

Abstract
INTRODUCTION
Soluble amyloid beta (Aβ) oligomers have been suggested as initiating Aβ related neuropathologic change in Alzheimer's disease (AD) but their quantitative distribution and chronological sequence within the AD continuum remain unclear.
METHODS
A total of 526 participants in early clinical stages of AD and controls from a longitudinal cohort were neurobiologically classified for amyloid and tau pathology applying the AT(N) system. Aβ and tau oligomers in the quantified cerebrospinal fluid (CSF) were measured using surface‐based fluorescence intensity distribution analysis (sFIDA) technology.
RESULTS
Across groups, highest Aβ oligomer levels were found in A+ with subjective cognitive decline and mild cognitive impairment. Aβ oligomers were significantly higher in A+T− compared to A−T− and A+T+. APOE ε4 allele carriers showed significantly higher Aβ oligomer levels. No differences in tau oligomers were detected.
DISCUSSION
The accumulation of Aβ oligomers in the CSF peaks early within the AD continuum, preceding tau pathology. Disease‐modifying treatments targeting Aβ oligomers might have the highest therapeutic effect in these disease stages.
Highlights
Using surface‐based fluorescence intensity distribution analysis (sFIDA) technology, we quantified Aβ oligomers in cerebrospinal fluid (CSF) samples of the DZNE‐Longitudinal Cognitive Impairment and Dementia (DELCODE) cohort
Aβ oligomers were significantly elevated in mild cognitive impairment (MCI)
Amyloid‐positive subjects in the subjective cognitive decline (SCD) group increased compared to the amyloid‐negative control group
Interestingly, levels of Aβ oligomers decrease at advanced stages of the disease (A+T+), which might be explained by altered clearing mechanisms
Keywords: Alzheimer's disease, APOE, AT(N) classification, Aβ, cerebrospinal fluid, oligomers, preclinical, prodromal, sFIDA, tau
1. BACKGROUND
Alzheimer's disease (AD) is a neuropathological disorder accompanied by abnormal protein deposits such as amyloid plaques and neurofibrillary tangles (NFTs) 1 which may occur up to 20 years before the onset of clinical symptoms. 2 , 3
Soluble amyloid beta (Aβ) oligomers are neuro‐ and synaptotoxic Aβ aggregates implicated in triggering AD‐related Aβ pathology that are derived from the sequential cleavage of the transmembrane amyloid precursor protein (APP). 4 , 5 A growing body of research indicates that deficient clearing mechanisms prevent Aβ oligomer degradation and facilitate the accumulation of Aβ species into insoluble plaques. 4 , 6 , 7 , 8 , 9 Furthermore, the formation of toxic Aβ oligomers and fibrils in the cerebrospinal fluid (CSF) has been associated with a decreased ratio of Aβ42/Aβ40, and an increased amyloid plaque burden as measured by positron emission tomography (PET). 10 , 11 Thus, increased Aβ oligomer levels and increasing Aβ plaque burden might act as a surrogate marker for deficient Aβ monomer clearance. Furthermore, in a series of experiments that included human brain autopsy and rat models, tau pathology measured by phosphorylated tau (pTau) in the CSF or by PET was found to act downstream of synaptic Aβ oligomer accumulation. 12 Consequently, examining potential differences in Aβ oligomer concentrations in different biomarker profiles across the AD spectrum might help identify individuals in the earliest stages of AD, thereby allowing to identify a unique window of opportunity for effective therapeutic intervention. 13 Yet, little is known how the levels of Aβ oligomers in the CSF correlate with the stages of AD, or whether they could serve as reliable biomarkers for disease progression. 14 In order to investigate the role of Aβ oligomer concentrations, the National Institute on Aging and the Alzheimer's Association (NIA‐AA) research framework provide a unified biological definition of Alzheimer's disease, capable of identifying early pathological changes and biomarker interactions related to the disease. 1 , 15
According to the NIA‐AA research framework, individuals can be placed on the Alzheimer continuum once pathological Aβ aggregation (ie, A+) arises, regardless of their cognitive status. 1 , 15 In the revised NIA‐AA research framework, six symptom stages of AD are defined, 16 with stages 1 and 2 characterizing individuals with preclinical AD that are cognitively unimpaired (CU). 15 , 16 In stage 1, experiencing subjective cognitive decline (SCD) has to be absent, whereas individuals can experience SCD or newly acquired neurobehavioral symptoms in stage 2. 16 Stages 3, 4, 5, and 6 correspond to mild cognitive impairment (MCI), and mild, moderate and severe AD dementia, respectively. Although the revised research framework is closely linked to the AT(N) (A, amyloid; T, tau; N, neurodegeneration) classification system for AD biomarkers, the presence of pTau pathology (ie, T+) is not essential to be placed on the AD continuum, as pTau pathology has been suggested to be a downstream event of amyloid pathology, and may therefore arise at later disease stages. 1 Consequently, individuals on the AD continuum can have varying biomarker profiles (eg, A+T− or A+T+), where A+T− indicates early pathologic change, whereas full‐blown AD pathology (ie, the presence of both pathological amyloid and pTau deposition; A+T+) might occur at a later time 1 .
Accordingly, the present study aims to elucidate the role of CSF Aβ and tau oligomers in individuals along the AD continuum stratified for different biomarker profiles (ie, A−T−, A−T+, A+T−, and A+T+), assuming that these different cross‐sectional profiles reflect the temporal evolution of AD, with subjects being in one of these biological disease stages. Using surface‐based fluorescence intensity distribution analysis (sFIDA), a platform technology for the quantitation of protein aggregates in biofluids, 17 , 18 , 19 , 20 , 21 we specifically aimed to investigate whether oligomer titers differ between biomarker profiles, and whether there is an association between amyloid positivity (ie, A+), oligomer concentrations, and diagnostic status. Lastly, as carrying the ε4 allele of the apolipoprotein E (APOE) gene is the most important genetic risk factor in sporadic AD, 22 we examined whether the concentrations of Aβ and tau oligomers are increased in APOE ε4 carriers.
2. METHODS
2.1. Samples and design
In the present study, demographic and clinical information and CSF baseline samples from 526 participants of the DZNE‐Longitudinal Cognitive Impairment and Dementia (DELCODE) study 23 were included. Details on the overall DELCODE study design, definition of patient groups including criteria for patient enrollment, and execution of cognitive, neuropsychological tests as well as biomaterial sampling, APOE‐genotyping, PET, magnetic resonance imaging (MRI) and CSF biomarker assessment are described in Jessen et al. 23 All participants provided their written informed consent. Only participants with a minimum of 18 points on the Mini‐Mental State Examination (MMSE) qualified for the AD group in the DELCODE study. 23 In short, participants were recruited from 10 memory clinics throughout Germany and allocated to 5 participant groups, that is, the cognitively unimpaired control (C) group, SCD, MCI, mild AD dementia, and healthy first‐degree relatives of AD patients (REL). To increase the number of controls in the present study, the REL were integrated into the control group. In order to classify participants according to the AT(N) classification system, we used cutoffs established within the DELCODE cohort, that is, Aβ42 ≤ 638.7 pg/mL, total tau (tTau) > 510.9 pg/mL, and pTau ≥ 73.65 pg/mL. 24 CSF samples were stored at −80°C and did not undergo a freeze‐thaw cycle. All 526 samples were blinded at the timepoint of sFIDA measurement.
