

Abstract
Hu Antigen R, also known as ELAVL1 (HuR), is a key posttranscriptional regulator in eukaryotic cells. HuR overexpression promotes several malignancies, including head and neck squamous cell carcinoma (HNSCC). However, its immune dysfunction-associated tumorigenesis pathways remain unknown. We examined HuR’s effects on oral malignancies and immune cell function in vitro and in vivo using oral carcinoma cells and transgenic HuR knockout (KO) mice. CRISPR/Cas9-mediated HuR deletion in mice syngeneic oral cancer cells eliminated colony formation and tumor development. HuR-KO tumors had a lower tumor volume, fewer CD4+CD25+FoxP3+ regulatory T cells, and more CD8+ T cells, suggesting that HuR may suppress the immune response during oral cancer progression. In contrast, HuR KO oral epithelial tissues are resistant to 4NQO-induced oral malignancies compared to control tumor-bearing mice. HuR KO mice showed fewer Tregs and greater IFN levels than WT tumor-bearing mice, suggesting anticancer activity. Finally, the HuR inhibitor pyrvinium pamoate lowers tumor burden by enhancing CD8+ infiltration at the expense of CD4+, suggesting anticancer benefits. Thus, HuR-dependent oral neoplasia relies on immunological dysfunction, suggesting that decreasing HuR may boost antitumor potential and offer a novel HNSCC therapy.
Keywords: Oral squamous cell carcinoma, RNA-Binding proteins, T-regulatory cells, Pyrvinium pamoate, Immune infiltration, Antitumor effects
1. Introduction
Head and neck squamous cell carcinoma is one of the most prevalent cancers in the world (HNSCC). In the US, the HNSCC accounts for about 4% of all cancer cases. A statistical forecast indicates that 54,540 people (39,290 males and 15,250 women) are expected to receive a head and neck cancer diagnosis in 2023 [1]. Approximately, 11,580 deaths (eight hundred and forty-four women and eight hundred males) are expected as a result of this disease [1]. Additionally, the 5-year survival rate drops to less than 50% due to the progression of invasive cancer and the diagnosis of stage III or IV. Thus, it is essential to determine the mechanisms and conditions that promote the development of oral premalignancy into invasive oral cancer. The immune system’s impairment, which makes it unable to stop malignant cell transformation, is linked to the genesis of HNSCC. The deregulation of immune cells in the context of oral cancer is caused by several mechanisms. Both effector and regulatory T-cells, which can have both pro- and anti-tumorigenic effects on various cancer types, including oral cancers, are involved in these mechanisms [2–4]. Furthermore, as demonstrated [5], the anticancer response of effector T-cells in HNSCC is solely dependent on their ability to circulate and infiltrate the tumor. Additionally, certain cancer-related variables render tumor-infiltrating lymphocytes (TILs) dormant, which promotes tumor growth by altering the tumor microenvironment. As a result, it has been noted that TILs become active as cancer advances. Nevertheless, it has been discovered that in HNSCC, TILs are functionally inactive [3]. Studies have shown that higher frequencies of CD4 and CD8 memory T-cells are present in people with HNSCC. But in HNSCC, CD4 memory T cells outnumber CD8 memory T cells and have a significantly greater frequency of lifespan [6]. More importantly, the protein factors responsible for the alteration in CD8 and CD4 levels and the activation of TILs are areas of HNSCC disease progression that have not been extensively studied.
Posttranscriptional gene regulation (PTR) is regulated by RNA-binding proteins (RBPs), which are frequently linked to various diseases. Hu-Antigen R (HuR) is an mRNA stabilizing protein belonging to the ELAV family of proteins. Its function in the immune system, cellular senescence, and embryonic development has been studied [7,8]. By controlling the stability and translation of mRNAs, which encode proteins involved in immune evasion, metastasis, local angiogenesis, cell proliferation, and survival, HuR plays a multifaceted role in the development of cancer [9,10]. HuR is crucial for preserving the integrity of the oral epithelium [11], averting cell death [12], and boosting drug resistance [13], as we have previously shown. Studies employing in vitro techniques have linked HuR to the development of oral cancer [14,15]. Furthermore, there is a significant lack of knowledge on HuR’s in vivo carcinogenic potential in oral cancer.
Our study provides a novel understanding of the complex interactions that occur between the development of HNSCC, immune system dysregulation, and the function of HuR in the development of oral cancer. Contrary to existing knowledge, our study delves into the previously unexplored area of HuR’s in vivo carcinogenic potential in oral cancer, shedding light on its multifaceted functions in preserving oral epithelium integrity, averting cell death, and boosting drug resistance. The pivotal discovery lies in our demonstration of HuR genetic deletion significantly inhibiting tumor development in both xenografts and 4NQO-induced oral neoplasia, presenting a novel avenue for targeted interventions. Additionally, our findings elucidate the unique immunosuppressive function of HuR in the occurrence of oral cancer, revealing a distinctive HuR-mediated immune metabolic control crucial for the emergence and metastasis of oral malignancies. Thus, this study establishes a foundation for further exploration and potential therapeutic strategies targeting HuR to prevent and treat oral cancer.
2. Materials and methods
2.1. Immunohistochemistry and western blot analysis
Frozen tissue sections from tongue tumors were cut using a cryostat and fixed in acetone for 5 min, washed in PBS for 5 min, and then processed for immunostaining. The primary antibodies directed against HuR (1:500, Santa Cruz), were diluted with DAKO antibody diluents, added to the slides, and incubated for 60 min at room temperature. A biotinylated link antibody plus streptavidin-biotin peroxidase kit (DAKO LSAB + System−HRP) was then used along with a 3,3’-diaminobenzidine chromogen and peroxide substrate to detect the bound antibody complexes. The slides were briefly counterstained with hematoxylin and dehydrated through graded alcohols to xylene. Finally, slides were cover-slipped with permanent mounting media and imaged. For Western blotting, protein extracts resolved by SDS/PAGE were analyzed as described by us [13].
2.2. Antibodies and cell lines
Most primary antibodies were from Cell Signaling Technologies except anti-HuR (Santa Cruz) and anti-β-actin (Sigma-Aldrich). Horseradish peroxidase-conjugated anti-mouse and anti-rabbit immunoglobulin Gs were procured from GE Healthcare Biosciences (Uppsala, Sweden). Tissue slides stained at CD8α (D4W2Z) XP® Rabbit mAb #98941 (Cell signaling technology) scanned at OPAL 480 with secondary rabbit HRP-760-4311 (Roche Diagnostics), CD4 #ab183685 (Abcam) scanned at OPAL 520 with rabbit HRP-760-4311 (Roche Diagnostics). MOC2 cells were a kind gift from Dr. Uppaluri, Harvard University, Boston, USA. The cells were cultured in IMDM/F12 (2:1) with 5% FCS, penicillin/streptomycin, 1% amphotericin, 5 ng/mL EGF (Millipore), 400 ng/mL hydrocortisone, and 5 μg/mL insulin as described [16].
2.3. Knockout of HuR in small animals and MOC2 cells
The Institutional Animal Care and Use Committee (IACUC) approved the procedures involving mice at the Medical University of South Carolina. An epithelial tissue-specific HuR knockout mouse was developed by backcrossing of HuRflox/flox (Strain#:021431, JAX) with K14Cre (Strain #018964, JAX) transgenic mouse. Specific HuR sgRNAs (Sigma mission; TRC N87 and TRC N93) and control sgRNA were used for the preparation of individual lentiviral particles. Cells were transduced with the lentiviral particles at an MOI (multiplicity of infection) of 25–50 in a medium supplemented with 8 μg/mL polybrene and incubated for 72 h. The puromycin selection of individual KO clones were selected and grown on appropriate culture conditions.
