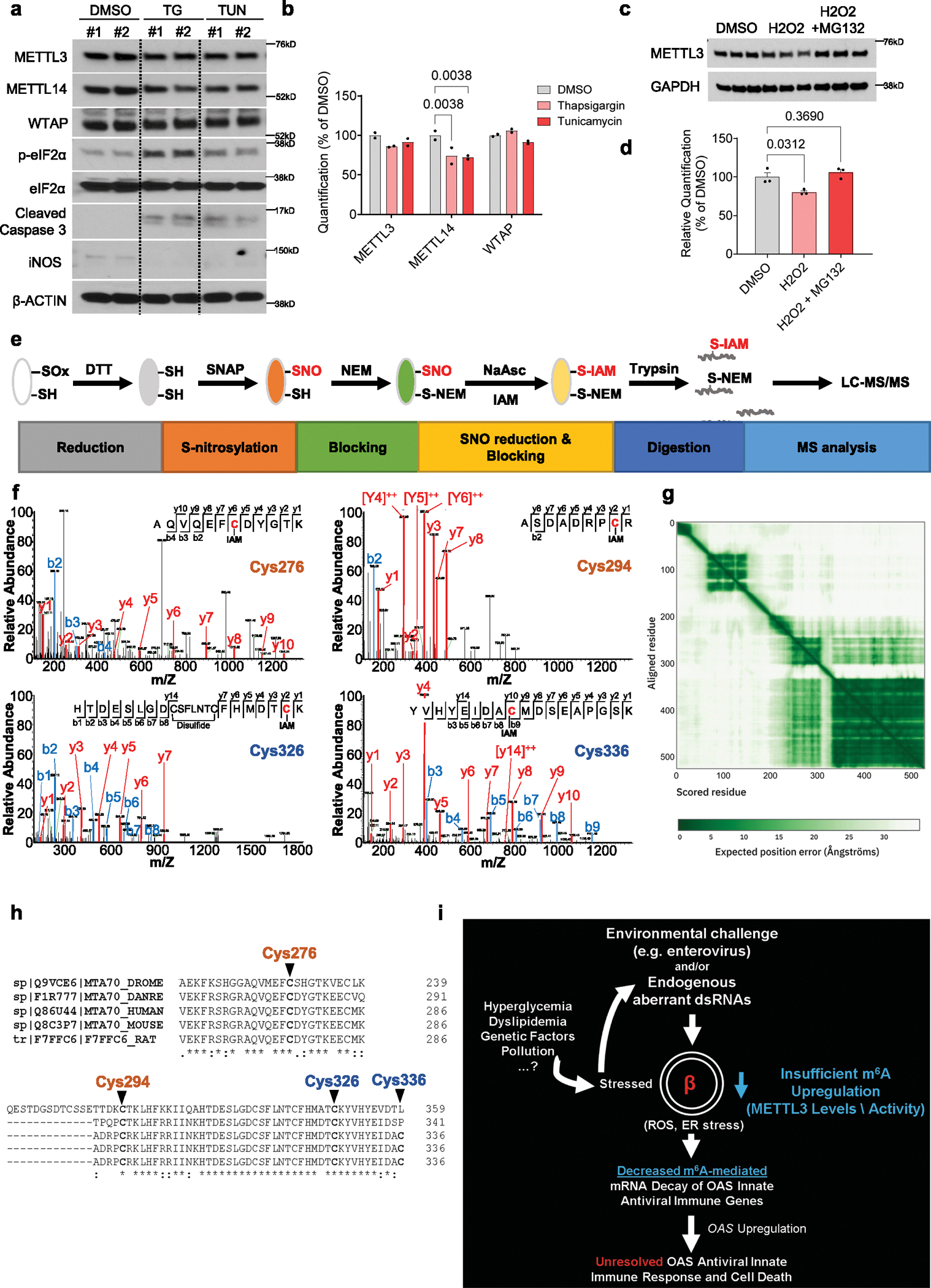
a, Western-blot analyses of indicated proteins in human islets cells treated with DMSO, 1 μM thapsigargin, or 2 μg/mL tunicamycin for 24h (n = 2 biological independent samples/group). b, Protein quantification of indicated proteins related to (a). c, Western-blot analyses of indicated proteins in EndoC-βH1 cells treated with DMSO, 25 μM H2O2, or 25 μM H2O2 + 5 μM MG132 for 24h (n = 3 independent experiments/group). d, Protein quantification of indicated proteins related to (c). e, Summary scheme of experimental approach to measure METTL3 SNO by LC-MS/MS. f, MS/MS spectra of S-nitrosylated METTL3 peptides and the identification of cysteines C276, C294, C326, and C336 (n = 2 replicates). g, Predicted aligned error related to Fig. 8i–k. The color at position (x, y) indicates AlphaFold’s expected position error at residue x, when the predicted and true structures are aligned on residue y. h, CLUSTAL (1.2.4) multiple sequence alignment of METTL3 in represented species. i, Conceptual model of the role of METTL3 in controlling the OAS innate antiviral immune response by regulating the formation of deleterious dsRNAs and/or controlling viral response. The METTL3 response is decreased by the presence of ER stress and increased ROS levels, resulting from several potential factors such as hyperglycemia, dyslipidemia, genetic, or other environmental mediators such as pollution. Data were expressed as means ± SEM. Statistical analysis was performed by Two-Way ANOVA with Holm-Sidak test in b, and One-Way ANOVA with Holm-Sidak test in d. Source unprocessed gel images and numerical data are available in source data.
