

Abstract
The atypical IκB family member Bcl-3 associates with p50/NF-κB1 or p52/NF-κB2 homodimers in nuclei, thereby either positively or negatively modulating transcription in a context-dependent manner. Previously we reported that Bcl-3 was critical for host resistance to Toxoplasma gondii. Bcl-3-deficient mice succumbed within 3–5 weeks after infection, correlating with an apparently impaired Th1-type adaptive immune response. However in which cell type(s) Bcl-3 functioned to assure resistance remained unknown. We now show that Bcl-3 expression in dendritic cells is required to generate a protective Th1-type immune response and confer resistance to T. gondii. Surprisingly, mice lacking Bcl-3 in dendritic cells were as susceptible as mice globally deficient for Bcl-3. Furthermore, early innate defenses were not compromised by the absence of Bcl-3, as initial production of IL-12 by dendritic cells and IFN-γ by NK cells were preserved. However, subsequent production of IFN-γ by CD4+ and CD8+ T-cells was compromised when dendritic cells lacked Bcl-3, and these mice succumbed at a time when T-cell-mediated IFN-γ production was essential for host resistance. These findings demonstrate that Bcl-3 is required in dendritic cells to prime protective T-cell-mediated immunity to T. gondii.
Keywords: Adaptive immunity, Bcl-3, Dendritic cells, IFN-γ, T-cells, T. gondii
Introduction
NF-κB transcription factors are dimers composed by various combinations of members of the Rel/NF-κB family, which comprises RelA [p65], RelB, c-Rel, p50 [NF-κB1], and p52 [NF-κB2]. Unlike the Rel factors, p50 and p52 lack transactivation domains and, in the form of homodimers, may antagonize transcription of some NF-κB target genes [1, 2]. Members of the IκB family regulate NF-κB complexes. This family comprises the classical IκBα, IκBβ, and IκBε inhibitors, the p105/NF-κB1, and p100/NF-κB2 precursors, and the atypical members IκBξ, IκBNS, and Bcl-3. The classical members and the precursors primarily retain and inhibit transactivating NF-κB factors in the cytoplasm and can be rapidly degraded in response to signals, liberating NF-κB activity. By contrast, atypical members are not typically subject to such induced degradation and instead modulate transcription in the nucleus. Bcl-3 exclusively binds homodimers of p50 or p52. Bcl-3 contains transactivation domains and may convert these homodimers into transactivating complexes, yet Bcl-3 may also promote their inhibitory capacity; consistent with such opposing activities, Bcl-3 is able to associate with histone acetylases as well as deacetylases. Bcl-3’s actions appear to be determined by the particular target gene and site, as well as the cellular context, which also involves multiple, not well-understood post-translation modifications of Bcl-3 [1, 3–5].
Various reports have documented a profound physiologic impact of Bcl-3. The gene encoding Bcl-3 is a partner in recurring chromosomal translocations in some B-cell tumors, resulting in its over-expression [6, 7], and various solid tumors express elevated levels of Bcl-3 [8]. Based on studies with mice with germline deficiency in Bcl-3, this regulator also appears to play critical roles in immune system development, host defense to certain pathogens and in autoimmune and inflammatory disease contexts [9–11]. Although the precise biologic functions and molecular mechanisms of Bcl-3 remain poorly understood, we recently demonstrated unique cell-specific roles in T-cells and dendritic cells [12, 13].
We previously showed that mice globally deficient in Bcl-3 (Bcl-3−/−) succumb to infection with Toxoplasma gondii, which correlated with an apparently defective Th1-type response [11]. Resistance in wild-type animals depends on an initial innate protective response that yields to an adaptive immune response by about 5–7 days. Several distinct cell types are known to contain the spread of this pathogen upon infection and/or to directly contribute to the generation of adaptive immunity, including, but not limited to dendritic cells (DCs), natural killer (NK) cells, other innate cells such as monocytes, macrophages, and of course T-cells. Protection critically depends on production of IFN-γ, a cytokine that directs production of NO and other mediators to severely restrict the growth of this parasite. During the early innate phase NK cells are the primary source of IFN-γ [14] following exposure to IL-12, which is rapidly produced by dendritic cells as they sense this pathogen [15]. Subsequently CD4+ Th1 cells become the primary source of IFN-γ during the initial phase of the adaptive response [16, 17]. Development of Th1 cells depends on both the antigen-presenting functions of DCs [18] as well as their production of IL-12, which directs Th1-specific differentiation of naïve T-cells [19–22]. Ultimately cytotoxic CD8 T-cells become the main producers of IFN-γ as infection enters the chronic phase and T. gondii becomes largely dormant, lying within cysts in brain.
We set out to identify cell type(s) in which Bcl-3 may exert critical functions required for host resistance to T. gondii. Given that DCs help orchestrate both the early innate response as well as the adaptive response, and given recent evidence that Bcl-3 in DCs contributes to antigen-mediated priming of T-cells in vitro [13], we examined whether the susceptibility of Bcl-3-deficient mice to T. gondii might be due, at least in part, to impaired functions of Bcl-3 in these cells. Surprisingly, we found that mice conditionally ablated for Bcl-3 in DCs were as susceptible to infection with T. gondii as with mice with germline deficiency in Bcl-3. Loss of Bcl-3 in DCs resulted in impaired CD4- and CD8-mediated IFN-γ production, while initial innate immunity was intact. Our findings reveal that Bcl-3 plays important roles in DCs during the priming/activation of the adaptive T-cell-mediated immune response to T. gondii, which is necessary for protection of the host, while Bcl-3 is dispensable for early innate functions carried out by DCs.
Results
Resistance to T. gondii in mice requires Bcl-3 expression in CD11c+ cells
As previously reported by us, Bcl-3-deficient mice were susceptible to T. gondii, succumbing between 3–5 weeks post infection (p.i.), correlating with an apparently insufficient Th1-type response [11]. To determine the cell type in which Bcl-3 might carry out critical functions to protect the host, we investigated mice in which Bcl-3 was conditionally ablated primarily in DCs [CD11c-Cre; Bcl-3fixffx] (Bcl-3-∆-DC) (conventional and monocyte-derived DCs, and certain macrophage subpopulations, such as alveolar macrophages, exhibit high levels of recombination; plasmacytoid DCs exhibit somewhat lower levels; low or no apparent recombination in many other cell types, including T-cells; Supporting Information Fig. 1A). We infected Bcl-3-∆-DC, WT (Bcl-3fix/fix), and Bcl-3-deficient (Bcl-3−/−) mice with T. gondii i.p., and monitored their health and immune responses for up to 40 days. Surprisingly, Bcl-3-∆-DC mice were as susceptible to T. gondii as mice globally deficient in Bcl-3, with almost all mutant mice succumbing between 3–5 weeks, while nearly all wild-type mice remained alive and healthy (Fig. 1A). Furthermore, Bcl-3-∆-DC and Bcl-3-deficient, but not WT mice incurred similarly severe weight losses by day 19 (Fig. 1B). Therefore, expression of Bcl-3 in CD11c+ cells (DCs) was absolutely required for protection from T. gondii.