2.4. Cell growth, survival assays, xenograft studies, and 4NQO-induced oral carcinogenesis
Detailed methods were described in the supplemental methods.
2.5. CD8+ and naive CD4+ T cell isolation and flow cytometry
Naive splenocytes and peripheral lymph nodes (LNs) were isolated from WT and HuR KO mice. CD4+ T cells were isolated using murine anti-CD4 (L3T4) MACS MicroBeads positive-column purification, following the manufacturer’s protocol (Miltenyi Biotec). Naive CD8+ and CD4+ T cells were isolated from peripheral LNs and spleens (SPs) of mice using a MACS Mouse Naive CD8+/CD4+ T Cell Isolation Kit, following the manufacturer’s protocol.
For cytokine staining, activated CD4+ T cells were restimulated with PMA (50 ng/ml), ionomycin(1 μg/ml), and brefeldin A (3 μg/ml) for 5 h in T cell media at a concentration of 1 × 106 cells per milliliter. Cells (1–5 × 106) were then blocked with 2% normal mouse serum and Fc blocker (CD16/32) in 100 μl of FACS buffer for 15 min on ice. Cells were stained with surface marker Abs for 30 min on ice and washed with 1 ml of FACS buffer three times. Fixation was done with 100 μl of 2% paraformaldehyde in PBS for 15 min at room temperature (RT), and cells were washed once with FACS buffer. Cells were permeabilized with 100 μl of 0.2% saponin for 10 min on ice, and cytokine Abs (IL-2, IL-4, IL-5, IL-13, and IFN-γ) were added and allowed to incubate on ice for 30 min. Cells were washed with 1 ml of FACS buffer three times and analyzed by flow cytometry.
2.6. Multiplex immunohistochemistry staining protocol, image acquisition and data analysis
Optimized multiplex immunofluorescence was performed using the OPAL™ multiplexing method. OPAL™ is based on Tyramide Signal Amplification (TSA) using the Roche Ventana Discovery Ultra Automated Research Stainer (Roche Diagnostics, Indianapolis, IN). Detailed methods were described in supplemental methods.
2.7. Statistical analysis
The tumor growth was analyzed by comparison analysis using Mann-Whitney U test (Nonparametric equivalent of independent samples t-test). For comparison of tumor growth in HuR knockdowns, a mixed between-within subject’s analysis of variance comparing scramble with the indicated HuR knockdown was performed. Two-sample t tests with equal variances were used to assess differences between the means. Results with P-values less than 0.05 and 0.01 were considered significant.
3. Results
3.1. Silencing HuR augments tumor growth and proliferation in oral carcinoma
High levels of cytoplasmic HuR expression are associated with increased cell survival and oral cancer growth and proliferation [12,14]. Despite this, how HuR exploits the tumor microenvironment to advance oral neoplasia remains poorly studied. Hence, we employed syngeneic MOC2 oral carcinoma cells to investigate the relationship between HuR and the tumor microenvironment. We first knocked out HuR with two distinct guide RNAs and then selected stably knockout clones for further analysis (Fig. 1A). HuR is necessary for cancer cell survival, as evidenced by the fact that its deletion (KO) considerably slowed the growth of cancer cells relative to WT cells (Fig. 1B). Next, we quantified the rate at which HuR-deficient cells formed colonies. Fig. 1C shows that HuR knockout cells failed to form colonies, indicating that HuR plays a crucial role in clonogenicity and unregulated cell division. Finally, we implanted wild-type (WT) and HuR KO cells in C57BL/6J mice flanks to determine the WT and HuR KO cell’s tumor-forming efficiency. HuR KO cell tumorigenicity was evaluated by tracking tumor development over 18 days and taking measurements daily or every other day. We observed that HuR is essential for developing oral tumors (Fig 1D), since WT cells generated larger tumors than the two HuR KO tumor cells. Altogether, our findings demonstrate that HuR is an essential protein for the growth of oral tumors [17].
Fig. 1.
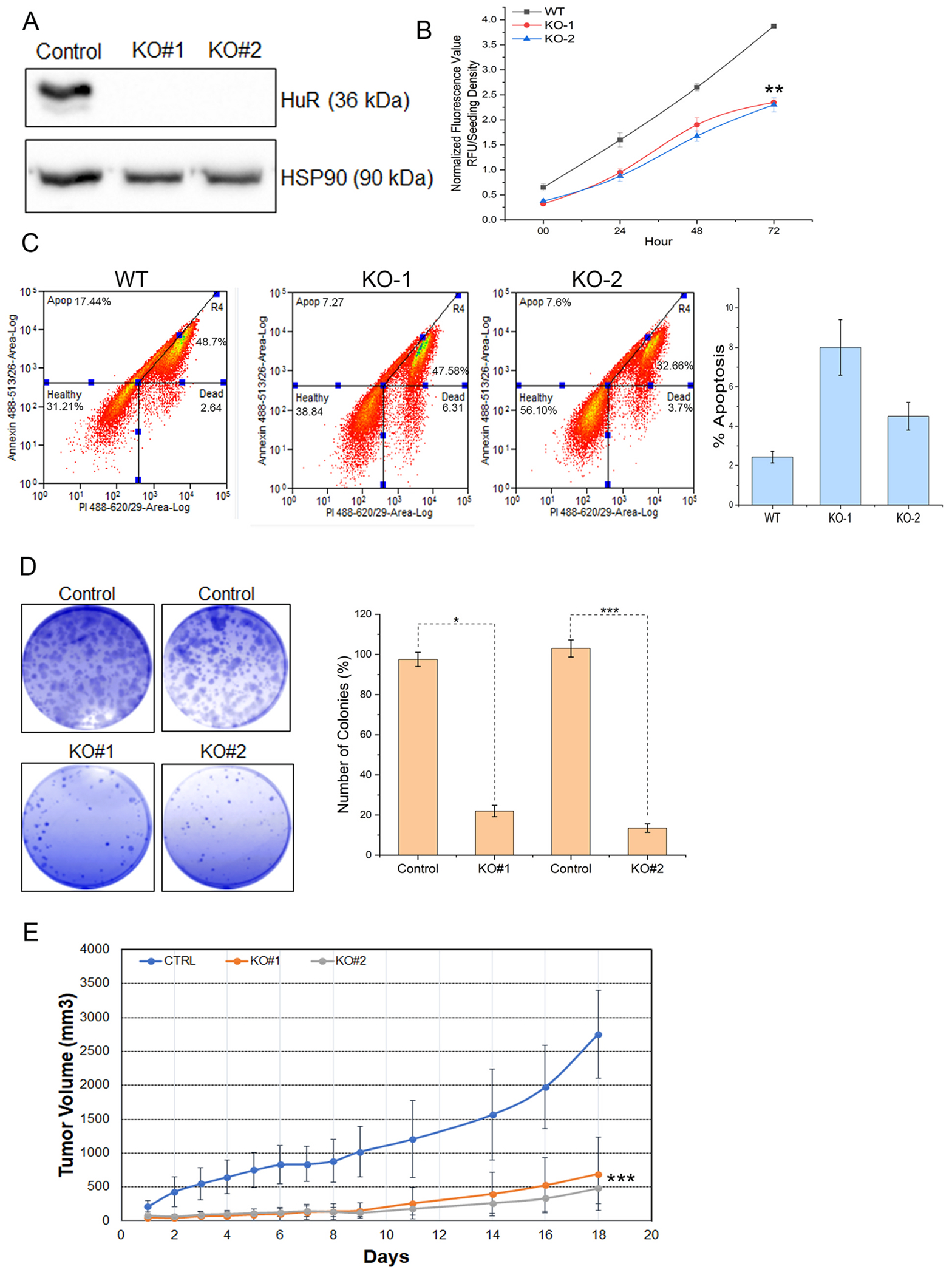
Genetic silencing of HuR abolishes oral tumors. A. Western blot analysis of proteins extracted from MOC2 cells transduced with lentiviral particles carrying two guide RNAs (sigma mission: TRCN87) and (TRCN93) against endogenous HuR. HSP90 serves as a loading control. B. Cell viability of WT and HuR KO MOC2 cells for 72 h. Data presented as the mean ± SD of three experiments. **p < 0.001. C. The panel depicts the colony-forming efficiency from clonogenic assays of MOC2 cells that knocked out HuR and controlled sgRNAs for 72h. The bar plot represents the means ± SD from three independent experiments. *p < 0.05 and ***p < 0.0001. D. In vivo, tumor growth of MOC2 cells and HuR KO clones 1 and 3 in syngeneic xenografts in C57BL/6J immunocompetent mice. Tumor volume (mm3) represents an average of 6 tumors± SEM.
