

Abstract
Reactive oxygen species (ROS) are formed by virtually all tissues. In normal concentrations they facilitate many physiologic activities, but in excess they cause oxidative stress and tissue damage. Local antioxidant enzyme synthesis in cells is regulated by the cytoplasmic KEAP-1/Nrf2 complex, which is stimulated by ROS, to release Nrf2 for entry into the nucleus, where it upregulates antioxidant gene expression. Major antioxidant enzymes include glutathione peroxidase (GPx), catalase (CAT), superoxide dismutases (SOD), hemoxygenases (HO), and peroxiredoxins (Prdx). Notably, the pancreatic islet β-cell does not express GPx or CAT, which puts it at greater risk for ROS damage caused by postprandial hyperglycemia. Experimentally, overexpression of GPx in β-cell lines and isolated islets, as well as in vivo studies using genetic models of type 2 diabetes (T2D), has demonstrated enhanced protection against hyperglycemia and oxidative stress. Oral treatment of diabetic rodents with ebselen, a GPx mimetic that is approved for human clinical use, reproduced these findings. Prdx detoxify hydrogen peroxide and reduce lipid peroxides. This suggests that pharmacologic development of more potent, β-cell–specific antioxidants could be valuable as a treatment for oxidative stress due to postprandial hyperglycemia in early T2D in humans.
Article Highlights
We examined evidence that postprandial hyperglycemia in early type 2 diabetes (T2D) causes oxidative stress.
We addressed the question of whether cessation of high-fat diet and return to a normal diet in T2D rodents cause improved glucose control and less oxidative stress.
Our findings are that oxidative stress is associated with functional and structural changes in β-cells and that this is met with an intrinsic response: translocate cytoplasmic Nrf2 to the nucleus to initiate gene expression of antioxidants.
The results of our work imply that early treatment of T2D with avoidance of a high-fat diet and the use of antioxidants might be helpful in restoring β-cell function.
Brief Overview of Endogenous Antioxidants
Key antioxidant enzymes in metabolic tissues include glutathione peroxidase (GPx) (1) (Fig. 1), which together with the glutamylcysteine ligase catalytic subunit (GCLC) synthesizes glutathione (GSH) to protect cells from oxidative damage caused by excessive levels of reactive oxygen species (ROS), specifically, intracellular peroxides and hydrogen peroxide (H2O2). Other antioxidants include the superoxide dismutases, SOD-1 and SOD-2, which metabolize superoxide radicals to form H2O2. SOD-1 is found primarily in the cytoplasm and is termed copper/zinc-SOD. SOD-2 is found principally in the mitochondria and is termed manganese-SOD. Another enzyme, catalase (CAT), is located in peroxisomes and metabolizes H2O2 to form water and oxygen, but it cannot metabolize lipid peroxides. Hemoxygenases (HO-1, HO-2), like GPx, catabolize intracellular peroxides and H2O2. A related group of antioxidants, the peroxiredoxins (Prdx), also detoxify hydrogen peroxide and reduce lipid peroxides (2,3).
Figure 1.
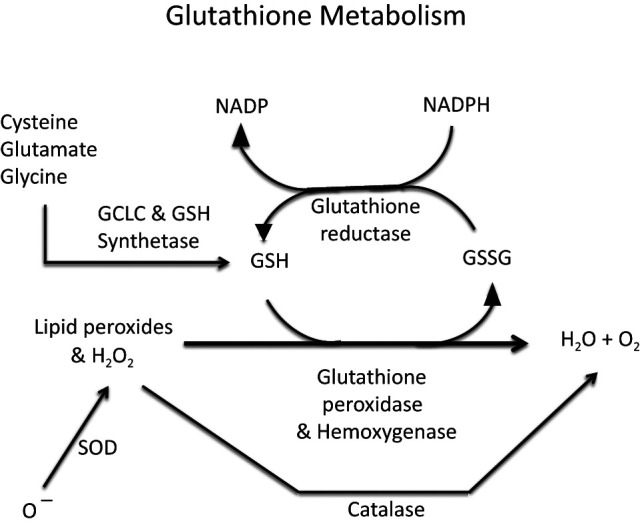
Glutathione metabolism principally regulated by the enzymes GCLC and GSH synthetase.