2.2. Oligomer measurement using sFIDA
We previously developed sFIDA to specifically and sensitively measure protein oligomers and aggregates in biofluids. 17 , 18 , 19 sFIDA uses a sandwich‐like biochemistry employing the same capture and detection antibodies with linear epitopes to reliably measure the whole fraction of Aβ or tau oligomers and larger, but still soluble, aggregates even in the presence of excessive monomers. Due to the use of fluorescence microscopy and sophisticated image analysis, sFIDA yields single particle sensitivity.
2.3. Synthesis of protein conjugated silica nanoparticles
For assay development and as assay control we used our previously developed silica nanoparticle (SiNaP) standard standard. 25 , 26 Aβ SiNaPs were coated with amino acids 1−15 of the Aβ protein (peptides and elephants, Henningsdorf, Germany) as described in Blömeke et al. 19 Shortly, SiNaPs were synthetized using the Stöber process and afterwards aminated using (3‐aminopropyl)triethoxysilane (APTES, Sigma‐Aldrich, St. Louis, MO, USA). 25 , 26 In the next step, activated maleimidohexanoic acid (MIHA, abcr GmbH, Karlsruhe, Germany) was allowed to react covalently with the amines. Finally, Aβ1−15 functionalized with cysteamine at the C‐terminus was added to react with the maleimide groups of the particles.
Tau SiNaPs were coated with full‐length tau protein (2N4R). Here, we used a different approach which was previously described in Hülsemann et al. 25 In this approach, synthesized and aminated SiNaPs were further functionalized with succinic anhydride (Sigma‐Aldrich). To enable reaction, the pellet of aminated SiNaPs was redispersed in 0.1 M succinic acid anhydride in N,N‐dimethylformamide anhydrous (DMF) and incubated for 2 hours at 70°C and afterwards for 2 days at room temperature (RT) when stirring. The carboxylated SiNaPs were washed three times with ddH2O by centrifugation (7 minutes at 10,000 × g) and redispersion. For biofunctionalization, carboxylated SiNaPs were activated with 20 mM 1‑ethyl‑3‑(3‑dimethylaminopropyl)carbodiimide (EDC; Sigma‐Aldrich) and 5 mM N‑hydroxysuccinimide (NHS; Sigma‐Aldrich) in a buffer of 0.1 M 2‐(N‐Morpholino)ethanesulfonic acid (MES; Carl Roth, Karlsruhe, Germany) while shaking for 1 hour at RT. After two washing steps (centrifugation at 18,200 × g for 10 minutes) and redispersion in phosphate‐buffered saline (PBS), full‐length tau protein was added. The next day, biofunctionalized SiNaPs were washed twice with ddH2O as described before.
SiNaPs were characterized based on particle size and shape as previously described. 19 , 25 Size and shape of the particles were determined using transmission electron microscopy while concentrations were determined using inductively coupled plasma‐mass spectrometry (ICP‐MS).
RESEARCH IN CONTEXT
Systematic review: Aβ oligomers are the most toxic Aβ species. They disrupt synaptic communication and may thereby initiate neurodegeneration in Alzheimer's disease (AD). In order to elucidate the temporal order of amyloid (A) and tau (T) pathology along the AD continuum, we quantified cerebrospinal fluid (CSF) Aβ and tau oligomers in different disease stages.
Interpretation: CSF Aβ oligomers were significantly higher in participants with subjective cognitive decline (stage 2) and mild cognitive impairment (stage 3) classified as A+. Interestingly, A+T+ showed comparably lower Aβ oligomer levels, which might be due to increased binding to amyloid plaques over time.
Future directions: Further research examining the underlying mechanisms of the rise and fall of Aβ oligomers along the AD continuum is needed.
2.4. Labeling of antibodies
To detect oligomers in samples, we labeled the antibodies Nab228 (Sigma‐Aldrich) and Tau12 (BioLegend, San Diego, CA, USA) with the fluorescent dyes CF633 (Sigma‐Aldrich) and CF488A (Sigma‐Aldrich) according to manufacturer's protocol. The principle of reaction, the purification, and the determination of concentration and degree of labeling were previously described. 19
2.5. Assay protocol
The biochemical principle of sFIDA has been reported elsewhere. 27 , 28 In the present study, we used Nunc MicroWell 384‐well plates for each experiment (Thermo Fisher Scientific, Waltham, MA, USA) functionalized with N‐terminal monoclonal antibodies Nab228 (Sigma‐Aldrich) and Tau12 (BioLegend) at a concentration of 2.5 µg/mL in 0.1 M NaHCO3. After overnight incubation at 4°C, we washed the plates five times with 80 µL tris‐buffered saline (TBS)‐T (1×TBS; Serva Electrophoresis, Duisburg, Germany) containing 0.05% Tween20 (AppliChem, Darmstadt, Germany) and afterwards five times with TBS (405 LS Microplate Washer, BioTek, VT, USA). To block remaining binding sides of the glass surface, 1% bovine serum albumin (BSA; AppliChem) in TBS with 0.03% ProClin (Sigma‐Aldrich) were incubated for 1.5 h at RT. After washing five times with TBS‐T and TBS as described above, we diluted a mix of Aβ and tau SiNaPs in PBS (Sigma‐Aldrich) containing 0.5% BSA and 0.05% Tween (dilution factor 1:2) and applied 20 µL of each dilution and 20 µL of undiluted samples to the plate. The plate was incubated for 2 hours at RT and thereafter washed five times with TBS. Then 20 µL of fluorescent detection antibodies Nab228 CF633 (0.156 µg/mL) and Tau12 CF488A (0.312 µg/mL) diluted in TBS were applied and incubated for 1 hour at RT. Finally, the plate was washed five times with TBS and the buffer was exchanged against TBS‐ProClin. In order to obtain a sufficient number of replicates, each dilution and all samples were applied in a quadruple determination.
2.6. Image data acquisition
For imaging of the assay surface, we used total internal reflection microscopy (TIRF‐M; Leica, Wetzlar, Germany) 27 (excitation: 635 nm, emission filter: 705/22 nm; excitation: 488 nm, emission filter: 525/36 nm; exposure time: 1500 ms; gain: 800). In total, 25 images per well were measured with 1000 × 1000 pixels each.