Supplementary Material
Acknowledgements
The authors thank F. Bosch (Universitat Autònoma de Barcelona) and her lab members for discussions about AAV design and in vivo administration. We thank A. Op de Beeck (ULB Center for Diabetes Research, Université Libre de Bruxelles) for helpful discussions related to viral infections. We thank E. Dirice (New York Medical College) for discussions related to co-culture of islets and splenocytes. We thank B. Kunkemoeller (Brigham and Women’s Hospital) for assistance with fluorescence-activated cell sorting analysis and E. Mandato (Dana-Farber Cancer Institute) and A. Ferreira (Brigham and Women’s Hospital) for discussions regarding site mutagenesis. The authors thank the Joslin Islet Isolation Core, Joslin Flow Cytometry Core and Joslin Bioinformatics Core (P30 DK36836). This work is supported by NIH grants R01 DK67536 (R.N.K.), UC4 DK116278 (R.N.K. and C.H.), RM1 HG008935 (C.H.) and R01 DK122160 (W.-J.Q. and R.N.K.). Portions of the mass spectrometry work were performed in the Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, a national scientific user facility sponsored by the Department of Energy under contract DE-AC05-76RL0 1830. R.N.K. acknowledges support from the Margaret A. Congleton Endowed Chair and C.H. is a Howard Hughes Medical Institute Investigator. D.F.D.J. acknowledges support from Mary K. Iacocca Junior Postdoctoral Fellowship, American Diabetes Association grant #7-21-PDF-140 and NIH K99 DK135927. The authors sincerely thank the families of the human islet donors.
Footnotes
Competing interests
R.N.K is on the scientific advisory board of Novo Nordisk, Biomea and REDD. C.H. is a scientific founder and a member of the scientific advisory board of Accent Therapeutics. The remaining authors have no conflicts of interest.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41556-024-01368-0.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41556-024-01368-0.
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Data availability
m6A-seq and RNA-seq data in human islets and EndoC-βH1 cells have been deposited with the National Center for Biotechnology Information Gene Expression Omnibus under accession code GSE228267. RNA-seq in NOD mouse β- and non-β-cells sorted by fluorescence-activated cell sorting have been deposited under the accession code GSE228219. RNA-seq data in EndoC-βH1 cells harbouring OAS overexpression have been deposited under the accession code GSE244273. Mass spectrometry data on METTL3 SNO have been deposited with Mass Spectrometry Interactive Visual Environment (MassIVE) under the accession code MSV000091547. Source data are provided with this paper. All other data supporting the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Crowl JT, Gray EE, Pestal K, Volkman HE & Stetson DB Intracellular nucleic acid detection in autoimmunity. Annu. Rev. Immunol. 35, 313–336 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lang KS, Burow A, Kurrer M, Lang PA & Recher M The role of the innate immune response in autoimmune disease. J. Autoimmun. 29, 206–212 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Kallionpaa H et al. Innate immune activity is detected prior to seroconversion in children with HLA-conferred type 1 diabetes susceptibility. Diabetes 63, 2402–2414 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Carry PM et al. Changes in the coexpression of innate immunity genes during persistent islet autoimmunity are associated with progression of islet autoimmunity: Diabetes Autoimmunity Study in the Young (DAISY). Diabetes 71, 2048–2057 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Apaolaza PS et al. Islet expression of type I interferon response sensors is associated with immune infiltration and viral infection in type 1 diabetes. Sci. Adv. 7, eabd6527 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lundberg M, Krogvold L, Kuric E, Dahl-Jorgensen K & Skog O Expression of interferon-stimulated genes in insulitic pancreatic islets of patients recently diagnosed with type 1. Diabetes 65, 3104–3110 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Hornung V, Hartmann R, Ablasser A & Hopfner KP OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat. Rev. Immunol. 14, 521–528 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pedersen K et al. Genetic predisposition in the 2′-5′A pathway in the development of type 1 diabetes: potential contribution to dysregulation of innate antiviral immunity. Diabetologia 64, 1805–1815 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Field LL et al. OAS1 splice site polymorphism controlling antiviral enzyme activity influences susceptibility to type 1 diabetes. Diabetes 54, 1588–1591 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Tessier MC et al. Type 1 diabetes and the OAS gene cluster: association with splicing polymorphism or haplotype? J. Med. Genet. 43, 129–132 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonnevie-Nielsen V, Buschard K & Dyrberg T Differential responsiveness to interferon-alpha in beta-cells and non-beta cells. Diabetes 45, 818–821 (1996). [DOI] [PubMed] [Google Scholar]