3.2. HuR represses T-cell infiltration in oral tumors
The immune cell infiltration defines the oncogenicity of tumor growth and proliferation. Both CD4+ and CD8+ T cell effector subsets play an important role in shaping an anti-tumor immune response [18]. Thus, to determine if HuR downregulation played any role in the immune infiltration in the oral tumors, we used the tumors that were collected from the MOC2 syngeneic tumor experiment detailed in Fig. 1E, and characterized for CD8+ and CD4+ T cell infiltration by immunofluorescence and flow cytometry. The data in Fig. 2A confirms the HuR loss in HuR KO tumors. Compared to WT tumors, HuR is significantly reduced in HuR KO tumors, as seen by the immunohistochemistry (IHC) panel. The same panel of tissues were stained with antibodies against CD8, and CD4 antigens, respectively. We observed that WT tumors are characterized by a high level of HuR expression as well as considerable infiltration of CD4+ T cells noted by the positive immunofluorescence staining in Fig. 2A. Further FACS-based analysis showed that the distribution of the CD4 and CD8 T cell fraction was not much different in the spleen and tumor-draining lymph nodes harvested from the mice implanted with the WT vs. HuR-KD MOC2 tumors (Fig. 2B, upper and middle panel). However, significantly higher numbers of both CD4+ and CD8+ tumor-infiltrating T cells were obtained from the HuR-KD tumors than from WT tumors (Fig. 2B, lower panel). Since CD4+ T cells could also harbor pro-tumorogenic regulatory T cell (Treg) subsets characterized by the expression of FoxP3 [19], we determined if there are differences in Treg that correlate to the expression of HuR in the tumors. Fig. 2C shows that while there were no differences in the percentage of Treg’s that could be tracked in spleen or lymph nodes of the mice with WT or HuR-D tumors (Fig. 2C, upper and middle panel), there was indeed a significant reduction in Treg’s at the tumor site itself when HuR was silenced (Fig. 2C, lower panel). Importantly, the decrease in pro-tumorigenic myeloid-derived suppressor cells (MDSC) [20] was also noted in the HuR-KD tumors as compared to WT tumors (Fig. 2D). This data establishes that inhibiting the expression of HuR also reduces the pro-tumorigenic phenotype by creating a conducive tumor microenvironment where the differentiation of lymphoid and myeloid population to Treg’s or MDSC is constrained.
Fig. 2.
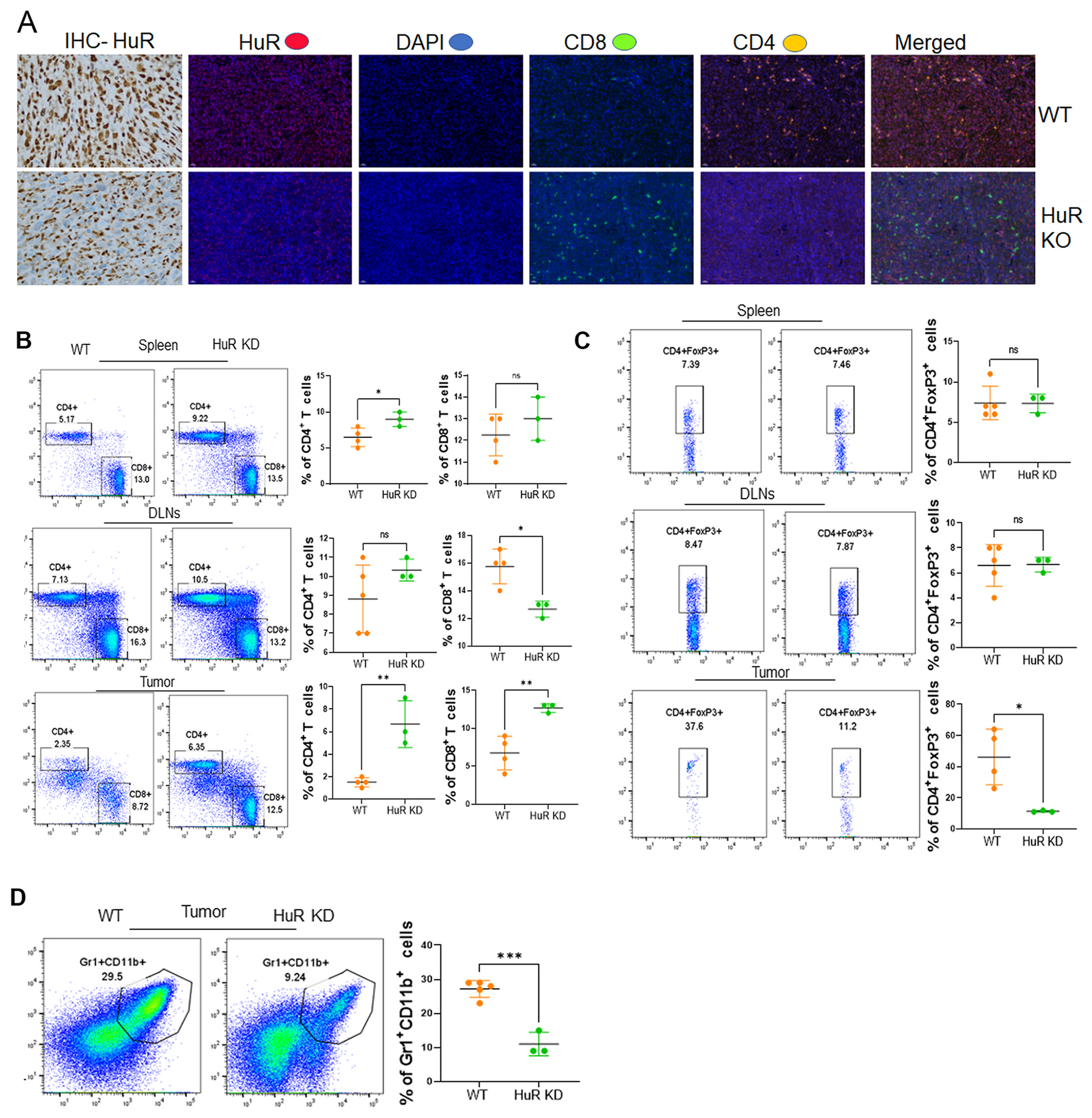
HuR silencing promotes effector T-cell immune infiltration in oral tumors. A. WT and HuR KO tumor tissues subjected to Immunohistology and Multiplex IHC staining of HuR indicated proteins CD4 and CD8 were co-stained with DAPI imaged with 20× magnification. B. The mice with WT vs. HuR-KD MOC2 tumors had their spleens and lymph nodes removed for FACS analysis to determine CD4 and CD8 T cell distribution. C. Treg cells present in WT vs. HuR-KD MOC2 tumors were determined by intranuclear staining for the transcription factor FoxP3. D. Myeloid-derived suppressor cells (MDSC) were quantified by CD11b + Gr + cells.