Figure 1.
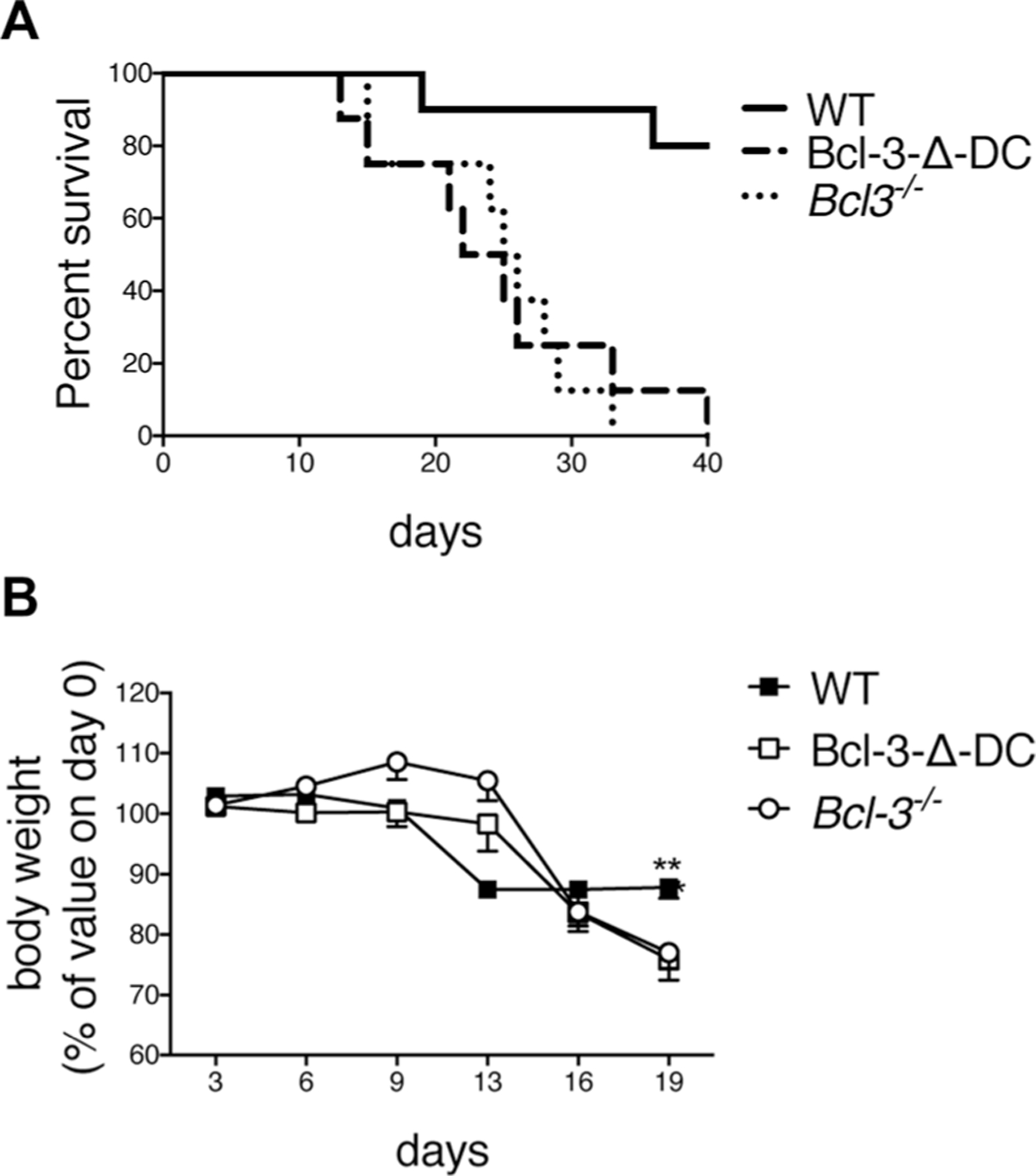
Bcl-3 is required in DCs for resistance to T. gondii. (A) WT, Bcl-3−/−, and Bcl-3-∆-DC mice were infected with 20 cysts of T. gondii (ME49 strain) and survival was monitored. WT n = 8; Bcl-3-∆-DC n = 8; Bcl-3−/− n = 10. (B) Body weight of individual mice from (A) that remained alive during the time of infection, shown as mean ± SEM; n = 5–6 mice/group. **p<0.01. Results in (A) were analyzed by the log-rank Mantel–Cox test. Unpaired, two-tailed Student’s t-test was used for (B). Data shown are from one experiment, representative of two experiments performed.
Innate immunity to T. gondii is not impaired by loss of Bcl-3 in DCs
To explore whether T. gondii susceptibility of mice with global or DC-specific deficiency in Bcl-3 might be due to already compromised early innate production of IL-12 by DCs, we measured IL-12p40 and IL-12p70 three days post infection. IL-12p40 and IL-12p70 protein levels were induced to a similar degree in WT, Bcl-3-∆-DC, and Bcl-3-deficient mice, as measured with Elisa assays of serum and fluid from the peritoneal cavity (Fig. 2A). This suggested that early production of IL-12 in mice was not significantly affected by the lack of Bcl-3 in DCs. In further support of this conclusions, we observed normal production of this cytokine in serum immediately after injection of mutant mice with T. gondii extracts (soluble T. gondii antigen; STAg) (Supporting Information Fig. 1B) or upon in vitro stimulation of splenic DCs with various TLR ligands, or upon similar stimulations of cells isolated instead from the peritoneal cavity 7 days p.i. (Supporting Information Fig. 1C and D); furthermore, IL-12 production by stimulated bone-marrow-derived DCs was unaffected by loss of Bcl-3 [13]. To confirm that innate immunity was not impaired in other ways, we also measured NK-cell activity. These cells respond to the initial wave of IL-12 to become the main source of early innate IFN-γ, which is essential to protect the host until adaptive immune responses are established [15, 23]. WT, Bcl-3-∆-DC, and Bcl-3-deficient mice were infected with T. gondii, and splenic NK cells were assayed 3 days later for intracellular IFN-γ production after stimulation with anti-NK1.1, PMA/Ionomycin or no stimulation (Fig. 2B and data not shown). Regardless of which mice NK cells were derived from, infection led to similar levels of IFN-γ production. By all of these measures, innate DC-mediated immune responses were intact. We additionally determined that recruitment of the inflammatory monocyte-derived population that gives rise to IL-12-producers in the peritoneal cavity 1 week p.i. was not impaired in the absence of Bcl-3 either (Supporting Information Fig. 1E). These results further suggested that loss of resistance to T. gondii exhibited by Bcl-3-∆-DC and Bcl-3-deficient mice might be due to impaired adaptive immunity.
Figure 2.