- 12.Li M, Zheng DJ, Field LL & Bonnevie-Nielsen V Murine pancreatic beta TC3 cells show greater 2′, 5′-oligoadenylate synthetase (2′5′AS) antiviral enzyme activity and apoptosis following IFN-alpha or poly(I:C) treatment than pancreatic alpha TC3 cells. Exp. Diabetes Res. 2009, 631026 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dan M, Zheng D, Field LL & Bonnevie-Nielsen V Induction and activation of antiviral enzyme 2′,5′-oligoadenylate synthetase by in vitro transcribed insulin mRNA and other cellular RNAs. Mol. Biol. Rep. 39, 7813–7822 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Zeng C et al. RNase L contributes to experimentally induced type 1 diabetes onset in mice. J. Endocrinol. 223, 277–287 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frye M, Harada BT, Behm M & He C RNA modifications modulate gene expression during development. Science 361, 1346–1349 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dominissini D et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Meyer KD et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149, 1635–1646 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JH et al. Enhancer RNA m6A methylation facilitates transcriptional condensate formation and gene activation. Mol. Cell 81, 3368–3385 e3369 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lasman L et al. Context-dependent functional compensation between Ythdf m6A reader proteins. Genes Dev. 34, 1373–1391 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou Z, Sepich-Poore C, Zhou X, Wei J & He C The mechanism underlying redundant functions of the YTHDF proteins. Genome Biol. 24, 17 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang P, Doxtader KA & Nam Y Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol. Cell 63, 306–317 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Du Y et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 46, 5195–5208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun HL et al. Stabilization of ERK-phosphorylated METTL3 by USP5 increases m6A methylation. Mol. Cell 80, 633–647 e637 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winkler R et al. m(6)A modification controls the innate immune response to infection by targeting type I interferons. Nat. Immunol. 20, 173–182 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Terajima H et al. N6-methyladenosine promotes induction of ADAR1-mediated A-to-I RNA editing to suppress aberrant antiviral innate immune responses. PLoS Biol. 19, e3001292 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao Y et al. m6A modification prevents formation of endogenous double-stranded RNAs and deleterious innate immune responses during hematopoietic development. Immunity 52, 1007–1021 e1008 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu W et al. N6-methyladenosine RNA modification suppresses antiviral innate sensing pathways via reshaping double-stranded RNA. Nat. Commun. 12, 1582 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makino S et al. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu 29, 1–13 (1980). [DOI] [PubMed] [Google Scholar]
- 29.De Jesus DF et al. m6A mRNA methylation regulates human beta-cell biology in physiological states and in type 2 diabetes. Nat. Metab. 1, 765–774 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prochazka M, Serreze DV, Frankel WN & Leiter EH NOR/Lt mice: MHC-matched diabetes-resistant control strain for NOD mice. Diabetes 41, 98–106 (1992). [DOI] [PubMed] [Google Scholar]
- 31.Carrero JA, Calderon B, Towfic F, Artyomov MN & Unanue ER Defining the transcriptional and cellular landscape of type 1 diabetes in the NOD mouse. PLoS ONE 8, e59701 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smelt MJ, Faas MM, de Haan BJ & de Vos P Pancreatic beta-cell purification by altering FAD and NAD(P)H metabolism. Exp. Diabetes Res 2008, 165360 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russell MA et al. HLA class II antigen processing and presentation pathway components demonstrated by transcriptome and protein analyses of islet beta-cells from donors with type 1 diabetes. Diabetes 68, 988–1001 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sledz P & Jinek M Structural insights into the molecular mechanism of the m6A writer complex. eLife 5, e18434 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dirice E et al. Increased beta-cell proliferation before immune cell invasion prevents progression of type 1 diabetes. Nat. Metab. 1, 509–518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eisenbarth GS Type I diabetes mellitus. A chronic autoimmune disease. N. Engl. J. Med. 314, 1360–1368 (1986). [DOI] [PubMed] [Google Scholar]