2.7. Quantification and statistical analyses
Quantification and general statistical analyses were carried out using Python 3.9.7 (Python software foundation, Wilmington, USA; packages: scipy version 1.7.3) and Origin 2020 (OriginLab Corporation, Northampton, USA). Data were further analyzed for normal distribution and in the case of not normally distributed data, nonparametric tests, for example, Spearman correlation or Mann–Whitney U‐test, were used for further analyses.
2.8. Image data analysis
For analysis of the images, the in‐house developed software tool sFIDAta was used including the detection and elimination of artefact‐containing images. 26 , 27 The analysis itself is based on the number of pixels above a defined cutoff value, which is defined as the pixel count. The cutoff is defined as the grayscale value at which the blank control exceeds a number of 100 pixels (0.01% of total pixels). The cutoff value is determined for each experiment individually. Moreover, 10% of images per well with the highest and lowest pixel counts were excluded from analysis to ensure that no artificial images influence the readouts. 28 , 29 Given pixel counts were calculated as the mean of the four sample wells.
2.9. Analytical validation
To assess intra‐assay variability of SiNaPs and samples, the coefficient of variation (CV) percentage of the four test replicates within the same run was calculated (Table S1). Furthermore, analytical selectivity of the assay was investigated applying different assay control setups as described previously. 18 , 19 , 30 In this study, we investigate if 2 pM of Aβ or tau SiNaPs interfere with the assay surface in the absence of capture antibodies (capture control), show false‐positive signals due to autofluorescence component of the used assay buffers (autofluorescence control, no detection probe), or show cross‐reactivity with anti‐α‐synuclein detection antibodies. In addition, the selectivity of the used assay antibodies was analyzed using equimolar concentrations of α‐synuclein‐coated SiNaPs. Since monomeric species of Aβ and tau are present in excess compared to synthetic oligomers and therefore can falsify the measurement signals, we have additionally compared equimolar concentrations of monomeric as well as oligomeric species, that is, 1 nM of synthetic Aβ oligomers 30 and 250 pM tau oligomers 19 (Figure S1). Afterward, the signal reduction of each assay control was calculated based on the normalized pixel counts according to Equation (1).
| (1) |
2.10. Data scaling
Since oligomer levels in samples were lower than suggested, not enough data points of the SiNaPs concentration series were available to create a suitable calibration curve. However, as it was not possible to use the raw data directly without distorting the statistical results, a scaling method was used to compensate for differences between the experiments. This method was based on the samples of the control group and was carried out separately for Aβ and tau oligomers. For each oligomer type, a cross‐plate median globalMedian of all control group samples was calculated. Subsequently, a separate medium plateMedian was formed for each plate p. Using these values, a scaling factor (Table S2) was calculated for each plate p according to Equation (2).
| (2) |
Finally, all measurement results of a plate were multiplied by the corresponding scaleFactorp . The scaled pixel counts for Aβ and tau oligomers, in the following referred to as Aβ and tau oligomer pixel counts, were used for all analyses. Figure S2 and Figure S3 show the data before and after scaling, respectively.
2.11. Descriptive analysis
Using Spearman correlations and two‐sided Mann–Whitney U‐tests, we examined the association of oligomer pixel counts with demographic variables and known risk factors for AD, including age, sex, and APOE status, 31 , 32 as well as associations with further CSF biomarkers.
2.12. Differences in Aβ and tau oligomer levels
To test for differences between participant groups, we performed a two‐sided Mann–Whitney U‐test. For this purpose, we regrouped the samples stepwise, based first on clinical diagnosis. However, symptoms used for the clinical diagnosis of AD can also be caused by other forms of dementia, which leads to a clinical diagnosis not necessarily being free of errors. Therefore, we subsequently subdivided the four clinically defined groups based on the amyloid status using Aβ42 biomarker data. Afterward, the samples were analyzed independently of their clinical diagnosis using the AT(N) classification. To this end, cutoffs established within the study of Jessen et al. 24 can be used to classify the participants according to AT(N) system. In the present study, the cut‐offs for Aβ42 (≤ 638.7 pg/mL), pTau (≥ 73.65 pg/mL) and tTau (> 510.9 pg/mL) positivity were applied to determine amyloid positivity or tau positivity or neurodegeneration. In addition, we also regrouped the samples based on APOE status with carrying at least one APOE ε4 allele defining APOE positivity (ε2/ε4, ε3/ε4, or ε4/ε4) since the APOE ε4 allele is an important genetic risk factor for AD. 22
2.13. Modeling Aβ oligomer levels in the AD continuum
In an effort to model levels of Aβ oligomers in the course of AD, a regression pipeline was designed to anticipate the Aβ oligomers utilizing Aβ42 and pTau monomers as features. The pipeline is made up of a standard scaler and a bagging model, the latter comprising three support vector regression models with radial basis function (RBF) kernels that are trained on various data subsets. Given the presumption that the relationship between the features and target might vary depending on the APOE ε4 status, three different instances of this pipeline were created using solely APOE ε4 carrier data, APOE ε4 noncarrier data, and all‐encompassing data.
To generate continuous oligomer curves describing the progression of AD with these pipelines, it is necessary to establish a probable trajectory of Aβ42 and pTau monomers throughout the AD stages. In order to exclude bias arising from disproportionate representation of AT groups, equal numbers of samples were randomly chosen from the A−T−, A+T−, and A+T+ sets. Following this, data were scaled employing a min‐max scaler, centered, and aligned to the antidiagonal. The trajectory was finally ascertained by applying a second‐degree polynomial regression, and subsequently reverse transformed. The resulting trajectory was utilized as input to predict oligomer curves for APOE ε4 carrier, APOE ε4 noncarrier, and all data by the three system instances, which were later refined by a moving average. Given their relationship to Aβ42 and pTau, these curves can be integrated into other models. With this feature in mind, the curves were deliberately superimposed on a biomarker model for AD progression. 33
All of the above was achieved using the scikit‐learn Python package, version 1.0.2.
3. RESULTS
The aim of the present study was to investigate Aβ and tau oligomer levels in human CSF samples of patients across the clinical and neurobiological continuum of AD.
3.1. Descriptive analysis of patient and control groups
In the present study, 526 CSF samples from DELCODE, that is, 137 samples from controls, 211 samples from SCD participants, 112 samples from MCI patients, and 66 samples from AD patients, were screened for Aβ and tau oligomer levels. Demographic and clinical information for these four groups on age, gender, neuropsychological tests, amyloid‐, tau‐, and APOE ε4‐status is available in Table 1.