3.3. Epithelial tissue-specific HuR knockout disintegrates the oral epithelium
Due to hematopoietic organ shrinkage, intestinal villi loss, and obstructive enterocolitis, a global knockout (KO) of HuR in mice is embryonically fatal [21]. To further understand how HuR influences oral carcinogenesis, we have created epithelial tissue-specific HuR knockout mice. We have inbred HuRflox/flox, and Keratin-14Cre mice (Fig. 3A), and the progeny were screened for HuR KO by PCR. This allowed us to determine the HuR KO in epithelial tissues precisely. Exons 2–5 are excluded by the lack of a 400–570 bp flox gene, as seen in Fig. 3B, proving that HuR exons with K14Cre have been knocked out. We took samples of the oral mucosa, skin, and tail tissues and checked the expression of HuR to evaluate the loss of HuR in epithelial tissues. The loss of HuR in all KO tissues compared to WT tissues is depicted in Fig. 3C, demonstrating that epithelial-specific KO of HuR is successful and that the mice have no further abnormalities in normal phenotype. By employing immunohistochemistry (IHC), we further revealed that the loss of HuR is exclusive to the keratin-14-expressing epithelium. According to the findings, the constitutive epithelial deletion of HuR in the basal epithelium of the tongue tissue exhibits basal epithelium disintegration and shows hyperplasia-like phenotype (Fig. 3D). A second warranty is necessary to understand why the lack of HuR in the normal mouse basal epithelial layer encourages the breakdown of the epithelium and lesions to appear. Next, IHC was used to examine further and validate the presence of HuR in the basal epithelial layer using the basal epithelial marker protein TAp63 (Fig. 3D). Additionally, mouse oral mucosa has been found to have HuR depletion (Fig. S1). Together, these results show that HuR is highly expressed in the basal epithelial layer, and HuR KO oral epithelium degrades and exhibits hyperplastic lesions in the basal epithelial layer, demonstrating that HuR is necessary to preserve basal epithelial layer cellularity and integrity.
Fig. 3. HuR KO causes oral epithelial disintegration.
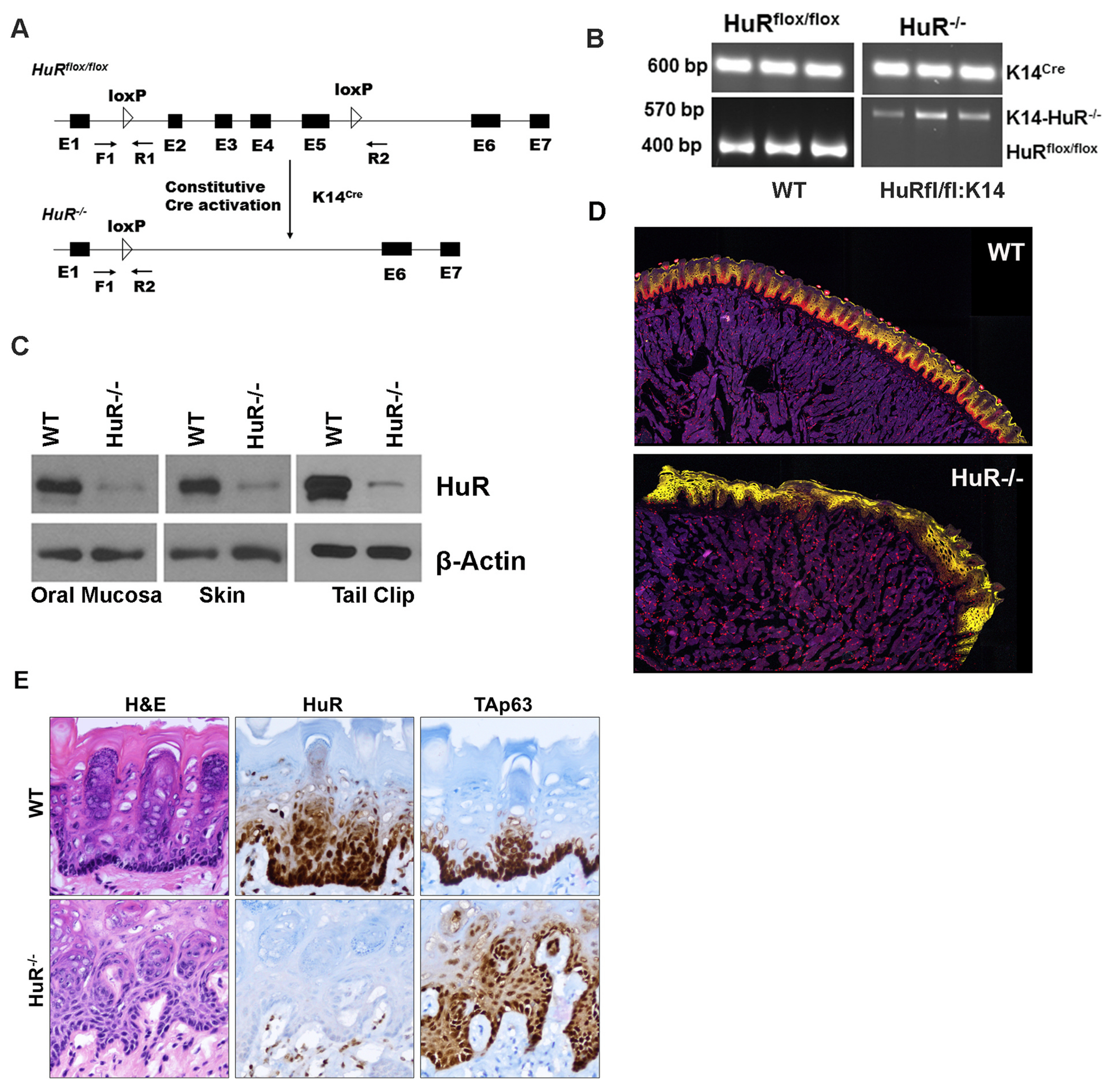
A. Schematics of the HuR KO schema and primers used to detect the Cre-loxP sites. Indicated primer sequences: F1-5’-AGGCAGATGAGCACATGTGA-3’: R1-5’-AGGCTCTGGGATGAAACCTA-3’ and R2-5’-TACTGAGATGTTCTGGGAGG-3’. B. PCR analysis of genomic DNA from the oral epithelium from the mouse mucosal layer indicating either floxed or deletion of HuR (Δ) or Cre bands. C. Western blot analysis of WT and KO mice tissue extracts using a HuR antibody. The same blot was reprobed with a β-actin mouse mAb as housekeeping control. D. Tissue sections from the WT and HuR KO stained Alexa-647 conjugated HuR antibody (red color). Yellow indicated PAN-CK (Alexa-532). E. Tongue tissue sections from WT and HuR-KO mouse tongue tissue sections with H&E, TAp63, and HuR IHC analysis. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.4. HuR aids the progression of oral tumors