- 38.Ramos-Rodriguez M et al. The impact of proinflammatory cytokines on the beta-cell regulatory landscape provides insights into the genetics of type 1 diabetes. Nat. Genet. 51, 1588–1595 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eizirik DL et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 8, e1002552 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benazra M et al. A human beta cell line with drug inducible excision of immortalizing transgenes. Mol. Metab. 4, 916–925 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.York IA, Brehm MA, Zendzian S, Towne CF & Rock KL Endoplasmic reticulum aminopeptidase 1 (ERAP1) trims MHC class I-presented peptides in vivo and plays an important role in immunodominance. Proc. Natl Acad. Sci. USA 103, 9202–9207 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thomaidou S et al. β-Cell stress shapes CTL immune recognition of preproinsulin signal peptide by posttranscriptional regulation of endoplasmic reticulum aminopeptidase 1. Diabetes 69, 670–680 (2020). [DOI] [PubMed] [Google Scholar]
- 43.Szymczak F et al. ADAR1-dependent editing regulates human β cell transcriptome diversity during inflammation. Front. Endocrinol. 13, 1058345 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu S-B et al. ADAR1p150 prevents MDA5 and PKR activation via distinct mechanisms to avert fatal autoinflammation. Mol. Cell 83, 3869–3884.e3867 (2023). [DOI] [PubMed] [Google Scholar]
- 45.Li X, Chen H & Epstein PN Metallothionein protects islets from hypoxia and extends islet graft survival by scavenging most kinds of reactive oxygen species. J. Biol. Chem. 279, 765–771 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Bensellam M, Laybutt DR & Jonas J-C Emerging roles of metallothioneins in beta cell pathophysiology: beyond and above metal homeostasis and antioxidant response. Biology 10, 176 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dor Y, Brown J, Martinez OI & Melton DA Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429, 41–46 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Rui J et al. Tet2 controls the responses of beta cells to inflammation in autoimmune diabetes. Nat. Commun. 12, 5074 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kulkarni A et al. 12-Lipoxygenase governs the innate immune pathogenesis of islet inflammation and autoimmune diabetes. JCI Insight 6, e147812 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nelson AS et al. Oral therapy with colonization factor antigen I prevents development of type 1 diabetes in non-obese diabetic mice. Sci. Rep. 10, 6156 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson CP, Yamamoto H, Peck AB & Humphreys-Beher MG Genetically programmed development of salivary gland abnormalities in the NOD (nonobese diabetic)-scid mouse in the absence of detectable lymphocytic infiltration: a potential trigger for sialoadenitis of NOD mice. Clin. Immunol. Immunopathol. 79, 50–59 (1996). [DOI] [PubMed] [Google Scholar]
- 52.Elkhateeb E et al. The role of mouse 2′,5′-oligoadenylate synthetase 1 paralogs. Infect. Genet. Evol. 45, 393–401 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Chen J, Stimpson SE, Fernandez-Bueno GA & Mathews CE Mitochondrial reactive oxygen species and type 1 diabetes. Antioxid. Redox Signal 29, 1361–1372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim YK, Sussel L & Davidson HW Inherent beta cell dysfunction contributes to autoimmune susceptibility. Biomolecules 11, 512 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tersey SA et al. Islet beta-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 61, 818–827 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mallone R & Eizirik DL Presumption of innocence for beta cells: why are they vulnerable autoimmune targets in type 1 diabetes? Diabetologia 63, 1999–2006 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Wang X et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rubio RM, Depledge DP, Bianco C, Thompson L & Mohr I RNA m6A modification enzymes shape innate responses to DNA by regulating interferon beta. Genes Dev. 32, 1472–1484 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Balzano-Nogueira L et al. Integrative analyses of TEDDY Omics data reveal lipid metabolism abnormalities, increased intracellular ROS and heightened inflammation prior to autoimmunity for type 1 diabetes. Genome Biol. 22, 39 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eizirik DL, Miani M & Cardozo AK Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 56, 234–241 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Sahin GS, Lee H & Engin F An accomplice more than a mere victim: the impact of β-cell ER stress on type 1 diabetes pathogenesis. Mol. Metab. 