

Abstract

A new series of cinnamide-fluorinated derivatives has been synthesized and characterized by using different spectroscopic and elemental microanalyses methods. All of the prepared p-fluorocinnamide derivatives were evaluated for their cytotoxic activity against the HepG2 liver cancerous cell line. The imidazolone derivative 6, which bears N-(N-pyrimidin-2-ylbenzenesulphamoyl) moiety, displayed antiproliferative activity against HepG2 liver cancerous cells with an IC50 value of 4.23 μM as compared to staurosporin (STU) (IC50 = 5.59 μM). In addition, compound 6 experienced epidermal growth factor receptor (EGFR) inhibitory activity comparable to palatinib. The cell cycle analysis by flow cytometry indicated that compound 6 arrested the cellular cycle of HepG2 cells at the G1 phase. Additionally, as demonstrated by the fluorescence-activated cell sorting (FACS) technique, compound 6 increased both early and late apoptotic ratios compared to control untreated HepG2 cells. Moreover, imidazolone compound 6 induced apoptosis via the intrinsic apoptotic pathway by decreasing the level of mitochondrial membrane polarization (MMP) compared to untreated HepG2 cells. Therefore, the new N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone derivative 6 could be considered a potential platform for further optimizing an antitumor agent against hepatocellular carcinoma.
1. Introduction
Cancer has remained a critical health issue around the world and is the second fatal cause after cardiovascular diseases.1,2 Protein phosphorylation is among the most abundant and important cellular signaling post-translational modification in the biological system.3 This reversible enzymatic reaction plays a pivotal role in nearly all signal translational pathways and is carried out by a special class of proteins called protein kinases.4,5 The functional annotation of this phosphorylation process in a growing number of human diseases and physiological conditions underscores the relevance of protein kinases in medicine.6,7 Protein kinases catalyze the phosphorylation of target proteins by transferring the γ-phosphate group of adenosine triphosphate (ATP) onto the side-chain alcohol group of serine, threonine, and tyrosine amino acid residues.8 Misregulation of protein kinase signaling by kinase hyperactivation or mutation commonly results in human diseases including cancer.9 Therefore, protein kinases are important pharmaceutical targets in cancer treatment.
Epidermal growth factor receptor (EGFR) was the first receptor to be sequenced and discovered to possess tyrosine kinase activity.10 EGFR has been shown to be aberrantly expressed or activated in a number of tumors.11,12 The downstream signaling effects of such aberration lead to impaired apoptosis, enhanced proliferation, angiogenesis, and metastatic spread.13,14 Therefore, the inhibition of the EGFR signaling pathway has become a valuable approach in the treatment of cancer.15 One effective approach to inhibit EGFR-mediated signaling is to compete with ATP for binding to the tyrosine domain of EGFR utilizing small molecules of tyrosine kinase inhibitors (TKIs).16 Gefitinib, erlotinib, and lapatinib represent the most explored TKIs in the clinical setting for the treatment of various types of cancers.17,18 In addition, there is an ongoing and persistent need to design new EGFR-TK inhibitors to develop candidates with reduced toxicity and enhanced tumor susceptibility to therapy.19−21
Cinnamide, also known as cinnamamide or cinnamic acid amide, has a wide distribution and application in the fields of natural products and medicinal chemistry with a wide range of pharmacological activities.22,23 It is important to note that cinnamide-based derivatives could be used as a promising framework for the development of anticancer agents.24 Over the past few years, the anticancer activities of numerous cinnamide derivatives have been increasingly reported to show potent anticancer activities on targets such as mitochondria, tubulin polymerization, and so on.25,26 Cinnamide is also frequently used as a potential pharmacological active fragment in the design of EGFR tyrosine kinase inhibitors.27 In addition, imidazolone and 1,2,4-triazinone derivatives were found to have anticancer activity against several human cancer cell lines.28,29
In parallel, organo-fluorine compounds have gained important and valuable results in the construction and development of biologically relevant fluorine-containing drugs.30 The high electronegativity of the fluorine atom in the aromatic part of these drug molecules plays an important role in changing the physicochemical properties and, hence, enhancing the biological potencies.31 In addition, it has been reported that compounds containing a fluoro substituent were highly active anticancer agents against different cancer cell lines.32 Several studies reported phenyl moiety with a 4-fluoro substituent as a key part of several potent EGFR-TK inhibitors with pronounced cytotoxic activity on several cancer cell lines.33−35
Continuing our investigations on the preparation of newer bioactive compounds with active biological properties on human cancerous cell lines,36−40 we have synthesized cinnamide-fluorinated compounds 2a–7 in 60–76% yield (Figure 1). The cytotoxic effects of these compounds on liver cancer HepG2 cell line were evaluated. In addition, the efficiency of the most active synthesized cinnamide derivatives was assessed on a cellular cycle for 48 h. Additionally, the EGFR-TK inhibition and staining assay of apoptotic impact were also accomplished.
Figure 1.
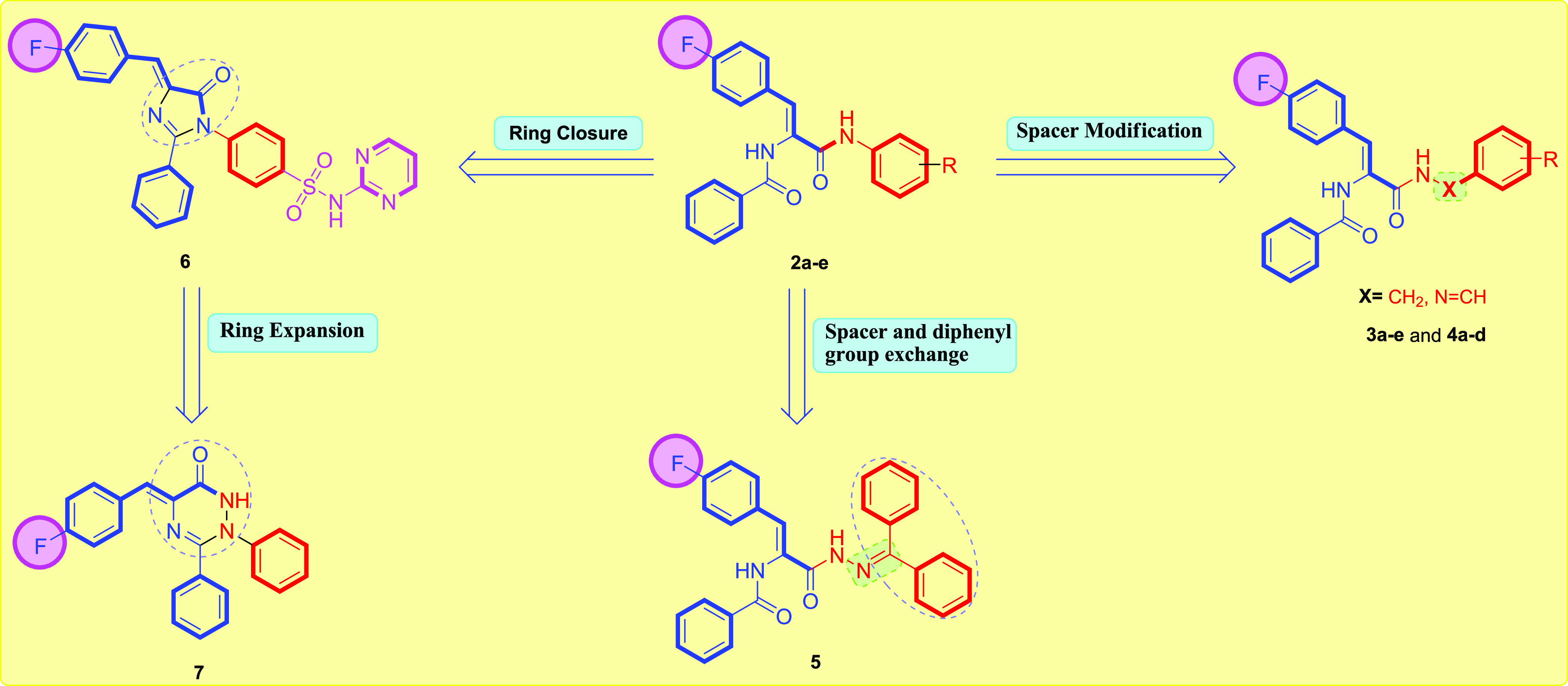
Design for the herein reported cinnamide-fluorinated derivatives 2a–7.