An experimental 4NQO-induced mouse oral carcinogenesis model revealed that carcinogenesis is a multistep process similar to human oral squamous cell carcinoma [22,23]. The establishment of numerous lesions under 4NQO increases the formation of malignant SCC, which resembles the genesis and progression of human oral cancer [24]. The initial pathological stage is flat squamous dysplasia, ranging from moderate to severe (Fig. S2). After 4NQO, which is discontinued at 16 weeks, tumors were collected on or after 28 weeks [25]. The second stage is papillary squamous tumors (papillomas), followed by the third, carcinoma in situ (non-invasive squamous cell carcinoma (SCC), and the fourth, invasive SCC. To identify the point at which HuR contributes to the promotion of oral cancer, we treated the WT (HuRflox/flox x K14Cre− (n = 17) and HuR KO (n = 12) with 4NQO and examined the lesions in the tongue following 4NQO removal (schema shown in Fig. 4A). As demonstrated in Fig. 4B, the total incidence of oral tumors are 59% in WT mice (10/17) and 0% in HuR-KO mice (0/12) (two-tailed P-value 0.0001). Mice tongue tissues were obtained and examined for tumor development using cross-eye inspection and IHC. As demonstrated in Fig. 4C, WT mice treated with 4NQO developed oral SCC and invasive oral SCC according to tumor size. Surprisingly, HuR-KO mice treated with 4NQO did not develop oral SCC and had hyperplastic lesions in the tongue tissues, as demonstrated by H&E staining (Fig. 4C). In addition, HuR-KO mice were resistant to developing oral carcinoma for a more extended period (>48 weeks) than WT mice (28 weeks), demonstrating that the absence of HuR prevented the growth of oral malignancies despite the presence of hyperplastic lesions or disintegrated basal epithelium. In this multistep oral carcinogenesis, IHC analysis also revealed increased expression of HuR and the proliferation marker ki67 (Fig. S2). Next, we determine the immune responses of WT and HuR in 4NQO-derived cancer, we collected the spleen and examined the expression of the immune cell population by flow cytometry. We analyzed peripheral T cells from WT and HuR-KO mice after 4NQO administration to characterize the T cell subsets. The HuR-KO recipients that exhibited no tumor growth were also low in pro-tumorigenic suppressive Tregs (as characterized by CD4+CD25+FoxP3+), as indicated in Fig. 4D, which shows 10–12% Treg in HuR-KO vs. 16–18% in WT. We also observed that the reduced Treg number correlated with a two-fold increase in the fraction of CD8+ T cells secreting IFNγ in the HuR-KO (Fig. 4E). These data indicate that inhibiting HuR in epithelial tumors reduces immunosuppression and allows effector T cells to maintain their anti-tumor functionality.
Fig. 4.
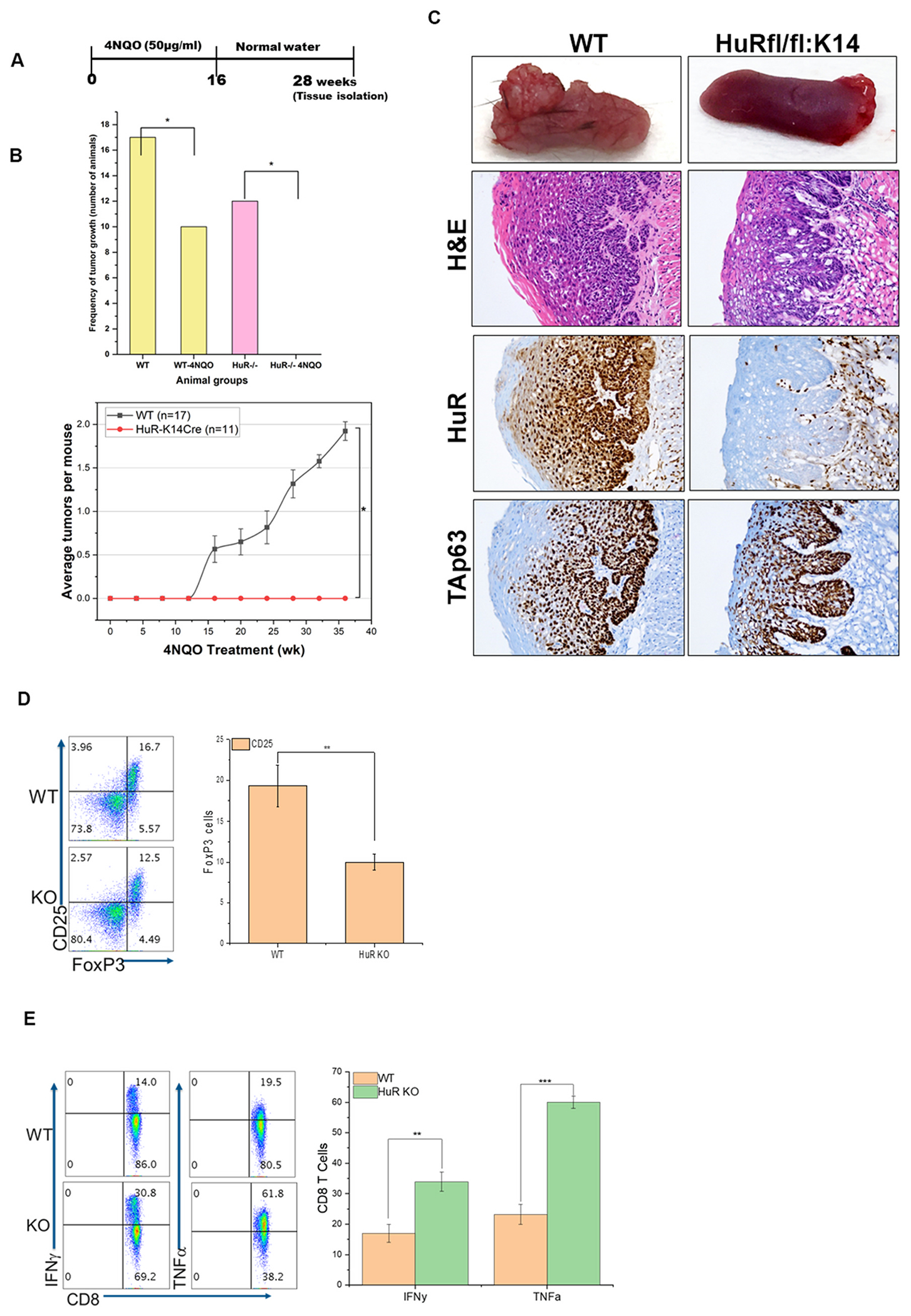
HuR KO blocks induction of 4NQO-derived oral carcinoma. A. Schematic of 4NQO treatment used to produce oral tumors in mice. B. Number WT and HuR-KO mice with oral tumors, *p-value <0.05. Bottom panel: Incidence of oral tumors after exposure to 4NQO. C. Tongue tissue sections from WT and HuR-KO mice with H&E and TAp63, and HuR staining. D-E. Decreased Tregs and increased IFNg function in 4NQO-exposed HuR-KO mice. 4NQO-treated WT and HuR-KO mice were evaluated for tumor growth and T cell subset populations. Representative data from five mice is shown. D. Expression of Treg signature transcription factor FoxP3 was determined by gating on CD4+CD25+ T cells. E. CD8+ T cells were stimulated with PMA/ionomycin for 4 h before intracellular cytokine IFNg was evaluated using FACS. **p-value <0.001, ***p-value <0.0001.