TABLE 1.
Demographic information and study characteristics of participants grouped by clinical diagnosis.
| Control | SCD | MCI | AD | |
|---|---|---|---|---|
| Number | 137 | 211 | 112 | 66 |
| Age, years (SD) | 68.2 (4.9) | 71.4 (5.8) | 72.3 (5.4) | 75.5 (6.3) |
| Female | 54% | 43% | 43% | 65% |
| MMSE (SD) | 29.3 (0.9) | 29.1 (1.1) | 27.5 (1.9) | 23.2 (3.1) |
| A+ | 27.7% | 40.8% | 72.3% | 92.4% |
| T+ | 7.3% | 15.2% | 37.5% | 68.2% |
| APOE ε4 a | 27.6% | 34.6% | 50.5% | 64.5% |
Abbreviations: A+, amyloid pathology above cut‐off; AD, Alzheimer's disease; APOE ε4, apolipoprotein E ε4 allele; MCI, mild cognitive impairment; MMSE, Mini‐Mental State Examination; SCD, subjective cognitive decline; SD, standard deviation; T+, tau pathology above cut‐off.
No APOE data were available for 17 participants.
To avoid misinterpretation of the statistical results due to demographic characteristics, we checked for correlations of age and gender with oligomer levels. Since the Shapiro–Wilk test showed that neither Aβ nor tau oligomer values were normally distributed (p = 9.22 × 10−42 and 1.44 × 10−34, respectively), nonparametric tests were used. No significant Spearman correlation was found between age and oligomer pixel counts (Aβ: r = .033, p = .446; tau: r = .016, p = .709). Results from two‐sided Mann–Whitney U‐tests showed that Aβ and tau oligomer pixel counts did not significantly differ between genders, even if the significance is nearly reached for Aβ oligomers (p = .063; tau p = .973). Furthermore, as age and gender are equally distributed for all combinations of groupings by AT(N) classification as well as APOE ε4 genotpye, it can be assumed that there is no respective bias for the analysis.
3.2. Clinically diagnosed MCI patients showed significantly higher levels of Aβ oligomers compared to controls
First, we investigated if Aβ and tau oligomer levels differ between clinical diagnosis groups, using two‐tailed Mann–Whitney U‐tests (Figure 1). In the case of MCI patients, significantly higher Aβ oligomer levels were found compared to the control group (p = .017, Figure 1A). Although Aβ and tau oligomer levels showed a highly significant correlation (Spearman r = .541, p = 2.7 × 10−41), no increase of tau oligomer levels regarding the disease stage was observed (Figure 1B).
FIGURE 1.
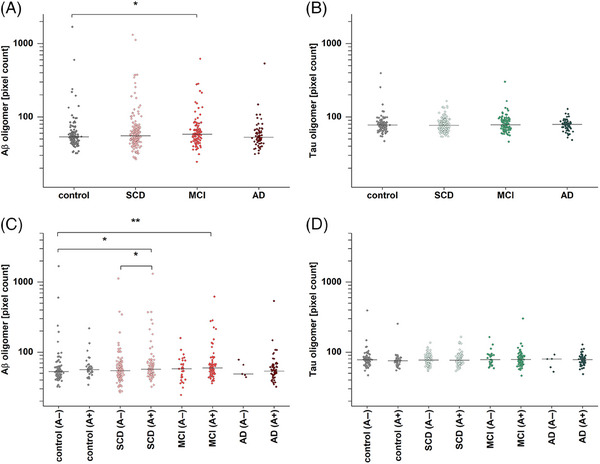
Amyloid beta (Aβ) and tau oligomer pixel count based on amyloid pathology (A+/A−). (A) Aβ oligomer pixel counts in mild cognitive impairment (MCI) are significantly increased compared to the controls (p = .017). (B) By contrast, no significant changes were detected for tau oligomer pixel count. (C) After dividing groups along amyloid status, significantly higher levels in subjective cognitive decline (SCD) (A+) and MCI (A+) compared to controls (A−) were observed (p = .014 and p = .0028, respectively). Furthermore, SCD (A+) is significantly elevated compared to SCD (A−) (p = .048). (D) Tau oligomers in subgroups show no significant differences when divided in A+ and A−. Effect sizes for the significantly differing groups are provided in Table S3, while receiver operating characteristic curves and area under the curve scores are presented in Figure S4 and Table S5. Horizontal lines indicate the median; y‐axis scales are logarithmic. A two‐sided Mann–Whitney U‐test (confidence interval = .05) was carried out to investigate differences between the groups. Abbreviation: AD, Alzheimer's disease. *p ≤ .05, **p ≤ .01.
Next, we divided the four clinical groups based on presence of amyloid pathology into amyloid negative (A−) and amyloid positive (A+) cases to analyze if differences in Aβ oligomer levels are due to underlying AD pathology. When comparing these refined groups with the A− control group, it became apparent that SCD (p = .014) and MCI (p = .003) participants with underlying amyloid pathology, but not those without evidence for AD, showed significantly increased Aβ oligomer levels (Figure 1C). Furthermore, SCD participants with amyloid positive states showed elevated Aβ oligomer levels compared to SCD individuals without diagnosed amyloid pathology (p = .048). However, even after the additional breakdown by A, there was no difference in tau oligomer levels (Figure 1D).
3.3. Differences in Aβ oligomer levels based on AT(N) profiles and APOE ε4 status
As the results of Section 3.2 indicate the importance of the underlying pathologies for increases of Aβ oligomers, we regrouped and analyzed the results based on the AT classification proposed by the NIA‐AA research framework and the currently established cutoff values by Jessen et al. for Aβ42 and pTau 24 (Figure 2). Furthermore, demographic information was regrouped based on AT classification (Table 2). Since again no differences in tau oligomer levels between the sample groups were observed, the subsequent analyses were only described with respect to Aβ oligomer levels.
FIGURE 2.
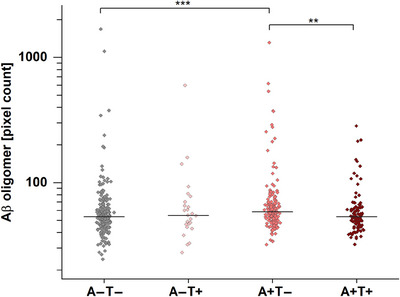
Amyloid beta (Aβ) oligomers in quantified cerebrospinal fluid (CSF) samples after AT (amyloid/tau) classification. Aβ oligomer pixel counts in CSF of A+T− patients are significantly increased compared to the reference group A−T− (p = 5.8 × 10−5) and to A+T+ (p = .0026). Effect sizes for the significantly differing groups are provided in Table S3, while receiver operating characteristic curves and area under the curve scores are presented in Figure S4 and Table S5. Horizontal lines indicate the median; y‐axis scale is logarithmic. A two‐sided Mann–Whitney U‐test (confidence interval = .05) was carried out to investigate differences between the groups. **p ≤ .01, ***p value ≤ .001.