2. Results and Discussion
2.1. Chemistry
Scheme 1 reports the synthetic strategy utilized to obtain the target molecules 2–7 from the starting 4-(4-fluorobenzylidene)-1,3-oxazol-5(4H)-one 1. The reaction of 1 with a series of aryl amine resulted in N-aryl-4-fluorocinnamide derivatives 2a–f. The 1H NMR spectrum of N-(4-hydroxyphenyl)-4-fluorocinnamide 2d as a representative example displayed three broad singlet peaks at δ 10.04, 9.92, and 9.23 ppm corresponding to the two amidic NH and phenyl OH protons, respectively. The 13C NMR spectrum of N-(4-hydroxyphenyl)-4-fluorocinnamide 2d exhibited two signals at δ 166.4 and 164.2 ppm assigned to the two carbonyl (C=O) groups. In addition, the reaction of 4-(4-fluorobenzylidene)-1,3-oxazol-5(4H)-one 1 with respective substituted benzyl amine yielded the corresponding 3a–c. The 1H NMR spectrum of N-(2-methoxybenzyl)-4-fluorocinnamide 3b, as an example, displayed an upfield singlet at δ 3.79 ppm for the methyl (CH3) group, in addition to a doublet peak at δ 4.36 ppm corresponding to methylene (CH2) protons. The 13C NMR spectrum of 3b exhibited two new signals at δ 55.7 and 38.2 ppm for the methoxy and methylene carbons, respectively.
Scheme 1. General Synthesis of Compounds 2–7.
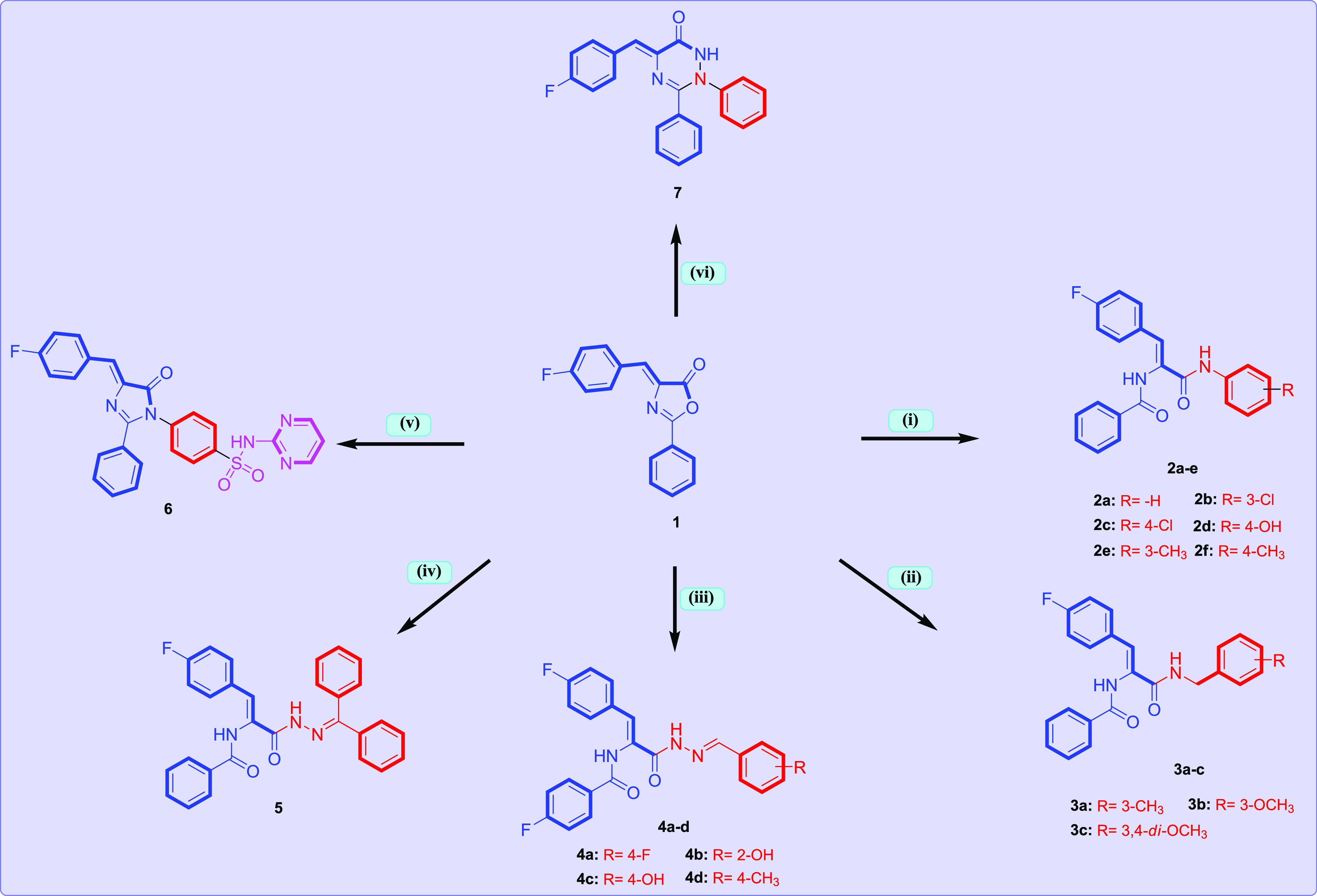
Condition: (i) respective aryl amine, AcOH, reflux 3–5 h; (ii) respective substituted benzyl amine, EtOH, reflux 1–2 h; (iii); respective N-arylidenehydrazine, EtOH, AcOH, reflux 4–5 h; (iv) N-(diphenylmethylene)hydrazine, NaOAc, AcOH, reflux 24 h; (v) sulfadiazine, NaOAc, AcOH, reflux 8 h; (vi) phenyl hydrazine, EtOH, AcOH, reflux 6 h.
Additionally, the N-arylidene-4-fluorocinnamic acid hydrazides 4a–d were prepared from the reaction of 4-(4-fluorobenzylidene)-1,3-oxazol-5(4H)-one 1 with respective N-arylidenehydrazine derivatives in refluxing pure ethanol for 4–5 h. Structures of compounds 4a–d were elucidated from their 1H NMR and 13C NMR spectral data. The 1H NMR spectrum of N-(4-hydroxyphenyl)-4-fluorocinnamic acid hydrazide 4c, as an example, exhibited three broad signals at δ 11.50, 10.07, and 9.91 ppm assigned to amidic NH and phenyl OH protons, respectively. In addition, signals of the two C=O groups were recorded in the 13C NMR spectrum of compound 4c at δ 166.4 and 162.3 ppm. Moreover, the N-(diphenylmethylene)-4-fluorocinnamic acid hydrazide 5 was prepared by the reaction of 1 with N-(diphenylmethylene)hydrazine in refluxing glacial acetic acid in the presence of NaOAc for 24 h. The 1H NMR spectrum of N-(diphenylmethylene)-4-fluorocinnamic acid hydrazide 5 displayed the presence of two singlet peaks at δ 10.04 and 9.39 ppm assigned to amidic NH and hydrazinyl NH protons, respectively, in addition to additional aromatic signals at δ 7.78–7.14 ppm due to the newly generated two phenyl moieties. The carbon skeleton of compound 5 was confirmed by the 13C NMR spectrum due to the presence of new signals at δ 166.6 and 165.4 ppm assigned to C=O, as well as additional carbon signals at the aromatic region due to the presence of two phenyl groups of the newly introduced benzophenone group.
Furthermore, the reaction of 1 with sulfadiazine in glacial acetic in the presence of sodium acetate to promote the reaction afforded the target N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6. The 1H NMR spectrum of 6 displayed a singlet peak at δ 12.04 ppm for the introduced sulfamoyl NH proton, in addition to new aromatic signals in the region at δ 8.52–7.09 ppm corresponding to the introduced N-pyrimidin-2-ylbenzenesulphamoyl moiety. The 13C NMR spectrum of N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6 confirmed the carbon skeleton due to the presence of additional aromatic signals corresponding to the newly introduced N-pyrimidin-2-ylbenzenesulphamoyl motif at δ 162.5–116.4 ppm. Finally, triaryl-1,2,4-triazinone 7 was obtained from the reaction of 4-(4-fluorobenzylidene)-1,3-oxazol-5(4H)-one 1 with phenyl hydrazine in pure ethanol. 1H NMR spectrum showed the presence of NH at δ 9.01 ppm. The 13C NMR spectrum of compound 7 showed the presence of new signals of the newly generated phenyl carbons in the region at δ 165.0–112.7 ppm.
2.2. Biology
2.2.1. Cytotoxic Activity against HepG2 Liver Cancerous Cell Line
The antiproliferative properties of the synthesized cinnamide derivatives 2a–7 were evaluated against the HepG2 (liver) cancerous cell line by the standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) technique using staurosporin (STU) as a reference standard. The results showed (Table 1) that the synthesized target cinnamide derivatives showed variable antiproliferative activity against the tested cell line. Regarding the activity of N-aryl-4-fluorocinnamides 2a–f toward HepG2 cells, N-phenyl-4-fluorocinnamides 2a revealed moderate cytotoxicity (IC50 = 53.20 μM), and it was the least active, suggesting that the relative substitution on the phenyl ring seemed to be critical for the antiproliferative activity of the N-aryl-4-fluorocinnamide derivatives 2a–f. In addition, the presence of an electron-withdrawing group, e.g., chlorine group in compounds 2b and 2c, decreased the cytotoxic activity compared with the electron-donating group, e.g., methyl group in compounds 2e and 2f. On the other hand, the N-(4-hydroxypheny)-4-fluorocinnamide derivative 2d revealed good cytotoxicity despite its electron-withdrawing character (IC50 = 7.81 μM), which may be attributed to its ability to form hydrogen bond with the target receptor. Additionally, N-(substiutedbenzyl)-4-fluorocinnamides 3a–c (IC50 ranges: 22.43–34.58 μM) were equipotent to the N-arylidene-4-fluorocinnamic acid hydrazides 4a–d (IC50 ranges: 19.57–38.12 μM). Moreover, the increase in lipophilicity due to the presence of diphenylmethylene moiety in compound 5 showed a distinct decrease in the cytotoxic activity (IC50 = 25.72 μM). Furthermore, the N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6 exerted impressive cytotoxic activity with an IC50 value of 4.23 μM. Finally, triaryl-1,2,4-triazinone derivative 7 displayed a noticeable increase in cytotoxicity (IC50 = 5.59 μM).