54, 101365 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou HL, Premont RT & Stamler JS The manifold roles of protein S-nitrosylation in the life of insulin. Nat. Rev. Endocrinol. 18, 111–128 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garban HJ, Marquez-Garban DC, Pietras RJ & Ignarro LJ Rapid nitric oxide-mediated S-nitrosylation of estrogen receptor: regulation of estrogen-dependent gene transcription. Proc. Natl Acad. Sci. USA 102, 2632–2636 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kroncke KD Zinc finger proteins as molecular targets for nitric oxide-mediated gene regulation. Antioxid. Redox Signal 3, 565–575 (2001). [DOI] [PubMed] [Google Scholar]
- 65.Huang J et al. Solution structure of the RNA recognition domain of METTL3–METTL14 N6-methyladenosine methyltransferase. Protein Cell 10, 272–284 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ravassard P et al. A genetically engineered human pancreatic β cell line exhibiting glucose-inducible insulin secretion. J. Clin. Investig. 121, 3589–3597 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.El Ouaamari A et al. SerpinB1 promotes pancreatic beta cell proliferation. Cell Metab. 23, 194–205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.El Ouaamari A et al. Compensatory islet response to insulin resistance revealed by quantitative proteomics. J. Proteome Res. 14, 3111–3122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dirice E et al. Soluble factors secreted by T cells promote beta-cell proliferation. Diabetes 63, 188–202 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.De Jesus DF et al. Parental metabolic syndrome epigenetically reprograms offspring hepatic lipid metabolism in mice. J. Clin. Invest. 130, 2391–2407 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen S, Zhou Y, Chen Y & Gu J fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang Z et al. RADAR: differential analysis of MeRIP-seq data with a random effect model. Genome Biol. 20, 294 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fisher RA in Breakthroughs in Statistics (eds Kotz S & Johnson NL) 66–70 (Springer, 1992). [Google Scholar]
- 78.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 79.Bray NL, Pimentel H, Melsted P & Pachter L Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Soneson C, Love MI & Robinson MD Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Robinson MD & Oshlack A A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Law CW, Chen Y, Shi W & Smyth GK voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McCarthy DJ, Campbell KR, Lun AT & Wills QF Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lun AT, Bach K & Marioni JC Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 17, 75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Scrucca L, Fop M, Murphy TB & Raftery AE mclust 5: clustering, classification and density estimation using Gaussian finite mixture models. R. J. 8, 289–317 (2016). [PMC free article] [PubMed] [Google Scholar]
- 86.Herwig R, Hardt C, Lienhard M & Kamburov A Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 11, 1889 (2016). [DOI] [PubMed] [Google Scholar]
- 87.Zhou Y et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Saito R et al. A travel guide to Cytoscape plugins. Nat. Methods 9, 1069–1076 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Snel B et al. STRING: known and predicted protein–protein associations, integrated and transferred across organisms. Nucleic Acids Res. 33, D433–D437 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Duan J et al. Stochiometric quantification of the thiol redox proteome of macrophages reveals subcellular compartmentalization and susceptibility to oxidative perturbations. Redox Biol. 36, 101649 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
m6A-seq and RNA-seq data in human islets and EndoC-βH1 cells have been deposited with the National Center for Biotechnology Information Gene Expression Omnibus under accession code GSE228267. RNA-seq in NOD mouse β- and non-β-cells sorted by fluorescence-activated cell sorting have been deposited under the accession code GSE228219. RNA-seq data in EndoC-βH1 cells harbouring OAS overexpression have been deposited under the accession code GSE244273. Mass spectrometry data on METTL3 SNO have been deposited with Mass Spectrometry Interactive Visual Environment (MassIVE) under the accession code MSV000091547. Source data are provided with this paper. All other data supporting the findings of this study are available from the corresponding author upon reasonable request.