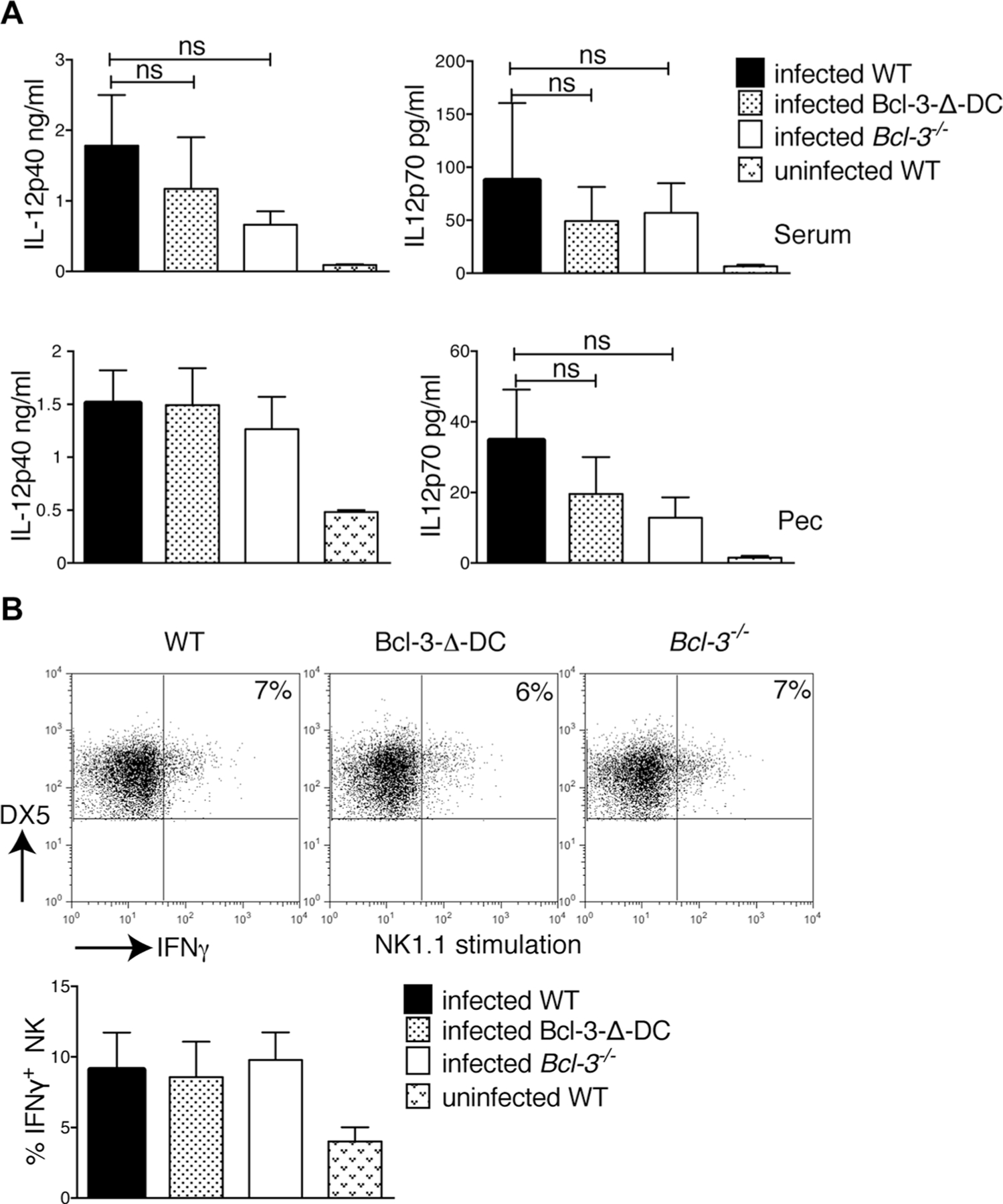
Innate immune response after T. gondii infection is independent of Bcl-3 expression in CD11c+ cells. (A) WT, Bcl-3−/−, and Bcl-3-∆-DC mice were infected with 20 cysts of T. gondii (ME49 strain). Three days p.i., the levels of IL-12-p40 (left) and IL-12p70 (right) in serum (top) and fluid of peritoneal cavity (PEC, bottom) were assessed by ELISA. Data are shown as mean ± SEM for n = 6 mice/group, pooled from 2 experiments; ns = not significant. (B) Mice were infected as in (A), splenocytes were isolated 3 days later and stimulated with plate-bound NK1.1 for 6 h. Cells were stained for DX5 and CD3, then for intracellular IFN-γ, and IFN-γ production by NK cells was assessed after gating on DX5+CD3− cells. Representative FACS analysis is shown (top) as well as the mean ± SEM; n = 8 mice/group pooled from 2 experiments. Statistical analysis performed by unpaired, two-tailed Student’s t-test.
Adaptive immunity to T. gondii infection depends on Bcl-3 expression in DCs
Next we investigated IFN-γ levels 1 week p.i., when T-cells are optimally activated and the main source of this critical cytokine. At this time IFN-γ was readily apparent in serum of WT, but largely absent in serum of Bcl-3-∆-DC and Bcl-3-deficient mice (Fig. 3A; levels were also reduced in the peritoneal cavity; Supporting Information Figure 1F). Importantly, there was pronounced parasitemia in the peritoneal cavity of both mutant, but not WT mice, correlating with the greatly reduced levels of IFN-γ (Fig. 3B). At 1 week p.i. we also observed a significant decrease in levels of both IL-12p40 and IL-12p70 in the serum of Bcl-3-∆-DC and Bcl-3-deficient mice compared to WT mice (Fig. 3C; levels also trended lower in peritoneal cavity; Supporting Information Fig. 1F).
Figure 3.