Table 1. Cytotoxic Activity of p-Fluorocinnamide Derivatives 2a–7 and STU against the HepG2 Liver Cancer Cell Linea.
| IC50 (μM) | |
|---|---|
| compound no | HepG2 |
| 2a | 53.20 ± 1.73 |
| 2b | 23.67 ± 1.32 |
| 2c | 18.12 ± 0.96 |
| 2d | 7.81 ± 0.34 |
| 2e | 16.51 ± 0.77 |
| 2f | 15.93 ± 0.92 |
| 3a | 34.58 ± 1.16 |
| 3b | 41.30 ± 1.52 |
| 3c | 22.43 ± 0.88 |
| 4a | 38.12 ± 1.88 |
| 4b | 27.33 ± 1.29 |
| 4c | 19.57 ± 0.33 |
| 4d | 31.60 ± 1.57 |
| 5 | 25.72 ± 1.04 |
| 6 | 4.23 ± 0.17 |
| 7 | 5.89 ± 0.19 |
| STU | 5.59 ± 0.27 |
Values are the mean of three independent replicates ± standard error (SE).
2.2.2. EGFR-TK Enzyme Assay
EGFR-TK is an essential kinase enzyme that is the cytocidal target for several structurally disparate classes of antineoplastic agents.41 EGFR has gained great interest in recent years because of its role in tumor development and as a therapeutic target.42 To further explore the antiproliferative effect of the prepared compounds, additional mechanistic studies were conducted by investigating the binding affinity of representative active N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6 to EGFR using Palatinib as a positive control. The results displayed a good IC50 value (IC50 of 0.13 μM on EGFR) for the tested N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6 compared to erlotinib (IC50 = 0.07 μM on EGFR) (Figure 2). The observed results indicated that the imidazolone derivative 6, which bears N-(N-pyrimidin-2-ylbenzenesulphamoyl) moiety, is favorable for EGFR-TK inhibition.
Figure 2.

Graphical representation of the IC50 values (μM) of N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6 and lapatinib on EGFR-TK enzyme.
2.2.3. Cell Cycle Analysis Using Flow Cytometry
The processes of new blood vessel formation and cellular divisions require the timely execution of a series of events known as the cellular cycle, during which a cell undergoes several discrete cellular states referred to as the G1, S, G2, and M phases.43 The propensity of a cell to either progress through the cell cycle or cease cell division and differentiate or enter quiescence is governed by the influence of stimuli such as chemotherapeutic agents.44 To investigate the molecular mechanism of cytotoxicity of compound 6, the effects of this compound at the IC50 concentration (4.23 μM) on the cell cycle progression of HepG2 cancer cells were evaluated by the fluorescence-activated cell sorting (FACS) technique. Since cell cycle arrest cause DNA fragmentation, the percentage of the pre-G1 apoptotic phase in cancer cells treated with compound 6 was measured. As displayed in Figure 3A,C, the treatment of HepG2 liver cancer cells with compound 6 for 48 h induced cell cycle arrest at the G1 phase and increases the percentage of cells from 50.7% in the control group to 64.2% in the cells treated with compound 6 at the IC50 concentration. In addition, the cells at the pre-G1 phase were increased from 2.5% in the control untreated cells to 55.5% in the cells treated with compound 6, which indicated that the cells were subjected to apoptosis (Figure 3B). The results confirmed that compound 6 possibly displayed antiproliferative potency through cell cycle arrest at the G1 phase and finally the induction of cellular apoptosis.
Figure 3.

(A) Quantitative analysis of the percentage of HepG2 cells in each cell cycle phase. (B) Quantitative analysis of the percentage of the pre-G1 phase in HepG2 cells treated for 48 h with compound 6 at its IC50 concentration and compared with untreated HepG2 control. (C) Cell cycle distribution by flow cytometry analysis in HepG2 cells treated for 48 h with compound 6 at its IC50 concentration and compared with untreated HepG2 control.
2.2.4. Apoptosis Assay
Apoptosis induction is a host defense mechanism against oncogenesis in that cells are actively killed by the immune system in order to prevent the propagation of the mutated, potentially malignant population of cells.45 The commitment of a cell to an apoptotic program can result from intracellular events by signals received from the extracellular milieu.46 The ability of cells to inappropriately evade or undergo programmed cellular death by using chemotherapeutic agents has been identified as a key feature of certain diseases such as cancer.47 Compound 6 was evaluated by the Annexin V-FITC/PI dual staining assay to characterize the mode of cellular death induced by these compounds. The assay can detect between live cells (Q1), early apoptosis (Q2), late apoptosis (Q3), and necrotic cells (Q4). In this study, HepG2 cells were treated with compound 6 for 48 h at the IC50 concentration (4.23 μM). The results showed an accumulation of total apoptotic cells (early and late apoptosis and necrosis) from 2.5% in the control untreated cells to 55.5% in the IC50 concentration-treated cells (Figure 4). The findings indicated that compound 6 can induce cellular apoptosis, and these results are in line with its antiproliferative activity in the tested HepG2 cell line.
Figure 4.

Flow cytometric analysis of Annexin V-FITC/PI to quantify the apoptosis induced by compound 6 at a concentration equal to the IC50 value in the HepG2 cell line for 48 h.
2.2.5. Mitochondrial Membrane Potential
As previously verified for several cytotoxic molecules, mitochondria can participate in the intrinsic apoptotic process by releasing proapoptotic factors after mitochondrial membrane polarization (MMP).48 In this regard, the effect of N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6 on mitochondria was studied by labeling whole cells with a green fluorescent dye. The obtained results shown in Figure 5 demonstrate that HepG2 cells incubated with N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6 at the IC50 concentration for 48 h underwent a notable decrease in mitochondrial membrane depolarization with respect to the control. In particular, the percentage of cells with depolarized mitochondrial membrane decreased by 2.84-fold compared with the untreated sample (control HepG2) following incubation for 48 h in the presence of N-(N-pyrimidin-2-ylbenzenesulphamoyl)-4-fluorocinnamide 6 at 4.23 μM; the IC50 concentration. In conclusion, a significantly low MMP impact was observed in accordance with the cytotoxic and cellular cycle data, as shown in Table 1 and Figure 5.
Figure 5.

Effect of N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6 on the mitochondrial membrane potential in HepG2 cancerous cells and compared with the untreated HepG2 cells.
2.2.6. Real-Time Polymerase Chain Reaction for Apoptotic Markers
In order to investigate the apoptotic induction pathway, the most active compound 6 was further screened for its activity against p53, Bax, and Bcl2 using real-time polymerase chain reaction (qRT-PCR). The data demonstrated in Figure 6 shows that N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6 increases the expression levels of apoptotic markers (p53 and Bcl2) compared with no treatment control cells. In this regard, N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6 boosted the levels of p53 and Bax with fold change values 4.7- and 3.7-fold, respectively, compared with no treatment control. On the other hand, the antiapoptotic marker Bcl2 declined after the treatment with N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6 with a 0.38-fold change value. In summary, compound 6 was able to cause the G1 cell cycle arrest by increasing the levels of p53 and Bax and decreasing the expression of antiapoptotic marker Bcl-2.
Figure 6.

Effect of N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6 on p53, Bax, and Bcl2.