TABLE 2.
Demographic information of participants based on the AT classification.
| Characteristic | A−T− | A−T+ | A+T− | A+T+ |
|---|---|---|---|---|
| number | 234 | 26 | 163 | 103 |
| Age, years (SD) | 69.62 (5.61) | 73.08 (5.15) | 71.61 (6.00) | 74.08 (5.78) |
| Female | 48.7% | 53.8% | 61.0% | 51.5% |
| MMSE (SD) | 29.1 (1.3) | 28.3 (2.9) | 28.2 (2.4) | 25.7 (3.4) |
| Aβ42, pg/mL (SD) | 960.9 (212.2) | 1218.9 (471.2) | 445.3 (122.9) | 419.8 (116.4) |
| Aβ42/40 ratio (SD) | 0.108 (0.015) | 0.094 (0.029) | 0.071 (0.023) | 0.046 (0.009) |
| pTau, pg/mL (SD) | 47.7 (10.9) | 102.2 (45.3) | 46.4 (16.3) | 108.3 (35.8) |
| tTau, pg/mL (SD) | 333.0 (120.2) | 696.8 (361.0) | 353.0 (153.3) | 849.8 (273.3) |
| APOE ε4 a | 20.0% | 23.1% | 48.1% | 73.0% |
| Clinical diagnosis in percentage of subgroups | ||||
| Control | 39.7% | 23.1% | 20.9% | 3.9% |
| SCD | 48.3% | 46.2% | 40.5% | 19.4% |
| MCI | 10.7% | 23.1% | 27.6% | 35.0% |
| AD | 1.3% | 7.7% | 11.0% | 41.7% |
| 100% | 100% | 100% | 100% |
Abbreviations: A+, amyloid pathology above cut‐off; Aβ, amyloid beta; AD, Alzheimer's disease; APOE ε4, apolipoprotein E ε4 allele; AT, amyloid/tau; MCI, mild cognitive impairment; MMSE, Mini‐Mental State Examination; SCD, subjective cognitive decline; SD, standard deviation; T+, tau pathology above cut‐off.
No APOE data were available for 17 participants.
Applying this classification, samples of participants with amyloid pathology without tau pathology (A+T−) had significantly increased levels of Aβ oligomers compared to A+T+ participants with amyloid and tau pathology (p = 5.8 × 10−5). This applied to participants with (A+T−N+) and without (A+T−N−), a concomitant non‐Alzheimer's pathologic change (A+T−N+: p = .032; A+T−N−: P = 1.8 × 10−4). Aβ oligomer levels of participants classified as A+T+ did not differ significantly from control group participants but compared to A+T− participants (p = .0026).
Because the APOE ε4 allele is an important genetic risk factor for AD, 22 we also investigated Aβ oligomer levels in APOE ε4 carriers. Here, significantly increased Aβ oligomer levels were found compared APOE ε4 noncarriers independent of the disease stage (p = .02). For further evaluation, receiver operating characteristic (ROC) curves, area under the curve (AUC) scores, and effect sizes were presented in Figure S4, Tables S3 and S4.
3.4. Aβ oligomer levels are elevated in early disease stages
To investigate how Aβ oligomers fit into the AD continuum, we established a regression model that predicts Aβ oligomer levels based on monomeric Aβ42 and pTau concentrations. This model was validated by normalized mean absolute error (Table S5) and by comparing the binned raw data and the binned regression model predictions (Figure S5). As displayed in Figure 3A, increased oligomer levels were only found at low Aβ42 and pTau concentrations in early AD stages. Furthermore, we also investigated if the observed increase and later decrease of Aβ oligomer levels depends on APOE ε4 status. To this end, we classified samples into APOE ε4 carriers (Figure 3B) and noncarriers (Figure 3C). The predicted Aβ oligomer levels in context of different APOE ε4 states indicated that the levels of Aβ oligomers are increased in APOE ε4 carriers, and that the peak shifted to higher pTau concentrations (red areas in Figure 3B,C). To provide a more specific statement about the probable level of the Aβ oligomer in the AD continuum, the prediction was reduced to the most likely trajectory of Aβ42 and pTau monomer through the AD stages, as described in 2.3.6 and displayed in Figure 4A. The model of Jack et al. 33 supplemented by the modeled Aβ oligomer curves revealed that in APOE ε4 noncarriers the peak is reached during the SCD stage, whereas in APOE ε4 carriers it is reached at the MCI stage (Figure 4B). Regardless of the APOE ε4 status, Aβ oligomer levels already decrease within the MCI stage.
FIGURE 3.
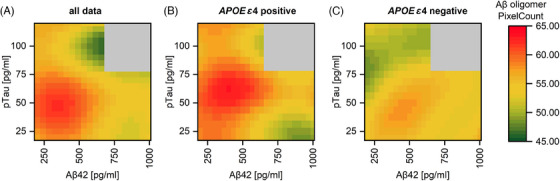
Regression model for the interrelationship of amyloid beta (Aβ) oligomers pixel count, Aβ42, and phosphorylated tau (pTau) in cerebrospinal fluid (CSF). Based on Aβ42, pTau, and Aβ oligomer levels in CSF, regression models for prediction of Aβ oligomers pixel count were performed. Highest Aβ oligomer levels can be expected in patients with low Aβ42 while pTau is relatively low in (A) all patients, (B) apolipoprotein E (APOE) gene ε4 allele carriers, and (C) APOE ε4 noncarriers. Aβ42 oligomer levels in APOE ε4‐positive participants are overall higher and shifted toward higher pTau concentrations compared to those who were APOE ε4 negative.
FIGURE 4.