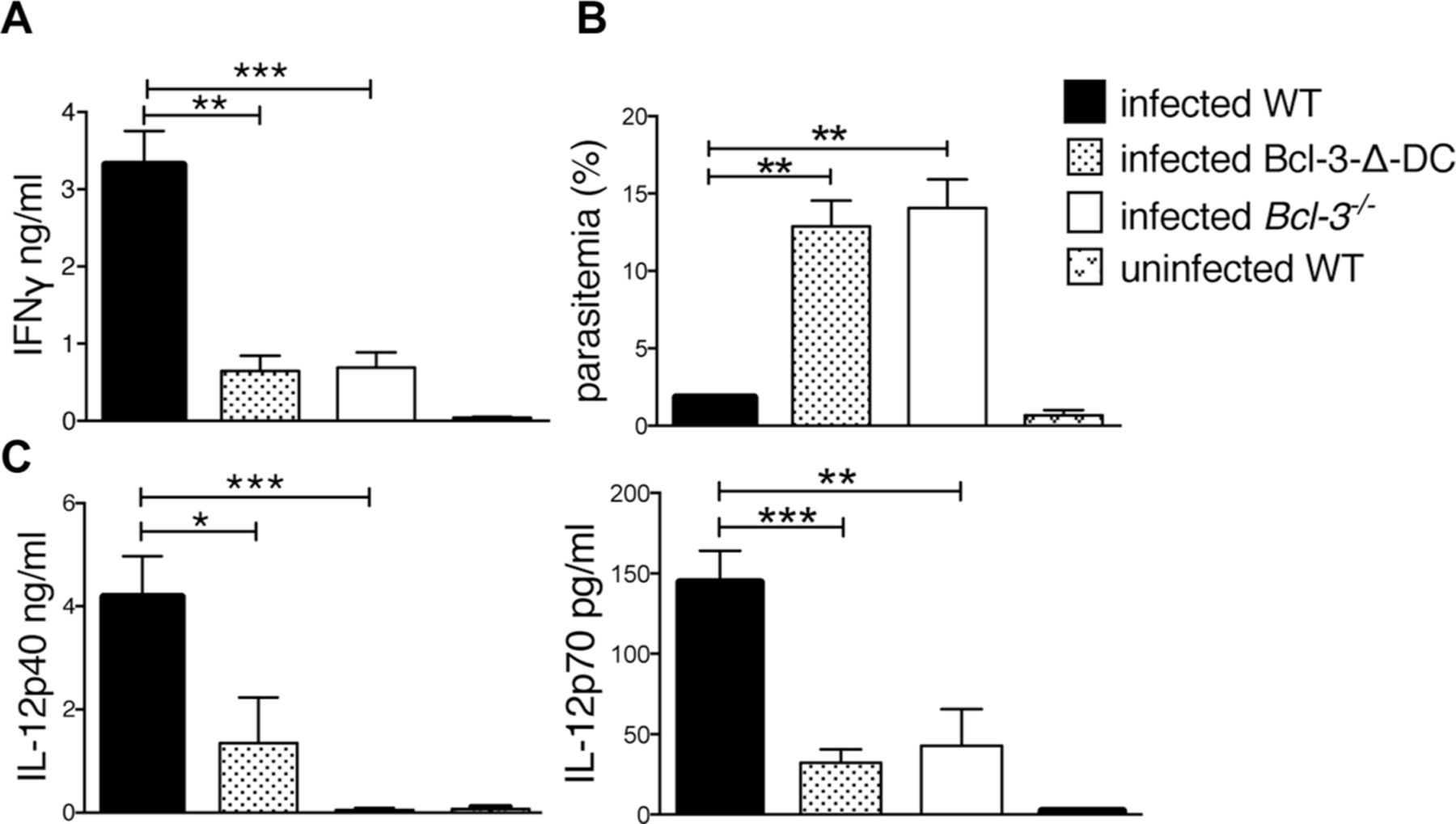
Bcl-3 expression in DCs is required for IFN-γ production in response to T. gondii infection. (A) WT, Bcl-3−/−, and Bcl-3-∆-DC mice were infected with T. gondii and IFN-γ was measured in serum from infected animals 7 days p.i. Mean ± SEM; n = 14 mice/group pooled from 2 experiments. (B) Measurement of % infected cells in the peritoneal cavity of infected mice. Mean ± SEM; n = 6 mice/group pooled from 2 experiments. (C) Levels of IL-12p70 and IL-12-p40 in serum were assessed by ELISA 5 and 7 days p.i. Data are shown as mean ± SEM for n = 6 mice/group, pooled from 2 experiments. *p<0.05, **p<0.01, ***p<0.0001, unpaired, two-tailed Student’s t-test.
Based on these findings we examined CD4+ T-cells 1 week post infection. Splenic and peritoneal cells were isolated from infected WT, Bcl-3-∆-DC, and Bcl-3-deficient mice and stimulated ex vivo with anti-CD3. Intracellular IFN-γ levels in CD4+ T-cells were significantly reduced when derived from infected Bcl-3-∆-DC and Bcl-3-deficient mice compared to infected WT mice (Fig. 4A). To more specifically test the antigen-specific response we isolated splenic and peritoneal CD4+ T-cells from infected WT, Bcl-3-∆-DC, and Bcl-3-deficient animals, incubated them with irradiated APCs in the presence or absence of T. gondii antigens (STAg) and measured the IFN-γ levels in the supernatants after 3 days with CBA. CD4+ T-cells isolated from spleens and peritoneal cavities of infected Bcl-3-∆-DC and Bcl-3-deficient mice produced significantly less IFN-γ than the corresponding control cells isolated from WT mice (Fig. 4B). The absence of Bcl-3 did not alter numbers of DCs in lymphoid organs under homeostatic conditions (prior to infection) [13], but did reduce numbers in draining lymph node by day 7 p.i., with a trend evident at d3 p.i. (Supporting Information Fig. 1G), which would be consistent with a shortened half-life of activated DCs [13] (see Discussion).
Figure 4.
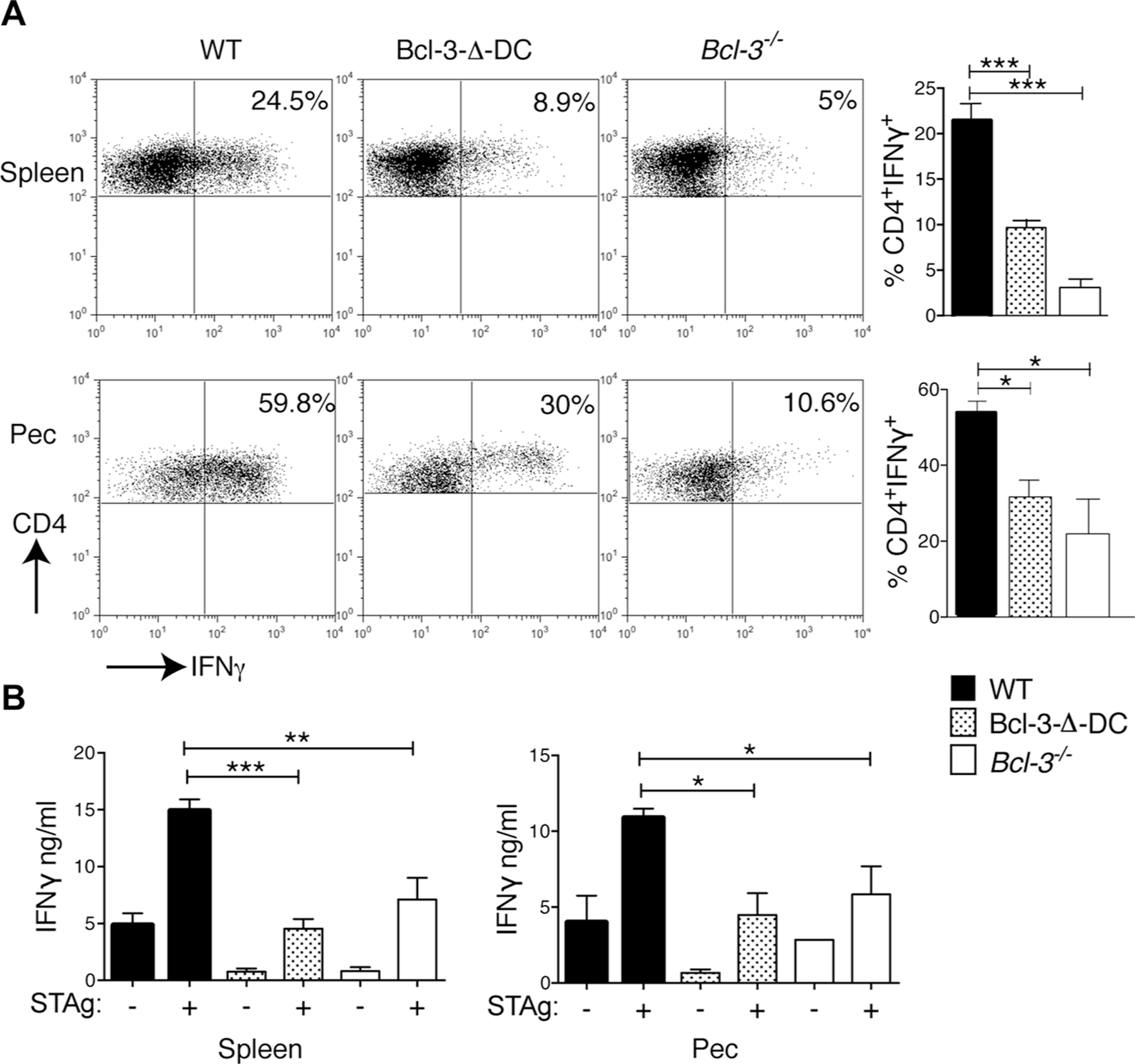