Uniquely, pancreatic β-cells contain very little GPx or CAT (4–6), which renders them especially vulnerable to oxidative stress. This is particularly germane to type 2 diabetes (T2D) wherein patients early in the disease are challenged by elevated levels of postprandial blood glucose that lead to excessive concentrations of glucose metabolites and ROS formation within β-cells via several different metabolic pathways (7) (Fig. 2).
Figure 2.
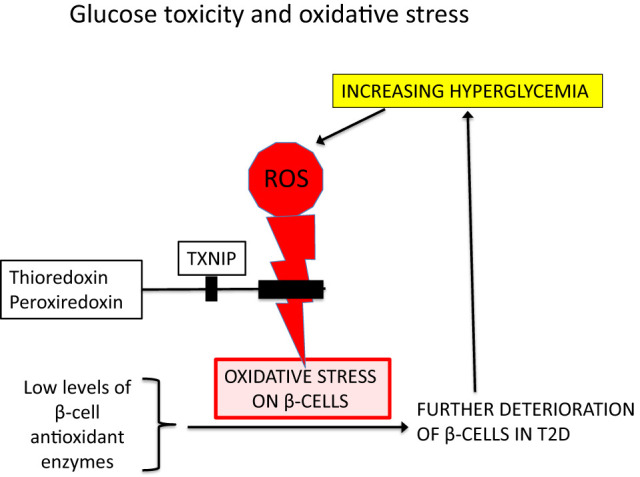
Glucose toxicity and oxidative stress. Accumulation of excess ROS occurs during chronic hyperglycemia, which causes progressive oxidative stress and further compromises already damaged β-cells that characteristically and uniquely do not express a full complement of antioxidant enzymes. A separate system features the antioxidant actions of thioredoxin and peroxiredoxin, which can be inhibited by cellular TXNIP (thioredoxin-interacting protein) (2).
In Vitro and In Vivo Studies of Antioxidant Effects on β-Cell Function
A full, exhaustive review of the many publications (8–15) that have linked oxidative stress, glucose toxicity, and use of antioxidant strategies for their amelioration would not be within the scope of this brief review. Rather, the intent of this article is to briefly review work from our group that suggests that earlier rather than later intervention of oxidative stress should be considered as a translational approach to management of T2D in humans. For example, Tanaka et al. (16) reported that in both HIT-T15 β-cells and in Zucker diabetic fatty (ZDF) rats, toxic effects of high glucose concentrations on Pdx-1 and MAFA gene expression were prevented by exogenous antioxidants. In the same study, treatment of diabetic ZDF rats with the antioxidant N-acetyl-l-cysteine or aminoguanidine prevented decreases in mRNAs for insulin, Pdx-1, and MAFA, insulin content, and glucose-induced insulin secretion. Both drugs prevented a rise in blood oxidative stress markers (8-hydroxy-2′-deoxyguanosine and malondialdehyde plus 4-hydroxy-2-nonenal). Studies by Harmon et al. (17) demonstrated that use of the drug troglitazone to treat ZDF animals fed a high-fat diet prevented hyperglycemia and preserved Pdx-1 gene expression and glucose-induced insulin secretion. These reports suggested that chronic oxidative stress is a likely mechanism for glucose toxicity that worsens the severity of T2D in these animals. Other studies, by Tanaka et al. (18), involved isolated human islets and the HIT-T15 β-cell line and were designed to test the hypothesis that high glucose concentrations cause the accumulation of intracellular ROS in β-cells and that decreasing or increasing the level of GPx would, respectively, augment or prevent the induction by ribose of β-cell functional defects. It was observed that glucose and ribose increase the intracellular oxidant load of islets and that decreasing endogenous provision of GSH by buthionine sulphoximine (an inhibitor of γ-glutamyl cysteine ligase synthetase, the enzyme that with GPx regulates GSH synthesis) (Fig. 1) enhances the deleterious effects of ribose. Tran et al. (19) observed that GCLC overexpression caused increases in GSH levels in β-cells, which protected them from the adverse effects of interleukin-1β–mediated ROS synthesis on glucose-stimulated insulin secretion. Investigators have demonstrated the protective effects of other antioxidant enzymes, primarily SOD and catalase, by overexpressing them in islets to test their efficacy in protection of β-cells against the adverse effects of streptozotocin and cytokines in models of type 1 diabetes (20–23). Harmon et al. (24) examined the effects of overexpressing a β-cell–specific GPx-1 transgene in C57BLKS/J mice fed a high-fat diet. Imaging confirmed that the GPx-1 gene localized to the β-cells only. GPx-1 transgenic mice were then backcrossed with db/db mice. Glucose levels in db/db-GPx(-) mice rose steadily over 20 weeks to 567 ± 14 mg/dL. Glucose levels in db/db-GPx(+) mice initially rose to 395 ± 48 mg/dL at the 10th week but thereafter reversed and decreased steadily to 195 ± 31 mg/dL by the 20th week. The explanation for this initial increase and then decrease in levels of glycemia in the GPx(+) transgenic mice was suggested to be that the transgene contained a glucose-sensitive insulin promoter, which was activated by progressive hyperglycemia in the animals. Further, islets from db/db-GPx(+) mice had larger β-cell volumes and greater insulin staining and granulation at 20 weeks compared with islets from db/db-GPx(-) mice. There were no differences in body weights of control and transgenic mice.