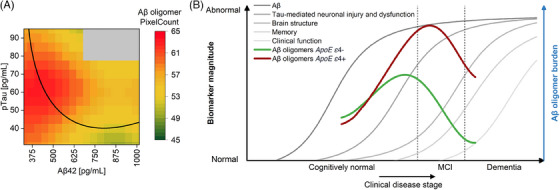
Hypothetical model of amyloid beta (Aβ) oligomers in cerebrospinal fluid (CSF) within the Alzheimer's disease (AD) continuum. (A) Along the trajectory of biomarkers during AD progression from high Aβ42 and low pTau (A−T−) to lowered Aβ42 first (A+T−) followed by elevated pTau (A+T+), Aβ oligomers start to rise until a turning point is reached. Soon after pTau starts to increase, Aβ oligomer concentrations decrease. This panel is a zoomed‐in portion of Figure 3B. Data are represented in a binned form. (B) Hypothetical changes of Aβ oligomers during disease progression are transferred to the model of AD biomarker changes according to Jack et al. 1 Apolipoprotein E (APOE) gene ε4 allele carriers show higher oligomer concentrations with a peak at a more advanced disease stage but still in the early stages of the disease. Due to the high age of the cohort (60+) and the absence of persons with advanced AD, it was not possible to cover the entire x‐axis with the curves. For validation of the model, Figure S5 shows a comparison between the measured oligomer level and the oligomer level determined by regression. Figure modified after Jack et al. 33
4. DISCUSSION
In the present study we measured Aβ oligomers in CSF aiming to allocate the presence of these most neurotoxic Aβ species within the neurobiological continuum of AD and thereby elucidate the temporal sequence of Aβ oligomers in AD pathology. Our work provides novel evidence for the importance of Aβ oligomers in early biological and symptomatic disease stages, as we found the highest levels of Aβ oligomers in participants with the clinical diagnosis of MCI. However, 59.8% of SCD and 27.7% of MCI participants did not have evidence for amyloid pathology and might therefore not suffer from AD. Stratification for amyloid positivity (ie, A+ vs A−) alone yielded significant differences within the SCD group and enhanced the discrimination of SCD and MCI to controls. Strikingly, stratification of participants by A/T biomarker profiles, thereby also taking tau pathology as reflected by CSF pTau into account, yielded significantly elevated Aβ oligomer levels in individuals with an A+/T− biomarker profile compared to participants with nonpathological AD biomarkers (A−/T−) on the one hand, and full‐blown AD neuropathological changes (A+/T+ profile) on the other. Consequently, Aβ oligomers in our cohort peak in early disease stages, where tau pathology is still inconspicuous (A+T−). Intuitively, one may assume that while Aβ aggregation is increasing in the brain during early disease stages, the Aβ oligomer level is also rising in the CSF. That is exactly what we observed in the present study when comparing A−T− with A+T− subjects. Rather surprisingly, we observed reduced Aβ oligomer levels in the more advanced disease stage A+T+ compared to the earlier A+T− stage. Deposition of oligomer species into plaques or breakdown of active clearance mechanisms from brain to CSF are just two of many possible explanations (Figure 5). To elucidate the relationship of Aβ oligomers, Aβ42, and pTau (Figure 3), we calculated a chronological sequence of Aβ oligomer levels based on the regression model and superimposed it to key biomarkers of AD as depicted in Figure 4. With respect to the limited range of disease stages within our cohort, lacking very early and advanced disease stages, no statements about oligomer levels over the whole spectrum of disease stages can be made. A limitation of the study is the low number of samples from stage 1 of AD to determine the age at which Aβ oligomer levels start to rise. According to our model, Aβ oligomers start to rise approximately at the beginning of stage 2 and reach their peak early in stage 3 before the oligomer level decreases again, which is in line with previous studies. 34 , 35 However, other studies, which did not include early stages, reported increased Aβ oligomer concentrations in CSF samples of demented patients compared to the control group 21 , 34 , 36 or no differences between dementia or MCI and controls. 37 In addition to differences in patient preanalytical variables such as freeze‐thaw cycles, storage period, or centrifugation of the samples, 38 different results may be caused by the choice of assay setups, especially regarding the selection of antibodies, 35 targeting different epitopes and oligomer structures. More recent publications focused on blood‐based detection reported increased Aβ oligomer levels in plasma over the course of the disease or a correlation with amyloid‐PET positivity. 39 , 40 , 41
FIGURE 5.
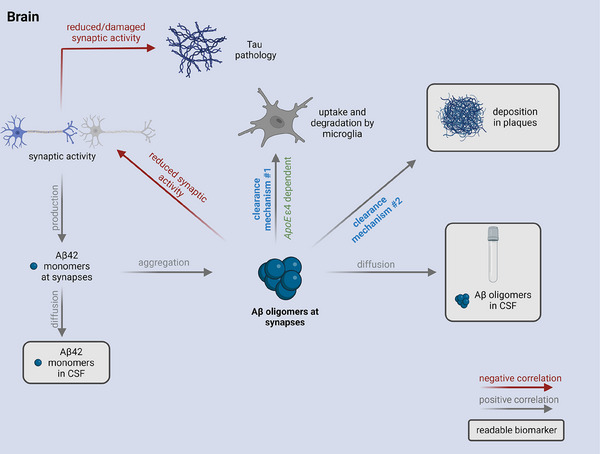
Proposed clearance mechanisms for amyloid beta (Aβ) oligomers and the influence on the use of Aβ oligomers as biomarker. Hypothetic scenario: Aβ monomer production at synapses is dependent on synaptic activity. At a certain time point, aggregation of Aβ monomers leads to the formation of toxic Aβ oligomers which can be cleared by different mechanisms. Aβ oligomers can be degraded by microglia (clearance mechanism #1), diffuse into cerebrospinal fluid (CSF), or be deposited into plaques (clearance mechanism #2) as soon as there are plaques. Formation of plaques in patients with amyloid pathology allows oligomers to be deposited there (clearance mechanism #2), which may well become the preferred fate of Aβ oligomers. Figure created with BioRender.com. APOE ε4, apolipoprotein E ε4 allele.