3.5. Inhibition of HuR by small molecule pyrvinium pamoate abrogates oral tumors and elicits an antitumor immune response
Several small molecule inhibitors for HuR have been proposed and established [26]. The anthelminthic drug pyrvinium pamoate (PP), for instance, has been shown to inhibit cytoplasmic HuR and exhibit antitumor activity in urothelial [27] and pancreatic malignancies (ClinicalTrials.gov Identifier: NCT0505533). Consequently, we wish to determine if PP inhibits HuR and exerts antitumor activity associated with alterations in immune infiltration. To determine the IC50 value of PP in MOC2 cells, we evaluated the viability of cells treated with escalating concentrations of PP As shown in Fig. 5A, PP inhibits the viability of MOC2 cells at an IC50 concentration of 1 M. We evaluated the clonogenicity of cells treated with various concentrations of PP for 48 h. At a PP concentration of 1 M, colony formation in MOC2 cells is reduced by 50% compared to 2.5 M, and 5 M eliminates colony formation (Fig. 5B). This finding suggests that PP may regulate cell proliferation and division in MOC2 cells. According to a previously published study, PP induces cell apoptosis in cancer cells by inhibiting the WNT pathway [28]. We, therefore, examined whether a 1 M concentration of PP induces cell apoptosis in MOC2 cells. As shown in Fig. 5C, greater than 50% of DMSO- and PP-treated cells underwent late apoptosis as shown by Annexin V and PI staining, whereas approximately 50% of PP-treated cells either underwent necrosis or died. This observation suggests that PP could inhibit the proliferation of oral cancer cells and induce apoptosis. Next, to ascertain the in vivo antitumor efficacy of the PP, MOC2 cells were implanted into the flank of B6 mice, and xenografts were developed. After the tumor reached 100mm3, we administered PP thrice weekly at a dosage of 1.5 mg/kg of body weight, as previously reported [29]. As depicted in Fig. 5D, tumors treated with DMSO grow significantly larger than those treated with PP, exhibiting substantially smaller tumors and slower growth. Finally, we confirmed whether immune cells infiltrate the tumors and show reduced tumor growth upon PP treatment. The tumors collected from WT and PP-treated tumors were subjected to multispectral IHC imaging and evaluated for immune cell infiltration. As shown in Fig. 5E–a reduced HuR expression in PP-treated tumors with low CD4 and high CD8 infiltration indicates that CD8+ may induce antitumor activity, which is associated with low tumor volume (Fig. 5D). Thus, PP could be an adjuvant therapeutic drug to inhibit HuR alleviating oral tumors.
Fig. 5.
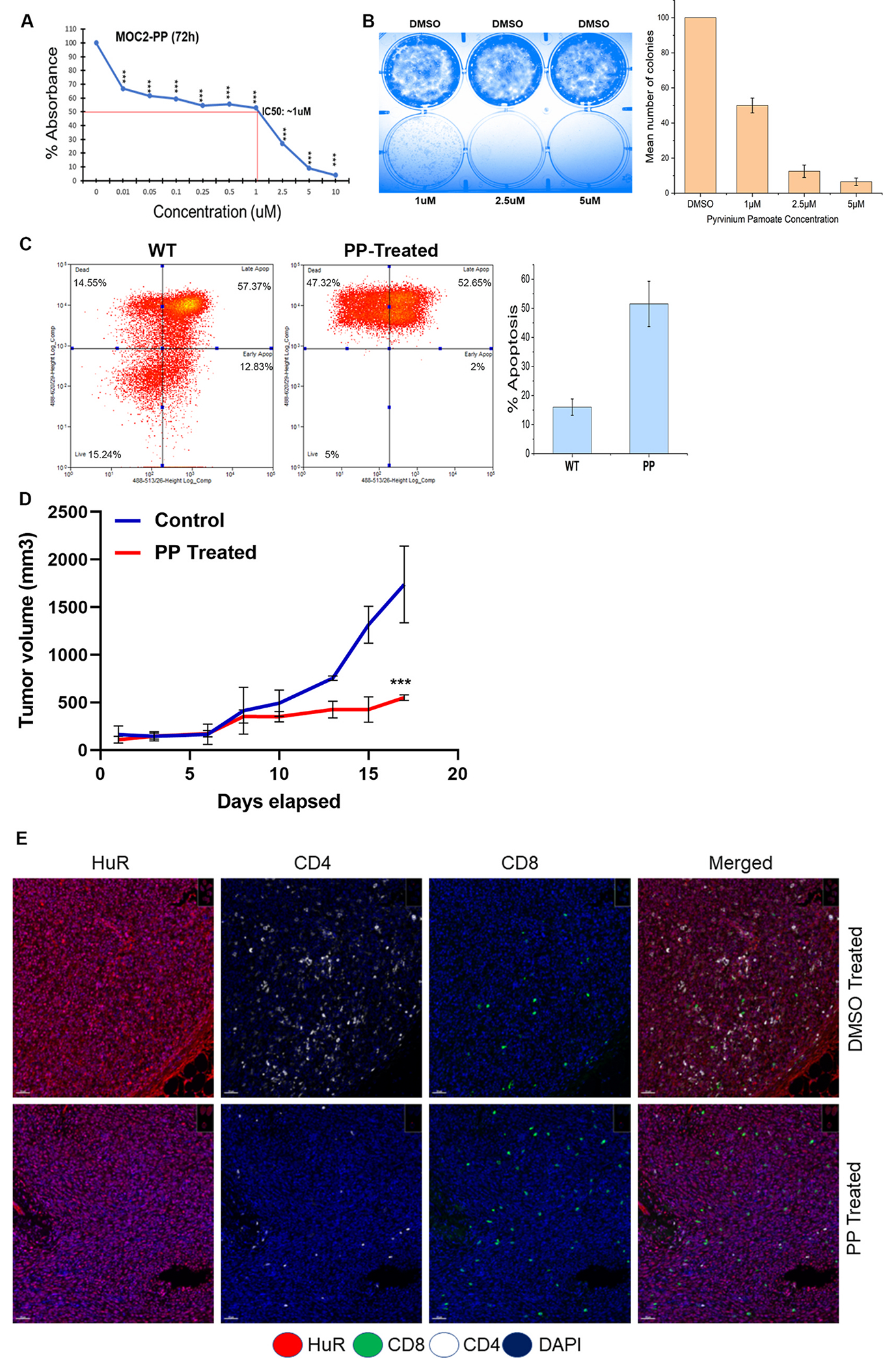
Treatment of HuR inhibitor PP reduces oral tumor burden and increases survival. A. IC50 value of PP is determined through cell viability assay in MOC2 cells treated with indicated concentration of PP. B. The images depict the colony-forming efficiency from clonogenic assays of MOC2 cells treated with the indicated concentration of PP for 72h. The bar plot represents the means ± SD from three independent experiments. C. Cell death measured by flow cytometry. After gating on live and dead cells from WT, PP-treated cells were analyzed, and X-axis denotes PI 488, and the Y-axis denotes Annexin 488. The bar graph indicates the percentage of apoptosis after PP treatment. N = 3. D. Mean tumor volume of control DMSO and PP treated mice group. N = 12. ***p-value <0.0001. E. DMSO and PP-treated tumor tissues subjected to Multiplex IHC staining of indicated proteins HuR, CD4, and CD8 were co-stained with DAPI imaged with 20× magnification.