Ebselen, an Oral GPx Mimetic
The experimental evidence cited above led to the conclusion that the inherent absence of GPx-1 in islet β-cells makes them especially vulnerable to excessive ROS caused by postprandial hyperglycemia. Even though endogenous hemoxygenases are present in islets and have functions similar to those of GPx, they do not by themselves appear to be sufficient to defend against hyperglycemia-induced excessive levels of ROS in β-cells. Therefore, Mahadevan et al. (25) used rodents to examine the effects of ebselen, a GPx mimetic that can be taken orally, to assess the potential clinical benefits of enhancing GPx activity for people with T2D. Ebselen [2-phenyl-1, 2-benzisoselenazol-3(2H)-one] is a nontoxic seleno-organic drug that is a lipid soluble and an orally bioavailable small molecule (26–28). It has been safely used in randomized double-blind control human trials for hearing loss and neurovascular disease (29–32). Mahadevan et al. (25) reported that ebselen prevented islet apoptosis and preserved β-cell mass and function in diabetic ZDF rats fed a high-fat diet. Eight-week treatment with ebselen alone ameliorated fasting hyperglycemia, improved glucose tolerance and postprandial levels of glucose and insulin, and maintained lower levels of HbA1c. It also prevented accumulation of 4-HNE in β-cells and increased β-cell mass twofold over that of age-matched untreated animals. This larger β-cell mass had robust insulin staining, whereas the nontreated animals had 50% fewer β-cells that stained poorly for insulin. Islet apoptosis was abundant in untreated animals and rare in ebselen-treated animals. Intranuclear Pdx-1 and MAF-A levels were enhanced in treated animals.
In marked contrast to favorable outcomes in this ebselen study in rodents, Beckman et al. (33) reported that use of this drug in humans with diabetes failed to favorably affect levels of blood glucose or HbA1c. However, there were major differences between these two studies. Consistent with the Mahadevan study, in the Beckman study the investigators reported elevated levels of markers of oxidative stress in subjects with diabetes but also that ebselen did not lower these levels. This suggests that the dosage of the drug used was not adequate to be efficacious. Moreover, the average duration of diabetes in the human subjects was 6 years ± 4 years SD. It remains to be seen whether ebselen given early in the course of T2D in humans might have beneficial effects on continual worsening of postprandial hyperglycemia.
Perspective
Historically, the diagnosis of early, mild T2D has usually been met with diet and exercise advice, with a wait and see approach by physicians before starting treatment with hypoglycemic agents. This strategy disregards the fact that people with early-onset T2D inevitably experience chronic oxidative stress via postprandial hyperglycemia that may establish a feed-forward and important component of continuing β-cell dysfunction over time. This raises the question of whether there might be a role for antioxidant drugs for early treatment for T2D. It is true that most clinically available antioxidants that have been tried to treat diabetes met with no success. However, this is perhaps not too surprising because such drugs are not β-cell specific and are in general only mildly antioxidant in nature.