We further investigated the effect of APOE ε4 positivity on Aβ oligomer levels. As can be seen in the regression model (Figure 3) and the hypothetical model of Aβ oligomer changes (Figure 4), APOE ε4 carriers showed higher oligomer levels. Notably, in APOE ε4 carriers, the Aβ oligomer level in CSF peaks further right of the peak in APOE ε4 noncarriers. This is not later in time, but further down in disease progression strengthening the view that APOE ε4 carriers start earlier into the Alzheimer's continuum. 22 , 42
Although Aβ and tau oligomers show a highly significant correlation, no differences of tau oligomers with respect to the biomarker profile or the clinical syndrome were observed, which is in line with previous findings from our lab. 19 In the present study, we investigated patients who were at mild disease stages when enrolled in the DELCODE study. 23 Presumably, tau oligomers in CSF might be increased and detectable in late stages of dementia. Indeed, a previous study from Sengupta et al. reported elevated tau oligomers only at moderate to severe dementia. 43
Our results emphasize the relevance of the biologically based definition of Alzheimer's disease, as Aβ oligomers, which are thought to be the major toxic species in the disease, are only increased in patients with abnormal Aβ42. Moreover, we have previously demonstrated that sFIDA is now a robust method to quantitate aggregates from tau, α‐synuclein, and Aβ in body fluids. 17 , 18 , 19 , 20 , 30 To further understand the differences between assay outcomes and investigate setups or antibodies which are best suited for diagnosis, it will be of great interest to measure a pool of samples with diverse Aβ oligomer assays. Probably, the combination of different oligomer biomarkers improves the understanding of the underlying pathology and the diagnostic accuracy as calculated by the probability analysis of Lewczuk et al. 44 In particular, longitudinal analysis of Aβ and tau oligomer concentrations over a longer period of time may support our model of Aβ oligomer concentration changes and add to our understanding about which patients at a predementia stage will develop AD in the future. Besides the diagnostic aspects, Aβ oligomers measured by sFIDA are promising biomarkers for clinical drug development to easily monitor their effects. 20
AUTHOR CONTRIBUTIONS
Victoria Kraemer‐Schulien, Lara Blömeke, and Marlene Pils developed the assay. Victoria Kraemer‐Schulien performed the experiments together with Lara Blömeke and Marlene Pils. Fabian Rehn, Victoria Kraemer‐Schulien, and Lara Blömeke analyzed the data and carried out the statistics. Lara Blömeke, Fabian Rehn, and Victoria Kraemer‐Schulien wrote the manuscript together with Oliver Peters, Marlene Pils, Oliver Bannach, and Dieter Willbold. Oliver Bannach, Dieter Willbold, and Oliver Peters supervised the project. Piotr Lewczuk and Johannes Kornhuber were involved in the establishment of the assay. Janine Kutzsche, Tuyen Bujnicki, Luisa‐Sophie Schneider, Silka D. Freiesleben, and Michael Wagner carefully revised the manuscript. Lukas Preis, Josef Priller, Eike J. Spruth, Slawek Altenstein, Anja Schneider, Klaus Fliessbach, Jens Wiltfang, Frank Jessen, Wenzel Glanz, Katharina Buerger, Daniel Janowitz, Robert Perneczky, Boris‐Stephan Rauchmann, Stefan Teipel, Ingo Kilimann, Doreen Goerss, Christoph Laske, Matthias H. Munk, Michael Ewers, Emrah Düzel, Andrea Lohse, Niels Hansen, Ayda Rostamzadeh, Enise I. Incesoy, Michaela Butryn, Carolin Sanzenbacher, Matthias Schmid were responsible for the management of the DELCODE study at the various sites. Oliver Peters, Frank Jessen, Annika Spottke, Nina Roy‐Kluth, Michael T. Heneka, Frederic Brosseron, Steffen Wolfsgruber, Luca Kleineidam, Melina Stark, Michaela Butryn, and Emrah Düzel were responsible for methodological core central data management and quality control of the DELCODE study.
CONFLICTS OF INTERESTS STATEMENT
Dieter Willbold and Oliver Bannach are co‐founders and shareholders of attyloid GmbH. This had no influence of the interpretation of the data. All other authors declare no competing interests related to this work. The sFIDA method is protected by patents EP3271724A1, EP3014279B1 and EP2794655B1. Author disclosures are available in the supporting information.
CODE AVAILABILITY STATEMENT
For image data analysis, we used the sFIDAta software tool which can be made available upon request.
CONSENT STATEMENT
All local institutional review boards and ethical committees approved the study protocol. All participants gave written informed consent before inclusion in the study. DELCODE is registered at the German clinical trials register (drks00007966) (04/may/2015).
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We received funding from the programs “Biomarkers Across Neurodegenerative Diseases I + II” of The Alzheimer's Association, Alzheimer's Research UK and the Weston Brain Institute (11084 and BAND‐19‐614337). We are also grateful for support from The Michael J. Fox Foundation for Parkinson's Research (14977, 009889), from the ALS Association, and from the Packard Center (19‐SI‐476). We further received funding from the Deutsche Forschungsgemeinschaft (INST 208/616‐1 FUGG, INST 208/794‐1 FUGG) and the Helmholtz Association (HVF0079).
Open access funding enabled and organized by Projekt DEAL.
Blömeke L, Rehn F, Kraemer‐Schulien V, et al. Aβ oligomers peak in early stages of Alzheimer's disease preceding tau pathology. Alzheimer's Dement. 2024;16:e12589. 10.1002/dad2.12589
Lara Blömeke, Fabian Rehn, and Victoria Kraemer‐Schulien contributed equally to this study.
Contributor Information
Oliver Bannach, Email: o.bannach@attyloid.com.
Dieter Willbold, Email: d.willbold@fz-juelich.de.
Oliver Peters, Email: oliver.peters@charite.de.
DATA AVAILABILITY STATEMENT
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
REFERENCES
- 1. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA Research framework: toward a biological definition of Alzheimer's disease. Alzheimer's Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jack CR Jr, Holtzman DM. Biomarker modeling of Alzheimer's disease. Neuron. 2013;80:1347‐1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013;12:357‐367. [DOI] [PubMed] [Google Scholar]
- 4. Tolar M, Hey J, Power A, Abushakra S. Neurotoxic soluble amyloid oligomers drive Alzheimer's pathogenesis and represent a clinically validated target for slowing disease progression. Int J Mol Sci. 2021;22(12):6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Larson ME, Lesné SE. Soluble Aβ oligomer production and toxicity. J Neurochem. 2012;120(supp l):125‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brinkmalm G, Hong W, Wang Z, et al. Identification of neurotoxic cross‐linked amyloid‐beta dimers in the Alzheimer's brain. Brain. 2019;142:1441‐1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gaspar RC, Villarreal SA, Bowles N, Hepler RW, Joyce JG, Shughrue PJ. Oligomers of beta‐amyloid are sequestered into and seed new plaques in the brains of an AD mouse model. Exp Neurol. 2010;223:394‐400. [DOI] [PubMed] [Google Scholar]
- 8. Iljina M, Garcia GA, Dear AJ, et al. Quantitative analysis of co‐oligomer formation by amyloid‐beta peptide isoforms. Sci Rep. 2016;6:28658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci. 2011;34:185‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Olsson B, Lautner R, Andreasson U, et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta‐analysis. Lancet Neurol. 2016;15:673‐684. [DOI] [PubMed] [Google Scholar]