3. Conclusions
In conclusion, a congeneric series of new p-fluorocinnamide derivatives 2a–7 were designed, synthesized, and structurally confirmed based on 1H NMR and 13C NMR spectroscopic methods as well as elemental microanalyses. The in vitro cytotoxic activity of the newly synthesized p-fluorocinnamide derivatives 2a–7 was evaluated against the liver cancerous HepG2 cell line. N-(N-Pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6 displayed the most potent cytotoxic activity with IC50 of 4.23 μM higher than that of STU as the reference compound (IC50 = 5.59 μM). Imidazolone compound 6 demonstrated potential inhibitory activity against EGFR with the IC50 value of 0.13 μM compared to palatinib (IC50 = 0.07 μM). Moreover, compound 6 was tested for its impact on the cellular cycle progression and induction of apoptosis in the HepG2 cell line. It induced cell cycle arrest at the G1 phase by 1.28-fold compared to untreated HepG2 control (from 50.7% in the control group to 64.2% in the compound 5-treated group). Also, compound 6 increased the ratio of both early and late apoptosis from 0.7% in the control untreated cells to 44.8% in the IC50 concentration-treated cells. Additionally, compound 6 decreased the level of MMP by 2.84-fold compared the control untreated HepG2 cells. Moreover, N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6 was found to induce apoptosis in HepG2 cells through the activation of p53 and Bax along with the downregulation of Bcl2, indicating that the intrinsic pathway was activated. In conclusion, N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone compound 6 showed potent cytotoxic activity in HepG2 cells with a novel mechanism. These findings will provide some valuable hints for further optimization to develop small active molecules for hepatocellular carcinoma therapy.
4. Experimental Section
4.1. General
4.2. Chemistry
4.2.1. General Method of the Synthesis of ((Z)-N-(3-Aryl)-1-(4-fluorophenyl)-3-oxoprop-1-en-2-yl)benzamides 2a–f
A mixture of 4-(4-fluorobenzylidene)-1,3-oxazol-5(4H)-one (0.14 g, 0.50 mmol) and the appropriate aryl amine (0.50 mmol) in (20 mL) glacial acetic acid was heated to reflux for 3–5 h. The solid separated on cooling was filtered off, dried, and crystallized from dimethylformamide (DMF)/water (3:1) to give pure 2a–f.
4.2.1.1. (Z)-N-(1-(4-Fluorophenyl)-3-oxo-3-(phenylamino)prop-1-en-2-yl)benzamide (2a)
Yield: 67%, mp 246–248 °C. Analysis: Calcd for C22H17FN2O2 (360.38): C 73.32, H 4.75, N 7.77%, Found: C 73.39, H 4.81, N 7.68%. 1H NMR (400 MHz, DMSO-d6) δ: 10.20 (s, 1H, NH), 10.13 (s, 1H, NH), 8.04 (d, J = 7.5 Hz, 2H), 7.73 (d, J = 8.4 Hz, 2H, Ar–H), 7.70 (d, J = 8.0 Hz, 2H, Ar–H), 7.61 (t, J = 7.0 Hz, 1H, Ar–H), 7.54 (t, J = 7.5 Hz, 2H, Ar–H), 7.33 (t, J = 7.7 Hz, 2H, Ar–H), 7.25 (t, J = 8.7 Hz, 2H, Ar–H), 7.16 (s, 1H, olefinic CH), 7.08 (t, J = 7.3 Hz, 1H, Ar–H). 13C NMR (DMSO, 101 MHz) δ: 166.5, 164.8, 163.6, 161.1, 139.8, 133.9, 132.3, 132.1, 132.0, 131.36, 129.0, 128.9, 128.4, 127.5, 123.9, 120.5, 116.1, 115.9 ppm.
4.2.1.2. (Z)-N-(3-(3-Chlorophenylamino)-1-(4-fluorophenyl)-3-oxoprop-1-en-2-yl)benzamide (2b)
Yield: 65%, mp 205–207 °C. Analysis: Calcd for C22H16ClFN2O2 (394.83): C 66.92, H 4.08, N 7.10%, Found: C 67.05, H 4.01, N 6.98%. 1H NMR (400 MHz, DMSO-d6) δ: 10.37 (s, 1H, NH), 10.16 (s, 1H, NH), 8.03 (d, J = 7.4 Hz, 2H, Ar–H), 7.92 (s, 1H, Ar–H), 7.71 (dd, J = 8.7, 5.7 Hz, 2H, Ar–H), 7.67 (d, J = 8.3 Hz, 1H, Ar–H), 7.62 (t, J = 7.3 Hz, 1H, Ar–H), 7.54 (t, J = 7.5 Hz, 2H, Ar–H), 7.37 (t, J = 8.1 Hz, 1H, Ar–H), 7.26 (t, J = 8.9 Hz, 2H, Ar–H), 7.15 (s, 1H, olefinic CH), 7.13 (d, J = 1.4 Hz, 1H, Ar–H) ppm.
4.2.1.3. (Z)-N-(3-(4-Chlorophenylamino)-1-(4-fluorophenyl)-3-oxoprop-1-en-2-yl)benzamide (2c)
Yield: 70%, mp 215–217 °C. Analysis: Calcd for C22H16ClFN2O2 (394.83): C 66.92, H 4.08, N 7.10%, Found: C 67.09, H 4.17, N 7.03%. 1H NMR (400 MHz, DMSO-d6) δ: 10.33 (s, 1H, NH), 10.13 (s, 1H, NH), 8.03 (d, J = 7.4 Hz, 2H, Ar–H), 7.77 (d, J = 8.8 Hz, 2H, Ar–H), 7.70 (dd, J = 8.5, 5.7 Hz, 2H, Ar–H), 7.61 (t, J = 7.2 Hz, 1H, Ar–H), 7.54 (t, J = 7.4 Hz, 2H, Ar–H), 7.39 (d, J = 8.8 Hz, 2H), 7.26 (t, J = 8.8 Hz, 2H, Ar–H), 7.15 (s, 1H, olefinic CH). 13C NMR (DMSO, 101 MHz) δ: 166.5, 164.9, 163.6, 161.2, 138.8, 133.7, 132.3, 132.2, 132.1, 131.2, 128.9, 128.9, 128.4, 127.6, 127.4, 122.0, 116.1, 115.9 ppm.
4.2.1.4. (Z)-N-(1-(4-Fluorophenyl)-3-(4-hydroxyphenylamino)-3-oxoprop-1-en-2-yl)benzamide (2d)
Yield: 74%, mp 236–238 °C. Analysis: Calcd for C22H17FN2O3 (376.38): C 70.20, H 4.55, N 7.44%, Found: C 70.33, H 4.41, N 7.60%. 1H NMR (400 MHz, DMSO-d6) δ: 10.04 (s, 1H, NH), 9.92 (s, 1H, NH), 9.23 (s, 1H, OH), 8.02 (d, J = 7.6 Hz, 2H, Ar–H), 7.68 (dd, J = 8.1, 5.9 Hz, 2H, Ar–H), 7.64–7.58 (m, 1H, Ar–H), 7.54 (d, J = 7.5 Hz, 2H, Ar–H), 7.51–7.44 (m, 2H, Ar–H), 7.24 (t, J = 8.7 Hz, 2H, Ar–H), 7.15 (s, 1H, olefinic CH), 6.72 (d, J = 8.7 Hz, 2H, Ar–H). 13C NMR (DMSO, 101 MHz) δ: 166.4, 164.2, 163.5, 161.1, 154.0, 134.0, 132.2, 132.0, 131.9, 131.5, 131.3, 128.8, 128.4, 127.4, 122.4, 116.1, 115.8, 115.4 ppm.
4.2.1.5. (Z)-N-(1-(4-Fluorophenyl)-3-oxo-3-(m-tolylamino)prop-1-en-2-yl)benzamide (2e)
Yield: 73%, mp 218–220 °C. Analysis: Calcd for C23H19FN2O2 (374.41): C 73.78, H 5.11, N 7.48%, Found: C 73.66, H 5.22, N 7.69%. 1H NMR (400 MHz, DMSO-d6) δ: 10.09 (s, 2H, 2NH), 8.03 (d, J = 7.6 Hz, 2H, Ar–H), 7.69 (dd, J = 8.5, 5.8 Hz, 2H, Ar–H), 7.61 (t, J = 7.2 Hz, 1H, Ar–H), 7.56 (s, 2H, Ar–H), 7.53 (d, J = 7.6 Hz, 2H, Ar–H), 7.25 (t, J = 8.7 Hz, 2H, Ar–H), 7.20 (d, J = 7.8 Hz, 1H, Ar–H), 7.15 (s, 1H, olefinic CH), 6.90 (d, J = 7.5 Hz, 1H, Ar–H), 2.30 (s, 3H, CH3). 13C NMR (DMSO, 101 MHz) δ: 166.5, 164.7, 163.6, 161.1, 139.6, 138.1, 133.9, 132.3, 132.1, 132.0, 131.3, 128.9, 128.8, 128.4, 127.5, 124.6, 121.1, 117.7, 116.1, 115.9, 21.7 (CH3) ppm.
4.2.1.6. (Z)-N-(1-(4-Fluorophenyl)-3-oxo-3-(p-tolylamino)prop-1-en-2-yl)benzamide (2f)
Yield: 76%, mp 242–244 °C. Analysis: Calcd for C23H19FN2O2 (374.41): C 73.78, H 5.11, N 7.48%, Found: C 73.83, H 5.17, N 7.38%. 1H NMR (400 MHz, DMSO-d6) δ: 10.08 (s, 1H, NH), 10.08 (s, 1H, NH), 8.02 (d, J = 7.4 Hz, 2H, Ar–H), 7.69 (dd, J = 8.8, 5.7 Hz, 2H, Ar–H), 7.64–7.58 (m, 3H, Ar–H), 7.53 (t, J = 7.4 Hz, 2H, Ar–H), 7.24 (t, J = 8.9 Hz, 2H, Ar–H), 7.15 (s, 2H, Ar–H), 7.12 (s, 1H, olefinic CH), 2.28 (s, 3H, CH3). 13C NMR (DMSO, 101 MHz) δ: 166.4, 164.6, 163.6, 161.1, 137.2, 133.9, 132.8, 132.3, 132.1, 132.0, 131.4, 129.4, 128.8, 128.4, 127.5, 120.6, 116.1, 115.9, 21.0 (CH3) ppm.