The potential for using oral GPx mimetics to provide treatment for early T2D is twofold. First, ebselen has no intrinsic hypoglycemic effects that might complicate treatment of early diabetes. This adverse effect has been observed with the use of some oral hypoglycemic agents, such as the sulfonylureas. Secondly, and more generally, there is evidence that treating postprandial hyperglycemia earlier rather than later in the course of T2D, using general approaches such as low-fat diet and weight control, has beneficial effects. Abebe et al. (34) reported that feeding a high-fat diet to normoglycemic ZDF rats over a matter of a few days caused a rise in intracellular markers of oxidative stress and frank diabetes. When the high-fat diet was replaced with a normal diet early in the course of the disease, β-cells intrinsically initiated self-repair structurally and functionally and the animals returned to a state of normoglycemia (Figs. 3–6). These beneficial effects were accompanied by appearance of intranuclear Nrf2 and the antioxidant enzyme HO-1 and reduction of an intracellular marker of oxidative stress (4-HNE). When the high-fat diet was replaced earlier but not later with the regular diet, the animals regained normoglycemia and their β-cells underwent structural repair with subsequent disappearance of intracellular Nrf2 and HO-1. These findings support the concept that use of antioxidant strategies early in T2D might be useful in arresting progression of the disease.
Figure 3.

KEAP1:Nrf2 complex. Illustration of the functional relationships between KEAP1 and Nrf2 under conditions of nonstress and oxidative stress is shown. Under quiescent conditions, Nrf2 is bound by KEAP1 in the cytoplasm, which ushers Nrf2 to proteosomes for degradation. In the face of oxidative stress, Nrf2 is released from KEAP1 and enters the nucleus, where it serves as a key activator for the promoter of antioxidant genes, such as HO-1. Adapted with permission from DeGroot’s Endocrinology (35).
Figure 6.

Electron microscopy. Recovery from structural damage in ZDF rat caused by high-fat diet and its reversal to normal structure after return to 2 weeks of regular diet is shown. Control data are shown in A–C; 9-day data in D–H; and HFD reversal data in I–K. Significant alterations can be noted in β-cells from animals fed with HFD, including dysmorphic secretory vesicles (arrowheads) (D), disorganized Golgi apparatus (asterisks) (E), numerous autophagic bodies (black arrowheads) (F), increased cytosolic free ribosomes (white arrowheads) (F), and significantly enlarged endoplasmic reticulum cisternae (arrowheads) (G and H).
Figure 4.
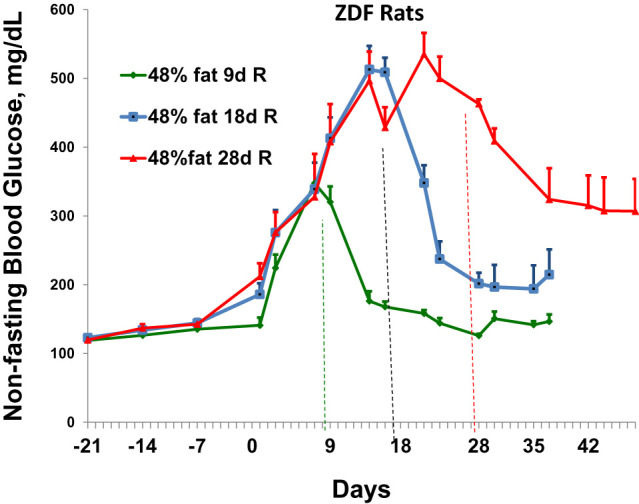
Assessment of the extent to which elevated nonfasting blood glucose levels caused by feeding of high-fat meals in ZDF rats can return to normal glycemia levels after reversal to normal diets for 2 weeks. The first three data points refer to nonfasting glucose levels during feeding with regular diet. Subsequent data refer to glucose levels after initiation of high-fat diet at time zero. The three different-colored lines after time zero represent the animals that were fed high-fat diets for specific times (9,18, or 28 days [d]) and then reversed (R) to normal diets for 2 weeks before sacrifice. Data represent four animals at each time point (mean ± SE).
Figure 5.