- 11. Lewczuk P, Lukaszewicz‐Zajac M, Mroczko P, Kornhuber J. Clinical significance of fluid biomarkers in Alzheimer's disease. Pharmacol Rep. 2020;72:528‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bilousova T, Miller CA, Poon WW, et al. Synaptic amyloid‐β oligomers precede p‐Tau and differentiate high pathology control cases. Am J Pathol. 2016;186:185‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hefti F, Goure WF, Jerecic J, Iverson KS, Walicke PA, Krafft GA. The case for soluble Aβ oligomers as a drug target in Alzheimer's disease. Trends Pharmacol Sci. 2013;34:261‐266. [DOI] [PubMed] [Google Scholar]
- 14. Hayden EY, Teplow DB. Amyloid β‐protein oligomers and Alzheimer's disease. Alzheimer's Res Ther. 2013;5:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kiselica AM. Empirically defining the preclinical stages of the Alzheimer's continuum in the Alzheimer's disease neuroimaging initiative. Psychogeriatrics. 2021;21:491‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Knopman DS, Haeberlein SB, Carrillo MC, et al. The National Institute on Aging and the Alzheimer's Association Research Framework for Alzheimer's disease: perspectives from the research roundtable. Alzheimer's Dement. 2018;14:563‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pils M, Rutsch J, Eren F, et al. Disrupted‐in‐schizophrenia 1 (DISC1) protein aggregates in cerebrospinal fluid are elevated in first‐episode psychosis patients. Psychiatry Clin Neurosci. 2023;77:665‐671. [DOI] [PubMed] [Google Scholar]
- 18. Schaffrath A, Schleyken S, Seger A, et al. Patients with isolated REM‐sleep behavior disorder have elevated levels of alpha‐synuclein aggregates in stool. NPJ Parkinson's Dis. 2023;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bloemeke L, Pils M, Kraemer‐Schulien V, et al. Quantitative detection of alpha‐synuclein and Tau oligomers and other aggregates by digital single particle counting. NPJ Parkinsons Dis. 2022;8:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kass B, Schemmert S, Zafiu C, et al. Abeta oligomer concentration in mouse and human brain and its drug‐induced reduction ex vivo. Cell Rep Med. 2022;3:100630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang‐Dietrich L, Funke SA, Kuhbach K, et al. The amyloid‐beta oligomer count in cerebrospinal fluid is a biomarker for Alzheimer's disease. J Alzheimer's Dis. 2013;34:985‐994. [DOI] [PubMed] [Google Scholar]
- 22. Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15:501‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jessen F, Spottke A, Boecker H, et al. Design and first baseline data of the DZNE multicenter observational study on predementia Alzheimer's disease (DELCODE). Alzheimer's Res Ther. 2018;10:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jessen F, Wolfsgruber S, Kleineindam L, et al. Subjective cognitive decline and stage 2 of Alzheimer disease in patients from memory centers. Alzheimer's Dement. 2022;19(2):487‐497. [DOI] [PubMed] [Google Scholar]
- 25. Hülsemann M, Zafiu C, Kühbach K, et al. Biofunctionalized silica nanoparticles: standards in amyloid‐beta oligomer‐based diagnosis of Alzheimer's disease. J Alzheimer's Dis. 2016;54:79‐88. [DOI] [PubMed] [Google Scholar]
- 26. Kühbach K, Hülsemann M, Herrmann Y, et al. Application of an amyloid beta oligomer standard in the sFIDA assay. Front Neurosci. 2016;10:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kravchenko K, Kulawik A, Hülsemann M, et al. Analysis of anticoagulants for blood‐based quantitation of amyloid beta oligomers in the sFIDA assay. Biol Chem. 2017;398:465‐475. [DOI] [PubMed] [Google Scholar]
- 28. Herrmann Y, Bujnicki T, Zafiu C, et al. Nanoparticle standards for immuno‐based quantitation of alpha‐synuclein oligomers in diagnostics of Parkinson's disease and other synucleinopathies. Clin Chim Acta. 2017;466:152‐159. [DOI] [PubMed] [Google Scholar]
- 29. Herrmann Y, Kulawik A, Kuhbach K, et al. sFIDA automation yields sub‐femtomolar limit of detection for Aβ aggregates in body fluids. Clin Biochem. 2017;50:244‐247. [DOI] [PubMed] [Google Scholar]
- 30. Pils M, Dybala A, Rehn F, et al. Development and implementation of an internal quality control sample to standardize oligomer‐based diagnostics of Alzheimer's disease. Diagnostics. 2023;13:1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vermunt L, Sikkes SAM, van den Hout A, et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer's disease in relation to age, sex, and APOE genotype. Alzheimer's Dement. 2019;15:888‐898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alzheimer's Association . 2022 Alzheimer's disease facts and figures. Alzheimer's Dement. 2022;18:700‐789. [DOI] [PubMed] [Google Scholar]
- 33. Jack CR, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. The Lancet Neurology. 2010;9:119‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Holtta M, Hansson O, Andreasson U, et al. Evaluating amyloid‐beta oligomers in cerebrospinal fluid as a biomarker for Alzheimer's disease. PLOS ONE. 2013;8:e66381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang T, O'Malley TT, Kanmert D, et al. A highly sensitive novel immunoassay specifically detects low levels of soluble Aβ oligomers in human cerebrospinal fluid. Alzheimer's Res Ther. 2015;7:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Savage MJ, Kalinina J, Wolfe A, et al. A sensitive Aβ oligomer assay discriminates Alzheimer's and aged control cerebrospinal fluid. J Neurosci. 2014;34:2884‐2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jongbloed W, Bruggink KA, Kester MI, et al. Amyloid‐beta oligomers relate to cognitive decline in Alzheimer's disease. J Alzheimer's Dis. 2015;45:35‐43. [DOI] [PubMed] [Google Scholar]
- 38. Dulewicz M, Kulczynska‐Przybik A, Mroczko P, Kornhuber J, Lewczuk P, Mroczko B. Biomarkers for the diagnosis of Alzheimer's disease in clinical practice: the role of CSF biomarkers during the evolution of diagnostic criteria. Int J Mol Sci. 2022:23:8598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Babapour Mofrad R, Scheltens P, Kim S, et al. Plasma amyloid‐beta oligomerization assay as a pre‐screening test for amyloid status. Alzheimer's Res Ther. 2021;13:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu L, Kwak H, Lawton TL, et al. An ultra‐sensitive immunoassay detects and quantifies soluble Aβ oligomers in human plasma. Alzheimer's Dement. 2022;18:1186‐1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shea D, Colasurdo E, Smith A, et al. SOBA: development and testing of a soluble oligomer binding assay for detection of amyloidogenic toxic oligomers. Proc Natl Acad Sci U S A. 2022;119:e2213157119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leoni V. The effect of apolipoprotein E (ApoE) genotype on biomarkers of amyloidogenesis, tau pathology and neurodegeneration in Alzheimer's disease. Clin Chem Lab Med. 2011;49:375‐383. [DOI] [PubMed] [Google Scholar]
- 43. Sengupta U, Portelius E, Hansson O, et al. Tau oligomers in cerebrospinal fluid in Alzheimer's disease. Ann Clin Transl Neurol. 2017;4:226‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lewczuk P, Wiltfang J, Kornhuber J, Verhasselt A. Distributions of Aβ42 and Aβ42/40 in the cerebrospinal fluid in view of the probability theory. Diagnostics. 2021;11:2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.