4.2.2. General Method of the Synthesis of (Z)-N-(1-(4-Fluorophenyl)-3-(3-substitutedbenzylamino)-3-oxoprop-1-en-2-yl)benzamides 3a–c
A mixture of 4-(4-fluorobenzylidene)-1,3-oxazol-5(4H)-one (0.14 g, 0.50 mmol) and the respective substituted benzyl amine (0.50 mmol) in (20 mL) pure ethanol was heated to reflux for 1–2 h. The solid separated on cooling the reaction mixture was filtered off, dried, and crystallized from ethanol/water (3:1) to give pure 3a–c.
4.2.2.1. (Z)-N-(1-(4-Fluorophenyl)-3-(3-methylbenzylamino)-3-oxoprop-1-en-2-yl)benzamide (3a)
Yield: 64%, mp 144–146 °C. Analysis: Calcd for C24H21FN2O2 (388.43): C 74.21, H 5.45, N 7.21%, Found: C 74.08, H 5.56, N 7.34%. 1H NMR (400 MHz, DMSO-d6) δ 9.97 (s, 1H, NH), 8.68 (t, J = 5.9 Hz, 1H, NH), 8.04 (d, J = 7.6 Hz, 2H, Ar–H), 7.64 (dd, J = 8.4, 5.9 Hz, 2H, Ar–H), 7.59 (d, J = 7.0 Hz, 1H, Ar–H), 7.53 (t, J = 7.4 Hz, 2H, Ar–H), 7.31 (s, 1H, olefinic CH), 7.22 (d, J = 4.6 Hz, 1H, Ar–H), 7.21 (d, J = 2.9 Hz, 1H, Ar–H), 7.18 (s, 1H, Ar–H), 7.16 (s, 1H, Ar–H), 7.12 (d, J = 7.5 Hz, 1H, Ar–H), 7.04 (d, J = 7.3 Hz, 1H, Ar–H), 4.36 (d, J = 6.0 Hz, 2H, NCH2), 2.30 (s, 3H, CH3). 13C NMR (DMSO, 101 MHz) δ: 166.4, 165.4, 163.6, 161.1, 140.1, 137.6, 134.1, 132.2, 132.0, 131.9, 131.3, 130.4, 128.8, 128.5, 128.4, 128.2, 127.6, 124.7, 116.0, 115.8, 43.0 (NCH2), 21.5 (CH3).
4.2.2.2. (Z)-N-(1-(4-Fluorophenyl)-3-(3-methoxybenzylamino)-3-oxoprop-1-en-2-yl)benzamide (3b)
Yield: 61%, mp 178–180 °C. Analysis: Calcd for C24H21FN2O3 (404.43): C 71.27, H 5.23, N 6.93%, Found: C 71.17, H 5.14, N 7.04%. 1H NMR (400 MHz, DMSO-d6) δ 9.99 (s, 1H, NH), 8.50 (t, J = 5.8 Hz, 1H, NH), 8.04 (d, J = 7.3 Hz, 2H, Ar–H), 7.65 (dd, J = 8.5, 5.7 Hz, 2H, Ar–H), 7.60 (d, J = 7.2 Hz, 1H, Ar–H), 7.53 (t, J = 7.3 Hz, 2H, Ar–H), 7.34 (s, 1H, olefinic CH), 7.29 (d, J = 7.1 Hz, 1H, Ar–H), 7.27–7.12 (m, 3H, Ar–H), 6.97 (d, J = 8.1 Hz, 1H, Ar–H), 6.92 (t, J = 7.4 Hz, 1H, Ar–H), 4.36 (d, J = 5.8 Hz, 2H, NCH2), 3.79 (s, 3H, OCH3). 13C NMR (DMSO, 101 MHz) δ: 166.5, 165.5, 163.6, 161.2, 156.8, 134.1, 132.2, 132.0, 131.9, 131.3, 130.3, 128.8, 128.4, 128.1, 127.7, 127.4, 120.5, 116.1, 115.9, 110.6, 55.7 (OCH3), 38.2 (NCH2).
4.2.2.3. (Z)-N-(3-(3,4-Dimethoxybenzylamino)-1-(4-fluorophenyl)-3-oxoprop-1-en-2-yl)benzamide (3c)
Yield: 68%, mp 184–186 °C. Analysis: Calcd for C25H23FN2O4 (434.46): C 69.11, H 5.34, N 6.45%, Found: C 69.19, H 5.22, N 6.54%. 1H NMR (400 MHz, DMSO-d6) δ: 9.99 (s, 1H, NH), 8.65 (t, J = 5.9 Hz, 1H, NH), 8.02 (d, J = 7.3 Hz, 2H, Ar–H), 7.63 (dd, J = 8.7, 5.7 Hz, 2H, Ar–H), 7.59 (d, J = 7.2 Hz, 1H, Ar–H), 7.53 (t, J = 7.4 Hz, 2H, Ar–H), 7.26 (s, 1H, olefinic CH), 7.21 (t, J = 8.9 Hz, 2H, Ar–H), 6.97 (s, 1H, Ar–H), 6.88 (d, J = 8.2 Hz, 1H, Ar–H), 6.83 (d, J = 8.1 Hz, 1H, Ar–H), 4.33 (d, J = 5.9 Hz, 2H, NCH2), 3.77 (s, 3H, OCH3), 3.72 (s, 3H, OCH3). 13C NMR (DMSO, 101 MHz) δ: 166.5, 165.4, 161.1, 149.1, 148.0, 134.0, 132.7, 132.2, 132.0, 131.9, 131.3, 131.3, 130.7, 128.8, 128.4, 119.5, 116.1, 115.8, 112.1, 111.6, 56.1 (OCH3), 55.9 (OCH3), 42.7 (NCH2) ppm.
4.2.3. General Method of the Synthesis of N-(Z)-3-((E)-2-(Arylhydrazinyl)-1-(4-Fluorophenyl)-3-oxoprop-1-en-2-yl)benzamides 4a–d
A suspension mixture of 4-(4-fluorobenzylidene)-1,3-oxazol-5(4H)-one (0.14 g, 0.50 mmol), the respective N-arylidenehydrazine (0.50 mmol), and (2 mL) glacial acetic acid in (20 mL) pure ethanol was heated to reflux on a water bath for 4–5 h. After reaction accomplishment, the mixture was cooled to room temperature and then poured onto ice-cold water (15 mL). The solid formed was filtered off and crystallized from DMF/water (3:1) to give pure N-(arylidene)-4-fluorocinnamic acid hydrazide 4a–d.
4.2.3.1. N-((Z)-3-((E)-2-(4-Fluorobenzylidene)hydrazinyl)-1-(4-fluorophenyl)-3-oxoprop-1-en-2-yl)benzamide (4a)
Yield: 61%, mp 227–229 °C. Analysis: Calcd for C23H17F2N3O2 (405.40): C 68.14, H 4.23, N 10.37%, Found: C 68.06, H 4.18, N 10.53%. 1H NMR (400 MHz, DMSO-d6) δ: 11.74 (s, 1H, NH), 10.12 (s, 1H, NH), 8.42 (s, 1H, CH = N), 8.03 (d, J = 7.4 Hz, 2H, Ar–H), 7.82–7.74 (m, 2H, Ar–H), 7.69 (dd, J = 8.1, 5.9 Hz, 2H, Ar–H), 7.65–7.58 (m, 1H, Ar–H), 7.54 (t, J = 7.3 Hz, 2H, Ar–H), 7.35 (dd, J = 16.6, 7.8 Hz, 1H, Ar–H), 7.29 (d, J = 11.1 Hz, 2H, Ar–H), 7.25 (s, 1H, Ar–H), 7.16 (s, 1H, olefinic CH). 13C NMR (DMSO, 101 MHz) δ: 166.4, 164.8, 163.6, 162.7, 160.9, 146.7, 133.8, 132.3, 132.1, 131.2, 130.0, 129.6, 128.9, 128.4, 128.0, 116.5, 116.3, 116.1, 115.9 ppm.