Pancreatic sections were double labeled for insulin (green fluorescence), 4-HNE (a marker for oxidative stress [red fluorescence]), and HO-1 (hemoxygenase-1) (red fluorescence). ZDF rats after 9 days of a high-fat diet showed intense cytoplasmic staining for 4-HNE in β-cells (B). Control data are shown in A, E, and I; 9-day data in B, F, J; and 9-day reversal of HFD data in C, G, and K. D, H, and L illustrate all data as a percentage of β-cells counted. This was reduced 2 weeks after a return to regular diets (C). Immunostaining for Nrf-2 (red fluorescence) revealed significant immunoreactivity both in the cytoplasm and in the nucleus of β-cells in ZDF rats fed high-fat diets for 9 days (F) as compared with ZDF controls (E). In contrast, 2 weeks after return to regular diets the immunoreactivity for Nrf2 was dramatically reduced (G). Similarly, pancreatic sections stained for HO-1 (red fluorescence) showed increased cytoplasmic and nuclear localization of HO-1 (J) in β-cells (green fluorescence), which after 9 days of HFD was greatly diminished 2 weeks after return to regular diet (K). Specificity of detected immunoreactivities was validated with incubation of tissue sections with control IgGs from each species (lower panels). Reprinted with permission from Abebe et al. (34).
Article Information
Funding. This work was funded by the Center for Scientific Review, National Institutes of Health (RO1 DK 38325-35).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Funding Statement
This work was funded by the Center for Scientific Review, National Institutes of Health (RO1 DK 38325-35).
References
- 1. Hopkins RZ, Li YR. Essentials of Free Radical Biology and Medicine. Cell Med Press, 2017 [Google Scholar]
- 2. Shalev A. Minireview: thioredoxin-interacting protein: regulation and function in the pancreatic β-cell. Mol Endocrinol 2014;28:1211–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stancill JS, Hansen PA, Mathison AJ, Schmidt EE, Corbett JA. Deletion of thioredoxin reductase disrupts redox homeostasis and impairs β-cell function. Function (Oxf) 2022;3:zqac034 [DOI] [PMC free article] [PubMed]
- 4. Grankvist K, Marklund SL, Täljedal IB. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem J 1981;199:393–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997;46:1733–1742 [DOI] [PubMed] [Google Scholar]
- 6. Tonooka N, Oseid E, Zhou H, Harmon JS, Robertson RP.. Glutathione peroxidase protein expression and activity in human islets isolated for transplantation. Clin Transplant 2007;21:767–772 [DOI] [PubMed] [Google Scholar]
- 7. Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem 2004;279:42351–42354 [DOI] [PubMed] [Google Scholar]
- 8. Kaneto H, Kajimoto Y, Miyagawa J, et al. Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 1999;48:2398–2406 [DOI] [PubMed] [Google Scholar]
- 9. Kawahito S, Kitahata H, Oshita S.. Problems associated with glucose toxicity: role of hyperglycemia-induced oxidative stress. World J Gastroenterol 2009;15:4137–4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kajimoto Y, Matsuoka T, Kaneto H, et al. Induction of glycation suppresses glucokinase gene expression in HIT-T15 cells. Diabetologia 1999;42:1417–1424 [DOI] [PubMed] [Google Scholar]
- 11. Johnson DA, Johnson JA. Nrf2--a therapeutic target for the treatment of neurodegenerative diseases. Free Radic Biol Med 2015;88:253–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo Y, Yu S, Zhang C, Kong A-NT. Epigenetic regulation of Keap1-Nrf2 signaling. Free Radic Biol Med 2015;88:337–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kanninen KM, Pomeshchik Y, Leinonen H, Malm T, Koistinaho J, Levonen AL. Applications of the Keap1-Nrf2 system for gene and cell therapy. Free Radic Biol Med 2015;88:350–361 [DOI] [PubMed] [Google Scholar]
- 14. Kang JS, Choi I-W, Han MH, et al. The cytoprotective effects of 7,8-dihydroxyflavone against oxidative stress are mediated by the upregulation of Nrf2-dependent HO-1 expression through the activation of the PI3K/Akt and ERK pathways in C2C12 myoblasts. Int J Mol Med 2015;36:501–510 [DOI] [PubMed] [Google Scholar]