Bcl-3 expression in DCs is required for CD4-mediated IFN-γ production in response to T. gondii infection. (A) WT, Bcl-3−/−, and Bcl-3-∆-DC mice were infected with T. gondii. Splenocytes (top) and cells from peritoneal cavity (PEC, bottom) were isolated 1 week later, stimulated for 6 h with plate-bound anti-CD3, and intracellular IFN-γ production by CD4+ cells was measured by flow cytometry. Representative plots are shown (left) and data are summarized as mean ± SEM; n = 8 mice/group pooled from 2 experiments (right). (B) CD4+ T-cells isolated from splenocytes (left) and PEC cells (right) of infected mice as in (A) were stimulated in the presence STAg and irradiated WT splenocytes for 72 h and IFN-γ measured with CBA. Data are shown as mean ± SEM; n = 8 mice/group pooled from 2 experiments. *p<0.05, **p<0.01, ***p<0.0001, unpaired, two-tailed Student’s t-test.
We additionally investigated the activation of CD4+ and CD8+ T-cells 18 days p.i., when CD8 T-cells feature more prominently in adaptive immunity. Splenocytes were isolated from infected WT, Bcl-3-∆-DC, and Bcl-3-deficient mice and stimulated with anti-CD3. Intracellular IFN-γ levels in CD8+ and CD4+ T-cells from Bcl-3-∆-DC and Bcl-3-deficient as compared to WT mice were severely reduced (Fig. 5A). Finally, CD8+ T-cells, isolated from WT and Bcl-3-∆-DC mice 18 days p.i., were incubated with BMDCs previously infected with T. gondii in order to specifically query the antigen-specific response. CD8+ T-cells isolated from Bcl-3-∆-DC mice produced significantly less IFN-γ when stimulated with T. gondii-infected BMDCs (Fig. 5B). We conclude that Bcl-3 plays a vital role in DCs required to effectively prime CD4 and CD8 T-cells during infection with T. gondii parasites and thereby assures host resistance.
Figure 5.
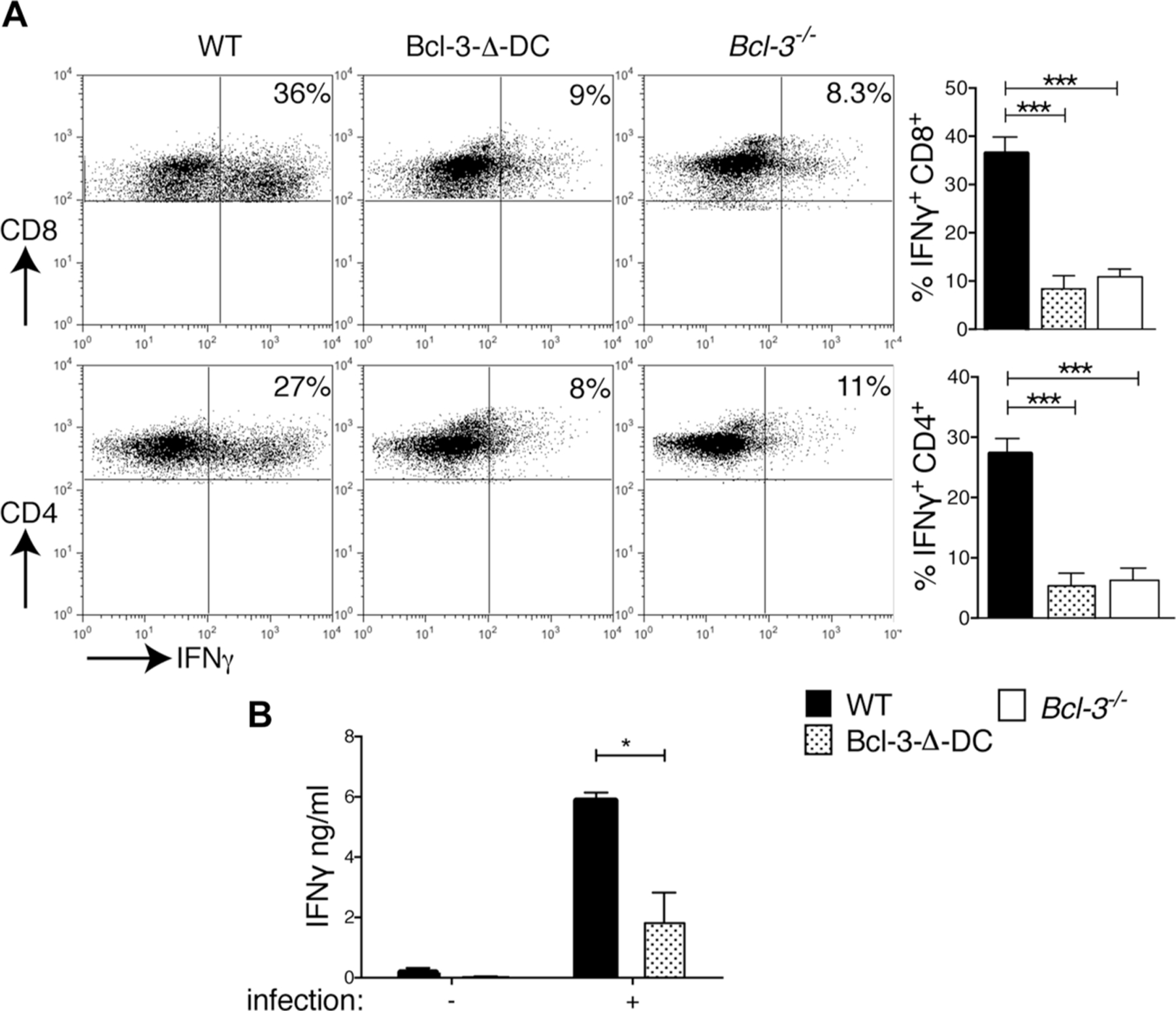
Bcl-3 expression in DCs is required for CD8-mediated IFN-γ production in response to T. gondii infection. (A) WT, Bcl-3−/−, and Bcl-3-∆-DC mice were infected with T. gondii; splenocytes were isolated 18 days later and stimulated for 6 h with plate-bound anti-CD3; and intracellular IFN-γ in CD4+ (bottom) and CD8+ (top) cells measured by flow cytometry. Representative plots are shown (left) and summarized as mean ± SEM of n = 8 mice/group pooled from 2 experiments (right). (B) Splenic CD8+ T-cells were isolated 18 days p.i., stimulated for 72 h with BMDCs previously infected with T. gondii parasites (or uninfected control BMDCs) and IFN-γ production was measured with CBA. Data are shown as mean ± SEM; n = 8 mice/group pooled from 2 experiments. *p<0.05, **p<0.01, ***p<0.0001, unpaired, two-tailed Student’s t-test.