4.2.3.2. N-((Z)-1-(4-Fluorophenyl)-3-((E)-2-(2-hydroxybenzylidene)hydrazinyl)-3-oxoprop-1-en-2-yl)benzamide (4b)
Yield: 69%, mp 253–255 °C. Analysis: Calcd for C23H18FN3O3 (403.41): C 68.48, H 4.50, N 10.42%, Found: C 68.59, H 4.63, N 10.23%. 1H NMR (400 MHz, DMSO-d6) δ: 11.96 (s, 1H, NH), 11.31 (s, 1H, OH), 10.15 (s, 1H, NH), 8.60 (s, 1H, CH = N), 8.04 (d, J = 7.4 Hz, 2H, Ar–H), 7.70 (dd, J = 8.5, 5.8 Hz, 2H, Ar–H), 7.66–7.59 (m, 1H, Ar–H), 7.55 (t, J = 7.5 Hz, 2H, Ar–H), 7.51 (d, J = 8.4 Hz, 1H, Ar–H), 7.35–7.27 (m, 2H, Ar–H), 7.26 (s, 1H, Ar–H), 7.23 (d, J = 2.4 Hz, 1H, Ar–H), 6.94 (d, J = 8.7 Hz, 2H, Ar–H and olefinic CH) ppm.
4.2.3.3. N-((Z)-1-(4-Fluorophenyl)-3-((E)-2-(4-hydroxybenzylidene)hydrazinyl)-3-oxoprop-1-en-2-yl)benzamide (4c)
Yield: 73%, mp 277–279 °C. Analysis: Calcd for C23H18FN3O3 (403.41): C 68.48, H 4.50, N 10.42%, Found: C 68.53, H 4.42, N 10.51%. 1H NMR (400 MHz, DMSO-d6) δ: 11.50 (s, 1H, NH), 10.07 (s, 1H, NH), 9.91 (s, 1H, OH), 8.31 (s, 1H, CH = N), 8.03 (d, J = 7.5 Hz, 2H, Ar–H), 7.68 (dd, J = 8.2, 5.9 Hz, 2H, Ar–H), 7.64–7.59 (m, 1H, Ar–H), 7.55 (s, 2H, Ar–H), 7.53 (s, 2H, Ar–H), 7.24 (t, J = 8.8 Hz, 2H, Ar–H), 7.15 (s, 1H, olefinic CH), 6.84 (d, J = 8.4 Hz, 2H, Ar–H). 13C NMR (DMSO, 101 MHz) δ: 166.4, 162.3, 161.1, 159.8, 148.2, 133.8, 132.3, 132.1, 132.0, 131.3, 130.2, 129.2, 128.9, 128.4, 127.8, 125.9, 116.2, 116.1, 115.9 ppm.
4.2.3.4. N-((Z)-1-(4-Fluorophenyl)-3-((E)-2-(4-methylbenzylidene)hydrazinyl)-3-oxoprop-1-en-2-yl)benzamide (4d)
Yield: 60%, mp 241–243 °C. Analysis: Calcd for C24H20FN3O2 (401.43): C 71.81, H 5.02, N 10.47%, Found: C 71.96, H 4.90, N 10.33%. 1H NMR (400 MHz, DMSO-d6) δ: 8.66 (s, 1H, CH = N), 8.46 (dd, J = 8.7, 5.9 Hz, 1H, Ar–H), 8.23–8.15 (m, 1H, Ar–H), 7.77 (d, J = 8.1 Hz, 3H, Ar–H), 7.73 (d, J = 8.1 Hz, 1H, Ar–H), 7.70–7.54 (m, 2H, Ar–H), 7.45–7.35 (m, 2H, Ar–H), 7.32 (d, J = 7.6 Hz, 4H, Ar–H and olefinic CH), 2.38 (s, 3H, CH3). 13C NMR (DMSO, 101 MHz) δ: 166.7, 162.5, 161.7, 158.3, 142.4, 141.8, 132.6, 131.7, 131.1, 130.2, 130.2, 130.0, 129.0, 128.8, 128.7, 128.4, 127.6, 116.7, 116.4, 21.6 (CH3) ppm.
4.2.4. General Method of the Synthesis of (Z)-N-(3-(2-(Diphenylmethylene)hydrazinyl)-1-(4-fluorophenyl)-3-oxoprop-1-en-2-yl)benzamide (5)
A suspension mixture of 4-(4-fluorobenzylidene)-1,3-oxazol-5(4H)-one (0.14 g, 0.50 mmol), N-(diphenylmethylene)hydrazine (0.10 g, 0.50 mmol), and NaOAc (0.05 g, 0.55 mmol) in (20 mL) glacial acetic acid was heated to reflux on an oil bath for 24 h. After reaction accomplishment, the reaction mixture was concentrated to half volume, then cooled, and left overnight. The formed precipitate was filtered off, dried, and crystallized from DMF/water (3:1) to give a pure 2-(diphenylmethylene)-4-fluorocinnamic acid hydrazide molecule 5.
Yield: 66%, mp 107–109 °C. Analysis: Calcd for C29H22FN3O2 (463.50): C 75.15, H 4.78, N 9.07%, Found: C 75.00, H 4.91, N 9.24%. 1H NMR (400 MHz, DMSO-d6) δ: 10.04 (d, J = 27.6 Hz, 1H, NH), 9.39 (s, 1H, NH), 7.78–7.70 (m, 2H, Ar–H), 7.68–7.59 (m, 3H, Ar–H), 7.51 (dd, J = 21.3, 7.5 Hz, 5H, Ar–H), 7.40 (d, J = 20.0 Hz, 4H, Ar–H), 7.30 (d, J = 6.8 Hz, 1H, olefinic CH), 7.26–7.14 (m, 5H, Ar–H). 13C NMR (DMSO, 101 MHz) δ: 166.6, 165.4, 161.5, 159.5, 137.9, 137.1, 135.6, 132.7, 132.4, 132.3, 131.5, 130.4, 129.9, 129.4, 129.3, 129.1, 128.9, 128.8, 128.6, 128.2, 127.8, 116.3, 116.1 ppm.
4.2.5. General Method of the Synthesis of (Z)-4-(4-(4-Fluorobenzylidene)-5-oxo-2-phenyl-4,5-dihydro-1H-imidazol-1-yl)-N-(pyrimidin-2-yl)benzenesulfonamide (6)
A suspension mixture of 4-(4-fluorobenzylidene)-1,3-oxazol-5(4H)-one (0.14 g, 0.50 mmol), sulfadiazine (0.13 g, 0.50 mmol), and NaOAc (0.05 g, 0.55 mmol) in (20 mL) glacial acetic acid was heated to reflux on an oil bath for 8 h. The formed solid on cooling was filtered off and crystallized from DMF/water (3:1) to give N-(N-pyrimidin-2-ylbenzenesulphamoyl)imidazolone 6.
Yield: 74%, mp 265–267 °C. Analysis: Calcd for C26H20FN5O4S (517.53): C 60.34, H 3.90, N 13.53%, Found: C 60.41, H 3.96, N 13.42%. 1H NMR (400 MHz, DMSO-d6) δ: 12.04 (s, 1H, NH), 8.52 (d, J = 4.9 Hz, 2H, Ar–H), 8.46 (dd, J = 8.8, 5.8 Hz, 2H, Ar–H), 8.04 (d, J = 8.6 Hz, 2H, Ar–H), 7.54–7.49 (m, 1H, Ar–H), 7.48 (d, J = 1.8 Hz, 1H, Ar–H), 7.47 (d, J = 1.8 Hz, 2H, Ar–H), 7.45 (d, J = 1.4 Hz, 1H, Ar–H), 7.39 (d, J = 3.7 Hz, 1H, Ar–H), 7.39–7.36 (m, 2H, Ar–H and olefinic CH), 7.35 (d, J = 2.3 Hz, 2H, Ar–H), 7.09 (t, J = 4.9 Hz, 1H, Ar–H). 13C NMR (DMSO, 101 MHz) δ: 169.6, 165.0, 162.5, 160.8, 158.8, 157.3, 140.6, 138.4, 138.3, 135.4, 135.3, 132.1, 131.2, 131.2, 129.4, 129.0, 128.9, 128.7, 128.5, 127.4, 116.7, 116.4 ppm.
4.2.6. General Method of the Synthesis of (Z)-5-(4-Fluorobenzylidene)-2,3-diphenyl-1,2-dihydro-1,2,4-triazin-6(5H)-one (7)
A suspension mixture of 4-(4-fluorobenzylidene)-1,3-oxazol-5(4H)-one (0.14 g, 0.50 mmol), phenyl hydrazine (0.06 g, 0.50 mmol), and (2 mL) glacial acetic acid in (20 mL) pure ethanol was heated to reflux on a water bath for 6 h. The solution mixture was cooled to room temperature and then poured on ice-cold water (15 mL). The solid formed was filtered off and crystallized from ethanol/water (70%) to give a pure triaryl-1,2,4-triazine molecule 7.