- 15. Tebay LE, Robertson H, Durant ST, et al. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med 2015;88:108–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tanaka Y, Gleason CE, Tran POT, Harmon JS, Robertson RP.. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci U S A 1999;96:10857–10862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harmon JS, Gleason CE, Tanaka Y, Oseid EA, Hunter-Berger KK, Robertson RP.. In vivo prevention of hyperglycemia also prevents glucotoxic effects on PDX-1 and insulin gene expression. Diabetes 1999;48:1995–2000 [DOI] [PubMed] [Google Scholar]
- 18. Tanaka Y, Tran PO, Harmon J, Robertson RP.. A role for glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity. Proc Natl Acad Sci U S A 2002;99:12363–12368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tran POT, Parker SM, LeRoy E, et al. Adenoviral overexpression of the glutamylcysteine ligase catalytic subunit protects pancreatic islets against oxidative stress. J Biol Chem 2004;279:53988–53993 [DOI] [PubMed] [Google Scholar]
- 20. Kubisch HM, Wang J, Luche R, et al. Transgenic copper/zinc superoxide dismutase modulates susceptibility to type I diabetes. Proc Natl Acad Sci U S A 1994;91:9956–9959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kubisch HM, Wang J, Bray TM, Phillips JP. Targeted overexpression of CU/Zn superoxide dismutase protects pancreatic β-cells against oxidative stress. Diabetes 1997;46:1563–1566 [DOI] [PubMed] [Google Scholar]
- 22. Benhamou PY, Moriscot C, Richard MJ, et al. Adenovirus-mediated catalase gene transfer reduces oxidant stress in human, porcine, and rat pancreatic islets. Diabetologia 1998;41:1093–1100 [DOI] [PubMed] [Google Scholar]
- 23. Xu B, Moritz JT, Epstein PN.. Overexpression of catalase provides partial protection to transgenic mouse beta cells. Free Radic Biol Med 1999;27:830–837 [DOI] [PubMed] [Google Scholar]
- 24. Harmon JS, Bogdani M, Parazzoli SD, et al. β-Cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology 2009;150:4855–4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mahadevan J, Parazzoli S, Oseid E, et al. Ebselen treatment prevents islet apoptosis, maintains intranuclear Pdx-1 and MafA levels, and preserves β-cell mass and function in ZDF rats. Diabetes 2013;62:3582–3588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Müller A, Cadenas E, Graf P, Sies H.. A novel biologically active seleno-organic compound--I. Glutathione peroxidase-like activity in vitro and antioxidant capacity of PZ 51 (ebselen). Biochem Pharmacol 1984;33:3235–3239 [DOI] [PubMed] [Google Scholar]
- 27. Brodsky SV, Gealekman O, Chen J, et al. Prevention and reversal of premature endothelial cell senescence and vasculopathy in obesity-induced diabetes by ebselen. Circ Res 2004;94:377–384 [DOI] [PubMed] [Google Scholar]
- 28. Maiorino M, Roveri A, Ursini F.. Antioxidant effect of ebselen (PZ 51): peroxidase mimetic activity on phospholipid and cholesterol hydroperoxides vs free radical scavenger activity. Arch Biochem Biophys 1992;295:404–409 [DOI] [PubMed] [Google Scholar]
- 29. Lynch E, Kil J.. Development of ebselen, a glutathione peroxidase mimic, for the prevention and treatment of noise-induced hearing loss. Semin Hear 2009;30:47–55 [Google Scholar]
- 30. Yamaguchi T, Sano K, Takakura K, et al. ; Ebselen Study Group . Ebselen in acute ischemic stroke: a placebo-controlled, double-blind clinical trial. Stroke 1998;29:12–17 [DOI] [PubMed] [Google Scholar]
- 31. Ogawa A, Yoshimoto T, Kikuchi H, et al. Ebselen in acute middle cerebral artery occlusion: a placebo-controlled, double-blind clinical trial. Cerebrovasc Dis 1999;9:112–118 [DOI] [PubMed] [Google Scholar]
- 32. Saito I, Asano T, Sano K, et al. Neuroprotective effect of an antioxidant, ebselen, in patients with delayed neurological deficits after aneurysmal subarachnoid hemorrhage. Neurosurgery 1998;42:269–277 [DOI] [PubMed] [Google Scholar]
- 33. Beckman JA, Goldfine AB, Leopold JA, Creager MA.. Ebselen does not improve oxidative stress and vascular function in patients with diabetes: a randomized, crossover trial. Am J Physiol Heart Circ Physiol 2016;311:H1431–H1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Abebe T, Mahadevan J, Bogachus L, et al. Nrf2/antioxidant pathway mediates β cell self-repair after damage by high-fat diet-induced oxidative stress. JCI Insight 2017;2:e92854 [DOI] [PMC free article] [PubMed]
- 35. Robertson PR (Ed.). DeGroot’s Endocrinology: Basic Science and Clinical Practice. 8th ed. Elsevier, Amsterdam, the Netherlands, 2023