Discussion
We previously reported that infection of Bcl-3-deficient animals with T. gondii resulted in death of the animals within 3–5 weeks, but in which cell type(s) Bcl-3 executed critical functions required for resistance remained unknown [11]. We now report the surprising finding that mice lacking Bcl-3 in DCs (Bcl-3-∆-DC) were as susceptible to infection with this pathogen as mice globally deficient in this regulator, succumbing during the exact same time interval. We demonstrate that Bcl-3 was required in DCs to generate a protective Th1-type adaptive immune response and thereby confer resistance to the host. At the time when adaptive immune responses begin to predominate in infected WT mice, the amount of the critical cytokine IFN-γ produced in mice lacking Bcl-3 in DCs was notably reduced; in line with this, the percentage of T. gondii antigen-specific, IFN-γ-producing CD4 and CD8 T-cells were significantly lower in these mutant mice. Innate host defense, which protects the host early during infection, appeared to be fully intact and thus independent of Bcl-3. The responses in mice lacking Bcl-3 in DCs phenocopied those in mice with germline ablation, indicating that host resistance was entirely dependent on functions of Bcl-3 in dendritic cells, with no evidence for significant roles of Bcl-3 in other cell types.
Our findings demonstrate that Bcl-3-deficient DCs were impaired in their ability to generate an adequate T-cell-mediated adaptive immune response during infection, while their contributions to the initial innate immunity appeared intact. In complementary studies we found that activated Bcl-3-deficient DCs were partially impaired in their ability to prime/expand T-cells to antigens in vitro, due in part to reduced survival [13], which may be reflected here by the reduced numbers of mutant DCs in draining lymph node 1 week after infection. The present findings strongly suggest that this partial defect of Bcl-3-deficient DCs had significant physiological consequences.
Nevertheless, it remained possible that the mutant DCs might also have been somewhat defective in carrying out critical innate functions early during infection in mice, specifically the production of IL-12, thereby potentially also compromising adaptive responses. This cytokine is absolutely required for resistance to T. gondii, not only because it directs T-cell differentiation to the Th1 phenotype, but also because it stimulates innate defenses, in particular early IFN-γ production by NK cells [19–22]. However, several lines of evidence indicate that the early, innate response was intact in Bcl-3-∆-DC mice, starting with the fact that mice succumbed after 3 weeks, not during the first 10 days, as is the case for mice lacking IL-12 or Batf3, or mice lacking MyD88 in DCs [[20–22, [24]]. Also, IL-12 was produced at near WT levels in both serum and in the peritoneal cavity of Bcl-3-∆-DC mice early after infection and DC/myeloid cells did not show any intrinsic defects in IL-12 production upon direct stimulation in vitro or in vivo. Furthermore, early NK-cell-mediated IFN-γ production, which follows initial exposure to IL-12 [14, 15], was unaffected by the absence of Bcl-3. Recently, circulating monocytes were reported to infiltrate the peritoneal cavity within days upon infection (inflammatory monocytes), where some differentiate into CD11c+, MHC II+, IL-12-producing DC-like cells [25]; our data showed that the absence of Bcl-3 did not affect the accumulation of these cells by 1 week post infection. In contrast to early times after infection, IL-12 levels were nevertheless reduced subsequently, by about 1 week p.i.; rather than reflecting a DC-intrinsic defect in IL-12 production, however, this reduction was potentially due to the aforementioned shortened half-life of activated DCs and the paucity of CD4+ T-cell-derived IFN-γ, a known amplifier of IL-12 [26, 27]. Disruption of this positive feedback loop may have contributed further to loss of resistance during persistent infection, thus amplifying the initial priming defect.
These data suggested that Bcl-3 was not directly involved in the production of IL-12 by DCs (conventional DCs and monocyte-derived DCs). It has recently been reported that TLR-11/12-mediated recognition of T. gondii may trigger IL-12 production not only in, e.g., CD8-α+ conventional DCs, but also in plasmacytoid dendritic cells (PDCs), and together with type I IFN-, PDCs may activate NK cells to produce IFN-γ [28, 29]. Among the various DC subtypes, PDCs are an exception as they express only low levels of CD11c and thus Bcl-3 was not likely to be fully ablated in these cells. Importantly though, mice globally deficient in Bcl-3 also exhibited fully intact innate production of IL-12 and IFN-γ, arguing against a requirement for Bcl-3 in the production of IL-12 by any of the DC subtypes.
Mice globally deficient in NF-κB1 or NF-κB2 have been reported to succumb to T. gondii infection within 4 and 5–8 weeks, respectively. Bcl-3 regulates gene transcription via homodimers of p50/NF-κB1 and p52/NF-κB2, which might suggest that mice deficient in these factors could exhibit similar defects. However, p50/NF-κB1 and p52/NF-κB2 do not only form homodimers but are also part of many other complexes with diverse functions in many cell types. It has in fact been proposed that increased mortality in the infected NF-κB1- and NF-κB2-deficient mice may be due to prominent roles of these factors in T-cells, also affecting their capacity to produce IFN-γ [30, 31]. It will be of future interest to investigate the relationships between Bcl-3 and the two factors in DCs.
In conclusion, adaptive CD4 and CD8 T-cell-mediated immunity was severely compromised in Bcl-3-∆-DC mice, even though DC-mediated initial innate immunity was intact and Th1-polarizing conditions preserved. Therefore, Bcl-3 was required in DCs to fully prime and activate T-cells, thereby establishing the adaptive Th1-type-dependent immunity necessary to resist T. gondii parasites.