Yield: 69%, mp 222–224 °C. Analysis: Calcd for C22H16FN3O (357.38): C 73.94, H 4.51, N 11.76%, Found: C 74.04, H 4.43, N 11.62%. 1H NMR (400 MHz, DMSO-d6) δ: 9.01 (s, 1H, NH), 8.47 (dd, J = 8.6, 5.9 Hz, 2H, Ar–H), 8.16 (d, J = 7.3 Hz, 2H, Ar–H), 7.61 (t, J = 7.3 Hz, 1H, Ar–H), 7.54 (t, J = 7.5 Hz, 2H, Ar–H), 7.39 (t, J = 8.9 Hz, 2H, Ar–H), 7.30 (s, 1H, olefinic CH), 7.21 (t, J = 7.8 Hz, 2H, Ar–H), 6.83 (t, J = 7.3 Hz, 1H, Ar–H), 6.72 (d, J = 7.9 Hz, 2H, Ar–H). 13C NMR (DMSO, 101 MHz) δ: 169.6, 165.0, 162.5, 161.5, 146.9, 136.4, 135.6, 135.5, 132.8, 131.2, 129.8, 129.2, 129.1, 128.0, 127.3, 120.6, 116.7, 116.4, 112.7 ppm.
4.3. Biological Studies
The experimental details for the performed biological assays, MTT assay, EGFR kinase, cell cycle analysis, apoptosis assay, mitochondrial membrane depolarization, and qRT-PCR for apoptotic markers are mentioned in the Supporting Information.
Acknowledgments
The authors extend their appreciation to the Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2024R155), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c00847.
All of the spectral analyses including 1H NMR and 13C NMR spectra for all prepared cinnamide-fluorinated compounds and detailed descriptions for Section 4.2 (PDF)
The authors extend their appreciation to the Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2024R155), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
The authors declare no competing financial interest.
Supplementary Material
References
- Siegel R. L.; Miller K. D.; Wagle N. S.; Jemal A. Cancer statistics, 2023. Ca-Cancer J. Clin. 2023, 73, 17–48. 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- Siobhan C.; Stuart D. R.; Duncan C. G.; Ruth E. L. Cardiovascular health: an important component of cancer survivorship. BMJ Oncol. 2023, 2, e000090 10.1136/bmjonc-2023-000090. [DOI] [Google Scholar]
- Pang K.; Wang W.; Qin J.-X.; Shi Z.-D.; Hao L.; Ma Y.-Y.; Xu H.; Wu Z.-X.; Pan D.; Chen Z.-S.; Han C. Role of protein phosphorylation in cell signaling, disease, and the intervention therapy. MedComm 2022, 3, e175 10.1002/mco2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi B.; Qiu M.; Liu P.; Wang Q.; Wen Y.; Li Y.; Li B.; Li Y.; He Y.; Zhao J. Protein post-translational modifications: A key factor in colorectal cancer resistance mechanisms. Biochim. Biophys. Acta, Gene Regul. Mech. 2023, 1866, 194977–194988. 10.1016/j.bbagrm.2023.194977. [DOI] [PubMed] [Google Scholar]
- Ahsan R.; Khan M. M.; Mishra A.; Noor G.; Ahmad U. Protein Kinases and their Inhibitors Implications in Modulating Disease Progression. Protein J. 2023, 42, 621–632. 10.1007/s10930-023-10159-9. [DOI] [PubMed] [Google Scholar]
- Pan S.; Chen R. Pathological implication of protein post-translational modifications in cancer. Mol. Aspects Med. 2022, 86, 101097 10.1016/j.mam.2022.101097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R.; Qian M.-P.; Cui Y.-Y. Protein kinases: The key contributors in pathogenesis and treatment of nonalcoholic fatty liver disease-derived hepatocellular carcinoma. Metabolism 2023, 147, 155665 10.1016/j.metabol.2023.155665. [DOI] [PubMed] [Google Scholar]
- Roskoski R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2024 update. Pharmacol. Res. 2024, 200, 107059 10.1016/j.phrs.2024.107059. [DOI] [PubMed] [Google Scholar]
- Sudhan D. R.; Guerrero-Zotano A.; Won H.; Ericsson P. G.; Servetto A.; Huerta-Rosario M.; Ye D.; Lee K.-m.; Formisano L.; Guo Y.; et al. Hyperactivation of TORC1 drives resistance to the pan-HER tyrosine kinase inhibitor neratinib in HER2-mutant cancers. Cancer cell 2020, 37, 183–199.e5. 10.1016/j.ccell.2019.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbah D. A.; Hajjo R.; Sweidan K. Review on epidermal growth factor receptor (EGFR) structure, signaling pathways, interactions, and recent updates of EGFR inhibitors. Curr. Top. Med. Chem. 2020, 20, 815–834. 10.2174/1568026620666200303123102. [DOI] [PubMed] [Google Scholar]
- Rodriguez S. M. B.; Kamel A.; Ciubotaru G. V.; Onose G.; Sevastre A.-S.; Sfredel V.; Danoiu S.; Dricu A.; Tataranu L. G. An Overview of EGFR Mechanisms and Their Implications in Targeted Therapies for Glioblastoma. Int. J. Mol. Sci. 2023, 24, 11110 10.3390/ijms241311110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J. J.; Ko A.; Kil S. H.; Mallen-St Clair J.; Shin D. S.; Wang M. B.; Srivatsan E. S. EGFR pathway targeting drugs in head and neck cancer in the era of immunotherapy. Biochim. Biophys. Acta, Rev. Cancer 2023, 1878, 188827–188839. 10.1016/j.bbcan.2022.188827. [DOI] [PubMed] [Google Scholar]
- Abo-El Fetoh M. E.; Abdel-Fattah M. M.; Afify H.; Ramadan L. A. A.; Mohamed W. R. Review on EGFR-ERK1/2 signaling cascade: implications on cell proliferation in health and disease. Egypt. Pharm. J. 2023, 22, 535–544. 10.4103/epj.epj_52_23. [DOI] [Google Scholar]
- Ramani S.; Samant S.; Manohar S. M. The story of EGFR: from signaling pathways to a potent anticancer target. Future Med. Chem. 2022, 14, 1267–1288. 10.4155/fmc-2021-0343. [DOI] [PubMed] [Google Scholar]
- Halder S.; Basu S.; Lall S. P.; Ganti A. K.; Batra S. K.; Seshacharyulu P. Targeting the EGFR signaling pathway in cancer therapy: What’s new in 2023?. Expert Opin. Ther. Targets 2023, 27, 305–324. 10.1080/14728222.2023.2218613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Vallentgoed W. R.; French P. J. Finding the Right Way to Target EGFR in Glioblastomas; Lessons from Lung Adenocarcinomas. Cancers 2018, 10, 489 10.3390/cancers10120489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P. Y.; Yeoh Y.; Low T. Y. A recent update on small-molecule kinase inhibitors for targeted cancer therapy and their therapeutic insights from mass spectrometry-based proteomic analysis. FEBS J. 2023, 290, 2845–2864. 10.1111/febs.16442. [DOI] [PubMed] [Google Scholar]
- Cristina Mendonça Nogueira T.; Vinicius Nora de Souza M. New FDA oncology small molecule drugs approvals in 2020: Mechanism of action and clinical applications. Bioorg. Med. Chem. 2021, 46, 116340–116352. 10.1016/j.bmc.2021.116340. [DOI] [PubMed] [Google Scholar]
- Zubair T.; Bandyopadhyay D. Small Molecule EGFR Inhibitors as Anti-Cancer Agents: Discovery, Mechanisms of Action, and Opportunities. Int. J. Mol. Sci. 2023, 24, 2651 10.3390/ijms24032651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasanvand Z.; Oghabi Bakhshaiesh T.; Peytam F.; Firoozpour L.; Hosseinzadeh E.; Motahari R.; Moghimi S.; Nazeri E.; Toolabi M.; Momeni F.; et al. Imidazo[1,2-a]quinazolines as novel, potent EGFR-TK inhibitors: Design, synthesis, bioactivity evaluation, and in silico studies. Bioorg. Chem. 2023, 133, 106383–106392. 10.1016/j.bioorg.2023.106383. [DOI] [PubMed] [Google Scholar]
- Hashem H. E.; Amr A. E.-G. E.; Nossier E. S.; Anwar M. M.; Azmy E. M. New Benzimidazole-, 1,2,4-Triazole-, and 1,3,5-Triazine-Based Derivatives as Potential EGFRWT and EGFRT790M Inhibitors: Microwave-Assisted Synthesis, Anticancer Evaluation, and Molecular Docking Study. ACS Omega 2022, 7, 7155–7171. 10.1021/acsomega.1c06836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaikwad N.; Nanduri S.; Madhavi Y. V. Cinnamamide: An insight into the pharmacological advances and structure–activity relationships. Eur. J. Med. Chem. 2019, 181, 111561–111574. 10.1016/j.ejmech.2019.07.064. [DOI] [PubMed] [Google Scholar]