4. Discussion
Here, we have shown that HuR is essential for the growth and progression of oral cancer via regulating the effector immune cells. Our findings also indicate that HuR is a crucial protein for maintaining the basal integrity of the oral epithelium. Yet, HuR deficiency renders the oral epithelial tumor resistant in a carcinogen-induced tumor model. Our results suggest that basal epithelial expression of HuR is critical for the integrity of oral epithelium since HuR expression is predominately found in the majority of the epithelial cells. Reports indicate that basal epithelial cellularity determines oral epithelial integrity [30]. Interestingly, our results showed that KO of HuR in the basal epithelium disintegrates and exhibits morphological alterations resembling hyperplasia in the oral epithelium. This observation is supported by the findings that HuR is required for intestinal epithelial regeneration following radiation-induced intestinal atrophy [31]. In addition, our results also showed that constitutive deletion of HuR results in an extended epithelium disintegration. By our earlier observation, HuR is essential for the integrity of the oral epithelium and oral mucositis, which is caused by ionizing radiation in mice that increases HuR cleavage and cell death [11]. We now demonstrate that specific-epithelial constitutive HuR KO destroys the mouse tongue tissue’s basal epithelial layer, proving that HuR is crucial in preserving healthy epithelial structure. To ascertain the role of HuR in basal epithelial regeneration, it is necessary to establish an inducible KO of HuR in the oral epithelium.
HuR has been observed to augment the growth and proliferation of cancer cells in various cultured cancer cell lines [12–15]. In this study, we have provided a comprehensive analysis of the in vivo role of HuR in a murine model of oral cancer, examining its potential implications within the tumor microenvironment. HuR has garnered significant attention as the most extensively investigated protein involved in the control of immune response, as evidenced by several studies [32–36]. The presence of HuR is necessary for the development of T-cells, as demonstrated by the observation that T-cell-specific HuR knockout mice exhibit thymic egress, leading to a decrease in the number of lymphocytes in the peripheral tissues [37]. The results of our study suggest a correlation between elevated levels of HuR and greater tumor size and infiltration of CD4+ Treg cells. This indicates that HuR may exert its influence on CD4+ Treg cells, which in turn can suppress the effector CD4+ and CD8+ T cell population infiltrating the tumor microenvironment that promotes tumor growth in the context of oral carcinogenesis. Thus, our findings provide support for the notion that HuR plays a crucial role in the process of immunosurveillance and the manipulation of immune responses in oral tumors [38].
Several small-molecule inhibitors have been discovered to target HuR and suppress its biological functions [34]. The results of our study indicate that the immune infiltration of effector T cells and the antitumor activity are enhanced by either knockout (KO) or inhibition of HuR using the small molecule PP (Fig. 5). The clinical significance of HuR inhibition in non-small cell lung cancer is demonstrated by the observed anti-cancer effects of the small molecule CMLD-2 targeting HuR [39]. The utilization of MS-444 [40], a widely recognized inhibitor of HuR, in the treatment of glioblastoma cells led to the inhibition of growth and the induction of apoptosis. These findings suggest that MS-444 possesses antitumor properties [41]. The small molecule PP, which has received approval from the FDA, has been demonstrated to effectively inhibit the growth of pancreatic ductal adenocarcinoma. This inhibition is achieved through the blocking of oxidative phosphorylation and fatty acid metabolism, as described by Ref. [29]. The findings from our observations suggest that the administration of PP treatment induces immune infiltration and decreases the tumor burden in oral cancer cells. These results demonstrate the potential of a HuR-targeted therapy as a promising approach for further preclinical investigations and, ultimately, the treatment of oral cancer [42–48].
Supplementary Material
Role of funding source
This work was supported by the National Institutes of Health (NIH Grant R01 DE030013) and supported in part by the Translational Science Shared Resource, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313).
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Mrinmoyee Majumder: Conceptualization, Data curation, Formal analysis, Methodology, and Writing - original draft. Harinarayanan Janakiraman: Conceptualization, Data curation, Formal analysis, Methodology, and Writing - original draft. Paramita Chakraborty: Data curation, and Formal analysis. Anitha Vijayakumar: Data curation, and Formal analysis. Sari Mayhue: Data curation, and Formal analysis. Hong Yu: Data curation, and Formal analysis. Toros Dincman: Data curation, and Formal analysis. Romeo Martin: Data curation, and Formal analysis. Elizabeth O Quinn: Data curation, and Formal analysis. Shikhar Mehrotra: Funding acquisition, Investigation, Supervision, Validation, Visualization, and Writing - original draft. Viswanathan Palanisamy: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing - original draft, and Writing - review and editing.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.oor.2024.100296.
References
- [1].Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA A Cancer J Clin 2023;73:17–48. [DOI] [PubMed] [Google Scholar]
- [2].Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Prim 2020;6:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Whiteside TL. Immunobiology of head and neck cancer. Cancer Metastasis Rev 2005;24:95–105. [DOI] [PubMed] [Google Scholar]
- [4].Cillo AR, Kurten CHL, Tabib T, Qi Z, Onkar S, Wang T, et al. Immune landscape of viral- and carcinogen-driven head and neck cancer. Immunity 2020;52:183–199 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Damasio MPS, Nascimento CS, Andrade LM, de Oliveira VL, Calzavara-Silva CE. The role of T-cells in head and neck squamous cell carcinoma: from immunity to immunotherapy. Front Oncol 2022,12:1021609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jewett A, Head C, Cacalano NA. Emerging mechanisms of immunosuppression in oral cancers. J Dent Res 2006;85:1061–73. [DOI] [PubMed] [Google Scholar]
- [7].Papadaki O, Milatos S, Grammenoudi S, Mukherjee N, Keene JD, Kontoyiannis DL. Control of thymic T cell maturation, deletion and egress by the RNA-binding protein HuR. J Immunol 2009;182:6779–88. [DOI] [PubMed] [Google Scholar]
- [8].Katsanou V, Milatos S, Yiakouvaki A, Sgantzis N, Kotsoni A, Alexiou M, et al. The RNA-binding protein Elavl1/HuR is essential for placental branching morphogenesis and embryonic development. Mol Cell Biol 2009;29:2762–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Abdelmohsen K, Gorospe M. Posttranscriptional regulation of cancer traits by HuR, vol 1. Wiley interdisciplinary reviews RNA; 2010. p. 214–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Grammatikakis I, Abdelmohsen K, Gorospe M. Posttranslational control of HuR function, vol. 8. Wiley interdisciplinary reviews RNA; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Talwar S, House R, Sundaramurthy S, Balasubramanian S, Yu H, Palanisamy V. Inhibition of caspases protects mice from radiation-induced oral mucositis and abolishes the cleavage of RNA-binding protein HuR. J Biol Chem 2014;289:3487–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Talwar S, Jin J, Carroll B, Liu A, Gillespie MB, Palanisamy V. Caspase-mediated cleavage of RNA-binding protein HuR regulates c-Myc protein expression after hypoxic stress. J Biol Chem 2011;286:32333–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Janakiraman H, House RP, Talwar S, Courtney SM, Hazard ES, Hardiman G, et al. Repression of caspase-3 and RNA-binding protein HuR cleavage by cyclooxygenase-2 promotes drug resistance in oral squamous cell carcinoma. Oncogene 2017;36:3137–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hasegawa H, Kakuguchi W, Kuroshima T, Kitamura T, Tanaka S, Kitagawa Y, et al. HuR is exported to the cytoplasm in oral cancer cells in a different manner from that of normal cells. Br J Cancer 2009;100:1943–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kakuguchi W, Kitamura T, Kuroshima T, Ishikawa M, Kitagawa Y, Totsuka Y, et al. HuR knockdown changes the oncogenic potential of oral cancer cells. Mol Cancer Res 2010;8:520–8. [DOI] [PubMed] [Google Scholar]