Materials and methods
Mice
Itgax-cre (CD11c-cre) (Jackson Laboratories), Bcl-3−/− [11] and Bcl-3fix/fix [13] were all on C57BL/6 backgrounds. Bcl-3 sufficient controls for conditional knockout mice were littermates. All mice were housed in NIAID Institute facilities and all experiments were done with approval of the NIAID Animal Care and Use Committee and in accordance with all relevant institutional guidelines.
Flow cytometry
Samples were stained at 4°C in the presence of Fc Block (2.4G2; BD Biosciences) in flow cytometry buffer (PBS 2% FBS). Antibodies used: allophycocyanin-conjugated anti-CD4 (RM4–5), fluorescein isothiocyanate anti-CD3ε (145–2C11), allophycocyanin anti-CD8 (53–6.7), phycoerythrin-cyanine7 anti-CD11c (HL-3) (BD Biosciences); phycoerythrin-cyanine7 anti-IFN-γ (XMG1.2) (eBioscience); allophycocyanin anti-CD8 (53–6.7), allophycocyanin anti-CD49b (DX5), allophycocyanin-cy7 MHC-II (M5/114.15.2) (Biolegend). Dead cells were excluded by Aqua Live/Dead fixable kit (Invitrogen). Stained cells were analyzed on a FACS CANTO and data analyzed with FlowJo software (BD Biosciences).
T. gondii infection
T. gondii cysts from the avirulent strain ME-49 were prepared from brains of infected C57BL/6 mice. For experimental infections, mice were inoculated i.p. with an average of 20 cysts/animal and monitored daily for survival.
Intracellular cytokine staining
To determine early IFN-γ production by NK cells, splenocytes were isolated from mice 3 days p.i. with T. gondii. Cells were stimulated with plate-bound NK1.1 in the presence of 10 μg/mL monensin (Sigma-Aldrich) for 6 h and then stained with APC-anti-DX5 and FITC-CD3ε. Cells were fixed with 1% formaldehyde, permeabilized with cytofix/cytoperm kit (BD) and intracellular IFN-γ levels were determined with flow cytometry after staining with PE-cy7-anti-IFN-γ antibody. To assess CD4- and/or CD8-mediated IFN-γ production, cells were isolated from PECs and spleens 7 and 18 days p.i., stimulated with anti-CD3 for 6 h, and then intracellular analysis for IFN-γ was performed as described above.
Cytokine measurement
For measurement of IFN-γ production in supernatants by antigen-activated CD4 T-cells ex vivo, single-cell suspensions were prepared from peritoneal fluid and spleen from mice 7 days p.i. with T. gondii. Splenic CD4+ T lymphocytes were purified by positive selection using MACS microbeads (CD4+ T-cell isolation kit Miltenyi Biotec) according to the manufacturer’s protocol. 2.5×105 purified CD4+ T-cells were subsequently co-cultured with 7.5 × 105 irradiated splenocytes for 72 h in the presence of 5 μg/mL STAg, and supernatants were removed for cytokine assays (cytometric bead analysis, CBA, BD Biosciences, CBA Mouse inflammation kit). To assess IFN-γ production in supernatants of antigen-activated CD8 T-cells, splenic CD8 T-cells were purified by positive selection from mice 18 days p.i., using MACS microbeads (CD8+ T-cell isolation kit Miltenyi Biotec) according to the manufacturer’s protocol. 105 CD8+ T cell were co-cultured with T. gondii-infected or uninfected 3×104 BMDCs and culture supernatants were harvested after 72 h for cytokine assays (CBA). For infection, BMDCs were incubated with irradiated (15 000 rads) T. gondii tachyzoites (RH88) at five parasites per DC ratio. After incubation for 12 h, free-floating parasite were removed by centrifugation at 900 rpm for 10 min and cells were plated in parallel with control uninfected BMDCs in 96-well round-bottom micro titer plates. To measure IL-12p40, Il-12p70, and IFN-γ production, serum and/or fluid from the peritoneal cavity were collected from noninfected and T. gondii-infected mice, or from mice 6 h after STAg (10 μg) injection and tested by ELISA (R&D Systems and BD Biosciences). To measure IL-12p40 production by splenic DCs, CD11c+ cells were purified with CD11c-selecting MACS microbeads (Miltenyi Biotec) and 105 cells were plated o.n. in the presence of STAg (5 μg/mL), LPS (100ng/mL, Invivogen), or poly I:C (100 μg/mL, Invivogen). To measure IL-12p40 production by inflammatory monocyte-derived cells, cells were isolated from peritoneal cavities of T. gondii-infected mice, and 3×105 cells were plated o.n. in the presence of LPS (100 ng/mL), CpG (1 μg/mL), Pam3CSK (1 μg/mL) or poly I:C (100 μg/mL).
Quantification of inflammatory monocytes and DCs
Single cell suspensions were prepared from peritoneal fluid or from collagenase D (Roche)-digested spleens and mesenteric LNs of T. gondii-infected mice; cells were then stained for CD11c and MHC-II and absolute numbers were determined via flow cytometry with CountBright Absolute Counting Beads (Invitrogen).
Parasitemia
To assess parasitemia, the percentage of infected cells was determined by counting after cytpospin of peritoneal fluid from infected mice.
Statistical analysis
Data were recorded as the mean ±SEM. Differences between groups were analyzed by unpaired, two-tailed Student’s t-tests. Results with a p value of 0.05 or less were considered significant (Prism; GraphPad Software). Survival studies were analyzed by the log-rank Mantel-Cox test. The number of independent data points (n) and the number of independent experiment is stated in figure legends.
Supplementary Material
Acknowledgments:
This research was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD.
Abbreviations:
- DC
dendritic cell
- BMDC
Bone marrow-derived DC
- PDC
plasmacytoid DC
- NK
natural killer (cell)
- PEC
peritoneal cavity
- STAg
soluble tachyzoite antigen
- CBA
cytometric bead array
- p.i.
post infection
- i.p.
intraperitoneal injection
Footnotes
Additional supporting information may be found in the online version of this article at the publisher’s web-site
Conflict-of-interest disclosure: The authors declare no commercial or financial conflict of interest.