- Come J. A. A.; Zhuang Y.; Li T.; Brogi S.; Gemma S.; Liu T.; da Silva E. R. In Vitro and In Silico Analyses of New Cinnamid and Rosmarinic Acid-Derived Compounds Biosynthesized in Escherichia coli as Leishmania amazonensis Arginase Inhibitors. Pathogens 2022, 11, 1020 10.3390/pathogens11091020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullah S.; Park Y.; Ikram M.; Lee S.; Park C.; Kang D.; Yang J.; Akter J.; Yoon S.; Chun P.; Moon H. R. Design, synthesis and anti-melanogenic effect of cinnamamide derivatives. Bioorg. Med. Chem. 2018, 26, 5672–5681. 10.1016/j.bmc.2018.10.014. [DOI] [PubMed] [Google Scholar]
- Yang K.; Li Y.; Tang Q.; Zheng L.; He D. Synthesis, mitochondrial localization of fluorescent derivatives of cinnamamide as anticancer agents. Eur. J. Med. Chem. 2019, 170, 45–54. 10.1016/j.ejmech.2019.03.001. [DOI] [PubMed] [Google Scholar]
- Fu X.-J.; Huang J.; Li N.; Liu Y.-H.; Liu Q.-G.; Yuan S.; Xu Y.; Chen Y.-F.; Zhao Y.-X.; Song J.; et al. Design, synthesis and biological evaluation of N-benzylaryl cinnamide derivatives as tubulin polymerization inhibitors capable of promoting YAP degradation with potent anti-gastric cancer activities. Eur. J. Med. Chem. 2023, 262, 115883–115895. 10.1016/j.ejmech.2023.115883. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Xu Z.; Liu Q.; Xia S.; Liu Z.; Liao Z.; Gou S. Design, synthesis and biological evaluation of cinnamamide-quinazoline derivatives as potential EGFR inhibitors to reverse T790M mutation. Bioorg. Chem. 2021, 117, 105420 10.1016/j.bioorg.2021.105420. [DOI] [PubMed] [Google Scholar]
- Avsar T.; Yigit B. N.; Turan G.; Altunsu D.; Calis S.; Kurt B.; Kilic T.; Yavuz Ergun M.; Durdagi S.; Acar M. Development of imidazolone based angiotensin II receptor type I inhibitor small molecule as a chemotherapeutic agent for cell cycle inhibition. All Life 2021, 14, 678–690. 10.1080/26895293.2021.1954098. [DOI] [Google Scholar]
- Zaki I.; Abdelhameid M. K.; El-Deen I. M.; Abdel Wahab A. H. A.; Ashmawy A. M.; Mohamed K. O. Design, synthesis and screening of 1, 2, 4-triazinone derivatives as potential antitumor agents with apoptosis inducing activity on MCF-7 breast cancer cell line. Eur. J. Med. Chem. 2018, 156, 563–579. 10.1016/j.ejmech.2018.07.003. [DOI] [PubMed] [Google Scholar]
- He J.; Li Z.; Dhawan G.; Zhang W.; Sorochinsky A. E.; Butler G.; Soloshonok V. A.; Han J. Fluorine-containing drugs approved by the FDA in 2021. Chin. Chem. Lett. 2023, 34, 107578–107588. 10.1016/j.cclet.2022.06.001. [DOI] [Google Scholar]
- Jayanetti K.; Takemura K.; Bendale H.; Garg A.; Ojima I. Recent advances in the strategic incorporation of fluorine into new-generation taxoid anticancer agents. J. Fluorine Chem. 2023, 267, 110106–110115. 10.1016/j.jfluchem.2023.110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabir G.; Saeed A.; Zahid W.; Naseer F.; Riaz Z.; Khalil N.; Muneeba; Albericio F. Chemistry and Pharmacology of Fluorinated Drugs Approved by the FDA (2016–2022). Pharmaceuticals 2023, 16, 1162 10.3390/ph16081162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.-M.; Xing M.; Zhao T.-T.; Ren Y.-J.; Yang X.-H.; Yang Y.-S.; Lv P.-C.; Zhu H.-L. Synthesis, molecular modeling and biological evaluation of cinnamic acid derivatives with pyrazole moieties as novel anticancer agents. RSC Adv. 2014, 4, 37197–37207. 10.1039/C4RA05257A. [DOI] [Google Scholar]
- Lawal H. A.; Uzairu A.; Uba S. QSAR, molecular docking, design, and pharmacokinetic analysis of 2-(4-fluorophenyl) imidazol-5-ones as anti-breast cancer drug compounds against MCF-7 cell line. J. Bioenerg. Biomembr. 2020, 52, 475–494. 10.1007/s10863-020-09858-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Otaibi F. A.; Bakhotmah D. A. Synthesis and Biological Evaluation of New Fluorine Compounds Bearing 4-Amino-1,2,4-triazino[4,3-b]-1,2,4-triazin-8-one and the Related Derivatives as CDK2 Inhibitors of Tumor Cell. Polycyclic Aromat. Compd. 2022, 42, 623–634. 10.1080/10406638.2020.1747098. [DOI] [Google Scholar]
- Fayad E.; Altalhi S. A.; Abualnaja M. M.; Alrohaimi A. H.; Elsaid F. G.; Abu Almaaty A. H.; Saleem R. M.; Bazuhair M. A.; Ahmed Maghrabi A. H.; Beshay B. Y.; Zaki I. Novel Acrylate-Based Derivatives: Design, Synthesis, Antiproliferative Screening, and Docking Study as Potential Combretastatin Analogues. ACS Omega 2023, 8, 38394–38405. 10.1021/acsomega.3c05051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Lateef H. M. A.; Saleem R. M.; Bazuhair M. A.; Maghrabi A. H. A.; Ali E. H. K.; Zaki I.; Masoud R. E. Design, synthesis and tubulin polymerization inhibition activity of newly synthesized hydrazone-linked to combretastatin analogues as potential anticancer agents. J. Mol. Struct. 2023, 1292, 136190–136202. 10.1016/j.molstruc.2023.136190. [DOI] [Google Scholar]
- Alshaya D. S.; Tawakul R. M.; Zaki I.; Almaaty A. H. A.; Fayad E.; Abd El-Aziz Y. M. Design, synthesis and antiproliferative screening of newly synthesized acrylate derivatives as potential anticancer agents. RSC Adv. 2023, 13, 23538–23546. 10.1039/D3RA03849A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abd El-Lateef H. M.; Ghany L. M. A.; Saleem R. M.; Maghrabi A. H. A.; Alahdal M. A. Y.; Ali E. H. K.; Beshay B. Y.; Zaki I.; Masoud R. E. Design, synthesis and antiproliferative screening of newly synthesized coumarin-acrylamide hybrids as potential cytotoxic and apoptosis inducing agents. RSC Adv. 2023, 13, 32547–32557. 10.1039/D3RA06644D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abd El-Lateef H. M.; Toson E. E. M.; Abu Almaaty A. H.; Saleem R. M.; Maghrabi A. H. A.; El-Sayed E. H.; Zaki I.; Youssef M. M. Synthesis, Characterization and Biological Evaluation of New Enamide Fluorinated-Schiff Base Derivatives as Potential Cytotoxic and Apoptosis-Inducing Agents. ChemistrySelect 2023, 8, e202303070 10.1002/slct.202303070. [DOI] [Google Scholar]
- Chu E.; Sartorelli A.. Cancer Chemotherapy. In Lange’s Basic and Clinical Pharmacology; McGraw-Hill Education, 2018; pp 948–976. [Google Scholar]
- Uribe M. L.; Marrocco I.; Yarden Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers 2021, 13, 2748 10.3390/cancers13112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallaert W.; Taylor S. R.; Kedziora K. M.; Taylor C. D.; Sobon H. K.; Young C. L.; Limas J. C.; Varblow Holloway J.; Johnson M. S.; Cook J. G.; Purvis J. E. The molecular architecture of cell cycle arrest. Mol. Syst. Biol. 2022, 18, e11087 10.15252/msb.202211087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews H. K.; Bertoli C.; de Bruin R. A. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. 10.1038/s41580-021-00404-3. [DOI] [PubMed] [Google Scholar]
- Gourisankar S.; Krokhotin A.; Ji W.; Liu X.; Chang C.-Y.; Kim S. H.; Li Z.; Wenderski W.; Simanauskaite J. M.; Yang H.; et al. Rewiring cancer drivers to activate apoptosis. Nature 2023, 620, 417–425. 10.1038/s41586-023-06348-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzaneh Behelgardi M.; Gholami Shahvir Z.; Asghari S. M. Apoptosis induction in human lung and colon cancer cells via impeding VEGF signaling pathways. Mol. Biol. Rep. 2022, 49, 3637–3647. 10.1007/s11033-022-07203-9. [DOI] [PubMed] [Google Scholar]
- Li X.; Hou Y.; Zhao J.; Li J.; Wang S.; Fang J. Combination of chemotherapy and oxidative stress to enhance cancer cell apoptosis. Chem. Sci. 2020, 11, 3215–3222. 10.1039/C9SC05997K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahsan N.; Shariq M.; Surolia A.; Raj R.; Khan M. F.; Kumar P. Multipronged regulation of autophagy and apoptosis: emerging role of TRIM proteins. Cell. Mol. Biol. Lett. 2024, 29, 1–35. 10.1186/s11658-023-00528-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.