- [16].Judd NP, Winkler AE, Murillo-Sauca O, Brotman JJ, Law JH, Lewis JS Jr, et al. ERK1/2 regulation of CD44 modulates oral cancer aggressiveness. Cancer Res 2012;72:365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Majumder M, Palanisamy V. RNA binding protein FXR1-miR301a-3p axis contributes to p21WAF1 degradation in oral cancer. PLoS Genet 2020;16:e1008580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ostroumov D, Fekete-Drimusz N, Saborowski M, Kuhnel F, Woller N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell Mol Life Sci 2018;75:689–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hariyanto AD, Permata TBM, Gondhowiardjo SA. Role of CD4(+)CD25(+)FOXP3(+) T(Reg) cells on tumor immunity. Immunol Med 2022;45:94–107. [DOI] [PubMed] [Google Scholar]
- [20].Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol 2016;37:208–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ghosh M, Aguila HL, Michaud J, Ai Y, Wu MT, Hemmes A, et al. Essential role of the RNA-binding protein HuR in progenitor cell survival in mice. J Clin Invest 2009;119:3530–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tang XH, Knudsen B, Bemis D, Tickoo S, Gudas LJ. Oral cavity and esophageal carcinogenesis modeled in carcinogen-treated mice. Clin Cancer Res 2004;10:301–13. [DOI] [PubMed] [Google Scholar]
- [23].Vitale-Cross L, Czerninski R, Amornphimoltham P, Patel V, Molinolo AA, Gutkind JS. Chemical carcinogenesis models for evaluating molecular-targeted prevention and treatment of oral cancer. Cancer Prev Res 2009;2:419–22. [DOI] [PubMed] [Google Scholar]
- [24].Hawkins BL, Heniford BW, Ackermann DM, Leonberger M, Martinez SA, Hendler FJ. 4NQO carcinogenesis: a mouse model of oral cavity squamous cell carcinoma. Head Neck 1994;16:424–32. [DOI] [PubMed] [Google Scholar]
- [25].Kanojia D, Vaidya MM. 4-nitroquinoline-1-oxide induced experimental oral carcinogenesis. Oral Oncol 2006;42:655–67. [DOI] [PubMed] [Google Scholar]
- [26].Majumder M, Chakraborty P, Mohan S, Mehrotra S, Palanisamy V. HuR as a molecular target for cancer therapeutics and immune-related disorders. Adv Drug Deliv Rev 2022;188:114442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Guo J, Lv J, Chang S, Chen Z, Lu W, Xu C, et al. Inhibiting cytoplasmic accumulation of HuR synergizes genotoxic agents in urothelial carcinoma of the bladder. Oncotarget 2016;7:45249–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Xu F, Zhu Y, Lu Y, Yu Z, Zhong J, Li Y, et al. Anthelmintic pyrvinium pamoate blocks Wnt/beta-catenin and induces apoptosis in multiple myeloma cells. Oncol Lett 2018;15:5871–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Schultz CW, McCarthy GA, Nerwal T, Nevler A, DuHadaway JB, McCoy MD, et al. The FDA-approved anthelmintic pyrvinium pamoate inhibits pancreatic cancer cells in nutrient-depleted conditions by targeting the mitochondria. Mol Cancer Therapeut 2021;20:2166–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jones KB, Klein OD. Oral epithelial stem cells in tissue maintenance and disease: the first steps in a long journey. Int J Oral Sci 2013;5:121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu L, Zhuang R, Xiao L, Chung HK, Luo J, Turner DJ, et al. HuR enhances early restitution of the intestinal epithelium by increasing Cdc42 translation. Mol Cell Biol 2017;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen J, Cascio J, Magee JD, Techasintana P, Gubin MM, Dahm GM, et al. Posttranscriptional gene regulation of IL-17 by the RNA-binding protein HuR is required for initiation of experimental autoimmune encephalomyelitis. J Immunol 2013;191:5441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chen J, Martindale JL, Abdelmohsen K, Kumar G, Fortina PM, Gorospe M, et al. RNA-binding protein HuR promotes Th17 cell differentiation and can Be targeted to reduce autoimmune neuroinflammation. J Immunol 2020;204:2076–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Christodoulou-Vafeiadou E, Ioakeimidis F, Andreadou M, Giagkas G, Stamatakis G, Reczko M, et al. Divergent innate and epithelial functions of the RNA-binding protein HuR in intestinal inflammation. Front Immunol 2018;9:2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gubin MM, Techasintana P, Magee JD, Dahm GM, Calaluce R, Martindale JL, et al. Conditional knockout of the RNA-binding protein HuR in CD4(+) T cells reveals a gene dosage effect on cytokine production. Mol Med 2014;20:93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rothamel K, Arcos S, Kim B, Reasoner C, Lisy S, Mukherjee N, et al. ELAVL1 primarily couples mRNA stability with the 3’ UTRs of interferon-stimulated genes. Cell Rep 2021;35:109178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Papadaki O, Milatos S, Grammenoudi S, Mukherjee N, Keene JD, Kontoyiannis DL. Control of thymic T cell maturation, deletion and egress by the RNA-binding protein HuR. J Immunol 2009;182:6779–88. [DOI] [PubMed] [Google Scholar]
- [38].Wang Z, Yin W, Zhu L, Li J, Yao Y, Chen F, et al. Iron drives T helper cell pathogenicity by promoting RNA-binding protein PCBP1-mediated proinflammatory cytokine production. Immunity 2018;49:80–92 e7. [DOI] [PubMed] [Google Scholar]
- [39].Muralidharan R, Mehta M, Ahmed R, Roy S, Xu L, Aube J, et al. HuR-targeted small molecule inhibitor exhibits cytotoxicity towards human lung cancer cells. Sci Rep 2017;7:9694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Meisner NC, Hintersteiner M, Mueller K, Bauer R, Seifert JM, Naegeli HU, et al. Identification and mechanistic characterization of low-molecular-weight inhibitors for HuR. Nat Chem Biol 2007;3:508–15. [DOI] [PubMed] [Google Scholar]
- [41].Wang J, Hjelmeland AB, Nabors LB, King PH. Anti-cancer effects of the HuR inhibitor, MS-444, in malignant glioma cells. Cancer Biol Ther 2019;20:979–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nguyen N, Bellile E, Thomas D, McHugh J, Rozek L, Virani S, et al. Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head Neck 2016;38:1074–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Spector ME, Bellile E, Amlani L, Zarins K, Smith J, Brenner JC, et al. Prognostic value of tumor-infiltrating lymphocytes in head and neck squamous cell carcinoma. JAMA Otolaryngol Head Neck Surg 2019;145:1012–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Seiwert TY, Zuo Z, Keck MK, Khattri A, Pedamallu CS, Strieker T, et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res 2015;21:632–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer 2018;18:139–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Thorne CA, Hanson AJ, Schneider J, Tahinci E, Orton D, Cselenyi CS, et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1alpha. Nat Chem Biol 2010;6:829–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Venerando A, Girardi C, Ruzzene M, Pinna LA. Pyrvinium pamoate does not activate protein kinase CK1, but promotes Akt/PKB down-regulation and GSK3 activation. Biochem J 2013;452:131–7. [DOI] [PubMed] [Google Scholar]
- [48].Wiegering A, Uthe FW, Huttenrauch M, Muhling B, Linnebacher M, Krummenast F, et al. The impact of pyrvinium pamoate on colon cancer cell viability. Int J Colorectal Dis 2014;29:1189–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.