References
- 1.Hayden MS and Ghosh S, Shared principles in NF-kappaB signaling. Cell 2008. 132: 344–362. [DOI] [PubMed] [Google Scholar]
- 2.Claudio E, Brown K and Siebenlist U, NF-kappaB guides the survival and differentiation of developing lymphocytes. Cell Death Differ. 2006. 13: 697–701. [DOI] [PubMed] [Google Scholar]
- 3.Siebenlist U, Brown K and Claudio E, Control of lymphocyte development by nuclear factor-kappaB. Nat. Rev. Immunol. 2005. 5: 435–445. [DOI] [PubMed] [Google Scholar]
- 4.Schuster M, Annemann M, Plaza-Sirvent C and Schmitz I, Atypical IkappaB proteins - nuclear modulators of NF-kappaB signaling. Cell Commun. Signal. 2013. 11: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang VY, Huang W, Asagiri M, Spann N, Hoffmann A, Glass C and Ghosh G, The transcriptional specificity of NF-kappaB dimers is coded within the kappaB DNA response elements. Cell Rep. 2012. 2: 824–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohno H, Takimoto G and McKeithan TW, The candidate proto-oncogene bcl-3 is related to genes implicated in cell lineage determination and cell cycle control. Cell 1990. 60: 991–997. [DOI] [PubMed] [Google Scholar]
- 7.Mathas S, Johrens K, Joos S, Lietz A, Hummel F, Janz M, Jundt F et al. , Elevated NF-kappaB p50 complex formation and Bcl-3 expression in classical Hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood 2005. 106: 4287–4293. [DOI] [PubMed] [Google Scholar]
- 8.Maldonado V and Melendez-Zajgla J, Role of Bcl-3 in solid tumors. Mol. Cancer 2011. 10: 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pene F, Paun A, Sonder SU, Rikhi N, Wang H, Claudio E and Siebenlist U, The IkappaB family member Bcl-3 coordinates the pulmonary defense against Klebsiella pneumoniae infection. J. Immunol. 2011. 186: 2412–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Wang H, Claudio E, Brown K and Siebenlist U, A role for the IkappaB family member Bcl-3 in the control of central immunologic tolerance. Immunity 2007. 27: 438–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franzoso G, Carlson L, Scharton-Kersten T, Shores EW, Epstein S, Grinberg A, Tran et al. , Critical roles for the Bcl-3 oncoprotein in T cell-mediated immunity, splenic microarchitecture, and germinal center reactions. Immunity 1997. 6: 479–490. [DOI] [PubMed] [Google Scholar]
- 12.Tang W, Wang H, Claudio E, Tassi I, Ha HL, Saret S and Siebenlist U, The oncoprotein and transcriptional regulator Bcl-3 governs plasticity and pathogenicity of autoimmune T-cells. Immunity 2014. 41: 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tassi I, Claudio E, Wang H, Tang W, Ha HL, Saret S, Ramaswamy M et al. , The NF-kappaB regulator Bcl-3 governs dendritic cell antigen presentation functions in adaptive immunity. J. Immunol. 2014. 193: 4303–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gazzinelli RT, Wysocka M, Hayashi S, Denkers EY, Hieny S, Caspar P, Trinchieri G and Sher A, Parasite-induced IL-12 stimulates early IFN-gamma synthesis and resistance during acute infection with Toxoplasma gondii. J. Immunol. 1994. 153: 2533–2543. [PubMed] [Google Scholar]
- 15.Trinchieri G, Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003. 3: 133–146. [DOI] [PubMed] [Google Scholar]
- 16.Yap GS, Shaw MH, Ling Y and Sher A, Genetic analysis of host resistance to intracellular pathogens: lessons from studies of Toxoplasma gondii infection. Microbes Infect. 2006. 8: 1174–1178. [DOI] [PubMed] [Google Scholar]
- 17.Munoz M, Liesenfeld O and Heimesaat MM, Immunology of Toxoplasma gondii. Immunol. Rev. 2011. 240: 269–285. [DOI] [PubMed] [Google Scholar]
- 18.John B, Harris TH, Tait ED, Wilson EH, Gregg B, Ng LG, Mrass P et al. , Dynamic imaging of CD8(+) T-cells and dendritic cells during infection with Toxoplasma gondii. PLoS Pathog. 2009. 5: e1000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reis e Sousa C, Hieny S, Scharton-Kersten T, Jankovic D, Charest H, Germain RN and Sher A, In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J. Exp. Med. 1997. 186: 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu CH, Fan YT, Dias A, Esper L, Corn RA, Bafica A, Machado FS and Aliberti J, Cutting edge: dendritic cells are essential for in vivo IL-12 production and development of resistance against Toxoplasma gondii infection in mice. J. Immunol. 2006. 177: 31–35. [DOI] [PubMed] [Google Scholar]
- 21.Mashayekhi M, Sandau MM, Dunay IR, Frickel EM, Khan A, Goldszmid RS, Sher A et al. , CD8alpha(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity 2011. 35: 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hou B, Benson A, Kuzmich L, DeFranco AL and Yarovinsky F, Critical coordination of innate immune defense against Toxoplasma gondii by dendritic cells responding via their Toll-like receptors. Proc. Natl. Acad. Sci. USA 2011. 108: 278–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldszmid RS, Bafica A, Jankovic D, Feng CG, Caspar P, Winkler-Pickett R, Trinchieri G and Sher A, TAP-1 indirectly regulates CD4+ T cell priming in Toxoplasma gondii infection by controlling NK cell IFN-gamma production. J. Exp. Med. 2007. 204: 2591–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gazzinelli RT, Hieny S, Wynn TA, Wolf S and Sher A, Interleukin 12 is required for the T-lymphocyte-independent induction of interferon gamma by an intracellular parasite and induces resistance in T-cell-deficient hosts. Proc. Natl. Acad. Sci. USA 1993. 90: 6115–6119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldszmid RS, Caspar P, Rivollier A, White S, Dzutsev A, Hieny S, Kelsall B et al. , NK cell-derived interferon-gamma orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity 2012. 36: 1047–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abdi K, Singh N and Matzinger P, T-cell control of IL-12p75 production. Scand. J. Immunol. 2006. 64: 83–92. [DOI] [PubMed] [Google Scholar]
- 27.Ma X, Chow JM, Gri G, Carra G, Gerosa F, Wolf SF, Dzialo R and Trinchieri G, The interleukin 12 p40 gene promoter is primed by interferon gamma in monocytic cells. J. Exp. Med. 1996. 183: 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koblansky AA, Jankovic D, Oh H, Hieny S, Sungnak W, Mathur R, Hayden MS et al. , Recognition of profilin by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity 2013. 38: 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pepper M, Dzierszinski F, Wilson E, Tait E, Fang Q, Yarovinsky F, Laufer TM et al. , Plasmacytoid dendritic cells are activated by Toxoplasma gondii to present antigen and produce cytokines. J. Immunol. 2008. 180: 6229–6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caamano J, Tato C, Cai G, Villegas EN, Speirs K, Craig L, Alexander J and Hunter CA, Identification of a role for NF-kappa B2 in the regulation of apoptosis and in maintenance of T cell-mediated immunity to Toxoplasma gondii. J. Immunol. 2000. 165: 5720–5728. [DOI] [PubMed] [Google Scholar]
- 31.Harris TH, Wilson EH, Tait ED, Buckley M, Shapira S, Caamano J, Artis D and Hunter CA, NF-kappaB1 contributes to T cell-mediated control of Toxoplasma gondii in the CNS. J. Neuroimmunol. 2010. 222: 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.